

Abstract
Pancreatic cancer is a genetic disease. Pancreatic cancers develop from one of three precursor lesions, pancreatic intraepithelial neoplasia (PanIN), intraductal papillary mucinous neoplasms (IPMNs), and mucinous cystic neoplasms (MCNs), and each arises in association with distinct genetic alterations. These alterations not only provide insight into the fundamental origins of pancreatic cancer but provide ample opportunity for improving early diagnosis and management of cystic precursors.
Keywords: GNAS, RNF43, cystic neoplasm, pancreatic cancer
INTRODUCTION
Every year in the United States, approximately 44,000 people are diagnosed with pancreatic adenocarcinoma (hereafter referred to as “pancreatic cancer”). It is among the most lethal of all cancers, with nearly 38,000 deaths per year and 5-year survival rates of 5%. To date, pancreatic cancer remains the 4th leading cause of cancer deaths among both men and women [1].
A number of genetic studies have proven instrumental in elucidating the key genes, and more recently molecular pathways, that drive the formation and progression of pancreatic cancer. Early studies identified several genes that are frequently altered in pancreatic cancers [2]. Subsequent analysis of the precursor lesions giving rise to pancreatic cancer indicated the temporal accumulation of these genetic alterations during pancreatic carcinogenesis [3]. Most recently, next generation sequencing technologies have revealed the genetic landscape of neoplasms that arise from preinvasive neoplasms of the pancreas. Such surveys are helping to shape the development of new, personalized approaches to clinical management and therapy for pancreatic cancer patients.
PANCREATIC CARCINOGENESIS
Precursors to Pancreatic Cancer
Three precursors to pancreatic cancer have been described [4,5]. The vast majority of pancreatic cancers develop from microscopic precursors called pancreatic intraepithelial neoplasia (PanIN), that originate in small terminal (<5 mm) pancreatic ducts [4] (Fig. 1). PanINs are further classifiable based on their degree of morphologic atypia. In PanIN-1 lesions transformation of the normal ductal epithelium into tall columnar cells with intracellular mucin is seen, whereas PanIN-2 lesions are notable for the development of cytologic atypia and nuclear crowding. PanIN-3 lesions are characterized by extensive nuclear crowding and nuclear atypia, pseudopapillary growth, mitotic figures, and intraluminal necrosis [4,6]. By contrast, intraductal papillary mucinous neoplasms (IPMNs) and mucinous cystic neoplasms (MCNs) are macroscopically visible cystic neoplasms. IPMNs arise in the mucin-producing main pancreatic duct or one of its branches (Fig. 2A,B) where they distend the duct system by intraductal growth of the neoplasm and by copious mucin production. MCNs are typically intraparenchymal and do not communicate with the pancreatic duct system (Fig. 2C,D). Histologically, MCNs are characterized by a mucinous epithelial lining in association with an underlying ovarian-like stroma (Fig. 3). A detailed discussion of the morphologic features of dysplasia in cystic precursors is beyond the scope of this review, but both IPMNs and MCNs also show a range of morphologic features ranging from low grade, to moderate, to high-grade dysplasia [5]. Ultimately, pancreatic cancer may arise from any of these above precursor lesions yet cancers arising in association with PanINs are much more common [7].
Fig. 1.
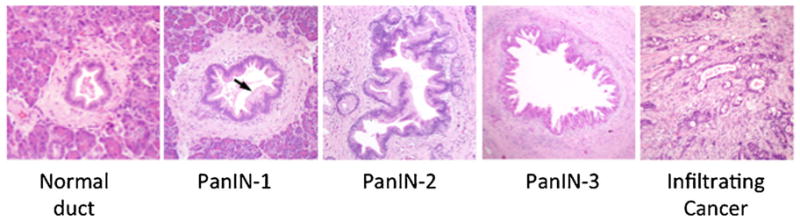
Morphologic progression model of pancreatic intraepithelial neoplasia. Shown from left to right are histological examples of a normal pancreatic duct, pancreatic intraepithelial neoplasia (PanIN), and pancreatic cancer. Normal ducts are characterized by a low cuboidal epithelium surrounded by a periductal fibrotic cuff. PanIN-1 lesions are differentiated from normal ductal epithelium by the presence of mucinous hyperplasia of the ductal cells (arrows) but without cytological atypia. PanIN-2 lesions are notable for the presence of nuclear enlargement, atypia, crowding and papillary infoldings of the epithelium. PanIN-3 lesions, synonymous to high-grade dysplasia/carcinoma in situ, show a complete loss of cell polarity (arrows) and marked cytological atypia in association with frequent mitotic figures and pseudopapillary growth of the neoplastic epithelium. PanIN-3 lesions may progress to invasive cancer that is characterized by poorly formed neoplastic glands with an infiltrative growth pattern admixed with abundant desmoplastic stroma.
Fig. 2.
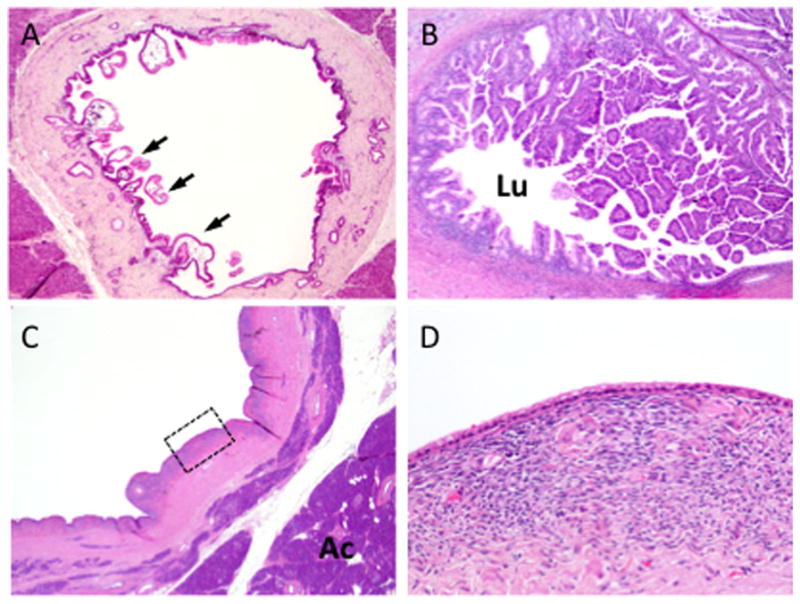
Morphologic features of cystic neoplasms of the pancreas. A: Intraductal papillary mucinous neoplasm (IPMN) of the main pancreatic duct. In this example the IPMN shows low-grade dysplasia, with scattered papillae seen at low power (arrows). B: IPMN with high-grade dysplasia. The neoplasm shows exuberant papillary growth that fills the lumen (Lu) of the main pancreatic duct. C: Low power view of a mucinous cystic neoplasm (MCN). The neoplasm is distinct from the surrounding pancreatic acinar tissue (Ac). D: High power view of the region outlined in panel C showing low cuboidal mucinous epithelial lining and underlying ovarian-like stroma.
Fig. 3.
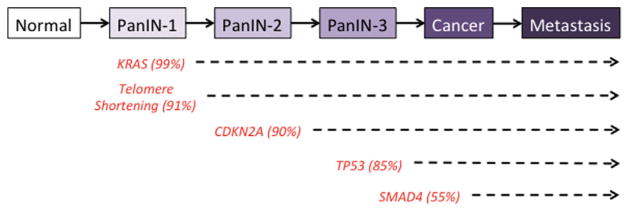
Genetic progression model of pancreatic carcinogenesis. The molecular alterations that accumulate during pancreatic carcinogenesis can be classified into early (telomere shortening and activating mutations in KRAS2), intermediate (inactivating mutations or epigenetic silencing of CDKN2A) and late (inactivating mutations of TP53 and SMAD4) events. Mutations in additional genes may also occur during PanIN formation but are not illustrated in this example.
The Genetic Progression Model
The genetic events that accumulate during carcinogenesis have been best described for PanINs (Fig. 3). Genetic events detected in PanIN-1 lesions include telomere shortening and activating mutations in KRAS [8,9]. PanIN-2 lesions exhibit CDKN2A loss while PanIN-3 lesions demonstrate genetic inactivation in TP53, SMAD4, and BRCA2 [8]. Of course, exceptions to these scenarios have been shown with occasional PanIN-1 having CDKN2A loss or PanIN-2 having TP53 inactivation [10]. While the morphological progression of IPMNs and MCNs from low to moderate to high-grade dysplasia is well known, the relationship of these morphologies to accumulating genetic abnormalities is less well characterized. However, as discussed later in this review the genetic events underlying IPMN and MCN formation are now being determined.
KRAS
One of the earliest and most universal genetic alterations observed in pancreatic cancer is activating mutations in the oncogene KRAS [11]. At least 99% of PanIN-1 lesions harbor mutations in KRAS, suggesting its activation is an important initiating step in carcinogenesis of most pancreatic cancers [10]. KRAS encodes a guanosine triphosphate (GTP)-binding protein, which functions as a key mediator in a variety of cellular processes, including cell survival, proliferation, and cell motility. In its inactive state, Ras is bound to GDP. Extracellular signaling through growth factor receptors triggers the removal of GDP from Ras, allowing GTP to bind. Inactivation of these active Ras–GTP complexes is accomplished when GTP is hydrolyzed. Activating mutations in the KRAS gene result in a loss of the intrinsic GTPase activity of the Ras protein, and consequently, constitutive signaling occurs even in the absence of extracellular signals [12]. Activated Ras feeds into a number of signaling pathways, including the RAF/mitogen-activated protein (MAP) kinase pathway, as well as into the phosphoinositide-3-kinase (PI3K)/AKT signaling pathway. Activating mutations in BRAF are observed in a subset of pancreatic cancers lacking KRAS mutations, resulting in aberrant Ras–Raf–MAPK signaling [13].
CDKN2A
The tumor suppressor gene CDKN2A is inactivated in over 90% of pancreatic ductal adenocarcinomas, with the vast majority of alterations arising as early as the PanIN-2 stage [14,15]. The CDKN2A gene possesses alternative splicing sites that result in the formation of several protein products. For example, the p14/ARF protein sequesters MDM2 helping to stabilize TP53 whereas the p16/INK4A protein acts to inhibit the formation of complexes between cyclins and cyclin-dependent kinases (CDKs) thus regulating progression through the G1 checkpoint in the cell cycle [16–18]. In about 40% of cases, inactivation of CDKN2A occurs through homozygous deletion. Intragenic mutation with subsequent loss of the second allele acts as a second mechanism for CDKN2A loss, accounting for another 40% of alterations. Methylation of the promoter region of CDKN2A, resulting in gene silencing, has also been observed [14].
TP53
Inactivation of the TP53 gene is observed in higher-grade, PanIN-3 lesions [19]. Up to 85% of pancreatic cancers have TP53 inactivation, with the most frequent mechanism of inactivation being intragenic mutation with loss of the second allele [20,21]. TP53 functions as an essential regulator of many interrelated cellular processes including apoptosis, cell cycle progression and DNA repair. In response to DNA damage, TP53 can promote the transcription of p21, a CDK inhibitor that can bind to cyclin–CDK complexes, leading to cell cycle arrest in the G1 phase. TP53 can also regulate transcription of both pro and anti-apoptotic genes. Loss of TP53 results in an overall increase in genomic instability, as cells are permitted to proliferate in the setting of otherwise catastrophic DNA damage [22].
SMAD4
As is seen with TP53, SMAD4 loss is observed in high-grade PanIN-3 lesions [23]. Inactivation of SMAD4, either by homozygous deletion or intragenic mutation with the loss of the second allele, occurs in ~55% of pancreatic ductal adenocarcinomas [24]. The Smad4 protein plays a key role in propagating extracellular signals through the transforming growth factor β (TGF-β) signaling pathway. TGF-β regulates cell proliferation and differentiation, and thus acts as a critical tumor suppressor in normal cells. Activation of this pathway begins with binding of a TGF-β ligand to type I and type II serine/threonine kinase cell surface receptors. This results in receptor dimerization and activation of the type I receptor leading to its phosphorylation of the Smad2 and Smad3 proteins. Smad4 complexes with phosphorylated Smad2/3 proteins and together they translocate into the nucleus where, in association with transcriptional cofactors, regulate the expression of genes involved in a variety of important cellular processes including cell cycle control, cell differentiation and growth [25]. Loss of SMAD4 and hence canonical TGF-β signaling in pancreatic ductal adenocarcinomas results in the loss of TGF-β-induced growth inhibition [26], and correlates with both poor prognosis and the development of widespread metastases in patients [25,27,28]. Of note, in a small subset of pancreatic cancers with intact SMAD4 genes, loss of the TGFBR1, TGFBR2, or ACVR1B receptors has been found [20,29].
OBSERVATIONS FROM EXOMIC SEQUENCING APPROACHES
Genetic alterations in KRAS, CDKN2A, TP53, and the TGF-β family were initially elucidated using candidate gene approaches, namely dideoxy (Sanger) sequencing. However, in recent years next generation sequencing methodologies have been used to examine the entire coding fraction of the genome (i.e., the “exome”) with the goal of identifying the entire compendium of somatic alterations in this tumor type [20]. This, in turn, has contributed to creating a more comprehensive genetic landscape by uncovering both gene “mountains” and “hills,” that is, genetic alterations that are observed in a high and low frequency of tumors, respectively [30].
The Landscape of Pancreatic Cancer
A total of 20,661 protein-coding genes have been analyzed in 24 pancreatic cancers [20], leading to identification of a total of 1,562 somatic alterations. The majority of these changes were found to be base substitutions; however, small insertions and deletions as well as alterations within the untranslated regions (UTRs) and at splice sites were also observed. Over 1,300 genes were found to contain at least one genetic alteration, with 148 genes containing two or more alterations. Copy number analysis was also utilized to interrogate gene deletion and amplification events, and revealed a total of 198 homozygous deletions and 144 focal high-copy amplifications in the 24 tumors analyzed. Genes within these regions of deletion or amplification included well-established tumor suppressors and oncogenes, respectively; however, additional genes that had not been previously associated with pancreatic ductal adenocarcinoma were also found.
A list of candidate cancer genes (“CAN” genes) was generated based on the set of genes harboring somatic alterations [20]. This analysis was based largely on passenger mutation rates, gene mutation type and frequency. Importantly, this list included each of the previously identified genes known to play a role in pancreatic carcinogenesis (KRAS, CDKN2A, TP53, and SMAD4) indicating the robustness of this approach. Genes not previously appreciated in this tumor type were also identified, for example MLL3. The significance and roles of each of these CAN genes remains to be determined in functional systems; however, new studies of these genes in various tumor types is shedding light onto emerging processes and pathways involved in tumorigenesis [31–33].
Core Signaling Pathways of Pancreatic Cancer
Further categorization of the somatic alterations data generated by whole exome sequencing revealed that they correspond to 12 core signaling pathways (Fig. 4). Many of these pathways consist of genes that have already been appreciated as players in pancreatic cancer formation and progression, such as DNA damage control (TP53), cell cycle regulation (CDKN2A), and TGF-β signaling (SMAD4). Most importantly, the authors were able to show that while most patients’ carcinomas exhibited a genetic alteration corresponding to each of the 12 core pathways, the specific gene mutated for a given pathway in each patient often varied. This finding may help account for both the heterogeneous nature of tumors, as well as offer insight into why agents targeting a specific gene in a pathway rarely result in a therapeutic advantage in more than a minor percentage of patients.
Fig. 4.

Core signaling pathways in pancreatic cancer. The 12 pathways and processes whose component genes were genetically altered in most pancreatic cancers based on whole exome sequencing [20] are shown in black, and the pathway more recently identified in pancreatic cancer in gray [31–34]. Therapeutic targeting of one or more of these pathways, rather than specific gene alterations that occur within a pathway, provides a new paradigm for treatment of pancreatic cancer. GTPase, guanosine triphosphatase; TGF-β, transforming growth factor β.
Chromatin Regulation in Pancreatic Cancer
In addition to the core pathways initially described above, more recent data indicate that chromatin regulation is an additional cellular process that plays a crucial role in pancreatic cancer [32,33]. There are two notable examples. The first is ARID1A that encodes a protein involved in the SWI/SNF (switch/sucrose non-fermentable) ATP-dependent chromatin remodeling complex [34]. Somatic mutations of ARID1A in pancreatic cancer were initially identified by whole exome sequencing [20], and since validated in a larger series of human tumors from a variety of organ sites [32]. Most mutations were truncating indicating this gene is tumor suppressive in nature, with the frequency of mutations being highest in colon (10%), stomach (10%), and pancreas (8%) [32]. Of interest, Shain et al. [33] recently reported that alterations of each individual SWI/SNF subunit occurred at modest-frequency in pancreatic cancer, but together they affected at least one-third of all pancreatic cancers, defining SWI/SNF as an additional mutational “mountain” for this tumor type.
The second example of a chromatin regulating gene is MLL3 (mixed-lineage leukemia 3) that encodes a histone methyltransferase and belongs to the TRX/MLL gene family [35]. Loss of the long arm of chromosome 7, where the MLL3 gene resides, is a frequent event in myeloid leukemia [35]. MLL3 was first implicated as a CAN gene in solid tumors by Sjoblom et al. [36] in colorectal and breast cancer, followed by Balakrishnan et al. [37] who identified somatic MLL3 alterations in glioblastoma and pancreatic ductal adenocarcinomas. Subsequently, Jones et al. [20] further identified MLL3 as a common mutational target in human pancreatic cancer by whole exome sequencing. More recently, a model utilizing the oncogenic LSL-KrasG12D mouse coupled with the Sleeping Beauty transposon system was created to screen for mutations that, in combination with Kras, drive pancreatic ductal adenocarcinoma tumorigenesis. From this screen, both previously implicated and novel cancer candidate genes were identified, including MLL3 [38]. Taken together, the data underscore the important and emerging role of chromatin remodeling in pancreatic ductal adenocarcinoma.
ALTERNATIVE ROUTES TO DUCTAL ADENOCARCINOMA: IPMNs AND MCNs
Early Genetic Studies of IPMNs and MCNs
The genetic alterations that underlie IPMN and MCN formation and progression have been much less characterized compared to PanINs [5]. Similar to PanIN lesions, mutations in KRAS, and TP53 have been reported although with lower frequencies [39–41]. BRAF mutations may also be seen [42]. SMAD4 expression may be lost in invasive pancreatic cancers that arise from IPMN lesions but is rarely lost in preinvasive IPMNs, unlike PanINs in which SMAD4 is lost in 30% of PanIN-3 lesions [23,43]. By contrast, genetic alterations in IPMNs not seen in PanIN lesions are inactivating mutations in the serine/threonine kinase STK11/LKB1 [44]. STK11/LKB1 functions as a tumor suppressor, with critical roles in apoptosis, metabolism, and cell polarity [45]. Germline inactivating mutations in this kinase occur in patients with Peutz–Jeghers syndrome [46], and patients with Peutz–Jeghers are at high risk of developing pancreatic cancer in association with an IPMN [47,48]. Activating mutations in PIK3CA that send oncogenic signals through AKT are found in 10% of IPMNs [42]. However, emerging evidence suggests that these mutations may be specific to a variant of intraductal neoplasia, intraductal tubulopapillary neoplasm (ITPN) [41].
MCNs, the least frequent precursor to pancreatic cancer, have even less well-defined genetic alterations. Mutations in KRAS and TP53 have been noted in higher-grade lesions [49] and loss of SMAD4 has been reported in infiltrating cancers that arise in association with MCNs with high-grade dysplasia [43].
New Insights From Whole Exome Sequencing
Unlike PanIN lesions, IPMNs and MCNs are often macroscopic and detectable by conventional imaging techniques. Both lesions can progress to invasive ductal adenocarcinoma; however, there is currently no definitive manner to differentiate these precursor lesions from each other, or other benign lesions not described in this review, except with surgical resection and subsequent histological evaluation [50,51]. Therefore, there is a definite need to better understand the genetic abnormalities that accompany the formation and progression of IPMNs and MCNs. To this end, several groups have recently reported the results of whole-exome sequencing on these two neoplasms [52,53].
Furukawa et al. [52] recently carried out whole-exome sequencing on DNA isolated from IPMNs and found somatic alterations in 17 genes, including the gene that encodes the guanine nucleotide-binding protein alpha stimulating activity polypeptide, GNAS. Over 40% of the IPMNs analyzed harbored a mutation in GNAS, and this mutation was always present at codon 201, indicating a hot spot of mutational activation. Additionally, 25% of the IPMN cases had mutations in both GNAS and KRAS, concurrently. Importantly, G-protein signaling was demonstrated in these neoplasms via expression analysis of the G-protein alpha subunit and a downstream target of this pathway, phosphorylated PKA substrates [52]. In a similar study, Wu et al. [53] analyzed IPMN cyst fluid for alterations in a defined set of genes that are frequently mutated in cancer. GNAS mutations were observed in 66% of the IPMNs sequenced, and over half contained concurrent mutations in both GNAS and KRAS. Moreover, because alterations in KRAS and GNAS were observed in both low-grade and high-grade IPMNs, the authors suggested that the identification of additional genomic alterations would be important for distinguishing the two.
In addition to analyses of cyst fluids, Wu et al. also sequenced IPMNs and MCNs samples directly as well as two additional types of cystic neoplasm, serous cystadenomas (SCAs) and solid pseudopapillary neoplasms (SPNs) [53] to define the genetic landscapes of these tumor types. Unique patterns of genetic alterations were observed for each cystic neoplasm (Table I). For example, the majority of IPMNs harbored mutations at codon 12 of KRAS and/or mutations at codon 201 in GNAS. However, IPMNs were also found to contain inactivating nonsense mutations in RNF43 that encodes a protein with E3 ubiquitin ligase activity [54]. MCNs, like IPMNs, also had alterations in RNF43 as well as in KRAS; however, no GNAS alterations were observed in MCNs. In sharp contrast to IPMN and MCN cystic neoplasms, SCAs and SPNs never contained mutations in RNF43, KRAS, or GNAS. Instead, SCAs contained mutations of the von-Hippel–Lindau (VHL) gene and SPNs exhibited alterations in CTNNB1, and these mutations were never seen in IPMNs or MCNs. Of note, the observed VHL mutations in SCA samples is interesting given that patients with VHL syndrome, a rare autosomal dominant familial disorder, are prone to developing SCAs [55]. The VHL genetic alterations observed in SCA lesions in this study were found to be identical to germline mutations in patients with VHL syndrome.
TABLE I.
Summary of Selected Somatic Alterations in Pancreatic Ductal Adenocarcinoma Precursor Lesions
| Gene | Genetic alteration | Pathway or regulatory process | Altered in PanINs | Altered in IPMNs | Altered in MCNs |
|---|---|---|---|---|---|
| Kras2 | Activating | GTPase-dependent signaling | Yes | Yes | Yes |
| CDKN2A | Inactivating | Cell cycle regulation | Yes | ||
| TP53 | Inactivating | DNA damage response | Yes | Yes | Yes |
| SMAD4 | Inactivating | TGF-β signaling | Yes | Yes | Yes |
| ARID1A | Inactivating | Chromatin remodeling | Yes | ||
| MLL3 | Inactivating | Chromatin remodeling | Yes | ||
| GNAS | Activating | G protein-mediated signaling | No | Yes | |
| RNF43 | Inactivating | Ubiquitin-dependent protein degradation | No | Yes | Yes |
These findings are important for two reasons. First, many of the genetic alterations identified in these cystic neoplasms encode proteins that either harbor intrinsic ubiquitin ligase activity or that interact with ubiquitin ligases indicating a pervasive role of ubiquitin-dependent pathways in neoplastic pancreatic cystic development [53]. As discussed, the RNF43 gene-encoded protein possesses E3 ubiquitin ligase activity, although further work will be required to determine which proteins are targeted by RNF43 for ubiquitination and that mediate its tumorigenic effects. Likewise, VHL recruits ubiquitin ligases to target HIF1α proteins for degradation [56] whereas CTNNB1 mutations inhibit phosphorylation marks of the protein product (β-catenin) that would cause its degradation by E3 ubiquitin ligases [57,58]. Second, these data suggest that analyzing a small panel of genes could aid in distinguishing these four cystic neoplasms from one another, and in turn, decrease the number of invasive resection procedures performed on otherwise benign lesions [59–61].
Summary and Implications
The wealth of data acquired from large-scale, whole-exome sequencing approaches of pancreatic cancer has strengthened our understanding of the signaling pathways and processes involved in the initiation and progression of this disease. In light of new data suggesting that a significant window of opportunity exists for detecting pancreatic cancers that arise from PanINs while still in the curative stage [62], increased efforts into the discovery of biomarkers for early detection of this disease are essential if we hope to improve patient survival. Until then, core signaling pathways and processes in pancreatic cancers offer options for therapeutic targeting based on the physiologic effects of the dysregulated pathways rather than single genes. Although less frequent, pancreatic cancer can arise from the cystic precursors IPMN and MCN. Recent sequencing efforts in these neoplasms have highlighted the unique genetic features of each that can be exploited for diagnosis and management, and the importance of ubiquitin-dependent pathways in cystic neoplasm development. Ultimately, elucidating which pathways to target in patients at various stages of disease progression will remain a challenge. Nonetheless, insights provided from examining tumor genomes provide an exciting avenue for new and more personalized cancer therapies for patients with this devastating disease.
Acknowledgments
Grant sponsor: National Institutes of Health; Grant numbers: CA140599. CA101955. CA62924.
Footnotes
The authors have no financial conflicts of interest to disclose related to this work.
References
- 1.Siegel R, Naishadham D, Jemal A. Cancer statistics, 2012. CA Cancer J Clin. 2012;62:10–29. doi: 10.3322/caac.20138. [DOI] [PubMed] [Google Scholar]
- 2.Bardeesy N, DePinho RA. Pancreatic cancer biology and genetics. Nat Rev Cancer. 2002;2:897–909. doi: 10.1038/nrc949. [DOI] [PubMed] [Google Scholar]
- 3.Maitra A, Hruban RH. Pancreatic cancer. Annu Rev Pathol. 2008;3:157–188. doi: 10.1146/annurev.pathmechdis.3.121806.154305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hruban RH, Adsay NV, Albores-Saavedra J, et al. Pancreatic intraepithelial neoplasia: A new nomenclature and classification system for pancreatic duct lesions. Am J Surg Pathol. 2001;25:579–586. doi: 10.1097/00000478-200105000-00003. [DOI] [PubMed] [Google Scholar]
- 5.Matthaei H, Schulick RD, Hruban RH, et al. Cystic precursors to invasive pancreatic cancer. Nat Rev Gastroenterol Hepatol. 2011;8:141–150. doi: 10.1038/nrgastro.2011.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hruban RH, Takaori K, Klimstra DS, et al. An illustrated consensus on the classification of pancreatic intraepithelial neoplasia and intraductal papillary mucinous neoplasms. Am J Surg Pathol. 2004;28:977–987. doi: 10.1097/01.pas.0000126675.59108.80. [DOI] [PubMed] [Google Scholar]
- 7.Gaujoux S, Brennan MF, Gonen M, et al. Cystic lesions of the pancreas: Changes in the presentation and management of 1,424 patients at a single institution over a 15-year time period. J Am Coll Surg. 2011;212:590–600. doi: 10.1016/j.jamcollsurg.2011.01.016. discussion 600–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Maitra A, Fukushima N, Takaori K, et al. Precursors to invasive pancreatic cancer. Adv Anat Pathol. 2005;12:81–91. doi: 10.1097/01.pap.0000155055.14238.25. [DOI] [PubMed] [Google Scholar]
- 9.van Heek NT, Meeker AK, Kern SE, et al. Telomere shortening is nearly universal in pancreatic intraepithelial neoplasia. Am J Pathol. 2002;161:1541–1547. doi: 10.1016/S0002-9440(10)64432-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kanda M, Matthaei H, Wu J, et al. Presence of somatic mutations in most early-stage pancreatic intraepithelial neoplasia. Gastroenterology. 2012;142:730–733. doi: 10.1053/j.gastro.2011.12.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Caldas C, Kern SE. K-ras mutation and pancreatic adenocarcinoma. Int J Pancreatol. 1995;18:1–6. doi: 10.1007/BF02825415. [DOI] [PubMed] [Google Scholar]
- 12.Schubbert S, Shannon K, Bollag G. Hyperactive Ras in developmental disorders and cancer. Nat Rev Cancer. 2007;7:295–308. doi: 10.1038/nrc2109. [DOI] [PubMed] [Google Scholar]
- 13.Calhoun ES, Jones JB, Ashfaq R, et al. BRAF and FBXW7 (CDC4, FBW7, AGO, SEL10) mutations in distinct subsets of pancreatic cancer: Potential therapeutic targets. Am J Pathol. 2003;163:1255–1260. doi: 10.1016/S0002-9440(10)63485-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schutte M, Hruban RH, Geradts J, et al. Abrogation of the Rb/p16 tumor-suppressive pathway in virtually all pancreatic carcinomas. Cancer Res. 1997;57:3126–3130. [PubMed] [Google Scholar]
- 15.Caldas C, Hahn SA, da Costa LT. Frequent somatic mutations and homozygous deletions of the p16 (MTS1) gene in pancreatic adenocarcinoma. Nat Genet. 1994;8:27–32. doi: 10.1038/ng0994-27. [DOI] [PubMed] [Google Scholar]
- 16.Larsson LG. Oncogene- and tumor suppressor gene-mediated suppression of cellular senescence. Semin Cancer Biol. 2011;21:367–376. doi: 10.1016/j.semcancer.2011.10.005. [DOI] [PubMed] [Google Scholar]
- 17.Rozenblum E, Schutte M, Goggins M, et al. Tumor-suppressive pathways in pancreatic carcinoma. Cancer Res. 1997;57:1731– 1734. [PubMed] [Google Scholar]
- 18.Kim WY, Sharpless NE. The regulation of INK4/ARF in cancer and aging. Cell. 2006;127:265–275. doi: 10.1016/j.cell.2006.10.003. [DOI] [PubMed] [Google Scholar]
- 19.DiGiuseppe JA, Redston MS, Yeo CJ, et al. p53-independent expression of the cyclin-dependent kinase inhibitor p21 in pancreatic carcinoma. Am J Pathol. 1995;147:884–888. [PMC free article] [PubMed] [Google Scholar]
- 20.Jones S, Zhang X, Parsons DW, et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science. 2008;321:1801–1806. doi: 10.1126/science.1164368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Redston MS, Caldas C, Seymour AB, et al. p53 mutations in pancreatic carcinoma and evidence of common involvement of homocopolymer tracts in DNA microdeletions. Cancer Res. 1994;54:3025–3033. [PubMed] [Google Scholar]
- 22.Vogelstein B, Kinzler KW. Cancer genes and the pathways they control. Nat Med. 2004;10:789–799. doi: 10.1038/nm1087. [DOI] [PubMed] [Google Scholar]
- 23.Wilentz RE, Iacobuzio-Donahue CA, Argani P, et al. Loss of expression of Dpc4 in pancreatic intraepithelial neoplasia: Evidence that DPC4 inactivation occurs late in neoplastic progression. Cancer Res. 2000;60:2002–2006. [PubMed] [Google Scholar]
- 24.Hahn SA, Schutte M, Hoque AT, et al. DPC4, a candidate tumor suppressor gene at human chromosome 18q21. 1. Science. 1996;271:350–353. doi: 10.1126/science.271.5247.350. [DOI] [PubMed] [Google Scholar]
- 25.Siegel PM, Massague J. Cytostatic and apoptotic actions of TGF-beta in homeostasis and cancer. Nat Rev Cancer. 2003;3:807–821. doi: 10.1038/nrc1208. [DOI] [PubMed] [Google Scholar]
- 26.Dai JL, Bansal RK, Kern SE. G1 cell cycle arrest and apoptosis induction by nuclear Smad4/Dpc4: Phenotypes reversed by a tumorigenic mutation. Proc Natl Acad Sci USA. 1999;96:1427– 1432. doi: 10.1073/pnas.96.4.1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tascilar M, Skinner HG, Rosty C, et al. The SMAD4 protein and prognosis of pancreatic ductal adenocarcinoma. Clin Cancer Res. 2001;7:4115–4121. [PubMed] [Google Scholar]
- 28.Iacobuzio-Donahue CA, Fu B, Yachida S, et al. DPC4 gene status of the primary carcinoma correlates with patterns of failure in patients with pancreatic cancer. J Clin Oncol. 2009;27:1806–1813. doi: 10.1200/JCO.2008.17.7188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Goggins M, Shekher M, Turnacioglu K, et al. Genetic alterations of the transforming growth factor beta receptor genes in pancreatic and biliary adenocarcinomas. Cancer Res. 1998;58:5329–5332. [PubMed] [Google Scholar]
- 30.Wood LD, Parsons DW, Jones S, et al. The genomic landscapes of human breast and colorectal cancers. Science. 2007;318:1108–1113. doi: 10.1126/science.1145720. [DOI] [PubMed] [Google Scholar]
- 31.Jones S, Wang TL, Shih Ie M, et al. Frequent mutations of chromatin remodeling gene ARID1A in ovarian clear cell carcinoma. Science. 2010;330:228–231. doi: 10.1126/science.1196333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jones S, Li M, Parsons DW, et al. Somatic mutations in the chromatin remodeling gene ARID1A occur in several tumor types. Hum Mutat. 2012;33:100–103. doi: 10.1002/humu.21633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shain AH, Giacomini CP, Matsukuma K, et al. Convergent structural alterations define SWItch/Sucrose NonFermentable (SWI/SNF) chromatin remodeler as a central tumor suppressive complex in pancreatic cancer. Proc Natl Acad Sci USA. 2012;109:E252–E259. doi: 10.1073/pnas.1114817109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang X, Nagl NG, Wilsker D, et al. Two related ARID family proteins are alternative subunits of human SWI/SNF complexes. Biochem J. 2004;383:319–325. doi: 10.1042/BJ20040524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ruault M, Brun ME, Ventura M, et al. MLL3, a new human member of the TRX/MLL gene family, maps to 7q36, a chromosome region frequently deleted in myeloid leukaemia. Gene. 2002;284:73–81. doi: 10.1016/s0378-1119(02)00392-x. [DOI] [PubMed] [Google Scholar]
- 36.Sjoblom T, Jones S, Wood LD, et al. The consensus coding sequences of human breast and colorectal cancers. Science. 2006;314:268–274. doi: 10.1126/science.1133427. [DOI] [PubMed] [Google Scholar]
- 37.Balakrishnan A, Bleeker FE, Lamba S, et al. Novel somatic and germline mutations in cancer candidate genes in glioblastoma, melanoma, and pancreatic carcinoma. Cancer Res. 2007;67:3545–3550. doi: 10.1158/0008-5472.CAN-07-0065. [DOI] [PubMed] [Google Scholar]
- 38.Mann KM, Ward JM, Yew CC, et al. Sleeping Beauty mutagenesis reveals cooperating mutations and pathways in pancreatic adenocarcinoma. Proc Natl Acad Sci USA. 2012;109:5934–5941. doi: 10.1073/pnas.1202490109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sessa F, Solcia E, Capella C, et al. Intraductal papillary-mucinous tumours represent a distinct group of pancreatic neoplasms: An investigation of tumour cell differentiation and K-ras, p53 and c-erbB-2 abnormalities in 26 patients. Virchows Arch. 1994;425:357–367. doi: 10.1007/BF00189573. [DOI] [PubMed] [Google Scholar]
- 40.Hong SM, Park JY, Hruban RH, et al. Molecular signatures of pancreatic cancer. Arch Pathol Lab Med. 2011;135:716–727. doi: 10.5858/2010-0566-ra.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yamaguchi H, Kuboki Y, Hatori T, et al. Somatic mutations in PIK3CA and activation of AKT in intraductal tubulopapillary neoplasms of the pancreas. Am J Surg Pathol. 2011;35:1812– 1817. doi: 10.1097/PAS.0b013e31822769a0. [DOI] [PubMed] [Google Scholar]
- 42.Schonleben F, Qiu W, Bruckman KC, et al. BRAF and KRAS gene mutations in intraductal papillary mucinous neoplasm/carcinoma (IPMN/IPMC) of the pancreas. Cancer Lett. 2007;249:242–248. doi: 10.1016/j.canlet.2006.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Iacobuzio-Donahue CA, Klimstra DS, Adsay NV, et al. Dpc-4 protein is expressed in virtually all human intraductal papillary mucinous neoplasms of the pancreas: Comparison with conventional ductal adenocarcinomas. Am J Pathol. 2000;157:755– 761. doi: 10.1016/S0002-9440(10)64589-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sahin F, Maitra A, Argani P, et al. Loss of Stk11/Lkb1 expression in pancreatic and biliary neoplasms. Mod Pathol. 2003;16:686–691. doi: 10.1097/01.MP.0000075645.97329.86. [DOI] [PubMed] [Google Scholar]
- 45.Jansen M, Ten Klooster JP, Offerhaus GJ, et al. LKB1 and AMPK family signaling: The intimate link between cell polarity and energy metabolism. Physiol Rev. 2009;89:777–798. doi: 10.1152/physrev.00026.2008. [DOI] [PubMed] [Google Scholar]
- 46.Su GH, Hruban RH, Bansal RK, et al. Germline and somatic mutations of the STK11/LKB1 Peutz-Jeghers gene in pancreatic and biliary cancers. Am J Pathol. 1999;154:1835–1840. doi: 10.1016/S0002-9440(10)65440-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lim W, Olschwang S, Keller JJ, et al. Relative frequency and morphology of cancers in STK11 mutation carriers. Gastroenterology. 2004;126:1788–1794. doi: 10.1053/j.gastro.2004.03.014. [DOI] [PubMed] [Google Scholar]
- 48.Giardiello FM, Brensinger JD, Tersmette AC, et al. Very high risk of cancer in familial Peutz-Jeghers syndrome. Gastroenterology. 2000;119:1447–1453. doi: 10.1053/gast.2000.20228. [DOI] [PubMed] [Google Scholar]
- 49.Thompson LD, Becker RC, Przygodzki RM, et al. Mucinous cystic neoplasm (mucinous cystadenocarcinoma of low-grade malignant potential) of the pancreas: A clinicopathologic study of 130 cases. Am J Surg Pathol. 1999;23:1–16. doi: 10.1097/00000478-199901000-00001. [DOI] [PubMed] [Google Scholar]
- 50.Waters JA, Schmidt CM. Intraductal papillary mucinous neoplasm— When to resect? Adv Surg. 2008;42:87–108. doi: 10.1016/j.yasu.2008.03.011. [DOI] [PubMed] [Google Scholar]
- 51.Basturk O, Coban I, Adsay NV. Pancreatic cysts: Pathologic classification, differential diagnosis, and clinical implications. Arch Pathol Lab Med. 2009;133:423–438. doi: 10.5858/133.3.423. [DOI] [PubMed] [Google Scholar]
- 52.Furukawa T, Kuboki Y, Tanji E, et al. Whole-exome sequencing uncovers frequent GNAS mutations in intraductal papillary mucinous neoplasms of the pancreas. Sci Rep. 2011;1:161. doi: 10.1038/srep00161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wu J, Jiao Y, Dal Molin M, et al. Whole-exome sequencing of neoplastic cysts of the pancreas reveals recurrent mutations in components of ubiquitin-dependent pathways. Proc Natl Acad Sci USA. 2011;108:21188–21193. doi: 10.1073/pnas.1118046108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sugiura T, Yamaguchi A, Miyamoto K. A cancer-associated RING finger protein, RNF43, is a ubiquitin ligase that interacts with a nuclear protein, HAP95. Exp Cell Res. 2008;314:1519– 1528. doi: 10.1016/j.yexcr.2008.01.013. [DOI] [PubMed] [Google Scholar]
- 55.Hammel PR, Vilgrain V, Terris B, et al. Pancreatic involvement in von Hippel-Lindau disease. The Groupe Francophone d’Etude de la Maladie de von Hippel-Lindau. Gastroenterology. 2000;119:1087–1095. doi: 10.1053/gast.2000.18143. [DOI] [PubMed] [Google Scholar]
- 56.Kaelin WG., Jr The von Hippel-Lindau tumour suppressor protein: O2 sensing and cancer. Nat Rev Cancer. 2008;8:865– 873. doi: 10.1038/nrc2502. [DOI] [PubMed] [Google Scholar]
- 57.Verheyen EM, Gottardi CJ. Regulation of Wnt/beta-catenin signaling by protein kinases. Dev Dyn. 2010;239:34–44. doi: 10.1002/dvdy.22019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Morin PJ, Sparks AB, Korinek V, et al. Activation of beta-catenin-Tcf signaling in colon cancer by mutations in beta-catenin or APC. Science. 1997;275:1787–1790. doi: 10.1126/science.275.5307.1787. [DOI] [PubMed] [Google Scholar]
- 59.Garcea G, Ong SL, Rajesh A, et al. Cystic lesions of the pancreas. A diagnostic and management dilemma. Pancreatology. 2008;8:236–251. doi: 10.1159/000134279. [DOI] [PubMed] [Google Scholar]
- 60.Tanaka M. Controversies in the management of pancreatic IPMN. Nat Rev Gastroenterol Hepatol. 2011;8:56–60. doi: 10.1038/nrgastro.2010.193. [DOI] [PubMed] [Google Scholar]
- 61.Allen PJ, Brennan MF. The management of cystic lesions of the pancreas. Adv Surg. 2007;41:211–228. doi: 10.1016/j.yasu.2007.05.014. [DOI] [PubMed] [Google Scholar]
- 62.Yachida S, Jones S, Bozic I, et al. Distant metastasis occurs late during the genetic evolution of pancreatic cancer. Nature. 2010;467:1114–1117. doi: 10.1038/nature09515. [DOI] [PMC free article] [PubMed] [Google Scholar]