

Abstract
Orexin A (OX) is a small excitatory neuropeptide hormone that stimulates feeding, wakefulness and energy expenditure via a pair of G-coupled protein receptors, namely orexin receptor-1 (OXR1) and orexin receptor-2 (OXR2). OX-deficient mice are sensitive to obesity despite being hypophagic. The obesogenic effect of OX-deletion is due to brown adipose tissue (BAT) dysfunction, a defect that originates during fetal growth. Brown preadipocytes in OX-null mice display undifferentiated histological appearance and fail to support both diet- and cold-induced thermogenesis. We show that the OXR1-null mice phenocopies the differentiation defect observed in the ligand-null mice indicating that OXR1 relays OX’s differentiation and thermogenic function. Consistent with this, OX fails to induce differentiation in cultured OXR1-null preadipocytes. Thus, OX signaling via OXR1 constitutes an important thermoregulatory mechanism that defends against cold and obesity.
Keywords: brown fat development, diet-induced thermogenesis, differentiation, obesity, orexin
Orexin A is an endogenous ligand for two G-protein coupled OX receptors, orexin-receptor 1 and 2 (OXR1 and OXR2).1,2 OX-deficiency is implicated in the pathogenesis of narcolepsy in human and animal models.1-5 A well-established body of evidence indicates that the neuropeptide is an important driver of energy expenditure (EE), wakefulness and feeding behavior, at least when acutely administered in the brain.6 Consistent with its appetite inducing function, loss of OX reduces energy intake by up to 30% in OX-deficient mice model.7 Paradoxically, OX-deficiency is also associated with obesity in humans and animals.7,8 Hypophagia and obesity can only coexist if EE is severely reduced. Indeed, reduction in EE has been observed in both human subjects and animal models of the disease.9,10 The underlying mechanism leading to diminished EE has not been clearly established but is thought to be due to reduced physical activity as a corollary to increased sleep duration during the active phase.7 This tenet holds true in animal models. However, the reduction in EE is striking as it is lowered by about 26% compared with their normal counterparts.11 Reduction of this magnitude cannot be explained by differences in physical activity alone and possibly reflects basic pathological alterations in other components of EE as well. Furthermore, total sleep duration and physical activity remain unchanged in narcoleptic human subjects,12 supporting the notion that decreased physical activity is not the cause of narcolepsy-associated obesity. The fact that narcoleptic patients are more obese than equally active subjects suffering from idiopathic hypersomnia strongly indicates that malfunction of other components of EE must be at the core of narcolepsy-associated obesity.8 In an attempt to understand the pathogenesis of weight gain in narcolepsy, we observed that OX-null mice were only marginally heavier than control animals on chow diet. On a high fat diet, OX-null mice become three times heavier compared with the control group in a matter of six weeks, despite consuming significantly less calories because they lacked the ability to activate diet-induced thermogenesis, a defense mechanism against weight-gain and obesity.10,13 Molecular characterization revealed that failure of thermogenesis was due to the inability of brown preadipocytes to differentiate into mature adipocytes in the absence of OX. Thus, OX signaling has the regulatory capacity to prime dissipation of excess calories as heat and therefore serves as a key internal anti-obesity mechanism. Without this energy burning mechanism, mammals become obese as excess calories are stored as fat.
We previously reported that OX-deficient mice rapidly gain weight on HFD. To investigate whether OX-deficiency-induced elevation in body weight correlated positively with adiposity, we fed OX-null and their wild-type littermates with HFD for six-weeks. Body composition analysis at the end of the study suggested that considerable weight gain in OX-null mice was accompanied by 150% increase in total fat mass compared with wild-type controls (Table 1). Lean mass did not differ significantly between the groups. These observations indicate that OX deficiency accelerates adiposity in rodents under conditions of calorific excess. Calorimetric measurements indicated that wild-type animals had increased their EE by about 14%. In contrast, this response was absent in OX-null mice, as has been noted before.10 At the end of the 6-week HFD regimen, OXKO mice exhibited a higher respiratory quotient (Fig. 1). OXKO mice had a RQ of 0.79 compared with 0.77 in controls (Fig. 2B) indicating preferential carbohydrate oxidation over fat.14,15 This inherent variability in substrate oxidation indicates that OX depletion reduces fat oxidation capacity in OX-null mice, resulting in increased fat accumulation. A high RQ is considered to be an indicator of positive energy balance and predictor of weight gain and obesity.14-16 It should be noted that we have focused exclusively on 24 h RQ, which may not reflect post-prandial fat oxidation. RQ measurements during fasting would be a more reliable indicator of endogenous free fatty acids as fuel in OX-null mice. Nevertheless, our data strongly suggest that fat oxidation capacity is compromised in OX-deficiency.
Table 1. Body composition analysis.
| Fat mass (g) | Lean mass (g) | %Body fat | |
|---|---|---|---|
| Wild-type (HFD) |
6.2 ± 0.8 |
19.4 ± 1.2 |
22.4 ± 1.3 |
| OX KO (HFD) | 15.4 ± 1.4 | 20.7 ± 1.1 | 37.3 ± 3.1 |
Body composition as measured by nuclear magnetic resonance (NMR) for fat mass, lean mass and body fat percentage) in 9-week-old OX-KO and wild-type littermates following 6 weeks of high-fat diet (HFD) consumption.
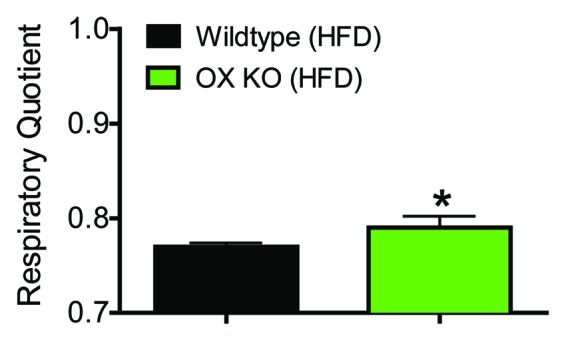
Figure 1. Reduced fat oxidation as indicated by increased respiratory quotient in OX KO mice. Respiratory quotient in 9-week-old OX-KO and wild-type littermates following 6 weeks of high fat diet (HFD) consumption (n = 6). Differences between groups were assessed by Student t-test.

Figure 2. OX-KO mice display cold intolerance. Core body temperature of 8–10-week-old OX KO and wild-type littermates during 6 h exposure to cold (4°C). (A) Expression changes in transcripts known to be important for brown adipose tissue (BAT) thermogenesis in OX KO mice. (B) Thermal images using an infrared camera, of the surface temperature of the interscapulary region of OX KO and wild-type mice following 6 h exposure to a temperature of 4°C for 6 h (C).
Thermogenesis increases during periods of overfeeding or high-fat consumption. This phenomenon is termed diet-induced thermogenesis (DIT) and considered a function of brown fat. Brown adipocytes in OX-null mutants are underdeveloped and therefore lack diet induced thermogenic response.10 In addition to DIT, BAT has a more established known function: thermogenic activity in response to cold.13 Thus studying the response of OX-null mice to cold was relevant. BAT is a highly vascularized organ filled with lipid droplets and loaded with mitochondria that express UCP1, which enables BAT to produce heat in cold environments. We postulated that OX-null mice will display heightened cold intolerance due to reduced triglyceride and mitochondrial content of brown adipocytes. To assess BAT functionality in OX-null mice, we studied their ability to defend their body temperature cold. When exposed to 4°C, core temperatures of both OX-null mice and wild-type littermates dropped. Temperature drop in Wt animals was gradual and after 4 h of cold exposure, they were able to increase thermogenesis and defend their body temperature (Fig. 2A). In contrast, temperature drop in OX-null mice was exaggerated after 4 h of cold exposure. Core temperatures in two of the six OX-null mice dropped to 20°C within 3 h and were excluded from the study. Within the time frame of the experiment, OX-null animals were unable to produce sufficient heat to defend their core temperature. Estimates suggest that thermal homeostasis in animals during acute cold stress requires a 4-fold increase in their metabolism over the basal metabolic rate.13,17 Failure of OX-null mice to successfully defend their body temperature suggests that metabolic capacity was reduced in these animals. We postulate that the defect in brown fat-derived thermogenesis in OX-null mice likely contributes to acute cold intolerance as has been demonstrated in UCP1-ablated mice.18,19 BAT activity is known to be stimulated by acute cold exposure in humans.20-22 However, it is noteworthy that thermogenesis in acute cold exposure is derived primarily by muscle shivering mechanism.17,23 We therefore suspect that cold intolerance of OX-null mice may also reflect a problem in either fatty acid release from white adipose tissue or fuel utilization by muscle/heart.
To investigate if brown fat thermogenic mechanisms were recruited in rodents upon acute cold exposure, we compared the mRNA expression of key factors involved in BAT metabolism at 25°C and 4°C. A six-hour cold-exposure in wild-type mice was accompanied by substantial increases in mRNA expression of various iBAT thermogenic genes including Ucp1, Cox8b and Pgc 1-α (Fig. 2B). These results are consistent with the idea that rodents recruit brown fat thermogenic mechanisms even in acute cold.24 We have reported that brown fat tissue in OX-deficient mice displays immature histologic appearance. This is associated with marked reduction in intracellular triglycerides and also in the expression of terminal brown fat differentiation markers.10 We observed that basal mRNA expression of Ucp-1, Cox, Pgc1-α and Tfam (mitochondrial transcription factor A) were substantially reduced in OX-null mice (at 25°C, Fig. 2B). These observations prompted us to postulate that OX-null mice may fail to upregulate thermogenic gene transcription in response to acute cold, as was observed in brown fat of wild-type mice. Surprisingly, following six hours cold exposure mRNA abundance of thermogenic genes were robustly elevated in OX-KO mice (Fig. 2B). Quantitative measurement of mRNA messages demonstrated that though the absolute levels were remarkably low, acute cold invoked a stronger induction of thermogenic gene expression in the brown adipocytes of OX-null mice than it did in control group (Fig. 2B). However, Ucp1 induction was significantly blunted in OX KO animals. These results imply that BAT in OX-deficient mice is inherently capable of sensing and appropriately mounting a transcriptional response with the exception for Ucp1. Ucp1 mRNA level in cold exposed OX-null mice was similar to that was typically observed at 25°C in wild-type rodents. This reduced Ucp1 abundance may not be enough to support heat production and is likely to contribute to cold intolerance in OX-null mice.
To assess the consequences of OX-deficiency on BAT thermogenesis during acute cold, we used infrared thermography, a non-invasive approach where restraint was not required. This approach involves measuring skin temperature with an infrared camera. Changes in temperature of the interscapulary skin overlying BAT are reflective of the BAT temperature,25 and has been validated as a noninvasive surrogate for assessing BAT thermogenesis.25-27 Real-time infrared thermography revealed that the surface temperature of the skin in the interscapulary skin directly above the iBAT was 33.5°C in wild-type mice but 29.4°C in OX-null mice following exposure to a temperature of 4°C for 6 h (Fig. 2C). Thus, following acute cold exposure intrascapular skin above iBAT was substantially lower in OX-deficient mice. This suggests that that iBAT thermogenic activity is reduced in these mice. A word of caution here is that the lower iBAT surface temperature could also originate from poor cardio vascular function in OX-null mice. Nevertheless, impaired Ucp1 expression coupled with dampened interscapulary skin temperature indicates that thermogenic response is impaired in Ox-deficiency. It is relevant to recall that intracellular triglycerides are severely reduced in OX-null mice.10 Since iBAT lipolysis is essential for thermogenesis in this tissue, which utilizes stored triglycerides to generate fuel for mitochondrial oxidation and uncoupling,13 it is likely that the lack of iBAT lipids is detrimental to both diet- and cold-induced thermogenesis. With a shortage of this triglyceride fuel in iBAT, there would be very little capacity for mitochondrial respiration and uncoupling. Attenuation of Ucp1 expression further exacerbates the BAT dysfunction in OX-null mice.
OX signals via two GPCRs, OXR1 and OXR2. Observations made on cultured mesenchymal stem cells and preadipocytes suggest that OXR1 drives OX’s brown fat differentiation functions.10 Contribution of the two receptors in mediating OX’s differentiation function in vivo is not clear. To investigate this, we first quantitatively measured relative abundance OXR1 and OXR2 mRNA abundance in interscapular brown fat. We noted that brown adipocytes expressed both the receptor types, the former being the more predominant of the two (Table 2), indicating that signaling through OXR1 may be more relevant for adipogenesis. To identify which of the two receptors relayed OX’s brown fat adipogenic function, we examined the iBAT histology of 6–8-week-old chow-fed wild-type mice, OX-null and OX- receptor null mice. Morphological examination following Hematoxilin and Eosin (H&E) staining revealed dense clusters of lipid droplets in Wt iBAT; in contrast, iBAT intracellular lipids were scarce in OX-null mice (Fig. 3A). Total intracellular triglyceride, as measured by glycerol release, was severely reduced by 88% (Fig. 3B). iBATs from OXR1 displayed a 70% reduction in triglycerol content (Fig. 3A and B). iBAT triglyceride was reduced in OXR2-null mice but not as severely as noted for OXR1-null BATs. These findings suggest that the OX-stimulated iBAT development and lipid droplet formation is dependent primarily on OXR1 signaling. We also observed that expression of key factors determining BAT differentiation and function was reduced. For example, PPAR-γ1 mRNA considered necessary for adipogenesis,28,29 was dimiminished by an order of magnitude in iBATs of OX-null mice. Pronounced reduction in iBAT lipid stores in the face of impaired PPAR-γ1/γ2, PGC-1α/β and UCP-1 mRNA expression (Fig. 3C) suggest that brown adipocytes are incapable of efficiently synthesizing triglycerides. We reasoned that if OXR1 mediated OX’s adipogenic signals, lack of OXR1 should mimic the tran scriptional defect observed for OX-null mice. We therefore measured mRNA levels of important BAT regulatory factors in OXR1 null mice and compared it to OX-null and wild-type control mice. As demonstrated in Figure 3C, mRNA abundance of these regulators in OX-null and OXR1-null mice was reduced by similar extent. Thus, disruption of OX signaling by ligand-or OXR1-ablation induces overlapping effects on triglyceride stores and iBAT gene expression. These observations indicate that OXR1 mediates OX’s adipogenic signals.
Table 2. mRNA expression of OXR1 and OXR2 in BAT.
| Transcript | Mean Ct value |
|---|---|
| Actin |
14.5 |
| OXR1 |
24.2 |
| OXR2 | 26.8 |
Ct values for β actin, OXR1 and OXR2 in brown adipose tissue from 8-week-old wild-type mice.

Figure 3. Reduced brown adipose tissue triglyceride content is receptor-1 dependent. Histological images of H&E-stained brown adipose tissue sections (A) and quantification of BAT triglyceride content (B) in 8–10-week-old wild-type littermates, OXR1-KO, OXR2-KO, and OX-KO mice. Thermogenic gene expression in brown adipose tissue of 8–10-week-old wild-type littermates and OXR1-KO mice (C). Histological images of H&E-stained brown adipose tissue sections from 1-day-old wildtype, OXR1, OXR2 and OX KO pups (D).
Previously, we reported that OX was necessary for BAT development.10 OX-deficient pups born from mating OX-null sibs and dams had severely reduced triglycerides in iBAT. We postulated that if OXR1 was solely responsible for mediating OX’s BAT developmental adipogenic signals, OXR1 but not OXR2 must phenocopy the defect observed in iBATs of OX-null mice. Morphological examination after H&E staining of wild-type neonatal iBATs showed that adipocytes were rich in fat droplets (Fig. 3D). As expected, fat droplets were drastically reduced in OX-null neonatal iBATs. Brown adipocytes for OXR1-null mice demonstrated a similar reduction in intracellular lipid stores. In contrast, OXR2-null brown adipocytes did not display any obvious reduction in lipid stores (Fig. 3D). These results strongly indicate that OXR1 but not OXR2 relays OX’s adipogenic signals during development.
Histological examination of iBAT in ligand and receptor-null mice indicated that (1) OXR1 was the primary driver in relaying OX’s differentiation function, and (2) OXR2 was not critical for adipogenesis. To demonstrate OX’s receptor-selective differentiation function, we compared the differentiation capacity of primary brown preadipocytes isolated from OX-receptor null mice and their wild-type littermates. Primary preadipocytes from wild-type littermates differentiated into adipocytes and accumulated lipids (Fig. 4). OX-treatment of wild-type preadipocytes markedly increased the basal adipogenesis. In contrast, preadipocytes from OXR1- null mice differentiated poorly. Exogenous OX failed to normalize this differentiation defect in OX1R-null preadipocytes. OXR2-ablated primary brown adipocytes however, did not display any measurable defect in basal differentiation capacity. Furthermore, OX-treatment substantially elevated the differentiation efficiency of OXR2-null preadipocytes as was observed for wild-type preadipocytes. Together, these data suggests that OXR1 is required for BAT development and differentiation. OXR2, on the other hand appears to be dispensable for in vivo and in vitro brown fat adipogenesis.
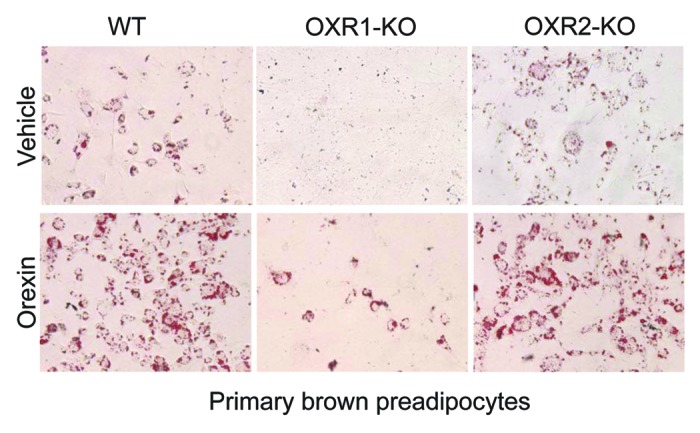
Figure 4. OX relays differentiation signals via OXR1 but not OXR2. Images of Oil-red-O stained primary brown adipocytes cultured from wild-type, OXR1-KO and OXR2-KO mice and subsequently treated with either orexin (OX) (100ng) or vehicle (PBS) for 7 consecutive days in the absence of adipogenic induction media (C). Lipids appear red in color.
Taken together our findings confirm that OX-induced adipogenesis and developmental differentiation is dependent on OXR1 signaling. We show that OX is crucial for both cold- and diet-induced thermogenesis. Attenuated diet-induced thermogenesis in OX-null mice is associated with elevated adiposity and exaggerated weight gain. Our findings also help to explain the high prevalence of obesity in narcoleptics and individuals with sleep disturbances. Our data also highlight the importance of iBAT function to whole body energy homeostasis and body-weight regulation. Increasing energy expenditure by enhancing iBAT function through developmental interventions thus presents a viable therapeutic approach to prevent or reduce obesity.
Acknowledgments
We thank Masashi Yanagisawa, UT Southwestern Medical Center for Orexin and Orexin-receptor knockout mice. We thank Marica Bordicchia for help with primary adipocyte isolation. We would like to thank metabolic phenotyping core members at Sanford Burnham Research Institute for helping us with calorimetric studies. We also acknowledge the histology core at SBMRI for assisting us with brown fat tissue histology. This work was partly supported by a fund made available by Marilyn Swan.
Footnotes
Previously published online: www.landesbioscience.com/journals/adipocyte/article/18965
References
- 1.Sakurai T, Amemiya A, Ishii M, Matsuzaki I, Chemelli RM, Tanaka H, et al. Orexins and orexin receptors: a family of hypothalamic neuropeptides and G protein-coupled receptors that regulate feeding behavior. Cell. 1998;92:573–85. doi: 10.1016/S0092-8674(00)80949-6. [DOI] [PubMed] [Google Scholar]
- 2.Chemelli RM, Willie JT, Sinton CM, Elmquist JK, Scammell T, Lee C, et al. Narcolepsy in orexin knockout mice: molecular genetics of sleep regulation. Cell. 1999;98:437–51. doi: 10.1016/S0092-8674(00)81973-X. [DOI] [PubMed] [Google Scholar]
- 3.Nishino S, Ripley B, Overeem S, Nevsimalova S, Lammers GJ, Vankova J, et al. Low cerebrospinal fluid hypocretin (Orexin) and altered energy homeostasis in human narcolepsy. Ann Neurol. 2001;50:381–8. doi: 10.1002/ana.1130. [DOI] [PubMed] [Google Scholar]
- 4.Lin L, Faroco J, Li R, Kadotani H, Rogers W, Lin X, et al. The sleep disorder canine narcolepsy is caused by a mutation in the hypocretin (orexin) receptor 2 gene. Cell. 1999;98:365–76. doi: 10.1016/S0092-8674(00)81965-0. [DOI] [PubMed] [Google Scholar]
- 5.Nishino S, Ripley B, Overeem S, Lammers G, Mignot E. Hypocretin (orexin) deficiency in human narcolepsy. Lancet. 2000;355:39–40. doi: 10.1016/S0140-6736(99)05582-8. [DOI] [PubMed] [Google Scholar]
- 6.Wang J, Osaka T, Inoue S. Energy expenditure by intracerebroventricular administration of orexin to anesthetized rats. Neurosci Lett. 2001;315:49–52. doi: 10.1016/S0304-3940(01)02322-9. [DOI] [PubMed] [Google Scholar]
- 7.Hara J, Beuckmann C, Nambu T, Willie J, Chemilli R, Sinton C, et al. Genetic ablation of orexin neurons in mice results in narcolepsy, hypophagia, and obesity. Neuron. 2001;30:345–54. doi: 10.1016/S0896-6273(01)00293-8. [DOI] [PubMed] [Google Scholar]
- 8.Kok SW, Overeem S, Visscher TL, Lammers GJ, Seidell JC, Pijl H, et al. Hypocretin deficiency in narcoleptic humans is associated with abdominal obesity. Obes Res. 2003;11:1147–54. doi: 10.1038/oby.2003.156. [DOI] [PubMed] [Google Scholar]
- 9.Chabas D, Foulon C, Gonzalez J, Nasr M, Lyon-Caen O, Willer JC, et al. Eating disorder and metabolism in narcoleptic patients. Sleep. 2007;30:1267–73. doi: 10.1093/sleep/30.10.1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sellayah D, Bharaj P, Sikder D. Orexin Is Required for Brown Adipose Tissue Development, Differentiation, and Function. Cell Metab. 2011;14:478–90. doi: 10.1016/j.cmet.2011.08.010. [DOI] [PubMed] [Google Scholar]
- 11.Zhang S, Zeitzer J, Sakurai T, Nishino S, Mignot E. Sleep/wake fragmentation disrupts metabolism in a mouse model of narcolepsy. J Physiol. 2007;581:649–63. doi: 10.1113/jphysiol.2007.129510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Middelkoop HA, Lammers G, Van Hilton B, Ruwhof C, Pijl H, Kamphuisen H. Circadian distribution of motor activity and immobility in narcolepsy: assessment with continuous motor activity monitoring. Psychophysiology. 1995;32:286–91. doi: 10.1111/j.1469-8986.1995.tb02957.x. [DOI] [PubMed] [Google Scholar]
- 13.Cannon B, Nedergaard J. Brown Adipose Tissue: Function and Physiological Significance. Physiol Rev. 2004;84:277–359. doi: 10.1152/physrev.00015.2003. [DOI] [PubMed] [Google Scholar]
- 14.Schutz Y. Abnormalities of fuel utilization as predisposing to the development of obesity in humans. Obes Res. 1995;3:173S–8S. doi: 10.1002/j.1550-8528.1995.tb00460.x. [DOI] [PubMed] [Google Scholar]
- 15.Weyer C, Pratley R, Salbe A, Bogardus C, Ravussin E, Tataranni P. Energy Expenditure, Fat Oxidation, and Body Weight Regulation: A Study of Metabolic Adaptation to Long- Term Weight Change. J Clin Endocrinol Metab. 2000;85:1087–94. doi: 10.1210/jc.85.3.1087. [DOI] [PubMed] [Google Scholar]
- 16.Zurlo F, Lilloja S, Esposito-Del Puente A, Nyomba BL, Raz I, Saad MF, et al. Low ratio of fat to carbohydrate oxidation as predictor of weight gain: study of 24-h RQ. Am J Physiol. 1990;259:E650–7. doi: 10.1152/ajpendo.1990.259.5.E650. [DOI] [PubMed] [Google Scholar]
- 17.Golozoubova V, Hohtola E, Matthias A, Jacobsson A, Cannon B, Nedergaard J. Only UCP1 can mediate adaptive nonshivering thermogenesis in the cold. FASEB J. 2001;15:2048–50. doi: 10.1096/fj.00-0536fje. [DOI] [PubMed] [Google Scholar]
- 18.Sheets AR, Fulop P, Derdak Z, Kassai A, Sabo E, Mark NM, et al. Uncoupling protein-2 modulates the lipid metabolic response to fasting in mice. Am J Physiol Gastrointest Liver Physiol. 2008;294:G1017–24. doi: 10.1152/ajpgi.00016.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Golozoubova V, Hohtola E, Matthias A, Jacobsson A, Cannon B, Nedergaard J. Only UCP1 can mediate adaptive nonshivering thermogenesis in the cold. FASEB J. 2001;15:2048–50. doi: 10.1096/fj.00-0536fje. [DOI] [PubMed] [Google Scholar]
- 20.Virtanen KA, Lidell M, Orava J, Heglind M, Westergren R, Niemi T, et al. Functional Brown Adipose Tissue in Healthy Adults. N Engl J Med. 2009;360:1518–25. doi: 10.1056/NEJMoa0808949. [DOI] [PubMed] [Google Scholar]
- 21.Saito M, Okamatsu-Ogura Y, Matsushita M, Watanabe K, Yoneshiro T, Nio-Kobayashi J, et al. High Incidence of Metabolically Active Brown Adipose Tissue in Healthy Adult Humans. Diabetes. 2009;58:1526–31. doi: 10.2337/db09-0530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.van Marken Lichtenbelt WD, Vanhommerig JW, Smulders NM, Drossaerts JM, Kemerink GJ, Bouvy ND, et al. Cold-Activated Brown Adipose Tissue in Healthy Men. N Engl J Med. 2009;360:1500–8. doi: 10.1056/NEJMoa0808718. [DOI] [PubMed] [Google Scholar]
- 23.Griggio MA. The participation of shivering and nonshivering thermogenesis in warm and cold-acclimated rats. Comp Biochem Physiol A Comp Physiol. 1982;73:481–4. doi: 10.1016/0300-9629(82)90189-X. [DOI] [PubMed] [Google Scholar]
- 24.Cannon B, Nedergaard J. Metabolic consequences of the presence or absence of the thermogenic capacity of brown adipose tissue in mice (and probably in humans) Int J Obes (Lond) 2010;34:S7–16. doi: 10.1038/ijo.2010.177. [DOI] [PubMed] [Google Scholar]
- 25.Haas B, Mayer P, Jennissen K, Scholz D, Diaz MB, Bloch W, et al. Protein Kinase G Controls Brown Fat Cell Differentiation and Mitochondrial Biogenesis. Sci Signal. 2009;2:ra78. doi: 10.1126/scisignal.2000511. [DOI] [PubMed] [Google Scholar]
- 26.Marks A, Vianna DM, Carrive P. Nonshivering thermogenesis without interscapular brown adipose tissue involvement during conditioned fear in the rat. Am J Physiol Regul Integr Comp Physiol. 2009;296:R1239–47. doi: 10.1152/ajpregu.90723.2008. [DOI] [PubMed] [Google Scholar]
- 27.Blumberg MS, Efimova IV, Alberts JR. Thermogenesis during ultrasonic vocalization by rat pups isolated in a warm environment: A thermographic analysis. Dev Psychobiol. 1992;25:497–510. doi: 10.1002/dev.420250704. [DOI] [PubMed] [Google Scholar]
- 28.Rosen ED, Walkey CJ, Puigserver P, Spiegelman BM. Transcriptional regulation of adipogenesis. Genes Dev. 2000;14:1293–307. [PubMed] [Google Scholar]
- 29.Barak Y, Nelson MC, Ong ES, Jones YZ, Ruiz-Lozano P, Chien PR, et al. PPAR[gamma] is required for placental, cardiac, and adipose tissue development. Mol Cell. 1999;4:585–95. doi: 10.1016/S1097-2765(00)80209-9. [DOI] [PubMed] [Google Scholar]