

Abstract
Inflammation accompanies obesity and its comorbidities—type 2 diabetes, non-alcoholic fatty liver disease and atherosclerosis, among others—and may contribute to their pathogenesis. Yet the cellular machinery that links nutrient sensing to inflammation remains incompletely characterized. The protein deacetylase sirtuin-1 (SirT1) is activated by energy depletion and plays a critical role in the mammalian response to fasting. More recently it has been implicated in the repression of inflammation. SirT1 mRNA and protein expression are suppressed in obese rodent and human white adipose tissue, while experimental reduction of SirT1 in adipocytes and macrophages causes low-grade inflammation that mimics that observed in obesity. Thus suppression of SirT1 during overnutrition may be critical to the development of obesity-associated inflammation. This effect is attributable to multiple actions of SirT1, including direct deacetylation of NFκB and chromatin remodeling at inflammatory gene promoters. In this work, we report that SirT1 is also suppressed by diet-induced obesity in macrophages, which are key contributors to the ontogeny of metabolic inflammation. Thus, SirT1 may be a common mechanism by which cells sense nutrient status and modulate inflammatory signaling networks in accordance with organismal energy availability.
Keywords: Sirtuin-1, Sirt1, metabolism, metabolic inflammation, obesity, adipose tissue inflammation
Inflammation in Obesity
Obesity has become a public health crisis in the United States and much of the world. More than a billion people worldwide are estimated to be overweight, and at least 30% of these, or 300 million, are obese.1 Obesity itself would be of little concern if it were not associated with the “metabolic syndrome,” a constellation of metabolic abnormalities that portend the development of type 2 diabetes mellitus, atherosclerosis and non-alcoholic fatty liver disease.2 The list of diseases associated with obesity is ever growing. This inspires the question of how obesity triggers these complications and what can be done to intervene in the progression from obesity to disease.
Low-grade, chronic inflammation is now widely recognized to be a salient feature of obesity and many of its accompanying pathologies. Studies of critically ill humans and animals have demonstrated that inflammation can profoundly alter metabolic function. Investigators have postulated that obesity-associated inflammation may induce similar metabolic shifts. As in other unhealthy states, the teleological role of the immune system is likely to clear the insult and return the organism to a healthy, functional state.3,4 However, during obesity, this restoration of tissue function does not occur. Rather, the elaboration of cytokines and other inflammatory mediators seems to lead to a downward spiral of tissue dysfunction in metabolic organs. Many scientific investigators and clinicians have been interested in the possibility that suppressing inflammation may lead to improvement of metabolic parameters in obese patients. Thus, substantial effort has been placed on understanding how obesity incites inflammation, with the hope that such knowledge will lead to the development of novel and much needed therapeutics.5
The components of the generic inflammatory response can be conceptually divided into four major categories: inducers (LPS, PolyIC), sensors (TLRs, NLRs), mediators (cytokines, eicosinoids) and effectors (cells that respond to inflammatory mediators),3 which are connected in a simple circuit (Fig. 1). In obesity, elevated free fatty acids, “metabolic endotoxemia,” local hypoxia, products of adipocyte death and endoplasmic reticulum stress have all been implicated as potential inducers of inflammation.6 These are thought to activate sensors such as NLRP3, TLRs, HIF-1α and NFκB within adipocytes, hepatocytes and tissue macrophages,5,7-10 leading to the elaboration of mediators such as tumor necrosis factor (TNF)-α, interleukin (IL)-1β and IL-6 and the recruitment of inflammatory cells to the white adipose tissue (WAT). The best appreciated of these are CD11c+ macrophages,11,12 although nearly every cell of the immune system—conventional T cells, innate-like lymphocytes, B cells and mast cells5—accumulates in obese WAT and has been implicated in the transition from healthy to inflamed fat. These cells release additional cytokines, which may act on adipocytes, myocytes or hepatocytes to induce insulin resistance and lipolysis.13 Conversely, a few cell types (Foxp3+CD4+ regulatory T cells and eosinophils, for example) may have a salutary influence on metabolic homeostasis in healthy WAT, and are reduced during obesity.
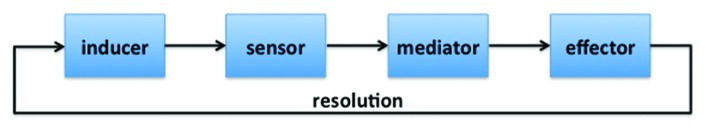
Figure 1. Four category model of inflammation, as adapted from Medzhitov 2010.
The inflammatory milieu of obesity is complex, featuring a panoply of elevated plasma and tissue cytokines, and infiltration of WAT with inflammatory cells. And while much progress has been made in identifying the inducers, sensors, mediators and effectors that participate in obesity-associated inflammation, many questions remain. One particularly enigmatic aspect of these investigations is that many of the reported inducers and sensors are present and active in physiologic states other than obesity, where they do not cause overt inflammation. Free fatty acids, for example, rise into the millimolar range during fasting, evidently without causing widespread activation of TLR4 or NLRP3. This suggests that something else—other than the simple presence or absence of a ligand for innate sensors—sets a context and contributes to the decision of whether or not to induce inflammation during obesity.
Nutrient Sensing Modulates the Inflammatory Response
The majority of studies examining the interaction between obesity and inflammation have concentrated on the pathophysiological role of the immune system in metabolism. Activation of the immune system in the setting of overnutrition is often assumed to be an accident that occurs when there is an overabundance of potential immunologic ligands such as gut-derived endotoxin, fatty acids or ceramides. However, some investigators believe that the intimate association between energy metabolism and immunity is deliberate. For example, the receptivity of inflammatory networks to activation may be regulated by nutrient availability because immune activation—particularly the pyrogenic and acute phase responses—inflicts a large energetic cost. In febrile humans, each 1°C increment in temperature raises basal metabolic rate (BMR) by 10–15%.14 As a result, during sepsis, BMR may be increased by 20–25%.15 When the bumblebee, Bombus terrestris, cannot prevent activation of the immune response under energy-limited conditions, high mortality ensues.16 Similarly, calorie restriction (CR) in mice leads to defective pathogen clearance and cytokine elaboration by peritoneal macrophages during infection, consequently reducing survival by 40%.17 Interestingly, the leptin-deficient ob/ob mouse, which lives in a state of simulated starvation, shows deficits in pathogen clearance similar to those of CR mice: an effect that can be rescued by exogenous leptin administration.18,19 Humans lacking leptin also exhibit lymphopenia and T-cell hyporesponsiveness.20 Opportunity costs are evident even at the level of individual tissues. Muscle protein wasting and the transcriptional suppression of many hepatic enzymes during sepsis provide amino acids and cellular machinery to support a dramatic increase in synthesis of acute phase proteins.21
Thus, the tight integration of metabolic and immune signaling seen in mammals may reflect an optimization process that reconciles the need for vigorous defense against pathogens with available energy supplies. For this reason, many organisms have evolved mechanisms to suppress the immune system during times of energy stress. As suggested above, soluble factors such as leptin may help communicate such signals between cells. Within cells, AMPK, mTOR and sirtuins have all been shown to participate in this communication. AMP-activated protein kinase (AMPK) and mammalian target of rapamycin (mTOR) sense energy depletion and repletion, respectively, and cooperate to permit an immune response only in the presence of adequate energy reserves. AICAR, a pharmacological activator of AMPK, suppresses LPS-mediated activation of NFκB while S6K1, a downstream target of mTOR, is required for leukotriene B4, TNF-α, IL-1β and IL-6 generation.20
SirT1 Opposes Inflammation in Metabolic Tissues
Sirtuin 1 (SirT1) is a NAD+-dependent protein deacetylase that coordinates the mammalian metabolic response to calorie restriction and fasting.22-25 During times of nutrient deficit, SirT1-mediated deacetylation of PGC1-α stimulates hepatic glucose production and fatty acid oxidation,22,26 while promoting metabolic efficiency through adiponectin production in adipose tissue.27
In addition to its metabolic effects, SirT1 can suppress inflammation.28,29 Overexpression of SirT1 (with Dnajc12) decreases hepatic expression of TNF-α and IL-6 in the setting of chronic high fat feeding,30 whereas liver-specific deletion of SirT1 increases hepatic NFκB activity.26 In 3T3-L1 adipocytes, reducing SirT1 levels with RNAi reduces inhibitory deacetylation of the NFκB subunit p65, and leads to increased NFκB activity at the TNF-α, IL-6, MCP-1, KC and IL-1β promoters.31 Yoshizaki et al. speculated that downregulation of SirT1 in adipose tissue of obese mice and humans “contributes to the heightened inflammatory state of adipose tissue in obesity.”
We found that suppression of SirT1 expression in vivo causes WAT inflammation and elevation of circulating TNF-α and IL-1β, resulting in anorexia and lipolysis. As in obese animals, WAT of fat-specific SirT1 KO mice shows aberrant CD11c+ macrophage recruitment and production of proinflammatory cytokines. In contrast, inflammation is reduced in WAT of high fat diet fed SirT1 overexpressing mice. Moreover, in two distinct human cohorts, WAT SirT1 mRNA expression correlated negatively with indices of macrophage infiltration.32 In our studies, the effects of SirT1 on cytokine expression were attributable to chromatin remodeling; in the absence of SirT1 deacetylase activity, H3K9 was hyperacetylated, increasing the accessibility of inflammatory cytokine promoters to NFκB. These findings are supportive of elegant recent work demonstrating a critical role for SirT1 in maintaining silent, loci-specific facultative heterochromatin at the TNF-α and IL-1β promoters.33,34
Macrophages Sense Nutritional Status through SirT1
Much argument still exists over where obesity-associated inflammation begins. A common view is that adipocytes initiate WAT cytokine production, and that macrophages simply propagate and amplify the original insult.35 However, tissue-resident macrophages, which abound in WAT, are specialized to act as sentinels for tissue pathology and are supremely sensitive to homeostatic threats.3 Moreover, macrophages, and in particular CD11c+ cells, are the primary producers of proinflammatory cytokines during metabolic disease.36 Thus, it is important to carefully assess the role of macrophages in initiating inflammation during metabolic stress.
In fact, recent work suggests that inflammation in macrophages is similarly influenced by SirT1 expression. Although we found that macrophages are not required for adipose tissue inflammation caused by SirT1 knockdown, we also found that suppression of SirT1 in peritoneal macrophages induces TNF-α mRNA expression. Similarly, RNAi targeting of SirT1 in RAW264.7 cells enhances LPS-elicited activation of the JNK and IKK pathways and increases NFκB DNA binding and cytokine secretion.31 Deletion of SirT1 in myeloid cells causes basal inflammation, NFκB hyperacetylation, and a heightened pro-inflammatory response to high fat feeding in liver and adipose tissue.37 Finally, the anti-inflammatory effects of AMPK activation in the presence of the inducers stearate and LPS require SirT1, specifically SirT1-mediated K310 deacetylation of NFκB p65.38 Thus, like adipocytes and hepatocytes, macrophages use SirT1 to determine immune responsiveness.
Building upon these observations, we sought to determine whether macrophages in vivo could also sense whole-body nutritional status using SirT1. To address this, we fed male mice high fat diet for 16 weeks, harvested peritoneal macrophages and isolated total RNA. Analysis of SirT1 expression by qPCR revealed a ~50% reduction in the high fat fed group, supporting the notion that decreases in SirT1 may also be involved in the pathogenesis of obesity-associated inflammation in macrophages in vivo (Fig. 2). These data raise the intriguing possibility that macrophages are autonomously capable of sensing whole body nutritional status, and can use this information to influence their propensity toward inflammation. Such a role could be important in determining the appropriateness of an immune response in cases of caloric deficit, in addition to calorie surfeit, such as we have discussed above.
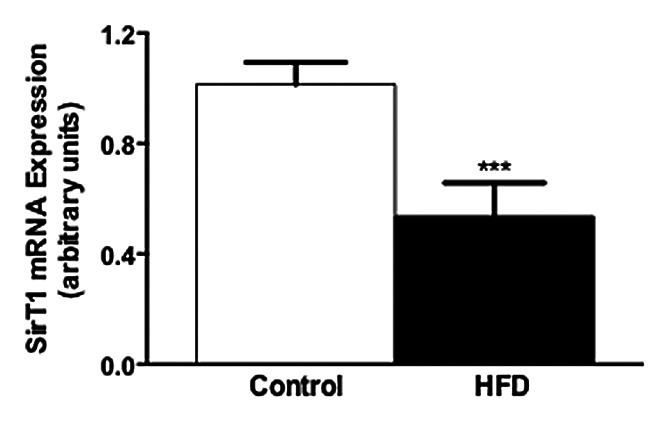
Figure 2. Suppression of SirT1 mRNA levels in peritoneal macrophages of male mice fed high-fat diet for 16 weeks (n = 5–6/group, p < 0.008).
Setting the Inflammatory Tone
Our studies and others suggest that SirT1 is a key participant in the inflammation that occurs in obesity through its actions in adipocytes and macrophages. Interestingly, unlike other proposed players in obesity-associated inflammation, SirT1 does not appear to act by providing novel ligands to stimulate immunologic receptors. Nor does it act as a sensor for an inflammatory ligand, a mediator to transduce inflammatory signals, or as a target for those mediators. Rather, SirT1 appears to act as a network regulator, which modifies the sensitivity of inflammatory circuits and thereby determines the outcome of circuit activation (Fig. 3). Network regulators like SirT1 may theoretically act at any point in the four-part model of inflammation. Given that microbial (e.g., LPS) and endogenous (i.e., fatty acid) inducers are ubiquitous, inflammation can thus result from increased sensitivity of sensors to inducers, increased elaboration of mediators in response to sensor activation, or increased response of effectors to mediators. This alteration of the circuit may be both quantitative and qualitative, changing not only the degree of inflammation (e.g., amount of cytokine produced), but also the type (e.g., ratio of “type 1” to “type 2” cytokines). Such a model could explain the ontology of metabolic inflammation.

Figure 3. (A) SirT1 intervenes in the four category model of inflammation by modifying the output of inflammatory mediators in response to sensor activation, or of effector activity in response to secreted mediators. NR, other network regulators. (B) Recent investigations suggest that SirT1 may influence inflammation through multiple means. For example, it is reported to deactylate NFκB, encouraging exit from the nucleus. Additionally, it may deacetylate multiple histones and thus change chromatin accessibility in a gene-specific manner. Thus, a similar stimulus (in this case LPS binding to TLR4), can lead to a quantitatively or qualitatively different inflammatory outcome. By the same mechanism, a different effector response may result after cytokine stimulation.
SirT1 acts on many substrates, including histones, FoxO, NFκB and p53. How acetylation alters the function of these proteins remains incompletely understood. In the case of NFκB, acetylation appears to be an important determinant of transcriptional activity once evoked.39 Deacetylation of FoxO may contribute to both its likelihood of translocating to the nucleus, and the gene targets that it chooses to activate.40 At various sites, the (de)acetylation of histones may either open/close chromatin directly, or simply make it a more attractive target for transcriptional machinery. Thus, deacetylation of targets by SirT1 is, in general, a mechanism by which cells alter transcriptional activity both quantitatively and qualitatively in response to changing nutrient status.33
Phenomena such as LPS tolerance, wherein the response to LPS is qualitatively and quantitatively altered by prior exposure, have clearly demonstrated that inflammatory circuits can be modified depending on the context in which they occur.41-43 While such modulation could theoretically occur through many means—ligand scavenging, receptor antagonism or desensitization, inhibitors of transcriptional activity—epigenetic modification of target genes may be a common methodology. Such mechanisms may change the threshold for activation, the system gain, or the transcriptional targets in order that the same amount of inducer provoke a different degree or type of inflammatory response.
We believe the circuit-modifying role of SirT1 is further evidence that the immune system and nutrition have a hard-wired and adaptive association in physiology, as well as pathophysiology. As discussed, suppression of inflammation in the undernourished state may be critical to resource allocation. It remains unclear, however, whether the converse should also be true: whether inflammation should be disinhibited in an overnourished state. In this vein, one possibility we find interesting is that “parainflammation”4 might play an important physiological role in regulating metabolism in response to environmental stressors. In this formulation, the immune system is viewed more as a “general manager” of tissue homeostasis as opposed to specialized system for pathogen disposal. Such speculations are supported by accumulating observations that animals with altered immune function spontaneously develop disordered food intake, metabolic rate, substrate utilization and adipose tissue depot size.32,44-46 We anticipate many surprises in this vigorous field over the next several decades, ultimately leading to an integrative perspective on the link between metabolism and immunology, as well as new therapeutic avenues.
Materials and Methods
Male C57BL6 mice were fed high-fat diet (60% kcal from fat, D12492, Research Diets) or regular chow (2018s, Harlan Teklad) for 16 weeks. Macrophages were harvested by peritoneal lavage, RNA was extracted using the RNeasy Kit (Qiagen), reverse transcribed and gDNA removed with the QuantiTect Kit (Qiagen), and transcript abundance assessed by real-time PCR on a 7500 Fast Real-Time PCR System (Applied Biosystems) and analyzed by ΔΔCt method with SirT1 primer sequences F: 5′-CACAAATACTGCCAAGATGTGAAT-3′, R: 5′-TCCAAAATATTACACTCTCCCCAGTA-3′.
Acknowledgments
The authors would like to thank Dr Gerald Shulman and members of his laboratory for formative discussions and the University of Iowa Department of Neurology for research support.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Footnotes
Previously published online: www.landesbioscience.com/journals/adipocyte/article/23437
References
- 1.Ford ES, Mokdad AH. Epidemiology of obesity in the Western Hemisphere. J Clin Endocrinol Metab. 2008;93(Suppl 1):S1–8. doi: 10.1210/jc.2008-1356. [DOI] [PubMed] [Google Scholar]
- 2.Cornier MA, Dabelea D, Hernandez TL, Lindstrom RC, Steig AJ, Stob NR, et al. The metabolic syndrome. Endocr Rev. 2008;29:777–822. doi: 10.1210/er.2008-0024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Medzhitov R. Inflammation 2010: new adventures of an old flame. Cell. 2010;140:771–6. doi: 10.1016/j.cell.2010.03.006. [DOI] [PubMed] [Google Scholar]
- 4.Medzhitov R. Origin and physiological roles of inflammation. Nature. 2008;454:428–35. doi: 10.1038/nature07201. [DOI] [PubMed] [Google Scholar]
- 5.Osborn O, Olefsky JM. The cellular and signaling networks linking the immune system and metabolism in disease. Nat Med. 2012;18:363–74. doi: 10.1038/nm.2627. [DOI] [PubMed] [Google Scholar]
- 6.Attie AD, Scherer PE. Adipocyte metabolism and obesity. J Lipid Res. 2009;50(Suppl):S395–9. doi: 10.1194/jlr.R800057-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Arkan MC, Hevener AL, Greten FR, Maeda S, Li ZW, Long JM, et al. IKK-beta links inflammation to obesity-induced insulin resistance. Nat Med. 2005;11:191–8. doi: 10.1038/nm1185. [DOI] [PubMed] [Google Scholar]
- 8.Hirosumi J, Tuncman G, Chang L, Görgün CZ, Uysal KT, Maeda K, et al. A central role for JNK in obesity and insulin resistance. Nature. 2002;420:333–6. doi: 10.1038/nature01137. [DOI] [PubMed] [Google Scholar]
- 9.Sabio G, Das M, Mora A, Zhang Z, Jun JY, Ko HJ, et al. A stress signaling pathway in adipose tissue regulates hepatic insulin resistance. Science. 2008;322:1539–43. doi: 10.1126/science.1160794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wen H, Gris D, Lei Y, Jha S, Zhang L, Huang MT, et al. Fatty acid-induced NLRP3-ASC inflammasome activation interferes with insulin signaling. Nat Immunol. 2011;12:408–15. doi: 10.1038/ni.2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, Ferrante AW., Jr. Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest. 2003;112:1796–808. doi: 10.1172/JCI19246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cinti S, Mitchell G, Barbatelli G, Murano I, Ceresi E, Faloia E, et al. Adipocyte death defines macrophage localization and function in adipose tissue of obese mice and humans. J Lipid Res. 2005;46:2347–55. doi: 10.1194/jlr.M500294-JLR200. [DOI] [PubMed] [Google Scholar]
- 13.Guilherme A, Virbasius JV, Puri V, Czech MP. Adipocyte dysfunctions linking obesity to insulin resistance and type 2 diabetes. Nat Rev Mol Cell Biol. 2008;9:367–77. doi: 10.1038/nrm2391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Roe CF, Kinney JM. The Caloric Equivalent of Fever. Ii. Influence of Major Trauma. Ann Surg. 1965;161:140–7. doi: 10.1097/00000658-196501000-00022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kreymann G, Grosser S, Buggisch P, Gottschall C, Matthaei S, Greten H. Oxygen consumption and resting metabolic rate in sepsis, sepsis syndrome, and septic shock. Crit Care Med. 1993;21:1012–9. doi: 10.1097/00003246-199307000-00015. [DOI] [PubMed] [Google Scholar]
- 16.Moret Y, Schmid-Hempel P. Survival for immunity: the price of immune system activation for bumblebee workers. Science. 2000;290:1166–8. doi: 10.1126/science.290.5494.1166. [DOI] [PubMed] [Google Scholar]
- 17.Jolly CA. Dietary restriction and immune function. J Nutr. 2004;134:1853–6. doi: 10.1093/jn/134.8.1853. [DOI] [PubMed] [Google Scholar]
- 18.Tschöp J, Nogueiras R, Haas-Lockie S, Kasten KR, Castañeda TR, Huber N, et al. CNS leptin action modulates immune response and survival in sepsis. J Neurosci. 2010;30:6036–47. doi: 10.1523/JNEUROSCI.4875-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lord GM, Matarese G, Howard JK, Baker RJ, Bloom SR, Lechler RI. Leptin modulates the T-cell immune response and reverses starvation-induced immunosuppression. Nature. 1998;394:897–901. doi: 10.1038/29795. [DOI] [PubMed] [Google Scholar]
- 20.Dixit VD. Adipose-immune interactions during obesity and caloric restriction: reciprocal mechanisms regulating immunity and health span. J Leukoc Biol. 2008;84:882–92. doi: 10.1189/jlb.0108028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ramadori G, Christ B. Cytokines and the hepatic acute-phase response. Semin Liver Dis. 1999;19:141–55. doi: 10.1055/s-2007-1007106. [DOI] [PubMed] [Google Scholar]
- 22.Rodgers JT, Lerin C, Haas W, Gygi SP, Spiegelman BM, Puigserver P. Nutrient control of glucose homeostasis through a complex of PGC-1alpha and SIRT1. Nature. 2005;434:113–8. doi: 10.1038/nature03354. [DOI] [PubMed] [Google Scholar]
- 23.Rodgers JT, Puigserver P. Fasting-dependent glucose and lipid metabolic response through hepatic sirtuin 1. Proc Natl Acad Sci U S A. 2007;104:12861–6. doi: 10.1073/pnas.0702509104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen D, Guarente L. SIR2: a potential target for calorie restriction mimetics. Trends Mol Med. 2007;13:64–71. doi: 10.1016/j.molmed.2006.12.004. [DOI] [PubMed] [Google Scholar]
- 25.Gillum MP, Erion DM, Shulman GI. Sirtuin-1 regulation of mammalian metabolism. Trends Mol Med. 2010;17:8–13. doi: 10.1016/j.molmed.2010.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Purushotham A, Schug TT, Xu Q, Surapureddi S, Guo X, Li X. Hepatocyte-specific deletion of SIRT1 alters fatty acid metabolism and results in hepatic steatosis and inflammation. Cell Metab. 2009;9:327–38. doi: 10.1016/j.cmet.2009.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Banks AS, Kon N, Knight C, Matsumoto M, Gutiérrez-Juárez R, Rossetti L, et al. SirT1 gain of function increases energy efficiency and prevents diabetes in mice. Cell Metab. 2008;8:333–41. doi: 10.1016/j.cmet.2008.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yeung F, Hoberg JE, Ramsey CS, Keller MD, Jones DR, Frye RA, et al. Modulation of NF-kappaB-dependent transcription and cell survival by the SIRT1 deacetylase. EMBO J. 2004;23:2369–80. doi: 10.1038/sj.emboj.7600244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ghosh HS, Spencer JV, Ng B, McBurney MW, Robbins PD. Sirt1 interacts with transducin-like enhancer of split-1 to inhibit nuclear factor kappaB-mediated transcription. Biochem J. 2007;408:105–11. doi: 10.1042/BJ20070817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pfluger PT, Herranz D, Velasco-Miguel S, Serrano M, Tschöp MH. Sirt1 protects against high-fat diet-induced metabolic damage. Proc Natl Acad Sci U S A. 2008;105:9793–8. doi: 10.1073/pnas.0802917105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yoshizaki T, Schenk S, Imamura T, Babendure JL, Sonoda N, Bae EJ, et al. SIRT1 inhibits inflammatory pathways in macrophages and modulates insulin sensitivity. Am J Physiol Endocrinol Metab. 2010;298:E419–28. doi: 10.1152/ajpendo.00417.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gillum MP, Kotas ME, Erion DM, Kursawe R, Chatterjee P, Nead KT, et al. SirT1 regulates adipose tissue inflammation. Diabetes. 2011;60:3235–45. doi: 10.2337/db11-0616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liu TF, Yoza BK, El Gazzar M, Vachharajani VT, McCall CE. NAD+-dependent SIRT1 deacetylase participates in epigenetic reprogramming during endotoxin tolerance. J Biol Chem. 2011;286:9856–64. doi: 10.1074/jbc.M110.196790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.McCall CE, El Gazzar M, Liu T, Vachharajani V, Yoza B. Epigenetics, bioenergetics, and microRNA coordinate gene-specific reprogramming during acute systemic inflammation. J Leukoc Biol. 2011;90:439–46. doi: 10.1189/jlb.0211075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Oh DY, Morinaga H, Talukdar S, Bae EJ, Olefsky JM. Increased macrophage migration into adipose tissue in obese mice. Diabetes. 2012;61:346–54. doi: 10.2337/db11-0860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Patsouris D, Li PP, Thapar D, Chapman J, Olefsky JM, Neels JG. Ablation of CD11c-positive cells normalizes insulin sensitivity in obese insulin resistant animals. Cell Metab. 2008;8:301–9. doi: 10.1016/j.cmet.2008.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schug TT, Xu Q, Gao H, Peres-da-Silva A, Draper DW, Fessler MB, et al. Myeloid deletion of SIRT1 induces inflammatory signaling in response to environmental stress. Mol Cell Biol. 2010;30:4712–21. doi: 10.1128/MCB.00657-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yang Z, Kahn BB, Shi H, Xue BZ. Macrophage alpha1 AMP-activated protein kinase (alpha1AMPK) antagonizes fatty acid-induced inflammation through SIRT1. J Biol Chem. 2010;285:19051–9. doi: 10.1074/jbc.M110.123620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chen LF, Mu Y, Greene WC. Acetylation of RelA at discrete sites regulates distinct nuclear functions of NF-kappaB. EMBO J. 2002;21:6539–48. doi: 10.1093/emboj/cdf660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Banks AS, Kim-Muller JY, Mastracci TL, Kofler NM, Qiang L, Haeusler RA, et al. Dissociation of the glucose and lipid regulatory functions of FoxO1 by targeted knockin of acetylation-defective alleles in mice. Cell Metab. 2011;14:587–97. doi: 10.1016/j.cmet.2011.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liu TF, Brown CM, El Gazzar M, McPhail L, Millet P, Rao A, et al. Fueling the flame: bioenergy couples metabolism and inflammation. J Leukoc Biol. 2012;92:499–507. doi: 10.1189/jlb.0212078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.McCall CE, Yoza B, Liu T, El Gazzar M. Gene-specific epigenetic regulation in serious infections with systemic inflammation. J Innate Immun. 2010;2:395–405. doi: 10.1159/000314077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Foster SL, Hargreaves DC, Medzhitov R. Gene-specific control of inflammation by TLR-induced chromatin modifications. Nature. 2007;447:972–8. doi: 10.1038/nature05836. [DOI] [PubMed] [Google Scholar]
- 44.Netea MG, Joosten LA, Lewis E, Jensen DR, Voshol PJ, Kullberg BJ, et al. Deficiency of interleukin-18 in mice leads to hyperphagia, obesity and insulin resistance. Nat Med. 2006;12:650–6. doi: 10.1038/nm1415. [DOI] [PubMed] [Google Scholar]
- 45.Matarese G, La Cava A. The intricate interface between immune system and metabolism. Trends Immunol. 2004;25:193–200. doi: 10.1016/j.it.2004.02.009. [DOI] [PubMed] [Google Scholar]
- 46.Wallenius V, Wallenius K, Ahrén B, Rudling M, Carlsten H, Dickson SL, et al. Interleukin-6-deficient mice develop mature-onset obesity. Nat Med. 2002;8:75–9. doi: 10.1038/nm0102-75. [DOI] [PubMed] [Google Scholar]