

Abstract
Our view of how adipose tissue metabolism is regulated recently experienced a change in focus and breadth, meaning that some of the key controlling factors were not fully in the picture. The catecholamines of the sympathetic nervous system are well-known activators of β-adrenergic receptors in adipocytes to increase lipolysis. They also drive energy expenditure in brown adipose tissue and, importantly, the “browning” of cells in white adipose depots. However, this is clearly not the whole story. In earlier work, we established a pathway from β-adrenergic receptors to p38 MAP kinase to drive the transcription of brown adipocyte genes and respiratory uncoupling. Now we recently discovered that cardiac natriuretic peptides (NPs) stimulate a similar “browning” of human and mouse adipocytes. NPs activate the guanylyl cyclase coupled NP receptor A and activation of protein kinase G. Importantly, this pathway also depends upon p38 MAPK. These two pathways work together, additively increasing expression of brown adipocyte marker genes, as well as reflexively controlling each other’s components. We discuss these findings and how the control of body fat by these cardiac hormones, in conjunction with the sympathetic nervous system, has implications for obesity as well as cardiovascular disease, including hypertension and heart failure.
Keywords: brown adipocyte, cyclic nucleotides, thermogenesis, uncoupling, gene expression
The pathway from the sympathetic nervous system to β-adrenergic receptors (βARs) and protein kinase A (PKA) was well established in earlier decades to be the key controlling element for activating the enzymatic apparatus to hydrolyze stored triglyceride in adipocytes1-3 (see upper part of Fig. 1). With the realization that brown adipose tissue was a thermogenic form of fat4-6 due to the unique “uncoupling protein” (UCP1) of the brown adipocyte7 (see ref. 8 for further details), it was subsequently shown that the βAR-PKA pathway was also responsible for the gene expression changes and expansion of brown adipocytes for respiratory uncoupling and energy expenditure. Interest in studying brown adipose tissue as a sink for excess calories has experienced several waves of enthusiasm and disdain over the decades, falling in and out of fashion depending upon whether it was considered clinically relevant or not. At the moment, brown adipocytes in humans are in vogue again as a result of radiology studies showing cold-stimulated bilateral glucose uptake in adult humans and biopsies from subjects demonstrating UCP1-positive brown adipocytes.9-13
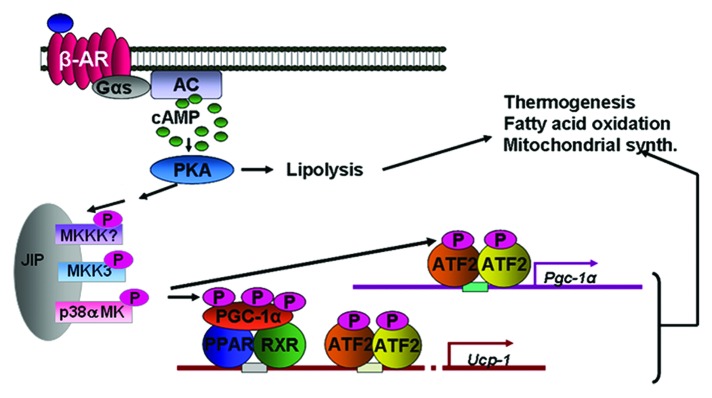
Figure 1. Catecholamines bind the βARs on adipocytes to activate the Gs and increase cAMP. cAMP activates PKA, which can phosphorylate proteins to allow lipolysis of stored triglycerides. βARs and PKA can also activate a protein kinase cascade, culminating in the activation of p38α MAPK, which phosphorylates key transcription factors to promote transcription of the UCP1 and PGC-1α genes, mitochondrial biogenesis and thermogenic energy expenditure.
Our lab has studied the βARs and their signaling mechanisms in adipocytes for several years, focusing on cAMP, PKA and downstream targets of the kinase such as enzymes and transcription factors.14,15 We became particularly interested in the transcriptional activation of the Ucp1 gene in response to β-agonist stimulation. Although the Ucp1 genes from various species had been isolated in the early 1990s and regulatory regions responsive to elevations in cAMP were identified,16-18 it seemed curious that relatively little progress had been made in identifying the transcription factors that controlled cAMP-dependent Ucp1 expression. It was only when we stumbled upon the PKA dependent activation of p38 MAPK19,20 that pieces of the puzzle began to be put into place. Discovering that p38 MAPK activation by βARs and PKA was necessary for driving Ucp1 gene transcription19,21 was a breakthrough in beginning to crack the code for how the SNS could stoke UCP1-dependent adaptive thermogenesis.
As shown in Figure 1, p38α MAPK (p38) is activated by a cascade downstream of PKA. The exact players in this cascade are not fully understood and their identity is a topic of ongoing work. However, functionally it is clear that p38 plays an important role at several levels: by phosphorylating PGC-1α, which boosts it co-activator function,22 as well as phosphorylating the CRE-binding factor ATF2 in order to drive expression of PGC-1α itself. There are several other known and suggested transcription factors involved in orchestrating Ucp1 gene transcription at this rich enhancer region of the gene (not shown here), but whether p38 or other kinases are involved in their control has not been fully investigated. Indeed the full picture of all the factors and the signals that promote their assembly at this gene has yet to be constructed.
Our recently published study demonstrating that cardiac NPs ANP (atrial NP) and BNP (B-type-NP) increase the expression of brown adipocyte genes and uncoupled respiration,23 which is the topic of this commentary, arose from intersecting interests: one of us (S.C.) was focused on the SNS and molecular signaling and the other (M.B.) comes from a more clinical research background on obesity and hypertension.
The discovery of the natriuretic peptides produced and secreted from the heart was a major change in the mind-set about the function of the heart: it was not only a contractile “pump” for blood circulation but a bona fide endocrine organ.24 The NP system is integral to the control of blood pressure through its stimulation of fluid and salt release by the kidney. Two receptors tightly control the response to the NPs: the signaling form of the receptor termed NPRA/NPR1/GC-A and the clearance form of the receptor termed NPRC/NPR3/ANPRC (Fig. 2). Almost two decades ago, NP receptors were unexpectedly found to be expressed in adipose tissue of both rats25 and humans26 and, interestingly, levels of NPRC in rat adipose tissue were found to be sharply decreased by fasting.27 Together, these were some of the first results to suggest that perhaps cardiac NPs have a metabolic role in adipocytes, including a putative role for adipose tissue in the clearance of these peptides from the circulation.28 It was then reported that ANP could increase lipolysis in human adipocytes, but that this property did not exist in rodent adipocytes.29 Why did this process not exist in rodent adipocytes? Experimental observations as well as inspecting the Novartis Institute gene expression database (www.biogps.org) showed that mouse adipose tissue expresses vastly more NPRC than human adipocytes. What if these levels could be modulated? Would that affect the response? Mice with targeted disruptions of the Npra and Nprc genes had been generated more than a decade earlier.30,31 However they had not been studied from the point of view of body composition and adipose tissue. We did notice in the literature, however, that mice with spontaneous loss-of-function mutations in the Nprc gene32 were referred to by names like “longjohn” and “strigosus,” the latter from the Latin meaning “long and emaciated.” We decided to obtain these mice, which proved valuable to our studies.
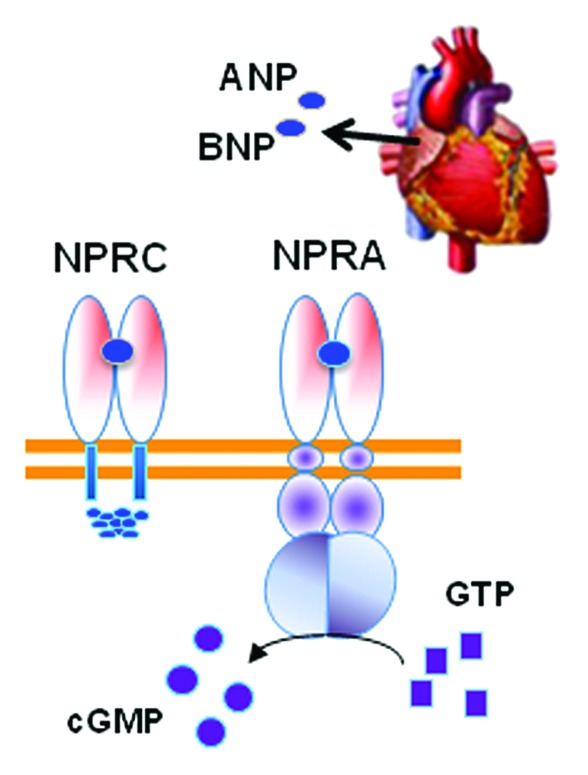
Figure 2. The cardiac hormones ANP and BNP released from the heart atria bind to the receptor NPRA to activate the intracellular guanylyl cyclase domain for the receptor to convert GTP to cGMP. Adipocytes also express NPRC that mainly removes NPs from circulation. The relative ratio of NPRA to NPRC is an important determinant of signal transduction.
We discovered that mice lacking NPRC were exceedingly lean and chock-full of brown adipocytes. By culturing their white and brown adipocytes it was easily observed that these murine cells could readily respond to ANP to increase lipolysis as well as turn on the brown adipocyte gene program. Our collaborator Nobu Takahashi provided his findings that Npra−/− mice tended to be obese. Thus we knew that it was not simply a primate-specific signaling mechanism, rather it is the ratio of NPRA/NPRC that dictates the response to NPs in adipocytes. As we went on to show and discuss below, the levels of these receptors and their response to NPs are quite dynamically regulated in mice as a normal course of their physiology. There may also be differences in responsiveness among different inbred strains of mice.
In order to better provide some physiological evidence of the role of NPs in driving both the “browning” of adipocytes within the white adipose tissue (WAT) depots and thermogenesis in brown adipose tissue (BAT), we set up a cold challenge experiment using wild-type mice. Part of the idea was that if fasting in mice was associated with a decrease in adipose tissue levels of NPRC,27 and blood pressure is increased in response to cold exposure,33-36 then maybe we would see an effect on the NP system in this experiment. This was indeed the case.23 In fact, blood levels of BNP were markedly higher in the mice kept at 5°C for 6 h. Perhaps this indicates that the SNS is promoting the secretion of these hormones from the heart as well as increasing the expression of their genes. How this cold-exposure maneuver has this effect mechanistically is not clear, but it may be related to the cold induced increase in blood pressure seen in rodents and humans.34,36-38 In these experiments we were also gratified to see that in both the interscapular BAT as well as in WAT depots, such as gonadal and inguinal, the expression levels of NPRA increased in response to the cold challenge, while NPRC levels were decreased. Together these results suggests, although certainly do not prove, that the SNS is modulating the NP system in order to set up conditions that favor enhanced signaling by the NPs to coordinate with the SNS to drive both fuel mobilization (lipolysis) as well as ramping up the machinery for adaptive thermogenesis. In conjunction with these mouse studies we used human multipotent adipocytes that can differentiate into a cell with both “white” and “brown” characteristics, and we quickly saw that the whole program of brown adipocyte gene expression and function was getting activated, and that the longer the stimulus the greater the response.23 The notion that the SNS and NP systems work together was further strengthened by the finding that low concentrations of ANP and the β-agonist isoproterenol work together in culture human adipocytes. So together with the in vivo data there seems to be a coordinated and dynamic regulation between the SNS and the NP systems. Whether the reciprocal regulation (i.e., NPs regulating SNS components in adipose tissue) exists needs to be investigated. Although we indeed observed that delivery of BNP itself via minipump (7 d) to mice resulted in increased energy expenditure and increased expression of all the brown adipocyte markers—especially in WAT,23 we did not examine whether this elevation also had effects on receptors and responses of the SNS. It will be important to understand this regulation in more detail as we tease apart the molecular mechanisms involved in this apparent cross-talk between these hormonal systems.
Since these cell culture and animal studies demonstrate the parallel pathways of the SNS and the NP system to regulate fat metabolism (Fig. 3), it may also shed some light on earlier perplexing studies in genetically modified mice. For example, Palmiter and colleagues reported that mice with targeted deletion of dopamine β-hydroxylase, which lack the ability to synthesize epinephrine and norepinephrine,39 were not prone to excess weight gain on a high-fat diet. The fact that they were also hypotensive40 might suggest that the NP system at some level is compensating in their adipocytes for the lack of SNS drive.

Figure 3. Model for parallel βAR and NPRA activation of p38 MAPK to trigger expression of the brown fat thermogenic gene program. Catecholamines binding to βARs on adipocytes increases cAMP (pink ovals), which binds the regulatory (R) subunits of PKA. The released catalytic (C) subunits (purple ovals) phosphorylate targets including HSL and perilipin (Peri A), to allow lipolysis of stored triglycerides. Lipolysis can also be activated by NPs. ANP and BNP bind to guanylyl cyclase (GC) receptor NPRA to increase cGMP (yellow circles). The cGMP produced by NPRA activates PKG (thin green and yellow ovals), whose substrate specificity for phosphorylation closely overlaps that of PKA. Thus, PKG can phosphorylate the same targets as PKA to elicit lipolysis. βAR and NPRA signaling can also activate a protein kinase cascade culminating in the activation of p38α MAPK (p38 MK). Thus, in response to β-agonist or NPs, p38α MAPK phosphorylates (light blue ovals) the transcriptional regulators ATF2 and PGC-1α. PGC-1α interacts with PPARγ and RXRα. These phosphorylated and activated factors are recruited to specific motifs within the UCP1 enhancer (PPRE, CRE2) to increase its gene expression (blue arrow). ATF2 also binds the CRE in the PGC-1α promoter to increase transcription (blue arrow) and increase the amount of PGC-1α. Reprinted with permission from the Journal of Clinical Investigation.52
Thinking more broadly about the implications of our findings, there is obviously a strong connection between obesity and hypertension. Earlier work on the NP system identified peptides that bound NPRC but not NPRA41 and conversely other reagents that favor interaction with NPRA were also identified.42 If therapeutic approaches could be developed that target NPRA—or attempt to block NPRC—these could serve to both promote fat oxidation and weight loss by increasing amounts of brown adipocytes, as well as reducing blood pressure. Obviously there is much additional work to be done in the basic research lab as well as in the clinical setting to test whether such an approach is viable. From another perspective, as we noted in our study, conditions such as heart failure could be a situation where one might want to interfere with adipose tissue NP signaling. This notion stems from the well-established observations of weight loss that occurs in heart failure, known as pathological cardiac cachexia. High levels of NPs are characteristic of heart failure and are used as diagnostic markers of disease severity.43,44 Cardiac cachexia is a commonly encountered clinical problem45,46 and is associated with a poor prognosis.47 Less well studied but deserving further investigation is the pathological weight loss associated with anorexia nervosa (AN), which in some reports is correlated with elevated levels of circulating NPs,48 and they are reported to also have increased diet-induced thermogenesis49 and large caloric requirements to gain weight.50,51 If such a correlation were strengthened, this might be a predisposing factor for AN, or a co-morbid state due to the excessive exercise that anorectics engage in. In either of these cases an attractive hypothesis that we are now pursuing—particularly for heart failure—is that the pathological weight loss associated with high circulating NPs and/or catecholamine levels may involve increased brown fat and energy expenditure.
For those of us who have focused for many years on the role of the SNS in regulating adipose tissue lipolysis and the expansion and activity of brown fat for adaptive thermogenesis, the realization that the NP system has been functioning in a parallel manner without our considering it is like discovering an elephant in the room. Now with this elephant clearly in our line of sight, our studies of adipose tissue metabolism will never be the same.
Acknowledgments
We thank fellow colleagues at SBMRI-Lake Nona for their continued support and sharing of ideas, comments and suggestions, as well as the members of the Collins lab for the team effort that made possible the work that is the subject of this commentary. We also thank Dr. Cynthia Nagle for her design of Figure 1.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Footnotes
Previously published online: www.landesbioscience.com/journals/adipocyte/article/22515
References
- 1.Weiss B, Davies JI, Brodie BB. Evidence for a role of adenosine 3′,5′-monophosphate in adipose tissue lipolysis. Biochem Pharmacol. 1966;15:1553–61. doi: 10.1016/0006-2952(66)90199-7. [DOI] [PubMed] [Google Scholar]
- 2.Schimmel RJ, Buhlinger CA, Serio R. Activation of adenosine 3′,5′-monophosphate-dependent protein kinase and its relationship to cyclic AMP and lipolysis in hamster adipose tissue. J Lipid Res. 1980;21:250–6. [PubMed] [Google Scholar]
- 3.Belfrage P, Fredrikson G, Nilsson NO, Strålfors P. Regulation of adipose tissue lipolysis: phosphorylation of hormones sensitive lipase in intact rat adipocytes. FEBS Lett. 1980;111:120–4. doi: 10.1016/0014-5793(80)80775-7. [DOI] [PubMed] [Google Scholar]
- 4.Ball EG, Jungas RL. On the action of hormones which accelerate the rate of oxygen consumption and fatty acid release in rat adipose tissue in vitro. Proc Natl Acad Sci U S A. 1961;47:932–41. doi: 10.1073/pnas.47.7.932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Smith R. Thermogenic activity of the hibernating gland in the cold-acclimatized rat. Physiologist. 1961;4:113. [Google Scholar]
- 6.Donhoffer S, Sardy F, Szegvari G. Brown Adipose Tissue and Thermoregulatory Heat Production in the Rat. Nature. 1964;203:765–6. doi: 10.1038/203765b0. [DOI] [PubMed] [Google Scholar]
- 7.Wirsén C, Hamberger B. Catecholamines in brown fat. Nature. 1967;214:625–6. doi: 10.1038/214625a0. [DOI] [PubMed] [Google Scholar]
- 8.Robidoux J, Martin TL, Collins S. Beta-adrenergic receptors and regulation of energy expenditure: a family affair. Annu Rev Pharmacol Toxicol. 2004;44:297–323. doi: 10.1146/annurev.pharmtox.44.101802.121659. [DOI] [PubMed] [Google Scholar]
- 9.Nedergaard J, Bengtsson T, Cannon B. Unexpected evidence for active brown adipose tissue in adult humans. Am J Physiol Endocrinol Metab. 2007;293:E444–52. doi: 10.1152/ajpendo.00691.2006. [DOI] [PubMed] [Google Scholar]
- 10.Cypess AM, Lehman S, Williams G, Tal I, Rodman D, Goldfine AB, et al. Identification and importance of brown adipose tissue in adult humans. N Engl J Med. 2009;360:1509–17. doi: 10.1056/NEJMoa0810780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.van Marken Lichtenbelt WD, Vanhommerig JW, Smulders NM, Drossaerts JM, Kemerink GJ, Bouvy ND, et al. Cold-activated brown adipose tissue in healthy men. N Engl J Med. 2009;360:1500–8. doi: 10.1056/NEJMoa0808718. [DOI] [PubMed] [Google Scholar]
- 12.Virtanen KA, Lidell ME, Orava J, Heglind M, Westergren R, Niemi T, et al. Functional brown adipose tissue in healthy adults. N Engl J Med. 2009;360:1518–25. doi: 10.1056/NEJMoa0808949. [DOI] [PubMed] [Google Scholar]
- 13.Zingaretti MC, Crosta F, Vitali A, Guerrieri M, Frontini A, Cannon B, et al. The presence of UCP1 demonstrates that metabolically active adipose tissue in the neck of adult humans truly represents brown adipose tissue. FASEB J. 2009;23:3113–20. doi: 10.1096/fj.09-133546. [DOI] [PubMed] [Google Scholar]
- 14.Collins S, Surwit RS. The beta-adrenergic receptors and the control of adipose tissue metabolism and thermogenesis. Recent Prog Horm Res. 2001;56:309–28. doi: 10.1210/rp.56.1.309. [DOI] [PubMed] [Google Scholar]
- 15.Collins S, Cao W, Robidoux J. Learning new tricks from old dogs: beta-adrenergic receptors teach new lessons on firing up adipose tissue metabolism. Mol Endocrinol. 2004;18:2123–31. doi: 10.1210/me.2004-0193. [DOI] [PubMed] [Google Scholar]
- 16.Cassard-Doulcier A-M, Gelly C, Fox N, Schrementi J, Raimbault S, Klaus S, et al. Tissue-specific and β-adrenergic regulation of the mitochondrial uncoupling protein gene: control by cis-acting elements in the 5′-flanking region. Mol Endocrinol. 1993;7:497–506. doi: 10.1210/me.7.4.497. [DOI] [PubMed] [Google Scholar]
- 17.del Mar Gonzalez-Barroso M, Pecqueur C, Gelly C, Sanchis D, Alves-Guerra MC, Bouillaud F, et al. Transcriptional activation of the human ucp1 gene in a rodent cell line. Synergism of retinoids, isoproterenol, and thiazolidinedione is mediated by a multipartite response element. J Biol Chem. 2000;275:31722–32. doi: 10.1074/jbc.M001678200. [DOI] [PubMed] [Google Scholar]
- 18.Kozak UC, Kopecky J, Teisinger J, Enerbäck S, Boyer B, Kozak LP. An upstream enhancer regulating brown-fat-specific expression of the mitochondrial uncoupling protein gene. Mol Cell Biol. 1994;14:59–67. doi: 10.1128/mcb.14.1.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cao W, Medvedev AV, Daniel KW, Collins S. β-Adrenergic activation of p38 MAP kinase in adipocytes: cAMP induction of the uncoupling protein 1 (UCP1) gene requires p38 MAP kinase. J Biol Chem. 2001;276:27077–82. doi: 10.1074/jbc.M101049200. [DOI] [PubMed] [Google Scholar]
- 20.Moule SK, Denton RM. The activation of p38 MAPK by the beta-adrenergic agonist isoproterenol in rat epididymal fat cells. FEBS Lett. 1998;439:287–90. doi: 10.1016/S0014-5793(98)01392-1. [DOI] [PubMed] [Google Scholar]
- 21.Cao W, Daniel KW, Robidoux J, Puigserver P, Medvedev AV, Bai X, et al. p38 mitogen-activated protein kinase is the central regulator of cyclic AMP-dependent transcription of the brown fat uncoupling protein 1 gene. Mol Cell Biol. 2004;24:3057–67. doi: 10.1128/MCB.24.7.3057-3067.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Puigserver P, Rhee J, Lin J, Wu Z, Yoon JC, Zhang CY, et al. Cytokine stimulation of energy expenditure through p38 MAP kinase activation of PPARgamma coactivator-1. Mol Cell. 2001;8:971–82. doi: 10.1016/S1097-2765(01)00390-2. [DOI] [PubMed] [Google Scholar]
- 23.Bordicchia M, Liu D, Amri EZ, Ailhaud G, Dessì-Fulgheri P, Zhang C, et al. Cardiac natriuretic peptides act via p38 MAPK to induce the brown fat thermogenic program in mouse and human adipocytes. J Clin Invest. 2012;122:1022–36. doi: 10.1172/JCI59701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.de Bold AJ. Atrial natriuretic factor: a hormone produced by the heart. Science. 1985;230:767–70. doi: 10.1126/science.2932797. [DOI] [PubMed] [Google Scholar]
- 25.Sarzani R, Paci VM, Dessì-Fulgheri P, Espinosa E, Rappelli A. Comparative analysis of atrial natriuretic peptide receptor expression in rat tissues. J Hypertens Suppl. 1993;11:S214–5. doi: 10.1097/00004872-199312050-00086. [DOI] [PubMed] [Google Scholar]
- 26.Sarzani R, Dessì-Fulgheri P, Paci VM, Espinosa E, Rappelli A. Expression of natriuretic peptide receptors in human adipose and other tissues. J Endocrinol Invest. 1996;19:581–5. doi: 10.1007/BF03349021. [DOI] [PubMed] [Google Scholar]
- 27.Sarzani R, Paci VM, Zingaretti CM, Pierleoni C, Cinti S, Cola G, et al. Fasting inhibits natriuretic peptides clearance receptor expression in rat adipose tissue. J Hypertens. 1995;13:1241–6. doi: 10.1097/00004872-199511000-00004. [DOI] [PubMed] [Google Scholar]
- 28.Dessì-Fulgheri P, Sarzani R, Tamburrini P, Moraca A, Espinosa E, Cola G, et al. Plasma atrial natriuretic peptide and natriuretic peptide receptor gene expression in adipose tissue of normotensive and hypertensive obese patients. J Hypertens. 1997;15:1695–9. doi: 10.1097/00004872-199715120-00074. [DOI] [PubMed] [Google Scholar]
- 29.Sengenès C, Zakaroff-Girard A, Moulin A, Berlan M, Bouloumié A, Lafontan M, et al. Natriuretic peptide-dependent lipolysis in fat cells is a primate specificity. Am J Physiol Regul Integr Comp Physiol. 2002;283:R257–65. doi: 10.1152/ajpregu.00453.2001. [DOI] [PubMed] [Google Scholar]
- 30.Matsukawa N, Grzesik WJ, Takahashi N, Pandey KN, Pang S, Yamauchi M, et al. The natriuretic peptide clearance receptor locally modulates the physiological effects of the natriuretic peptide system. Proc Natl Acad Sci U S A. 1999;96:7403–8. doi: 10.1073/pnas.96.13.7403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Knowles JW, Esposito G, Mao L, Hagaman JR, Fox JE, Smithies O, et al. Pressure-independent enhancement of cardiac hypertrophy in natriuretic peptide receptor A-deficient mice. J Clin Invest. 2001;107:975–84. doi: 10.1172/JCI11273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jaubert J, Jaubert F, Martin N, Washburn LL, Lee BK, Eicher EM, et al. Three new allelic mouse mutations that cause skeletal overgrowth involve the natriuretic peptide receptor C gene (Npr3) Proc Natl Acad Sci U S A. 1999;96:10278–83. doi: 10.1073/pnas.96.18.10278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kanayama N, Tsujimura R, She L, Maehara K, Terao T. Cold-induced stress stimulates the sympathetic nervous system, causing hypertension and proteinuria in rats. J Hypertens. 1997;15:383–9. doi: 10.1097/00004872-199715040-00009. [DOI] [PubMed] [Google Scholar]
- 34.Sun Z, Cade JR, Fregly MJ, Rowland NE. Effect of chronic treatment with propranolol on the cardiovascular responses to chronic cold exposure. Physiol Behav. 1997;62:379–84. doi: 10.1016/S0031-9384(97)00033-4. [DOI] [PubMed] [Google Scholar]
- 35.Cheng Y, Hauton D. Cold acclimation induces physiological cardiac hypertrophy and increases assimilation of triacylglycerol metabolism through lipoprotein lipase. Biochim Biophys Acta. 2008;1781:618–26. doi: 10.1016/j.bbalip.2008.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yuan K, Jin X, Park WH, Kim JH, Park BH, Kim SH. Modification of atrial natriuretic peptide system in cold-induced hypertensive rats. Regul Pept. 2009;154:112–20. doi: 10.1016/j.regpep.2009.02.011. [DOI] [PubMed] [Google Scholar]
- 37.Roukoyatkina NI, Chefer SI, Rifkind J, Ajmani R, Talan MI. Cold acclimation-induced increase of systolic blood pressure in rats is associated with volume expansion. Am J Hypertens. 1999;12:54–62. doi: 10.1016/S0895-7061(98)00213-1. [DOI] [PubMed] [Google Scholar]
- 38.Rintamäki H. Human responses to cold. Alaska Med. 2007;49(Suppl):29–31. [PubMed] [Google Scholar]
- 39.Thomas SA, Palmiter RD. Thermoregulatory and metabolic phenotypes of mice lacking noradrenaline and adrenaline. Nature. 1997;387:94–7. doi: 10.1038/387094a0. [DOI] [PubMed] [Google Scholar]
- 40.Swoap SJ, Weinshenker D, Palmiter RD, Garber G. Dbh(-/-) mice are hypotensive, have altered circadian rhythms, and have abnormal responses to dieting and stress. Am J Physiol Regul Integr Comp Physiol. 2004;286:R108–13. doi: 10.1152/ajpregu.00405.2003. [DOI] [PubMed] [Google Scholar]
- 41.Maack T, Suzuki M, Almeida FA, Nussenzveig D, Scarborough RM, McEnroe GA, et al. Physiological role of silent receptors of atrial natriuretic factor. Science. 1987;238:675–8. doi: 10.1126/science.2823385. [DOI] [PubMed] [Google Scholar]
- 42.Jin H, Li B, Cunningham B, Tom J, Yang R, Sehl P, et al. Novel analog of atrial natriuretic peptide selective for receptor-A produces increased diuresis and natriuresis in rats. J Clin Invest. 1996;98:969–76. doi: 10.1172/JCI118881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Maisel A, Mueller C, Adams K, Jr., Anker SD, Aspromonte N, Cleland JG, et al. State of the art: using natriuretic peptide levels in clinical practice. Eur J Heart Fail. 2008;10:824–39. doi: 10.1016/j.ejheart.2008.07.014. [DOI] [PubMed] [Google Scholar]
- 44.Noveanu M, Potocki M, Mueller C. Natriuretic peptides and their evolving clinical applications. Future Cardiol. 2008;4:593–8. doi: 10.2217/14796678.4.6.593. [DOI] [PubMed] [Google Scholar]
- 45.Sharma R, Anker SD. Cardiac cachexia is a world-wide problem. Int J Cardiol. 1999;71:113–4. doi: 10.1016/S0167-5273(99)00130-8. [DOI] [PubMed] [Google Scholar]
- 46.Anker SD, Chua TP, Ponikowski P, Harrington D, Swan JW, Kox WJ, et al. Hormonal changes and catabolic/anabolic imbalance in chronic heart failure and their importance for cardiac cachexia. Circulation. 1997;96:526–34. doi: 10.1161/01.CIR.96.2.526. [DOI] [PubMed] [Google Scholar]
- 47.Anker SD, Ponikowski P, Varney S, Chua TP, Clark AL, Webb-Peploe KM, et al. Wasting as independent risk factor for mortality in chronic heart failure. Lancet. 1997;349:1050–3. doi: 10.1016/S0140-6736(96)07015-8. [DOI] [PubMed] [Google Scholar]
- 48.Zastrow A, Wolf J, Giannitsis E, Katus H, Herzog W, Friederich HC, et al. Elevated myocardial enzymes and natriuretic peptides in anorexia nervosa: prototypic condition for the pathophysiology of cachexia? Cardiology. 2011;118:256–9. doi: 10.1159/000329512. [DOI] [PubMed] [Google Scholar]
- 49.Moukaddem M, Boulier A, Apfelbaum M, Rigaud D. Increase in diet-induced thermogenesis at the start of refeeding in severely malnourished anorexia nervosa patients. Am J Clin Nutr. 1997;66:133–40. doi: 10.1093/ajcn/66.1.133. [DOI] [PubMed] [Google Scholar]
- 50.Walker J, Roberts SL, Halmi KA, Goldberg SC. Caloric requirements for weight gain in anorexia nervosa. Am J Clin Nutr. 1979;32:1396–400. doi: 10.1093/ajcn/32.7.1396. [DOI] [PubMed] [Google Scholar]
- 51.Dempsey DT, Crosby LO, Pertschuk MJ, Feurer ID, Buzby GP, Mullen JL. Weight gain and nutritional efficacy in anorexia nervosa. Am J Clin Nutr. 1984;39:236–42. doi: 10.1093/ajcn/39.2.236. [DOI] [PubMed] [Google Scholar]
- 52.Whittle AJ, Vidal-Puig A. NPs -- heart hormones that regulate brown fat? J Clin Invest. 2012;122:804–7. doi: 10.1172/JCI62595. [DOI] [PMC free article] [PubMed] [Google Scholar]