

Abstract
RATIONALE
Protective effects of prostacyclin and its stable analog Iloprost are mediated by elevation of intracellular cAMP leading to enhancement of peripheral actin cytoskeleton and cell-cell adhesive structures. This study tested hypothesis that iloprost may exhibit protective effects against lung injury and endothelial barrier dysfunction induced by bacterial wall lypopolysacharide (LPS).
METHODS
Endothelial barrier dysfunction was assessed by measurements of transendothelial permeability, morphologically, and analysis of LPS-activated inflammatory signaling. In vivo, C57BL/6J mice were challenged with LPS with or without iloprost or 8-bromoadenosine-3′,5′-cyclic monophosphate (Br-cAMP) treatment. Lung injury was monitored by measurements of bronchoalveolar lavage protein content, cell count, and Evans blue extravasation.
RESULTS
Iloprost and Br-cAMP attenuated disruption of endothelial monolayer and suppressed activation of p38 mitogen activated protein (MAP) kinase, NFκB pathway, Rho signaling, ICAM1 expression, and neutrophil migration after LPS challenge. In vivo, iloprost was effective against LPS-induced protein and neutrophil accumulation in bronchoalveolar lavage fluid and reduced myeloperoxidase activation, ICAM-1 expression, and Evans blue extravasation in the lungs. Inhibition of Rac activity abolished barrier protective and anti-inflammatory effects of iloprost and Br-cAMP.
CONCLUSION
Iloprost-induced elevation of intracellular cAMP triggers Rac signaling, which attenuates LPS-induced NFκB and p38 MAPK inflammatory pathways and Rho-dependent mechanism of endothelial permeability.
Keywords: cytoskeleton, endothelium, permeability, lung, inflammation
INTRODUCTION
Acute respiratory distress syndrome is often associated with sepsis and remains a major cause of morbidity and mortality with an overall mortality rate of 30–40% [1, 2]. The acute phase of septic lung injury is characterized by increased vascular permeability, expression of adhesive surface molecules such as ICAM-1 by activated endothelial cell which promotes leukocyte adhesion and recruitment to the lung, impaired fluid balance, accumulation of protein-rich fluid in the air spaces, accumulation of activated neutrophils and inflammatory cytokines in the lung tissue (reviewed in [3, 4]). Altogether, these mechanisms may cause severe lung inflammation and pulmonary edema. However, little is known about intracellular processes, which determine lung endothelial cell (EC) barrier preservation and reduce inflammation in acute lung injury (ALI), and effective barrier-protective substances for ALI treatment remain to be identified.
Clinical observations and animal studies show beneficial effects of elevated intracellular cAMP concentrations in various lung pathologies [5, 6]. Prostaglandin I2, or prostacyclin, is a product of cyclooxygenase. Aerosolized prostacyclin shows marked protection against hyperoxic lung injury and lung damage caused by ischemia/reperfusion. Increased levels of prostacyclin stable metabolites have been associated with less severe respiratory distress [7, 8]. However, the effects of prostacyclin in septic models of lung injury were not investigated.
Our previous works described potent barrier-protective effects of prostacyclin in human pulmonary endothelium and in the in the animal model of ventilator-induced lung injury [9, 10]. Major biological effects of prostacyclin and its stable analog iloprost are associated with elevation of intracellular cAMP concentration, which triggers cAMP-activated protein kinase (PKA)-dependent [11, 12] and PKA-independent Epac-Rap1-Rac mechanisms of lung vascular endothelial barrier enhancement, which involve cAMP-induced activation of guanine nucleotide exchange factor Epac leading to activation of small GTPase Rap1 and Rac1 [9, 13, 14]. In addition to cAMP-mediated activation by Epac/Rap1, Rac1 may be additionally stimulated via PKA-dependent mechanisms, and recent report has described Rac1 as a hub coordinating barrier protective signaling from PKA and Epac-Rap1 [15].
In this study we used biochemical, molecular, and functional approaches to characterize iloprost effects the in vitro and in vivo models of LPS-induced lung injury. While recognizing potential role of PKA in iloprost-mediated effects, in this study we focused on Rac1 mechanism and investigated its role in protective effects of iloprost against septic inflammation.
MATERIALS AND METHODS
Cell culture and reagents
Human pulmonary artery endothelial cells (HPAEC) and cell culture medium were obtained from Lonza Inc (Allendale, NJ), and used at passages 5–8. Iloprost was obtained from Cayman (Ann Arbor, MI). Di-phospho-MLC, phospho-HSP27, IκBα, NFκB antibodies were obtained from Cell Signaling (Beverly, MA); phospho-VE-cadherin and ICAM1 from Santa Cruz Biotechnology (Santa Cruz, CA); VE-cadherin from BD Transduction Laboratories (San Diego, CA). NSC-23766 and 8-Bromo-adenosine-3′,5′-cyclic monophosphate (Br-cAMP) were purchased from Calbiochem (La Jolla, CA). All reagents for immunofluorescence were purchased from Molecular Probes (Eugene, OR). Unless specified, biochemical reagents were obtained from Sigma (St. Louis, MO).
Depletion of endogenous Rac1
Pre-designed Rac1-specific siRNA of standard purity was ordered from Ambion (Austin, TX). Transfection of EC with siRNA was performed as previously described [16]. Nonspecific, non-targeting siRNA was used as a control treatment. After 48 hrs of transfection cells were used for experiments or harvested for western blot verification of specific protein depletion.
Measurement of transendothelial electrical resistance
The cellular barrier properties were analyzed by measurements of transendothelial electrical resistance (TER) across confluent human pulmonary artery endothelial monolayers using an electrical cell-substrate impedance sensing system (Applied Biophysics, Troy, NY) as previously described [16].
Transwell permeability and migration assays
Permeability for FITC-labeled dextran across pulmonary EC monolayers grown on permeable filters was assessed in transwell assays using in Vitro Vascular Permeability Assay Kit (Chemicon International), according to the manufacturer’s instructions. Neutrophil chemotaxis was measured in a 96-well chemotaxis chamber (Neuroprobe, Gaithersburg, MD) as described previously [17]. Preliminary experiments have established that the number of cells (4 × 104 cells) used allow the optimal % cell migration without clogging the pores of transwell filter of the upper chamber. Data were expressed as % of migrated cells.
Immunofluorescence
Endothelial monolayers plated on glass cover slips were subjected to immunofluorescence staining as described previously [18]. Slides were analyzed using a Nikon video imaging system (Nikon Instech Co., Tokyo, Japan). Images were processed with Image J software (National Institute of Health, Washington, USA) and Adobe Photoshop 7.0 (Adobe Systems, San Jose, CA) software.
Differential protein fractionation and immunoblotting
Confluent HPAEC were stimulated with LPS with or without iloprost and nuclear fraction was isolated using S-PEK kit (EMD Chemicals, Gibbstown, NJ). Immunoblotting detection of proteins of interest was performed as described previously [16]. Protein extracts from mouse lungs or EC were separated by SDS-PAGE, transferred to polyvinylidene difluoride membranes, and the membranes were incubated with specific antibodies of interest. Equal protein loading was verified by reprobing membranes with β-actin or β-tubulin antibodies. Immunoreactive proteins were detected with the enhanced chemiluminescent detection system according to the manufacturer’s protocol (Amersham, Little Chalfont, UK).
Animal studies
All animal care and treatment procedures were approved by the University of Chicago Institutional Animal Care and Use Committee. Animals were handled according to the National Institutes of Health Guide for the Care and Use of Laboratory Animals. Detailed description of the experimental procedures is provided in the online data supplement.
Statistical analysis
Results are expressed as means ±SD of three to eight independent experiments. Stimulated samples were compared to controls by unpaired Student’s t-test. For multiple-group comparisons, one-way ANOVA and Tukey’s post hoc multiple-comparison test were used. P<0.05 was considered statistically significant.
RESULTS
Effects of iloprost on LPS-induced EC hyper-permeability
To test effects of iloprost on EC barrier dysfunction associated with septic inflammation, we used a model of LPS-induced EC permeability. LPS decreased transendothelial electrical resistance (TER), and iloprost treatment abolished this effect (Figure 1A). Protective effects of iloprost were largely related to elevation of intracellular cAMP levels, because similar protective effects were reproduced by EC treatment with stable cAMP analog, Br-cAMP (Figure 1B). Effects of iloprost and Br-cAMP against LPS-induced EC hyper-permeability were further tested in solute flux assay. Both iloprost and Br-cAMP attenuated LPS-induced permeability for FITC-labeled dextran (Figure 1C).
Figure 1. Effect of iloprost and Br-cAMP on LPS-induced EC permeability.

A and B- HPAEC plated on microelectrodes were pretreated with vehicle, iloprost (200 ng/ml) (A) or Br-cAMP (500 ng/ml) (B) for 15 min followed by stimulation with LPS (300 ng/ml), as shown by arrows. TER was monitored over 15 hours. C - Pulmonary EC were pre-incubated with iloprost (200 ng/ml) or Br-cAMP (500 ng/ml) for 15 min followed by LPS (300 ng/ml, 5 hr) challenge and measurements of permeability for FITC-labeled dextran. Permeability data are expressed as mean ± SD of eight independent experiments; *p<0.05.
Iloprost attenuates LPS-induced EC monolayer disruption
Effects of iloprost on LPS-induced EC disruption were monitored by analysis of actin cytoskeletal remodeling and changes in adherens junction integrity. LPS induced formation of actin stress fibers and cell contraction associated with appearance of paracellular gaps. These changes were reduced by EC pretreatment with iloprost or Br-cAMP (Figure 2A,B). Immunofluorescence analysis of adherens junction remodeling confirmed disruption of cell-cell contacts in response to LPS. In turn, iloprost and Br-cAMP pretreatment preserved continuous adherens junction pattern in LPS-challenged EC (Figure 2C). Iloprost also attenuated LPS-induced VE-cadherin phosphorylation at Y731 (Figure 2D) known to promote disassembly of adherens junction complexes [19, 20]. These results demonstrate potent protective effects of iloprost against LPS-induced EC barrier dysfunction mediated by cAMP.
Figure 2. Effect of iloprost and Br-cAMP on LPS-induced cytoskeletal remodeling and gap formation.


Endothelial monolayers were pretreated with vehicle, iloprost (200 ng/ml) or Br-cAMP (500 ng/ml) for 15 min and stimulated with LPS (300 ng/ml) for 5 hours. A - Analysis of actin cytoskeletal rearrangement was performed by immunofluorescence staining with Texas Red phalloidin. Paracellular gaps are marked by arrows. B - Quantitative analysis of paracellular gap formation in control and treated HPAEC. Data are expressed as mean ± SD of four independent experiments; *p<0.05, as compared to LPS alone. C - Immunofluorescence staining for VE-cadherin was performed to visualize adherens junctions. LPS-induced disruption of adherens junctions is shown by arrows. D – Western blot analysis of time-dependent VE-cadherin phosphorylation in HPAEC stimulated with LPS with or without iloprost pretreatment. β-Tubulin staining was used as normalization control.
Iloprost suppresses LPS-induced activation of inflammatory signaling
A number of signaling molecules, such as stress activated p38 MAP kinase, RhoA GTPase, and NFκB complex become activated by LPS [21, 22]. In the following experiments we tested whether iloprost was able to inhibit these barrier-disruptive pathways. LPS activated p38 MAP kinase leading to phosphorylation of p38 MAP kinase downstream target, heat shock protein-27 (HSP27), and increased phosphorylation of RhoA downstream target myosin light chain (MLC). LPS also triggered canonical inflammatory pathway and induced degradation of IκBα inhibitory subunit leading to activation of NFκB signaling. These effects were inhibited by iloprost or Br-cAMP (Figure 3A). LPS-induced activation of NFκB-dependent inflammatory gene expression requires nuclear translocation of NFκB p65 subunit. Immunofluorescence analysis (Figure 3B) and subcellular fractionation assays (Figure 3C) showed nuclear translocation of NFκB after LPS challenge, which was attenuated by iloprost or Br-cAMP.
Figure 3. Effects of iloprost and Br-cAMP on LPS-induced inflammatory signaling.

A - HPAEC were pretreated with vehicle, iloprost (200 ng/ml, 15 min) or Br-cAMP (500 ng/ml) for 15 min followed by stimulation with LPS (300 ng/ml) for 1 hr. Phosphorylation of p38 MAP kinase, HSP27 and MLC was detected by western blot with corresponding phospho-specific antibodies. Degradation of IκBα was detected using antibodies against non-phosphorylated protein. Equal protein loading was confirmed by determination of β-tubulin content in total cell lysates. B and C - Cells were pretreated with vehicle or iloprost followed by LPS challenge (300 ng/ml, 1 hr). NFκB nuclear translocation was assessed by immunofluorescence staining with specific antibody (B). Fractionation assay was performed and the content of NFκB in the nuclear fraction was determined by western blot analysis with specific antibodies. Determination of β-tubulin content in corresponding total cell lysates was used to ensure equal loading (C).
Activation of vascular endothelium by inflammatory agents stimulates neutrophil adhesion to the vascular EC lining followed by neutrophil transmigration through EC monolayer which leads to neutrophil recruitment to the inflamed lung tissue. In the following studies we evaluated effects of iloprost on endothelial activation. Western blot analysis of ICAM-1 expression, the endothelial surface molecule involved in neutrophil adhesion revealed time dependent increase in LPS-induced ICAM-1 expression (Figure 4A) attenuated by iloprost or Br-cAMP pretreatment (Figure 4B). Because LPS induces production of proinflammatory cytokines by vascular endothelium which promote neutrophil transmigration, we next evaluated effects of preconditioned medium collected from stimulated EC on directed neutrophil migration. LPS stimulation significantly increased neutrophil migration, whereas iloprost or Br-cAMP pretreatment suppressed this effect (Figure 4C). Additional experiments revealed LPS-induced IL-8 expression by pulmonary EC which was abolished by iloprost or Br-cAMP pretreatment (data not shown). Collectively, these data suggest potent protective effects of iloprost against activation of inflammatory signaling in pulmonary endothelium induced by LPS.
Figure 4. Effects of iloprost and Br-cAMP on LPS-induced activation of pulmonary endothelium.

A - Western blot analysis of time-dependent ICAM-1 expression in HPAEC induced by LPS (300 ng/ml). B - HPAEC were pretreated with vehicle, iloprost (200 ng/ml) or Br-cAMP (500 ng/ml) for 15 min followed by stimulation with LPS (300 ng/ml) for 4 hrs. ICAM-1 expression was detected by western blot with ICAM-1 antibody. β-Tubulin staining was used as normalization control. C - Cells were pretreated with vehicle, iloprost, or Br-cAMP followed by LPS stimulation (20 ng/ml, 4 hr). Neutrophil migration assay was performed as described in Methods. Data are expressed as mean ± SD of three independent experiments; *p<0.05, as compared to LPS alone.
Iloprost- and cAMP-stimulated Rac-1 signaling attenuates LPS-induced EC barrier dysfunction
Previous works demonstrated the role of iloprost-activated Rac1 signaling in EC barrier enhancement via specific peripheral cytoskeletal remodeling and increased assembly of cell-cell junctions [9, 13]. The following experiments tested involvement of Rac1 mechanism in suppression of inflammatory signaling by iloprost. SiRNA-induced Rac1 knockdown did not significantly change permeability response to LPS alone but attenuated protective effects of Br-cAMP and iloprost against LPS-induced permeability (Figure 5A,B).
Figure 5. Effects of siRNA-based Rac1 knockdown on iloprost- and Br-cAMP-induced EC barrier protection.


HPAEC were transfected with non-specific RNA or with Rac1-specific siRNA for 48 hrs. A - Cells plated on microelectrodes were treated with vehicle or Br-cAMP (500 ng/ml, 15 min) followed by LPS stimulation (300 ng/ml). TER was monitored over the time. B – Pooled data of TER experiments using cell pretreatment with Br-cAMP and iloprost prior to LPS treatment. Left panel - LPS effects on TER were compared in cells transfected with non-specific RNA or Rac1-specific siRNA. Right panel - Protective effect of iloprost or Br-cAMP against LPS-induced permeability in EC treated with non-specific RNA observed after 5 hrs was taken as 100%. Rac1 knockdown attenuated protective effects of iloprost (200 ng/ml) and Br-cAMP (500 ng/ml) against LPS-induced permeability. Data are expressed as mean ± SD of three to five independent experiments; *p<0.05, as compared to non-specific RNA. C - Cells were treated with vehicle or Br-cAMP followed by LPS stimulation (300 ng/ml, 5 hrs). Cytoskeletal remodeling was assessed by immunofluorescence staining of F-actin with Texas Red phalloidin. Paracellular gaps are marked by arrows. D - Quantitative analysis of paracellular gap formation in control and Rac-depleted HPAEC. Data are expressed as mean ± SD of three independent experiments; *p<0.05, as compared to LPS alone.
The role of Rac1 in EC barrier protective effects elicited by iloprost-induced cAMP elevation was further assessed by analysis of cytoskeletal remodeling in control and Rac1-depleted HPAEC preincubated with iloprost or Br-cAMP and stimulated with LPS. Similarly to non-transfected EC (Figure 2), pretreatment with iloprost or Br-cAMP attenuated LPS-induced stress fiber formation and disruption of monolayer integrity in cells transfected with non-specific RNA whereas Rac1 knockdown inhibited these protective effects (Figure 5C,D). Next, we used pharmacological inhibitor NSC-23766 as alternative approach to inhibit Rac1 function. NSC-23766 suppressed protective effects of Br-cAMP or iloprost against EC hyper-permeability (Figure 6A,B), formation of stress fibers and paracellular gaps in LPS-challenged EC monolayers (Figure 6C,D).
Figure 6. Effects of pharmacological Rac1 inhibition on iloprost- and Br-cAMP-induced EC barrier protection.


A - HPAEC were treated vehicle or Rac inhibitor NSC-23766 (100 μM, 30 min) followed by LPS (300 ng/ml) challenge with or without Br-cAMP pretreatment (500 ng/ml). TER was monitored over the time. B – Pooled data of TER experiments shown above. Protective effect of Br-cAMP against LPS-induced permeability in EC without NSC-23766 treatment observed after 5 hrs was taken as 100%. Data are expressed as mean ± SD of three independent experiments; *p<0.05. C - Control or NSC-23766-preincubated HPAEC were treated with vehicle, iloprost (200 ng/ml, 15 min) or Br-cAMP (500 ng/ml, 15 min) followed by LPS challenge (300 ng/ml, 5 hrs). Cytoskeletal remodeling was assessed by immunofluorescence staining of F-actin with Texas Red phalloidin. D - Quantitative analysis of paracellular gap formation. Data are expressed as mean ± SD of three independent experiments; *p<0.05, as compared to LPS alone.
Next, the role of Rac1 in the anti-inflammatory cAMP effects was evaluated in experiments with molecular or pharmacological inhibition of Rac1. Rac1 knockdown also abolished protective effects of Br-cAMP against LPS-induced activation of p38 MAP kinase, Rho and NFκB signaling (Figure 7A) monitored by phosphorylation of HSP27 and MLC, and IκBα degradation, respectively. Subcellular fractionation studies showed that inhibitory effect of Br-cAMP on LPS-induced accumulation of NFκB p65 subunit in the nuclear fraction was abolished by NSC-23766 (Figure 7B). Moreover, inhibition of Rac1 activity by NSC-23766 also attenuated protective effect of Br-cAMP against LPS-induced ICAM-1 expression, a hallmark of inflammatory activation of endothelial cells. (Figure 7C).
Figure 7. Effects of Rac1 inhibition on anti-inflammatory effects of Br-cAMP.

A - HPAEC transfected with non-specific or Rac1-specific siRNA for 48 hrs were treated with vehicle or Br-cAMP (500 ng/ml, 15 min) followed by LPS stimulation (300 ng/ml, 1 hr). Phosphorylation of pHSP27 and MLC was detected by western blot with corresponding phospho-specific antibodies. Degradation of IκBα was detected using antibodies against non-phosphorylated protein. Equal protein loading was confirmed by determination of β-tubulin content in total cell lysates. Rac1 depletion was verified by western blot with Rac antibody. B - HPAEC were treated as described above, and NFκB translocation to the nuclear fraction was monitored by western blot. Determination of β-tubulin content in corresponding total cell lysates was used to ensure equal loading. C - HPAEC preincubated with vehicle or Rac inhibitor NSC-23766 (100 μM, 30 min) were treated with Br-cAMP (500 ng/ml, 15 min) followed by stimulation with LPS (300 ng/ml) for 4 hr. ICAM-1 expression was detected by western blot with ICAM-1 antibody. β-Tubulin staining was used as normalization control.
Iloprost suppresses LPS-induced lung barrier dysfunction and inflammation in vivo
Protective effects of iloprost were further tested in the septic model of acute lung injury induced by intratracheal instillation of LPS. Intravenous injection of iloprost or Br-cAMP significantly reduced BAL total cell count, neutrophil count and decreased protein content in LPS-treated mice (Figure 8A). Severity of lung injury and inflammation was also monitored by measurements of myeloperoxidase (MPO) activity in lung tissue samples. LPS significantly increased MPO activity measured in tissue homogenates, while iloprost or Br-cAMP treatment attenuated this effect (Figure 8A).
Figure 8. Effects of iloprost on LPS-induced lung inflammation and barrier lung inflammation and barrier dysfunction.

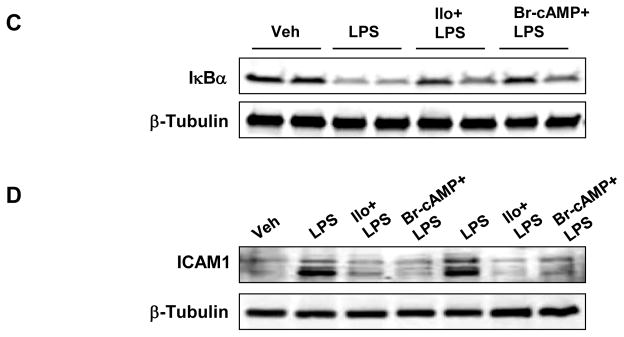
C57BL/6J mice were challenged with vehicle or LPS (0.63 mg/kg, i/t) with or without concurrent iloprost (20 μg/kg, i/v, at 0 and 90 min after LPS administration) or Br-cAMP treatment (20 μg/kg, i/v, at 0 and 90 min after LPS administration). Control animals were treated with sterile saline solution or iloprost alone. A - Cell count and protein concentration were determined in bronchoalveolar lavage fluid collected 18 hrs after treatments. Myeloperoxidase (MPO) activity was measured in lung tissue homogenates as described in the Methods. Data are expressed as mean ± SD, n=6–10 per condition; *p<0.05, as compared to LPS treatment. B - Evans blue dye (30 ml/kg, i/v) was injected 2 hr before termination of the experiment. Lung vascular permeability was assessed by Evans blue accumulation in the lung tissue. The quantitative analysis of Evans blue labeled albumin extravasation was performed by spectrophotometric analysis of Evans blue extracted from the lung tissue samples; n=4 per condition; *p<0.05, as compared to LPS treatment. C and D - IκBα degradation and ICAM1 expression after LPS challenge with or without iloprost or Br-cAMP treatment were determined in lung tissue homogenates by western blot analysis with specific antibodies. Equal protein loading was confirmed by membrane re-probing with β-tubulin antibodies.
Effects of iloprost on the lung vascular leak induced by LPS were evaluated by measurements of Evans blue extravasation into the lung tissue. Iloprost significantly reduced LPS-induced Evans blue accumulation in the lung parenchyma (Figure 8B). In consistence with cell culture studies, iloprost or Br-cAMP treatment inhibited LPS-induced IκBα degradation (Figure 8C) and ICAM1 expression (Figure 8D) in the lung, detected by western blot analysis of lung tissue homogenates. These results further support anti-inflammatory and barrier-protective effects of iloprost against septic inflammation and vascular endothelial barrier dysfunction in cell culture and the animal models of LPS-induced lung injury.
DISCUSSION
Clinical studies focused on testing of endogenous arachidonic acid metabolites generated by cyclooxygenase, which were increased in patients with sepsis and were related to changes in pulmonary hemodynamics and gas exchange [23–26]. The results showed that in ARDS but not in sepsis patients clear of pulmonary organ failure, a changing balance of prostacyclin and thromboxane A2 modulated gas exchange via interference with hypoxic pulmonary vasoconstriction [23]. Attempts to reduce levels of prostacyclin and thromboxane by treatment with COX inhibitor ibuprofen led to decreased fever, tachycardia, oxygen consumption, and lactic acidosis, but neither prevented the development of shock or the acute respiratory distress syndrome, nor improved survival [27].
Other studies used administration of pharmacological prostacyclin analogs to reach rapid prostacyclin-induced improvement of pulmonary hemodynamics, blood gas parameters, and vascular tone. Studies using this approach also showed that inhaled iloprost improved gas exchange due to a decrease of pulmonary shunt as a long-term effect suggesting potential effects of iloprost on reduction of lung oedema formation [28]. However, specific effects of pharmacologic prostacyclin administration on inflammatory parameters in ALI/ARDS have not been systematically analyzed. Our study addressed this important point and investigated protective effects of prostacyclin on endothelial barrier regulation inflammatory signaling associated with inflammatory innate immune responses in ALI.
Previous studies described barrier enhancing effects of prostacyclin synthetic analogs in endothelial cultures and demonstrated protective effects in vivo in aseptic models of lung injury including mechanical ventilation at high tidal volume and thrombin related activating peptide (TRAP) [9, 10, 13, 15]. These effects were linked to attenuation of RhoA-dependent mechanisms of cytoskeletal remodeling and endothelial hyper-permeability caused by elevation of intracellular cAMP [9, 15]. However, effects of prostacyclin in septic models of acute lung injury and inflammation remain unclear. The main finding of this study is protective effect of iloprost in the LPS model of ALI and pulmonary endothelial barrier dysfunction. The results of this study show that iloprost treatment suppressed both, the LPS-induced cytoskeletal mechanisms of EC barrier dysfunction and LPS-induced inflammatory signaling.
Our results also show the critical role of Rac1-dependent mechanisms in the improvement of LPS-induced lung injury and attenuation of inflammatory signaling and vascular leak by Iloprost. On the other hand, Rac activation has been reported in LPS-stimulated macrophages and neutrophils [29, 30]. In these cells, Rac activation triggered NADPH oxidase activity leading to ROS production and oxidative stress, one of major pathologic mechanisms of septic lung injury. Increased ROS production may also stimulate NFκB inflammatory signaling cascade and cytokine production leading to exacerbation of tissue inflammation, vascular leakiness, and worsening of lung injury [31]. In contrast, activation of Rac1 signaling in lung vascular endothelium by barrier protective agonists causes enhancement of EC monolayer integrity and plays a critical role in lung vascular barrier protection [16, 32–34]. Apparent discrepancy between these reported effects may be explained by diversity of Rac functions in different cells and differential roles of Rac1 and Rac2 isoforms in pathologic conditions. For example, inhibition of Rac2 function in Rac2−/− neutrophils impairs adhesion, chemotaxis, phagocytosis, and ROS production [Roberts, 1999 #2983; Gu, 2003 #2984], while knockout of Rac1 in neutrophils has no effect on ROS [35]. In turn, Rac1 is a key player in preservation of vascular endothelial barrier in the models of acute lung injury [15, 16, 36]. Rac1 and Rac2 functions are also independently regulated by distinct guanine nucleotide exchange factors (GEFs). Rac1 activation in endothelial cells mediated by guanine nucleotide exchange factors Tiam1, Vav2 and β-PIX is associated with endothelial barrier enhancement [9, 37, 38]. Moreover, Tiam1 and Vav2 are major activators of Rac1 signaling induced by cAMP elevation [12, 13]. In turn, Rac2 in neutrophils is preferentially activated by P-REX1 and Vav1 [30, 39], and double knockout of P-REX-1 and Vav1 in neutrophils dramatically reduces Rac activity and impairs pathogen-induced ROS-formation, adhesion and chemotaxis [39]. Taken together, these data illustrate cell-specific and context specific mechanisms of Rac functions in endothelial and immune cells and emphasize the complexity of Rac roles in development and attenuation of lung injury.
Role of RhoA signaling in control of NFκB activity remains an intriguing question. Sustained NFκB activation in cells and tissues was observed in genetic model of upregulated Rho activity [40]. That study showed that sustained activation of Rho-NFκB cascade mediates chronic skin inflammation. Other studies also indirectly indicate that activated Rho may stimulate NFκB activity [41], although precise mechanisms of such activation remain to be elucidated. Thus, previous reports [40, 42] and the results of this study suggest that prolonged Rho activation induced by LPS may be implied in additional stimulation of NFκB signaling in the model of inflamed lungs. Previously reported sustained Rho activation by LPS [42] may also stimulate p38 MAP kinase pathway [43, 44], and RhoA effector PKNα has been recently proposed as a novel transducer of RhoA signaling to p38 stress MAP kinase cascade [45]. Resulting activation of p38 MAPK pathway upregulates pro-inflammatory gene expression and contributes to increased EC permeability via p38-induced phosphorylation of actin regulatory protein HSP27.
Our results show that protective effects of iloprost against LPS-induced lung dysfunction were associated with Rac1-dependent preservation of pulmonary EC barrier integrity and attenuation of LPS-induced IκBα degradation, NFκB nuclear localization, and p38 MAP kinase activity. Such attenuation of inflammatory signaling by iloprost suppressed LPS-induced ICAM-1 expression in cultured human pulmonary EC and LPS-challenged lungs, and decreased infiltration of activated neutrophils in the lungs, which was associated with reduced inflammation and improved lung injury parameters in vivo. In addition to effects on inflammatory signaling, iloprost attenuated LPS-induced EC barrier disruption by inhibiting LPS-induced MLC phosphorylation, actin cytoskeletal remodeling and formation of paracellular gaps controlled by RhoA pathway of endothelial barrier disruption. Both barrier protective and anti-inflammatory effects of iloprost involved elevation of cAMP and required Rac1 activity, as demonstrated in experiments with Rac1 knockdown performed in this study. Based on the results of this study and published data, we speculate that iloprost-induced, cAMP-activated Rac1 signaling contributes to lung protection in septic conditions by intercepting the Rho-mediated cytoskeletal mechanisms of pulmonary EC barrier dysfunction and suppressing the p38 stress kinase and NFkB-dependent inflammatory cascades.
Thus, the results of this study demonstrate beneficial effects of prostacyclin analogs in models of septic lung injury and suggest an importance for further clinical testing of iloprost as potential therapeutic treatment of lung inflammation and barrier dysfunction.
Supplementary Material
Acknowledgments
Supported by National Heart, Lung, and Blood Institutes grants HL87823, HL076259, and HL058064 for KGB; HL089257, HL107920 and the American Heart Association Midwest Affiliate Grant-in-Aid for AAB
References
- 1.Ricard JD, Dreyfuss D, Saumon G. Ventilator-induced lung injury. Eur Respir J Suppl. 2003;42:2s–9s. doi: 10.1183/09031936.03.00420103. [DOI] [PubMed] [Google Scholar]
- 2.Matthay MA, Zimmerman GA, Esmon C, Bhattacharya J, Coller B, Doerschuk CM, Floros J, Gimbrone MA, Jr, Hoffman E, Hubmayr RD, Leppert M, Matalon S, Munford R, Parsons P, Slutsky AS, Tracey KJ, Ward P, Gail DB, Harabin AL. Future research directions in acute lung injury: summary of a National Heart, Lung, and Blood Institute working group. Am J Respir Crit Care Med. 2003;167(7):1027–1035. doi: 10.1164/rccm.200208-966WS. [DOI] [PubMed] [Google Scholar]
- 3.Goodman RB, Pugin J, Lee JS, Matthay MA. Cytokine-mediated inflammation in acute lung injury. Cytokine Growth Factor Rev. 2003;14(6):523–535. doi: 10.1016/s1359-6101(03)00059-5. [DOI] [PubMed] [Google Scholar]
- 4.Dreyfuss D, Ricard JD. Acute lung injury and bacterial infection. Clin Chest Med. 2005;26(1):105–112. doi: 10.1016/j.ccm.2004.10.014. [DOI] [PubMed] [Google Scholar]
- 5.Olschewski H, Simonneau G, Galie N, Higenbottam T, Naeije R, Rubin LJ, Nikkho S, Speich R, Hoeper MM, Behr J, Winkler J, Sitbon O, Popov W, Ghofrani HA, Manes A, Kiely DG, Ewert R, Meyer A, Corris PA, Delcroix M, Gomez-Sanchez M, Siedentop H, Seeger W. Inhaled iloprost for severe pulmonary hypertension. N Engl J Med. 2002;347(5):322–329. doi: 10.1056/NEJMoa020204. [DOI] [PubMed] [Google Scholar]
- 6.Matthay MA, Robriquet L, Fang X. Alveolar epithelium: role in lung fluid balance and acute lung injury. Proc Am Thorac Soc. 2005;2(3):206–213. doi: 10.1513/pats.200501-009AC. [DOI] [PubMed] [Google Scholar]
- 7.Howard LS, Morrell NW. New therapeutic agents for pulmonary vascular disease. Paediatr Respir Rev. 2005;6(4):285–291. doi: 10.1016/j.prrv.2005.09.006. [DOI] [PubMed] [Google Scholar]
- 8.Ueno Y, Koike H, Annoh S, Nishio S. Anti-inflammatory effects of beraprost sodium, a stable analogue of PGI2, and its mechanisms. Prostaglandins. 1997;53(4):279–289. doi: 10.1016/s0090-6980(97)89601-3. [DOI] [PubMed] [Google Scholar]
- 9.Birukova AA, Zagranichnaya T, Alekseeva E, Fu P, Chen W, Jacobson JR, Birukov KG. Prostaglandins PGE2 and PGI2 promote endothelial barrier enhancement via PKA- and Epac1/Rap1-dependent Rac activation. Exp Cell Res. 2007;313(11):2504–2520. doi: 10.1016/j.yexcr.2007.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Birukova AA, Fu P, Xing J, Birukov KG. Rap1 mediates protective effects of iloprost against ventilator induced lung injury. J Appl Physiol. 2009;107(6):1900–1910. doi: 10.1152/japplphysiol.00462.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yuan SY. Protein kinase signaling in the modulation of microvascular permeability. Vascul Pharmacol. 2002;39(4–5):213–223. doi: 10.1016/s1537-1891(03)00010-7. [DOI] [PubMed] [Google Scholar]
- 12.Bos JL. Epac proteins: multi-purpose cAMP targets. Trends Biochem Sci. 2006;31(12):680–686. doi: 10.1016/j.tibs.2006.10.002. [DOI] [PubMed] [Google Scholar]
- 13.Fukuhara S, Sakurai A, Sano H, Yamagishi A, Somekawa S, Takakura N, Saito Y, Kangawa K, Mochizuki N. Cyclic AMP potentiates vascular endothelial cadherin-mediated cell-cell contact to enhance endothelial barrier function through an Epac-Rap1 signaling pathway. Mol Cell Biol. 2005;25(1):136–146. doi: 10.1128/MCB.25.1.136-146.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kooistra MR, Corada M, Dejana E, Bos JL. Epac1 regulates integrity of endothelial cell junctions through VE-cadherin. FEBS Lett. 2005;579(22):4966–4972. doi: 10.1016/j.febslet.2005.07.080. [DOI] [PubMed] [Google Scholar]
- 15.Birukova AA, Burdette D, Moldobaeva N, Xing J, Fu P, Birukov KG. Rac GTPase is a hub for protein kinase A and Epac signaling in endothelial barrier protection by cAMP. Microvasc Res. 2010;79(2):128–138. doi: 10.1016/j.mvr.2009.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Birukov KG, Bochkov VN, Birukova AA, Kawkitinarong K, Rios A, Leitner A, Verin AD, Bokoch GM, Leitinger N, Garcia JG. Epoxycyclopentenone-containing oxidized phospholipids restore endothelial barrier function via Cdc42 and Rac. Circ Res. 2004;95(9):892–901. doi: 10.1161/01.RES.0000147310.18962.06. [DOI] [PubMed] [Google Scholar]
- 17.Duan Y, Learoyd J, Meliton AY, Clay BS, Leff AR, Zhu X. Inhibition of Pyk2 blocks airway inflammation and hyperresponsiveness in a mouse model of asthma. Am J Respir Cell Mol Biol. 2010;42(4):491–497. doi: 10.1165/rcmb.2008-0469OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Birukova AA, Fu P, Xing J, Yakubov B, Cokic I, Birukov KG. Mechanotransduction by GEF-H1 as a novel mechanism of ventilator-induced vascular endothelial permeability. Am J Physiol Lung Cell Mol Physiol. 2010;298(6):L837–848. doi: 10.1152/ajplung.00263.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Potter MD, Barbero S, Cheresh DA. Tyrosine phosphorylation of VE-cadherin prevents binding of p120- and beta-catenin and maintains the cellular mesenchymal state. J Biol Chem. 2005;280(36):31906–31912. doi: 10.1074/jbc.M505568200. [DOI] [PubMed] [Google Scholar]
- 20.Dejana E, Orsenigo F, Lampugnani MG. The role of adherens junctions and VE-cadherin in the control of vascular permeability. J Cell Sci. 2008;121(Pt 13):2115–2122. doi: 10.1242/jcs.017897. [DOI] [PubMed] [Google Scholar]
- 21.Lu YC, Yeh WC, Ohashi PS. LPS/TLR4 signal transduction pathway. Cytokine. 2008;42(2):145–151. doi: 10.1016/j.cyto.2008.01.006. [DOI] [PubMed] [Google Scholar]
- 22.Bogatcheva NV, Dudek SM, Garcia JG, Verin AD. Mitogen-activated protein kinases in endothelial pathophysiology. J Investig Med. 2003;51(6):341–352. doi: 10.1136/jim-51-06-30. [DOI] [PubMed] [Google Scholar]
- 23.Suchner U, Katz DP, Furst P, Beck K, Felbinger TW, Senftleben U, Thiel M, Goetz AE, Peter K. Effects of intravenous fat emulsions on lung function in patients with acute respiratory distress syndrome or sepsis. Crit Care Med. 2001;29(8):1569–1574. doi: 10.1097/00003246-200108000-00012. [DOI] [PubMed] [Google Scholar]
- 24.Arons MM, Wheeler AP, Bernard GR, Christman BW, Russell JA, Schein R, Summer WR, Steinberg KP, Fulkerson W, Wright P, Dupont WD, Swindell BB. Effects of ibuprofen on the physiology and survival of hypothermic sepsis. Ibuprofen in Sepsis Study Group. Crit Care Med. 1999;27(4):699–707. doi: 10.1097/00003246-199904000-00020. [DOI] [PubMed] [Google Scholar]
- 25.Lassus P, Viinikka L, Ylikorkala O, Pohjavuori M, Andersson S. Pulmonary prostacyclin is associated with less severe respiratory distress in preterm infants. Early Hum Dev. 2002;67(1–2):11–18. doi: 10.1016/s0378-3782(01)00244-4. [DOI] [PubMed] [Google Scholar]
- 26.Hucklenbruch C, Hinder F, Berger C, Ertmer C, Lange M, Westphal M, Van Aken H, Ellger B, Dirk Stubbe H. Effects of inhaled aerosolized iloprost and inhaled NO on pulmonary circulation and edema formation in ovine lung injury. Shock. 2008;30(1):75–80. doi: 10.1097/SHK.0b013e31815dd1ad. [DOI] [PubMed] [Google Scholar]
- 27.Bernard GR, Wheeler AP, Russell JA, Schein R, Summer WR, Steinberg KP, Fulkerson WJ, Wright PE, Christman BW, Dupont WD, Higgins SB, Swindell BB. The effects of ibuprofen on the physiology and survival of patients with sepsis. The Ibuprofen in Sepsis Study Group. N Engl J Med. 1997;336(13):912–918. doi: 10.1056/NEJM199703273361303. [DOI] [PubMed] [Google Scholar]
- 28.Dembinski R, Brackhahn W, Henzler D, Rott A, Bensberg R, Kuhlen R, Rossaint R. Cardiopulmonary effects of iloprost in experimental acute lung injury. Eur Respir J. 2005;25(1):81–87. doi: 10.1183/09031936.04.10085504. [DOI] [PubMed] [Google Scholar]
- 29.Maitra U, Singh N, Gan L, Ringwood L, Li L. IRAK-1 contributes to lipopolysaccharide-induced reactive oxygen species generation in macrophages by inducing NOX-1 transcription and Rac1 activation and suppressing the expression of antioxidative enzymes. J Biol Chem. 2009;284(51):35403–35411. doi: 10.1074/jbc.M109.059501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Miletic AV, Graham DB, Montgrain V, Fujikawa K, Kloeppel T, Brim K, Weaver B, Schreiber R, Xavier R, Swat W. Vav proteins control MyD88-dependent oxidative burst. Blood. 2007;109(8):3360–3368. doi: 10.1182/blood-2006-07-033662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fu P, Birukova AA, Xing J, Sammani S, Murley JS, Garcia JG, Grdina DJ, Birukov KG. Amifostine reduces lung vascular permeability via suppression of inflammatory signalling. Eur Respir J. 2009;33(3):612–624. doi: 10.1183/09031936.00014808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Birukova AA, Alekseeva E, Mikaelyan A, Birukov KG. HGF attenuates thrombin-induced permeability in the human pulmonary endothelial cells by Tiam1-mediated activation of the Rac pathway and by Tiam1/Rac-dependent inhibition of the Rho pathway. FASEB J. 2007;21(11):2776–2786. doi: 10.1096/fj.06-7660com. [DOI] [PubMed] [Google Scholar]
- 33.Garcia JG, Liu F, Verin AD, Birukova A, Dechert MA, Gerthoffer WT, Bamberg JR, English D. Sphingosine 1-phosphate promotes endothelial cell barrier integrity by Edg-dependent cytoskeletal rearrangement. J Clin Invest. 2001;108(5):689–701. doi: 10.1172/JCI12450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Singleton PA, Dudek SM, Ma SF, Garcia JG. Transactivation of sphingosine 1-phosphate receptors is essential for vascular barrier regulation. Novel role for hyaluronan and CD44 receptor family. J Biol Chem. 2006;281(45):34381–34393. doi: 10.1074/jbc.M603680200. [DOI] [PubMed] [Google Scholar]
- 35.Glogauer M, Marchal CC, Zhu F, Worku A, Clausen BE, Foerster I, Marks P, Downey GP, Dinauer M, Kwiatkowski DJ. Rac1 deletion in mouse neutrophils has selective effects on neutrophil functions. J Immunol. 2003;170(11):5652–5657. doi: 10.4049/jimmunol.170.11.5652. [DOI] [PubMed] [Google Scholar]
- 36.Maniatis NA, Kotanidou A, Catravas JD, Orfanos SE. Endothelial pathomechanisms in acute lung injury. Vascul Pharmacol. 2008;49(4–6):119–133. doi: 10.1016/j.vph.2008.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Malliri A, van Es S, Huveneers S, Collard JG. The Rac exchange factor Tiam1 is required for the establishment and maintenance of cadherin-based adhesions. J Biol Chem. 2004;279(29):30092–30098. doi: 10.1074/jbc.M401192200. [DOI] [PubMed] [Google Scholar]
- 38.Birukova AA, Malyukova I, Mikaelyan A, Fu P, Birukov KG. Tiam1 and betaPIX mediate Rac-dependent endothelial barrier protective response to oxidized phospholipids. J Cell Physiol. 2007;211(3):608–617. doi: 10.1002/jcp.20966. [DOI] [PubMed] [Google Scholar]
- 39.Lawson CD, Donald S, Anderson KE, Patton DT, Welch HC. P-Rex1 and Vav1 cooperate in the regulation of formyl-methionyl-leucyl-phenylalanine-dependent neutrophil responses. J Immunol. 2011;186(3):1467–1476. doi: 10.4049/jimmunol.1002738. [DOI] [PubMed] [Google Scholar]
- 40.Perez-Moreno M, Davis MA, Wong E, Pasolli HA, Reynolds AB, Fuchs E. p120-catenin mediates inflammatory responses in the skin. Cell. 2006;124(3):631–644. doi: 10.1016/j.cell.2005.11.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Perona R, Montaner S, Saniger L, Sanchez-Perez I, Bravo R, Lacal JC. Activation of the nuclear factor-kappaB by Rho, CDC42, and Rac-1 proteins. Genes Dev. 1997;11(4):463–475. doi: 10.1101/gad.11.4.463. [DOI] [PubMed] [Google Scholar]
- 42.Xiaolu D, Jing P, Fang H, Lifen Y, Liwen W, Ciliu Z, Fei Y. Role of p115RhoGEF in lipopolysaccharide-induced mouse brain microvascular endothelial barrier dysfunction. Brain Res. 2011;1387:1–7. doi: 10.1016/j.brainres.2011.02.059. [DOI] [PubMed] [Google Scholar]
- 43.Matoba K, Kawanami D, Ishizawa S, Kanazawa Y, Yokota T, Utsunomiya K. Rho-kinase mediates TNF-alpha-induced MCP-1 expression via p38 MAPK signaling pathway in mesangial cells. Biochem Biophys Res Commun. 2010;402(4):725–730. doi: 10.1016/j.bbrc.2010.10.093. [DOI] [PubMed] [Google Scholar]
- 44.Shatanawi A, Romero MJ, Iddings JA, Chandra S, Umapathy NS, Verin AD, Caldwell RB, Caldwell RW. Angiotensin II-induced vascular endothelial dysfunction through RhoA/Rho kinase/p38 mitogen-activated protein kinase/arginase pathway. Am J Physiol Cell Physiol. 2011;300(5):C1181–1192. doi: 10.1152/ajpcell.00328.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cariolato L, Cavin S, Diviani D. A-kinase anchoring protein (AKAP)-Lbc anchors a PKN-based signaling complex involved in alpha1-adrenergic receptor-induced p38 activation. J Biol Chem. 2011;286(10):7925–7937. doi: 10.1074/jbc.M110.185645. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.