

Abstract
Due to divergence, genetic variation is generally believed to be high among distantly related strains, low among closely related ones and little or none within the same classified clonal groups. Several recent genome-wide studies, however, revealed that significant genetic variation resides in a considerable number of genes among strains with identical MLST (Multilocus sequence typing) types and much of the variation was introduced by homologous recombination. Recognizing and understanding genomic variation within clonal bacterial groups could shed new light on the evolutionary path of infectious agents and the emergence of particularly pathogenic or virulent variants. This commentary presents our recent contributions to this line of work.
Keywords: homologous recombination, horizontal gene transfer, prophage, multilocus sequence typing, pathogenic adaptation, phylogenomics
Introduction
Nucleotide sequences diverge over time due to the combined effects of point mutation and homologous recombination. Recombination events cause changes to regions of contiguous bases in single events and were generally assumed to be rare in bacteria. However, there is growing evidence that homologous recombination has a significant impact on sequence diversification during bacterial genome evolution. A recent analysis on the MLST (Multilocus sequence typing) data of 46 bacterial and two archaeal species revealed 27 (56%) species in which homologous recombination contributed to more nucleotide changes than point mutation.1 The rapid genetic change introduced by homologous recombination could facilitate ecological adaption and drive pathogenesis in bacterial pathogens.2-5
Currently, the MLST scheme, using DNA fragments from seven housekeeping genes,6 has been routinely used to characterize bacterial isolates.7 The standard MLST scheme has also been extended to construct fine-scale relationships and further subdivide identical multilocus sequence types (STs) using more loci or a large amount of shared genomic sequences.8-12 Given the common occurrence of homologous recombination, it becomes crucial to investigate the genome-wide extent of homologous recombination, which could also benefit the construction of the strain history and tracking the spread of emerging pathogens.
Identification and Quantification of Nonvertically Acquired Genes via Recombination within Identical STs
Identifying recombinational exchanges in closely related strains is challenging as recombinational exchanges involved in a small number of nucleotides may be mistaken as point mutations. Guttman and Dykhuizen (1994) have successfully examined the clonal divergence of E. coli strains in the ECOR group A by considering the divergence time and mutation rate and showed that recombination has occurred at a rate 50-fold higher than the mutation rate in four loci.13 Feil et al. (2000) estimated the ancestral allele for the isolates that differ only one locus out of the seven MLST loci and assigned recombination based on the number of derived nucleotides from the ancestral allele and on whether the nucleotides are novel in the population.14
We adopted a new approach (illustrated in Fig. 1) to identify recombinant genes in Neisseria meningitidis strains with identical STs,15 which does not require the estimation of divergence time and ancestral alleles and can be applied on any two strains with identical STs. In brief, nucleotide substitution was assumed to follow a binomial distribution and an upper bound of genome-wide divergence () by point mutation was calculated for no observed substitution in all nucleotide sites of the seven MLST loci. The estimated maximum genome-wide divergence was then used as a benchmark to compute a P-value for the observed nucleotide changes of each gene in the genome to be explained by point mutation. Genes that have more than the expected number of nucleotide changes at a significance level of 0.001 were deemed as recombinant genes. Our results revealed that up to 19% of commonly present genes in N. meningitidis strains with identical STs have been affected by homologous recombination.15
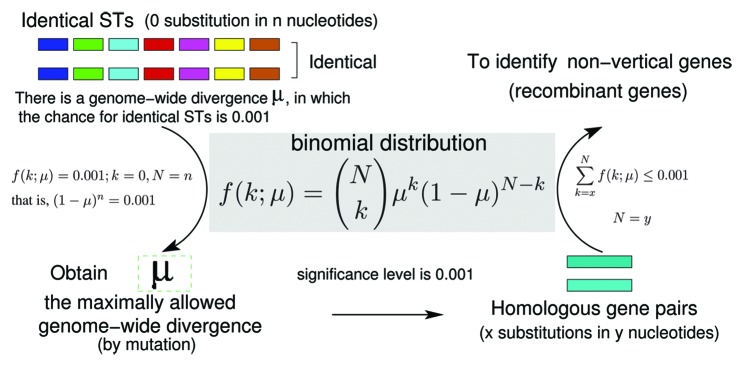
Figure 1. Inference of homologous recombination in strains with identical STs. Under a binomial distribution of nucleotide substitution, there is a probability for no nucleotide change in the seven MLST loci. That is (1-μ)n = 0.001, here n is the number of nucleotides in the seven MLST loci and μ is the upper bound of genome-wide nucleotide divergence (μ) at 0.001 significance level given no change in the seven MLST loci. At genome-wide divergence μ, genes that have more than the expected number of nucleotide changes at 0.001 significance level were deemed as nonvertically acquired genes.
In another study on E. coli O104 (ST678) genomes, we visualized recombinant genes by plotting the pairwise DNA distance of orthologous genes along the genome and identified 167 genes in three gene clusters that have likely undergone homologous recombination.16 A reanalysis on the orthologs between E. coli ON2010 and 55989 (labeled as Ec55989 thereafter to avoid unnecessary confusion) genomes using both pairwise DNA distance and the P-values as described in ref. 15 yielded remarkably similar results (Fig. 2). In fact, the use of nucleotide divergence between two genomes for homologous recombination detection has been successful in other studies,5,17 one of which was on two E. coli ST131 strains. It has been observed that a higher portion (at least 9%) of core genes in the E. coli ST131 genomes than in the E. coli ST678 genomes (Fig. 2) are affected by homologous recombination.5 The findings in both N. meningitidis and E. coli showed extensive genomic variation within identical STs. Since many bacterial species have a comparable or higher level of recombinogenicity than N. meningitidis or E. coli,1 extensive genomic variation within identical STs should be expected in many bacterial species.

Figure 2. Inferring genes involved in homologous recombination by comparing orthologs between two E. coli strains ON2010 and Ec55989. (A) DNA distance was measured using DNADIST of the PHYLIP package.29 (B) P-values were calculated based on the maximum genome-wide divergence given the seven identical MLST loci as illustrated in Figure 1. For simplicity, P -values smaller than 0.0001 were shown as 0.0001. Genes located in the prophage regions were colored in blue. Please note that more genes (4207 genes in total) were examined here than in our previous study16 (3794 genes), since our previous study focused on the genes present in both the O104 strains and the IAI1 strain.
It is important to note that the high genomic variation discovered within identical STs5,15,16 should not be interpreted as artifacts of these studies. The high level of genomic variation within identical STs could, instead, be explained by that many non-vertical genes within identical STs are deleterious or transiently adaptive and undergo fast rates of evolution.18 In fact, the ratio of recombination to mutation rates was higher in the comparison of clonally related strains13,14 than of relatively broadly sampled strains from the corresponding species.1 Such a discrepancy between the estimated recombination-mutation ratios highlights the need for a population genetics framework for the study of recombination and bacterial genome evolution.19
Genomic Regions Involved in Recombination
Among the three gene clusters of recombinant genes we identified in E. coli O104,16 one gene cluster contained 125 genes and was likely involved in direct chromosomal homologous recombination specific to the ON2010 strain. These 125 genes were found in 20 different functional categories and 70 of them were found in all the studied 57 E. coli and Shigella genomes. This is consistent with the conclusion that genes from all functional categories are subject to DNA exchange.20 Furthermore, the nearest phylogenetic neighbors of these genes were not clustered in a single phylogenetic group. We hypothesized that extensive recombination with a broad spectrum of strains has taken place in one genome, and this highly mosaic genome then recombined with the precursor to the ON2010 genome.
The other two gene clusters of recombinant genes in E. coli O104 were located in the prophage regions, but the genes in these two gene clusters were identical between ON2010 and Ec55989 genomes.16 It is noteworthy that the reanalysis with more single-copy genes (with details in Fig. 2) revealed 5 prophage genes involved in recombination. These prophage genes are not present in all O104 strains and the outgroup IAI1 strain. This could be explained by frequent recombination of the prophage genes with infecting phages or different prophages from other bacterial chromosomes. Since all examined O104 genomes are of conserved genome synteny, our observations support the argument that homologous (legitimate) recombination drives module exchange between phages.21 Together, these findings suggest that homologous recombination takes place frequently in both core genes and dispensable genes.
Phylogenomic Consequence
As the cost of sequencing drops, the characterization of bacterial isolates has utilized more shared genes or loci and shifted toward phylogenomic analysis.8-12,22 Quite often, multiple gene alignments were concatenated into a single super-alignment, from which phylogenies were reconstructed using a variety of methodologies. Such a data set, also known as a supermatrix, has been demonstrated to solve previously ambiguous or unresolved phylogenies,23 even in the presence of a low amount of horizontal gene transfer in the data set.24 Unfortunately, the supermatrix approach becomes very sensitive to recombination when applied to strains with identical STs due to limited genuine sequence diversity. The concatenated sequences of 3794 genes in the E. coli O104 strains16 were overwhelmed by the phylogenetic signal of the 125 recombinant genes, as many other genes are identical among the E. coli O104 strains (Fig. 2).
The accuracy and robustness of the constructed evolutionary relationships can be improved by the exclusion of recombinogenic and incongruent sequences.8,25 In fact, the removal of the 125 recombinant genes from the E. coli O104 data set16 has resulted in consistent phylogenetic relationships of O104 strains by different phylogenetic approaches. One interesting finding of our E. coli O104 study is that the number of identical loci implemented in BIGSdb26 was less sensitive to homologous recombination than the concatenated sequences of all loci.16 This could be explained by the fact that recombination has affected a relatively small number of genes but introduced a substantial amount of diversity in the ON2010 genome. It is further noteworthy that supertrees, another widely used approach for phylogenomic analysis22 are not suitable for characterizing strains with identical MLST types, as many individual genes are identical or nearly identical and contain no or very limited phylogenetic information for each individual gene tree.
Homologous Recombination and Pathogenic Adaptation
Homologous recombination can bring the beneficial mutations arising in different genomes together and have a strong impact on ecological adaptation.4,27 One well-known example was the recombination in the penA genes during the emergence of penicillin resistance in N. meningitidis.28 Variation of the penA gene corresponding to different levels of penicillin susceptibility has also been observed between N. meningitidis strains with the same MLST types.15 Furthermore, genetic variation within the same MLST types has been evident in the capsule gene cluster and genes used for vaccine target in N. meningitidis.15 These observations suggest a strong relationship between homologous recombination and pathogenic adaptation involved in antibiotic resistance, capsule biosynthesis and vaccine efficacy.
Recombination-mediated pathogenic adaptation was also evident in E. coli. Recombination has affected fimH which encodes mannose-specific type 1 fimbrial adhesin, resulting in distinct fluoroquinolone-resistance profiles in ST131 strains.5 A survey of the fimH gene on the 57 E. coli and Shigella genomes16 revealed that ON2010 was the only E. coli O104 genome containing a fimH blast hit > 10% of length (Fig. 3). Except one nucleotide, the fimH sequence in ON2010 was identical with E24377A and S88. On the ON2010 genome scaffold, fimH is upstream adjacent to a fructuronic acid transporter gene gntP, which is universally present in all E. coli and Shigella genomes. The gntP gene in ON2010 was also found to be involved in homologous recombination (Fig. 2), and most importantly, the most similar sequences to the ON2010 gntP were also in E24377A and S88 (data not shown). The shared origin between the adjacent fimH and gntP genes in ON2010 suggested that patchily distributed genes involved in pathogenesis could be introduced by homologous recombination of the conserved flanking genes.

Figure 3. Sequence alignment of fimH. Only informative sites are shown with coordinates at the top. The ON2010 sequence and its most similar sequences (differing by one nucleotide) are shown in light green.
Acknowledgments
The author would like to thank Drs Gabriel Moreno-Hagelsieb, Markus Friedrich and Edward Golenberg for critically reading earlier versions of the manuscript. This work was supported by funds from Wayne State University.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Footnotes
Previously published online: www.landesbioscience.com/journals/mge/article/23463
References
- 1.Vos M, Didelot X. A comparison of homologous recombination rates in bacteria and archaea. ISME J. 2009;3:199–208. doi: 10.1038/ismej.2008.93. [DOI] [PubMed] [Google Scholar]
- 2.Hughes AL, Friedman R. Nucleotide substitution and recombination at orthologous loci in Staphylococcus aureus. J Bacteriol. 2005;187:2698–704. doi: 10.1128/JB.187.8.2698-2704.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Marri PR, Paniscus M, Weyand NJ, Rendón MA, Calton CM, Hernández DR, et al. Genome sequencing reveals widespread virulence gene exchange among human Neisseria species. PLoS One. 2010;5:e11835. doi: 10.1371/journal.pone.0011835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Didelot X, Méric G, Falush D, Darling AE. Impact of homologous and non-homologous recombination in the genomic evolution of Escherichia coli. BMC Genomics. 2012;13:256. doi: 10.1186/1471-2164-13-256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Paul S, Linardopoulou EV, Billig M, Tchesnokova V, Price LB, Johnson JR, et al. Role of homologous recombination in adaptive diversification of extra-intestinal Escherichia coli. J Bacteriol. 2013;195:231–42. doi: 10.1128/JB.01524-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Maiden MC, Bygraves JA, Feil E, Morelli G, Russell JE, Urwin R, et al. Multilocus sequence typing: a portable approach to the identification of clones within populations of pathogenic microorganisms. Proc Natl Acad Sci U S A. 1998;95:3140–5. doi: 10.1073/pnas.95.6.3140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Maiden MC. Multilocus sequence typing of bacteria. Annu Rev Microbiol. 2006;60:561–88. doi: 10.1146/annurev.micro.59.030804.121325. [DOI] [PubMed] [Google Scholar]
- 8.Ogura Y, Ooka T, Iguchi A, Toh H, Asadulghani M, Oshima K, et al. Comparative genomics reveal the mechanism of the parallel evolution of O157 and non-O157 enterohemorrhagic Escherichia coli. Proc Natl Acad Sci U S A. 2009;106:17939–44. doi: 10.1073/pnas.0903585106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Touchon M, Hoede C, Tenaillon O, Barbe V, Baeriswyl S, Bidet P, et al. Organised genome dynamics in the Escherichia coli species results in highly diverse adaptive paths. PLoS Genet. 2009;5:e1000344. doi: 10.1371/journal.pgen.1000344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Luo C, Walk ST, Gordon DM, Feldgarden M, Tiedje JM, Konstantinidis KT. Genome sequencing of environmental Escherichia coli expands understanding of the ecology and speciation of the model bacterial species. Proc Natl Acad Sci U S A. 2011;108:7200–5. doi: 10.1073/pnas.1015622108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rasko DA, Webster DR, Sahl JW, Bashir A, Boisen N, Scheutz F, et al. Origins of the E. coli strain causing an outbreak of hemolytic-uremic syndrome in Germany. N Engl J Med. 2011;365:709–17. doi: 10.1056/NEJMoa1106920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bennett JS, Jolley KA, Earle SG, Corton C, Bentley SD, Parkhill J, et al. A genomic approach to bacterial taxonomy: an examination and proposed reclassification of species within the genus Neisseria. Microbiology. 2012;158:1570–80. doi: 10.1099/mic.0.056077-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Guttman DS, Dykhuizen DE. Clonal divergence in Escherichia coli as a result of recombination, not mutation. Science. 1994;266:1380–3. doi: 10.1126/science.7973728. [DOI] [PubMed] [Google Scholar]
- 14.Feil EJ, Smith JM, Enright MC, Spratt BG. Estimating recombinational parameters in Streptococcus pneumoniae from multilocus sequence typing data. Genetics. 2000;154:1439–50. doi: 10.1093/genetics/154.4.1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hao W, Ma JH, Warren K, Tsang RS, Low DE, Jamieson FB, et al. Extensive genomic variation within clonal complexes of Neisseria meningitidis. Genome Biol Evol. 2011;3:1406–18. doi: 10.1093/gbe/evr119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hao W, Allen VG, Jamieson FB, Low DE, Alexander DC. Phylogenetic incongruence in E. coli O104: understanding the evolutionary relationships of emerging pathogens in the face of homologous recombination. PLoS One. 2012;7:e33971. doi: 10.1371/journal.pone.0033971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Didelot X, Achtman M, Parkhill J, Thomson NR, Falush D. A bimodal pattern of relatedness between the Salmonella Paratyphi A and Typhi genomes: convergence or divergence by homologous recombination? Genome Res. 2007;17:61–8. doi: 10.1101/gr.5512906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hao W, Golding GB. The fate of laterally transferred genes: life in the fast lane to adaptation or death. Genome Res. 2006;16:636–43. doi: 10.1101/gr.4746406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fraser C, Hanage WP, Spratt BG. Recombination and the nature of bacterial speciation. Science. 2007;315:476–80. doi: 10.1126/science.1127573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhaxybayeva O, Gogarten JP, Charlebois RL, Doolittle WF, Papke RT. Phylogenetic analyses of cyanobacterial genomes: quantification of horizontal gene transfer events. Genome Res. 2006;16:1099–108. doi: 10.1101/gr.5322306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Clark AJ, Inwood W, Cloutier T, Dhillon TS. Nucleotide sequence of coliphage HK620 and the evolution of lambdoid phages. J Mol Biol. 2001;311:657–79. doi: 10.1006/jmbi.2001.4868. [DOI] [PubMed] [Google Scholar]
- 22.Köser CU, Ellington MJ, Cartwright EJ, Gillespie SH, Brown NM, Farrington M, et al. Routine use of microbial whole genome sequencing in diagnostic and public health microbiology. PLoS Pathog. 2012;8:e1002824. doi: 10.1371/journal.ppat.1002824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Delsuc F, Brinkmann H, Philippe H. Phylogenomics and the reconstruction of the tree of life. Nat Rev Genet. 2005;6:361–75. doi: 10.1038/nrg1603. [DOI] [PubMed] [Google Scholar]
- 24.Lapierre P, Lasek-Nesselquist E, Gogarten JP. The impact of HGT on phylogenomic reconstruction methods. Brief Bioinform. 2012 doi: 10.1093/bib/bbs050. [DOI] [PubMed] [Google Scholar]
- 25.Leigh JW, Susko E, Baumgartner M, Roger AJ. Testing congruence in phylogenomic analysis. Syst Biol. 2008;57:104–15. doi: 10.1080/10635150801910436. [DOI] [PubMed] [Google Scholar]
- 26.Jolley KA, Maiden MC. BIGSdb: Scalable analysis of bacterial genome variation at the population level. BMC Bioinformatics. 2010;11:595. doi: 10.1186/1471-2105-11-595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Moradigaravand D, Engelstädter J. The effect of bacterial recombination on adaptation on fitness landscapes with limited peak accessibility. PLoS Comput Biol. 2012;8:e1002735. doi: 10.1371/journal.pcbi.1002735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bowler LD, Zhang QY, Riou JY, Spratt BG. Interspecies recombination between the penA genes of Neisseria meningitidis and commensal Neisseria species during the emergence of penicillin resistance in N. meningitidis: natural events and laboratory simulation. J Bacteriol. 1994;176:333–7. doi: 10.1128/jb.176.2.333-337.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Felsenstein J. PHYLIP (phylogeny inference package). Version 3.2. Cladistics. 1989;5:164–6. [Google Scholar]