

Abstract
Sum frequency generation (SFG) vibrational spectroscopy has been developed into an important technique to study surfaces and interfaces. It can probe buried interfaces in situ and provide molecular level structural information such as the presence of various chemical moieties, quantitative molecular functional group orientation, and time dependent kinetics or dynamics at such interfaces. This paper focuses on these three most important advantages of SFG and reviews some of the recent progress in SFG studies on interfaces related to polymer materials and biomolecules. The results discussed here demonstrate that SFG can provide important molecular structural information of buried interfaces in situ and in real time, which is difficult to obtain by other surface sensitive analytical techniques.
1. Introduction
1.1 Importance
Interfacial properties largely depend on interfacial molecular structures. Understanding polymer interfacial structures is particularly important because interfacial properties influence many applications of polymer materials. Elucidation of polymer interfacial structures provides an understanding of interfacial molecular mechanisms for coating protection, adhesion, lubrication, friction, wettability, and biocompatibility. Understanding the molecular structures of biomolecules such as phospholipids, peptides, proteins, and DNA at interfaces is also important because their interfacial structures can impact many biological interactions and many chemical, biological, and medical applications. For example, understanding the molecular behavior of biomolecules at interfacial membrane environments can help elucidate molecular mechanisms of various biological functions such as cell adhesion, enzymatic catalytic reaction, and cell signaling. Interfacial studies on biomolecules can also help to develop biosensors and biochips with better sensitivity and stability. However, up to date many interfacial studies have been focused on macroscopic examinations and/or performance tests. Molecular level understanding of surface and interfacial structures has not been well developed until the recent emergence of appropriate techniques.
1.2 Surface sensitive techniques
Many surface sensitive techniques have been developed and applied to various surface studies. X-ray photoelectron spectroscopy (XPS) is a technique that irradiates sample surfaces with a beam of high energy, monochromatic X-rays and then measures the kinetic energy of emitted photoelectrons.1–3 The binding energy of the emitted electrons can then be deduced to determine the elemental composition of the top surface layers (usually 1–10 nm thick). Secondary ion mass spectrometry (SIMS) is a technique that bombards a surface with a focused primary ion beam. Sputtered secondary ions are then collected and analyzed to examine the composition of the surface.4–7 XPS and SIMS both require high vacuum to operate and cannot be used to study many biological interfaces which involve aqueous media. Atomic force microscopy (AFM) is a high-resolution scanning probe microscopy which uses a sharp tip to interact with a sample surface.8–11 AFM can provide a three dimensional surface profile without the need for sample pretreatment or high vacuum environment. However, it is difficult for AFM to measure molecular structures or to probe buried solid/solid interfaces. Surface plasmon resonance (SPR) is a laser based interfacial sensitive technique which can study buried interfaces in situ. It is sensitive to local refractive index changes at a thin metal film surface due to the adsorption of various materials such as biomolecules or nanoparticles to the surface.12–16 A linear relationship is often observed between the adsorbed mass and the resulting refractive index change in the SPR experiment which can then be used in a variety of biosensor applications. Ellipsometry is an optical technique used to study thin film dielectric properties.17–19 The change of polarization of polarized input light is measured after interaction with the sample. Ellipsometry is especially sensitive for measuring the chirality of materials and is contactless.20, 21 Although both SPR and ellipsometry can provide in situ measurements, they cannot provide molecular structural information.
Vibrational spectroscopies can provide molecular structural information about surfaces and interfaces. For example, molecular composition, orientation and time dependent dynamics at surfaces can be studied by using infrared light to characterize intrinsic vibrational modes of surface molecules. One important surface vibrational spectroscopy is attenuated total reflectance-Fourier transform infrared (ATR-FTIR) spectroscopy.22–26 The surface selectivity of ATR-FTIR is provided by the penetration depth of the evanescent wave which has the same order of magnitude as the IR wavelength. By applying different polarized incident light beams, interfacial molecular orientations can be derived in ATR-FTIR measurements. However, the surface sensitivity of ATR-FTIR is poor; sometimes in order to probe surface/interfacial structures, it is necessary to subtract large signal contribution from the bulk media, which can lead to error. Another surface specific vibrational technique is surface-enhanced Raman spectroscopy (SERS), which enhances the Raman scattering of molecules adsorbed on rough metal substrates (usually gold or silver).27–30 The enhancement factor can be as high as 1014~1015 which allows SERS to detect single molecules.31–33 However, it is difficult to apply SERS to study other surfaces and interfaces. Buried solid/solid interfaces in particular are difficult to study using above surface sensitive techniques.
1.3 Sum frequency generation (SFG) vibrational spectroscopy
In the last few decades, sum frequency generation (SFG) vibrational spectroscopy has been developed into a powerful analytical technique to study surfaces and interfaces.34–42 SFG is a nonlinear optical vibrational spectroscopy which can probe interfaces that are accessible to laser light. It has been extensively shown that SFG can provide in situ measurements on buried interfaces in real time. Furthermore, selection rule provides SFG intrinsic sub-monolayer interfacial selectivity.43–48 By applying different polarization combinations of the input/output laser beams, SFG can also be used to determine molecular orientations at interfaces.38, 49–53 SFG experiments do not require high vacuum to perform (as in XPS and SIMS experiments). Compared to AFM, SPR, and ellipsometry techniques, vibrational spectroscopic signatures can provide more detailed molecular structural information on surfaces. SFG spectroscopy also provides in situ measurement of molecular presence and orientation with great sensitivity at buried interfaces which cannot be obtained using ATR-FTIR or SERS techniques. SFG spectroscopy has been combined with other spectroscopic techniques such as IR,54–56 Raman,54, 57, 58 XPS,59–61 ultraviolet-visible (UV-vis)62 absorption and nuclear magnetic resonance (NMR),63 as well as microscopic techniques such as AFM64–67 and nonlinear imaging68, 69 for surface and interfacial studies. Other analytical techniques such as SPR and quartz crystal microbalance (QCM) have also been combined with SFG to depict clearer pictures of surface or interfacial molecular behavior.70–74 The combined techniques can provide more detailed structural information for surfaces or interfaces.
In this paper, we will review some of the recent progress in SFG studies on various interfaces involving polymers and biomolecules. The number of recent publications in this research field is vast, so we cannot cover all the topics in this review. Here we will focus on three most important advantages of SFG studies at interfaces. We will also only select a portion of the papers that illustrate these unique features of SFG. There are many other SFG review papers focusing on various topics published in the past years. 40, 45, 53, 73, 75–94
2. Introduction of IR + visible sum frequency generation (SFG) vibrational spectroscopy
As a surface sensitive nonlinear optical technique, SFG has been developed for more than twenty years. Agrowing number of research groups currently use the technique to study different systems. The theoretical background of SFG has been developed in early publications.35, 38, 47, 50, 95 New SFG data analysis methods have also been developed.96–102 In this review, we will not discuss all aspects of SFG theory but will only focus on some characteristics which provide SFG uniqueness in interfacial molecular level studies.
SFG is a second order nonlinear optical process. It occurs when two pulsed laser beams, one with a tunable IR frequency ωIR, and the other with a fixed visible frequency ωVIS, spatially and temporally overlap at an interface. A new signal beam is generated at a specific direction given by phase matching conditions with a frequency ωSFG = ωIR + ωVIS. The intensity of this sum frequency beam is resonantly enhanced when the tunable IR frequency equals a vibrational transition of a molecule. Therefore, SFG signal intensity plotted against the input IR frequency provides a vibrational spectrum. SFG can provide molecular level structural information because molecular vibrational modes are fingerprints of molecules. In narrowband SFG spectroscopy (usually using picosecond laser systems), an SFG vibrational spectrum is obtained by detecting SFG signal intensity at each IR input frequency and continuously tuning the IR frequency.40, 99 Recently, broadband SFG spectroscopy has been developed based on state-of-the-art femtosecond laser systems. In a broadband SFG system, the IR generated by the laser has a broad spectrum which covers the molecular vibrational signatures in a wide wavelength range (up to several hundreds of wave numbers). Overlapping this broad frequency IR beam and a narrow band visible beam, multiple vibrational features of the molecule at interfaces are enhanced simultaneously, generating spectrally separated SFG signal for different vibrational modes.37, 48, 57 With a spectrometer and a charge-coupled device (CCD) camera, these multiple molecular features at the interface can be obtained at the same time.
In addition to standard SFG which uses a frequency tunable IR beam and a fixed frequency visible beam, double resonance SFG (DRSFG) has been also developed. DRSFG uses a frequency tuneable IR beam and a frequency tuneable UV/visible beam to overlap at the interface to probe electronic and vibrational transitions of interfacial molecules.103–105 In this review, we will not discuss DRSFG in detail but will focus on the SFG experiments using regular IR+visible picosecond narrowband SFG spectrometers. Furthermore, SFG can also be combined with non-surface specific vibrational techniques such as coherent anti-Stokes Raman (CARS) scattering spectroscopy to characterize both surface and bulk structures of the same specimen in the same environment,106 but the details will not be discussed here.
The interfacial sensitivity of SFG is provided by the selection rule which is different from linear vibrational spectroscopy (e.g. IR spectroscopy or Raman spectroscopy). SFG is a second order nonlinear optical process in which the signal intensity is proportional to the square of the second order nonlinear optical susceptibility of the material χ(2) under the electric dipole approximation. χ(2) is a third rank tensor which changes sign under inversion operation: χ(2) (−r)=−χ(2) (r).107, 108 For materials with inversion symmetry, the relation χ(2)(−r)=χ(2)(r) holds. Comparing these two relations, we know χ(2)(r)= 0. This demonstrates that no SFG signal will be generated if the material has inversion symmetry under the electric dipole approximation. SFG signal can only be generated from a medium with no inversion symmetry. Most bulk materials have inversion symmetry and therefore do not generate SFG signal. However, at surfaces or interfaces where the centro-symmetry is broken, χ(2)(−r)≠ χ(2)(r), so the SFG process can occur. In the sample systems reviewed in this paper, signals contributed from surfaces or interfaces dominate the SFG spectra and bulk signal usually can be neglected. SFG can probe surfaces or interfaces which are accessible by visible and IR light in situ and in real time with sub-monolayer sensitivity.40, 91 SFG signals characterize the molecular vibrational modes of functional groups at interfaces, which can be used to probe the interfacial presence, coverage, and orientation of various functional groups. SFG can be applied to investigate interfacial structures at a molecular level even with very low interfacial coverage. Compared to other surface sensitive techniques, SFG can provide more detailed structural information about interfaces such as molecular orientation and orientation distribution.38, 51, 87 SFG can also provide interfacial molecular chirality information by utilizing polarized laser beams, which is impossible to obtain using other techniques such as circular dichroism (CD) spectroscopy.89, 109, 110
SFG signal intensity can be expressed as:38
| (1) |
Here IIR and Ivis are intensities of the input IR and visible beams, respectively. is the effective second order nonlinear optical susceptibility, which can be expressed as the sum of a nonresonant term and a resonant term:38
| (2) |
Here is the nonresonant contribution from the sample. The resonant contribution can be modeled as the sum of Lorentzians with signal strength or amplitude Aq, frequency ωq, and line width Γq. Equation (2) can be used to fit SFG spectrum in the experiment to obtain quantitative vibrational strength comparisons of different functional groups.
For an isotropic interface in the x-y plane, the effective second order nonlinear optical susceptibility components can be related to the second order nonlinear optical susceptibility components of the sample in the lab-fixed coordinating system:38
| (3) |
| (4) |
| (5) |
| (6) |
In these expressions, is the local nonlinear second order optical susceptibility of the material at the interface defined in the lab-fixed coordination.38 θIR and θvis are the incident angles of the input IR and visible beams vs. the surface normal, respectively. The angle θSFG is the output angle of SFG signal vs. the surface normal. Lii (i= x, y, z) is the Fresnel coefficient which is a function of beam input angles and the refractive indices of materials forming the interface.38 ωSF, ωvis and ωIR are frequencies of the sum frequency beam, the visible beam and the IR beam, respectively. Moreover, ssp, sps, pss and ppp are different polarization combinations of SFG measurement (ssp indicates s polarized signal, s polarized visible beam, and p polarized IR beam).
The measured SFG second order nonlinear optical susceptibility components defined in the lab-fixed coordination system can be related to the molecular hyperpolarizability components through molecular orientations considering the coordinate transformation.47
| (7) |
In this expression, N is the surface number density and ε0 is the vacuum permittivity. R is the transformation matrix from the molecular frame (a,b,c) to the lab frame (x,y,z). Hyperpolarizability component is the product of dipole transition moment component μk and the Raman tensor element αij, . The angle brackets here mean ensemble average, indicating that the macroscopic susceptibility is the ensemble average of the hyperpolarizability of each molecule projected to the lab frame multiplied by the total molecule density and divided by vacuum permittivity. R is usually a function of three angles, azimuthal angle φ, twist angle ψ, and tilt angle θ.47 Therefore, we have:
| (8) |
For an isotropic surface, the azimuthal angle can be averaged between 0 to 2π. Then the expression is reduced to
| (9) |
If the distribution of twist angle is considered to be random, then
| (10) |
Orientation analysis of different functional groups such as methyl (CH3),38, 50, 51 methylene (CH2),111, 112 aromatic C-H stretch,113–117 α-helical,55, 99 and β-sheet100, 109 has been reported and will be discussed later in this review.
3. Molecular presence and order can be monitored using SFG in situ
As discussed above, one of the unique properties of SFG is in situ measurement of buried interfaces, which allows minimum disturbance of the interfacial molecules compared to other analytical techniques. Therefore, such measurements can best reveal the interfacial molecular structures which can then be correlated to interfacial mechanisms and properties. SFG has been applied to study different interfaces in situ involving polymers and biomolecules, which will be discussed below.
3.1 Polymer interfaces
Polymer materials have wide applications in different environments and thus it is important to elucidate their interfacial structures and properties in different conditions. One common condition is polymer/water interface since many polymeric materials are extensively used in aqueous environments. For example, polymers such as poly(methacrylate)s, silicones, and polyurethanes are routinely used as implants in the human body118 and marine vessels use anti-biofouling polymer materials to reduce or prevent biofouling in the ocean.119–122 Therefore, it is important to characterize polymer surface structures in aqueous environments to understand and to help improve their performance. Extensive research has shown that SFG is a powerful tool to probe and analyze buried polymer/water interfaces. To the best of our knowledge, the first SFG study of polymer/water interface was carried out in 1997.123 In this work, Somorjai and his co-workers applied SFG to study the surface structure of polyurethane with poly(dimethylsiloxane) (PDMS) grafted as end groups in water. The results indicated that the investigated polymer surfaces underwent significant surface restructuring when transferred from air to water. In air, the hydrophobic PDMS segments covered most of the surface. In water, the hydrophilic polyurethane backbone tended to cover the surface while the hydrophobic PDMS end groups retreated from the surface. Since then, SFG has been widely applied to study surface structures of various polymers in water.
3.1.1 Poly(methacrylate)s
A report using SFG to study structural behavior of poly(2-hydroxyethyl)methacrylate (pHEMA) in hydrated and dehydrated states was published in 1999 by Chen et al.124 It was shown that in water, ethylene glycol groups which have stronger polarity tend to migrate to the polymer/water interface, contributing SFG signal at 2854 cm−1. At the same interface, methyl symmetric stretching and Fermi resonance signals at 2880 and 2945 cm−1 were observed which suggested that ordered methyl groups were also present at the interface. In air, the pHEMA surface was only covered by methyl groups, forming a hydrophobic conformation.
In 2001, the surface restructuring behavior of poly(methacrylate)s in water was studied in our group using SFG.125 It was found that surfaces of poly(methacrylate)s with different side chain lengths exhibited different restructuring behaviors when in contact with water. For poly(methyl methacrylate) (PMMA) which has the shortest side chain, a strong symmetric C-H stretching resonant signal at 2955 cm−1 (Figure 1a) suggested that ordered ester methyl groups dominated the PMMA/air interface. When contacted with water, the intensity of the dominating 2955 cm−1 ester methyl symmetric stretch signal decreased, which was mainly due to the change of refractive index, not the surface structural change. This shows that the PMMA surface is more or less similar in air and in water. For poly(n-butyl methacrylate) (PBMA), the ssp spectrum in air is dominated by a methyl symmetric stretching mode and a Fermi resonance at 2875 and 2940 cm−1, respectively (Figure 1b). In water, the ssp spectrum was dominated by a methyl asymmetric stretch at 2960 cm−1, while a methylene group asymmetric stretch signal at 2910 cm−1 was also detected. This suggests that PBMA undergoes surface restructuring when contacted with water. During water contact, ordered methylene groups segregate to the surface, while the different methyl signals detected from the PBMA surface in air and in water indicate the different orientations of surface methyl groups in different environments. The detailed orientation analysis on surface methyl groups will be discussed in the section 4 of this review paper. The ssp SFG spectrum collected from the poly(n-octyl methacrylate) (POMA) surface in air is dominated by contributions from both methylene and methyl groups, which indicates that both types of groups tend to adopt some surface order in air. While in contact with water, no SFG signal was observed, which suggested that functional groups at the POMA/water interface are randomly oriented. This study indicates that poly(methacrylate)s with different side chain lengths exhibit different surface restructuring behaviors in water. The surfaces are dominated with different surface functional groups with different orders.
Figure 1.
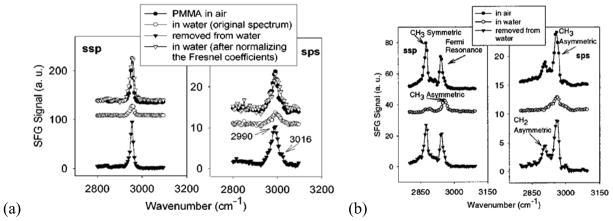
(a) SFG spectra of PMMA before, during, and after contacting water, left: ssp, right: sps. (b) SFG spectra of PBMA before, during, and after contacting water, left: ssp, right: sps. Reprinted with permission from ref. 125. (2001 American Chemical Society)
3.1.2 Poly(dimethyl siloxane) (PDMS)
PDMS and other silicone materials are widely used in applications to minimize biofouling through fouling release mechanisms.119, 120, 126 In situ studies on silicone materials in water are important in understanding such fouling release mechanisms and in developing novel coatings to control biofouling. SFG has been applied to study molecular surface structures of various types of PDMS materials in air and water.127 Four model PDMS materials including tetraethoxysilane-cured hydroxyl-terminated PDMS (TEOS-PDMS), platinum-cured vinyl-terminated PDMS (Pt-PDMS), platinum-cured vinyl-terminated poly(diphenylsiloxane)-co-poly(dimethylsiloxane) (PDPS-co-PDMS), and PDMS-co-polystyrene (PDMS-co-PS) have been investigated using SFG in air and water.127 The results suggested that all the above PDMS surfaces in air and in water are dominated by ordered methyl groups.127 Weak SFG signal generated by –Si-CH2-CH2- has been detected at 2865 and 2920 cm−1 from Pt-PDMS surface, indicating that in addition to the methyl group, these cross-linking groups were also present with order on the surface. SFG signal of the aromatic C-H stretching modes has also been detected from the PDMS-co-PS surface, showing that phenyl groups were also present on the copolymer surface. Moreover, surface restructuring of methyl groups for all the above PDMS samples has been observed after contacting with water and a non-polar solvent FC-75. This research demonstrated that SFG is sensitive to detect small surface structural differences on various PDMS surfaces in air and in water.
PDMS chain conformation in spread monolayers and multi-layers at the air/water interface has also been characterized using SFG.128 PDMS methyl groups were found to be completely disordered when the surface density was low. At higher surface densities, two adjacent methyl groups point into the air, with one nearly parallel to the interface and the other directed more to the surface normal. At even higher surface density (the first collapse regime), PDMS form odd-numbered multi-layers.
Fouling release coatings with biocide moieties covalently bound to a PDMS matrix have been developed as a new contact-active material for the control of marine biofouling through both fouling release and anti-fouling mechanisms. In order to design and optimize such coatings, it is necessary to study their surface structures and ensure that both the active biocide moieties (for anti-biofouling) and PDMS functional groups (for fouling release) are present on the surfaces and at the interfaces. SFG has been applied to study the surface and interfacial segregation of biocide moieties (e.g., triclosan (TCS) or tetradecyldimethyl (3-trimethoxysilylpropyl) ammonium chloride (C-14 QAS)) tethered to a PDMS matrix.129 PDMS coatings containing various amounts of TCS (0 wt % to 25 wt %) have been studied in air, in water, and after contact with water and exposed to air again. In air, SFG spectra showed that Si-CH3 group dominates the surface of all PDMS incorporated with TCS samples, similar to pure PDMS as discussed before. In water, SFG methyl intensities for all samples were weak, indicating TCS moieties may segregate more to the interface to reduce PDMS interfacial coverage. Samples were then removed from water and dried. Compared to the original surfaces, SFG methyl intensities collected at dried surfaces showed no significant change with TCS contents smaller than 8.75%, while a decrease in PDMS SFG methyl signal was observed for the PDMS samples with TCS contents higher than 8.75%. The SFG PDMS signal intensity decrease indicated that the surface segregation of TCS moieties occurred for the PDMS coating with higher than 8.75% TCS. This reduced the surface coverage of PDMS methyl groups, resulting in weaker PDMS methyl signals. These spectral features were well correlated to the anti-fouling activity testing experiments.129 The incorporation of more than 8.75% TCS moieties was needed to ensure adequate TCS interface segregation and good anti-fouling property. For the study of C-14 QAS, signal from biocides on surfaces can be directly detected for the PDMS materials in air and in water. SFG results can be correlated to the anti-fouling activities qualitatively.129
Various quaternary ammonium salt (QAS) moieties with different chain lengths (chain lengths of R1 and R2) have been chemically bound to PDMS as antimicrobial/anti-biofouling coatings.130 R1 is the longest aliphatic chain attached to quaternary ammonium nitrogen atom, R2 is the aliphatic chain separating the trimethoxy silane and the nitrogen atom. The surfaces and interfaces of such materials in air, water and nutrient growth medium (NGM) have been investigated using SFG. The results show that QAS molecules segregate to the coating surface in air for all the samples, while chain lengths of R1 and R2 affect surface structures. Surface restructuring of all samples was observed in water and NGM. Interfacial structures of different materials probed by SFG were well correlated to their antimicrobial activities. It was shown that the chain lengths of R1 and R2 can greatly affect the coating surface behavior in contact with NGM. When the R2 is short and the R1 is relatively long, the R1 chain can shield the quaternary ammonium nitrogen atom from water molecules, resulting in a hydrophobic surface which is affected by NGM less. In this case, QAS moieties can segregate to the interface and reduce biofouling (as shown in Figure 2a). When both R2 and R1 are short, the R1 chain cannot effectively protect the quaternary nitrogen atom from exposure. In this case, the coating surface is strongly affected by NGM. QAS moieties were not able to effectively segregate to the interfaces to reduce microbial biofilm formation. When R2 is long, it is more mobile, which may interact with R1 more easily. This may lead to the exposure of the quaternary nitrogen atom to water. In this case, NGM can effectively interact with the positively charged nitrogen atom, which may result in inhibition of interactions of microorganisms with the coating surface and reduce biofouling (as shown in Figure 2b).
Figure 2.
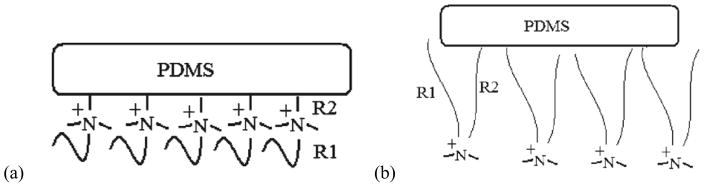
(a) Schematic for the molecular surface structure with N(R2)=3 in water (N(R2) is the number of carbon atoms for R2). (b) Schematic for the molecular surface structure with N(R2)=11 in water. Reprinted with permission from ref. 130. (2010 American Chemical Society)
Other compounds such as cationic, anionic, and zwitterionic polyelectrolytes (PEs) have also been tethered to a PDMS matrix through surface-initiated ultraviolet (UV) polymerization.131 SFG has been applied to examine the surface and interfacial molecular structures of such materials in situ. The studies show that –Si-CH3 groups segregate to PDMS surfaces along with grafted PEs in air for all material. In water, only the surface-tethered PE groups could be detected, indicating that the polymer surface restructured at the water interface. SFG has also been used to investigate PDMS surfaces that had been exposed to ultraviolet (UV) and oxygen plasma.132
In this section, we show that PDMS materials are widely used for fouling release. SFG can be used to examine interfacial structures of different PDMS materials in water in situ, which may help to develop molecular level understanding of PDMS fouling release mechanism and help to rational design fouling release materials.
3.1.3 Polymer/Silane Interfaces
Another type of important interface to study is the buried polymer/polymer interface. Polymer materials such as polyurethanes, silicones, and epoxies are widely used as adhesives in industry and everyday life. Adhesion properties of these polymer adhesives are determined by their interfacial structures. Therefore, characterizing interfacial structures between adhesives and different substrates, especially polymers, is important for understanding adhesion mechanisms. The related research can also help to develop better adhesives or adhesion promoters (small molecules added to the polymer adhesive to enhance its adhesion performance).
Silane molecules are widely used as adhesion promoters in industry. In order to study their adhesion promotion behavior at polymer/adhesive interfaces, it is necessary to investigate the molecular structures of the polymer/silane interfaces first. Silane behaviors at different polymer interfaces have been systemically investigated using SFG. For example, Chen et al. studied buried polymer/silane interfacial molecular structures using SFG.133 In this study, the polymers investigated include deuterated polymethyl methacrylate (d-PMMA) and deuterated polystyrene (d-PS), while the silanes examined includen-octadecyltrichlorosilane (OTCS), n-octadecyltrimethoxysilane (OTMS), and (3-aminopropyl)trimethoxysilane (ATMS). The SFG results show that silane molecules adopt different molecular conformations at different polymer/silane interfaces due to different interfacial molecular interactions. At the d-PMMA/OTCS interface, methyl end groups in OTCS favorably interact with the PMMA surface dominated by ester methyl groups and tend to order at the interface. However, OTMS methoxy head groups tend to interact with PMMA ester methyl groups and show some order at the d-PMMA/OTMS interface. At the d-PS/OTCS interface, methylene group from the OTCS backbone dominated the SFG spectra. At the d-PS/OTMS interface, both methylene backbone and methoxy headgroups from the silane could be detected at the interface. A schematic representation of the polymer/silane interfaces determined from the above results using SFG is shown in Figure 3. Different from the methyl terminated silanes discussed above, after the ATMS molecules contacted a polymer surface, the polymer/ATMS interface lost order quickly, leading to the disappearance of the SFG signal. Different interfacial behaviors of ATMS compared to OTMS and OTCS are likely due to the nature of the amino endgroup.
Figure 3.

Schematic demonstrations of different silane molecules at different polymer/silane interfaces. The boxes represent the bulk silane liquid. The molecular segments including headgroup, backbone, and endgroup of silane molecules are described by the figure legend. Reprinted with permission from ref. 133. (2003 American Chemical Society)
Loch et al. studied hydrogen bond formation at polyethylene terephthalate (PET)/silane interfaces.134 Ester carbonyl groups on a PET surface were found to form hydrogen bonds with amino endgroups on ATMS based on a slightly red-shift in SFG signal contributed by the PET ester carbonyl stretch. The shift was not observed at the interfaces between PET and (3-glycidoxypropyl)-trimethoxysilane (γ-GPS) or between PET and n-butyltrimethoxysilane (BTMS). The latter two silanes, γ-GPS and BTMS, do not have hydrogen bond donors, therefore they cannot form hydrogen bonds with PET. Absolute orientation studies of the functional groups at the PET/ATMS interface using nonresonant SFG signal generated from a TiO2 thin film suggested that the methoxy headgroups of ATMS oriented toward the silane bulk while the amino endgroups were oriented toward the interface. Differently, at the PET/BTMS interface, the BTMS methoxy headgroups face toward the interface.
SFG was also utilized to monitor silane diffusion into polymer matrices. Silanes such as ATMS, N-(2-aminoethyl)-3-amino-propyltrimethoxysilane (AATMS), and γ-GPS were observed to diffuse into polymer films.135,136 The details of these results will be discussed later in this paper (section 5.1).
It was reported that the mixture of γ-GPS and methylvinylsiloxanol (MVS) (mixture abbreviation: SAPM) could improve the adhesion of an addition-curing silicone elastomer to plastics and metal substrates better than γ-GPS alone.137 To further understand the adhesion mechanism, SFG has been applied to study the interface between PET with deuterated ethylene glycol subunits (d4-PET) and SAPMs with different γ-GPS/MVS mixing ratios.138 When SAPM with a γ-GPS/MVS ratio of 1:1 (w/w) was used, silane methoxy headgroups adopted greater net orientational order along the surface normal than that at d4-PET/γ-GPS interface. This provided insight into the correlation between strong adhesion and specific molecular features such as methoxy group ordering at the interfaces for curable silicone adhesives.
We have shown that SFG can be used to study the behavior of different silanes at various polymer interfaces. Molecular level information about silane functional groups at polymer interfaces can be examined in situ. The SFG study of silane behaviors at polymer/silane interfaces can help to understand adhesion promoting mechanisms by correlating molecular structure at buried interfaces to adhesion properties. However, it is necessary to study the polymer/adhesive (incorporated with silanes or silane-MVS mixtures) interfaces directly, as in real conditions.
3.1.4 Polymer/adhesive interfaces
SFG has been used to study silicone adhesives with incorporated silane molecules which more closely resemble industrial conditions.139, 140 The silane behavior at interfaces of cured and uncured silicone has been studied using SFG. Three different silanes: γ-GPS, OTMS and (tridecafluoro-1,1,2,2-tetrahydroctyl) trimethoxysilane (TDFTMS), with the same methoxy headgroups but different backbone and endgroups were used to incorporate into silicone matrix in contact with polymer substrate PET.139 For the PET/silane or PET/(silane+MVS) interfaces, γ-GPS methoxy groups were found to be ordered and the presence of MVS increased the interfacial order. However, MVS tended to decrease the order of methoxy group of OTMS18C while no change was observed for TDFTMS case at the PET/silane interfaces. After incorporating the three silanes or their mixtures into silicone, before curing, γ-GPS+MVS is the only mixture that shows significant methoxy group order at the PET/uncured PDMS interface. After curing PDMS, the methoxy group was shown to order at the PET/γ-GPS incorporated silicone interface and the PET/(γ-GPS+MVS) incorporated silicone interface. The other two silanes and silane MVS mixtures did not order at the silicone/PET interfaces. The γ-GPS+MVS mixture is a known adhesion promoter for silicone to PET polymer. Therefore, the adhesion mechanism is related to the methoxy group ordering, which was only present at interfaces between PET and γ-GPS or γ-GPS+MVS mixture incorporated silicone elastomer.
Following the above silane endgroup study, silane headgroups have also been compared to correlate the headgroup effect to the adhesion mechanism.140 Three different methoxysilanes including γ-GPS, (3-glycidoxypropyl) methyl-dimethoxysilane (γ-GPMS), and (3-glycidoxypropyl) dimethyl-methoxysilane (γ-GPDMS) which have the same endgroup and backbone, but different headgroups were investigated. At the PET/silane and PET/(silane+MVS) interfaces, it was found that MVS greatly increased the methoxy group ordering in γ-GPS at the interface, slightly affected methoxy group ordering in γ-GPMS at the interface, and disordered the methoxy group ordering in γ-GPDMS at the interface. When mixed with silicone elastomer and cure the samples, the SFG signal probed from methoxy groups at the PET/PDMS (incorporated with γ-GPS alone) interface was stronger than that at the PET/PDMS (incorporated with γ-GPS+MVS mixture) interface. For γ-GPMS case, the SFG signal from methoxy groups at the PET/PDMS incorporated with silane with or without MVS interfaces were similar. No methoxy SFG signal could be detected for the addition of γ-GPDMS and γ-GPDMS+MVS into PDMS at the PET/PDMS interface after curing. Strong adhesion was related to the interfacial segregation and ordering of methoxy groups at PET/silicone interfaces. The methoxy signal strength also decreased in the curing process, which was related to interfacial chemical reactions and/or diffusion, leading to stronger adhesion. A related study on the ethoxysilane headgroup effect on PET/PDMS adhesion found a quantitative correlation between headgroup methyl signal change and adhesion strength. 141
Epoxy materials are widely used as packaging materials in microelectronic industry and silane materials are extensively used to enhance adhesion of epoxy adhesives. SFG studies have been applied to study silane incorporated into both model epoxies and commercial epoxies.142 Bisphenol A digylcidyl ether (BADGE) was investigated at PET interfaces with and without silane adhesion modifiers. The molecular structures at PET/BADGE and PET/BADGE silane mixture interfaces were similar before curing. After curing, small amounts of certain silane molecules altered the interfacial structures. OTMS(18C) methyl endgroup tended to become highly ordered at the PET/BADGE interface. The ordering of methyl groups at the interfaces was correlated with weak adhesion while disordered interfacial structures were correlated with strong adhesion. The results were further supported by studies of two commercial epoxies at different polymer interfaces including PET and polystyrene (PS).142 Molecular level studies on interfacial structures of epoxy materials provide in-depth understanding of interfacial properties and performance of packaging materials.
The above studies show that SFG can be used to understand interfacial molecular structures of polymers and adhesive materials in situ, which may lead to a better understanding of adhesion mechanisms. It will also help to develop adhesive and adhesion promoters with better performance.
3.2 Biological interfaces
The importance, limitations and potential applications of using SFG to study biological interfaces were demonstrated in an early publication.143 SFG studies on biological interfaces have grown tremendously in recent years alongside rapid developments in the field. With the advantage of in situ detection, SFG can be used to characterize low concentrations of different biomolecules such as lipids, peptides and proteins at various interfaces. For example, SFG can be used to study membrane orientations of antimicrobial peptides at a peptide solution concentration close to or even below the minimum inhibitory concentration.144, 145
3.2.1 SFG studies on protein adsorption
Protein adsorption is an important process that occurs on biomedical device surfaces. SFG has been developed as a unique tool to study protein adsorption since it can provide in situ interfacial molecular understanding. In 2002, Wang et al. reported an SFG study of bovine serum albumin (BSA) adsorption at different substrates including silica, PS and PMMA.146 SFG spectra were collected from adsorbed proteins exposed to different environments including air, water, benzene, carbon tetrachloride, and FC-75 (a hydrophobic fluorinated solvent from 3M).146 It was found that adsorbed BSA molecule can exhibit different structures at different interfacial environments. This study was focused on SFG signals detected in the C-H stretching frequency range (between 2800 to 3100 cm−1). The results indicated that the adsorbed BSA tended to form a hydrophobic configuration between two hydrophobic media, whereas a hydrophilic configuration was observed between two hydrophilic media. At the interface of two media with different hydrophobicities, BSA configuration prefers to have hydrophobic groups aligned. These results show that the hydrophobic effect is a significant factor directing BSA adsorption.
A thin film model was used to interpret SFG spectra of BSA adsorption in more detail.147 An adsorbed protein layer is usually several to tens of nanometers thick, which is different from a traditional sharp interface between two materials. In the thin film model, SFG signal is considered to be generated from the entire protein layer adsorbed at the interface. Nonlocal contributions to SFG spectra of interfacial adsorbed protein layer usually can be ignored.147, 148
SFG studies have also been carried out to investigate effects of protein solution pH and protein interfacial coverage on protein interfacial structures. It was demonstrated that SFG C-H stretching signals collected from the BSA solution/air interface and BSA solution/d-PS buried interface were similar at each protein solution pH. Although the detected SFG spectra were different at different protein solution pH values, the contributions from the C-H stretching signals were very similar. The differences were due to the interferences between the C-H signal from BSA and the O-H signal from water. At different protein solution pH values, the interfacial O-H stretching signals from water were very different, but the protein C-H stretching signals were not.147 However, SFG spectra in the C-H stretching frequency range were different at the BSA solution/different substrate interfaces, due to different interfacial interactions.148 Related studies in the C-H stretching frequency region have also been reported on BSA as a function of surface coverage at the air/solution interface.149 It has also been demonstrated that SFG can study isotope labeled proteins for their adsorption.150 Polarization mapping, a data collection and analysis method based on SFG spectroscopy, has been developed to more reliably fit the SFG spectra detected from complex samples such as interfacial proteins and polymers.97
SFG has also been used to study adsorption behavior of peptides, serving as a model for complicated proteins.71, 151–153 As an example, Mermut et al. published a study which combines QCM, AFM, and SFG to elucidate the molecular adsorption behavior of a 14-amino acid amphiphilic peptide LK14 on hydrophilic silica and hydrophobic PS surfaces.71 The results suggest that LK14 peptide have different adsorption behavior at surfaces with different hydrophobicities.
We have shown that SFG can be applied to study protein or peptide adsorption on various substrates including polymer materials in situ. However, SFG signals in C-H region are usually contributed from protein side chains. In order to characterize more details of protein structures at interfaces, it is necessary to examine protein backbones.
Amide I (mainly contributed from the C=O stretching) SFG study can provide deeper understanding of protein structures at interfaces because it can directly probe protein backbone structure and will not have interference with any water O-H stretching signal which makes spectral analysis on C-H stretching signals difficult.147 In 2003, SFG detection of interfacial protein amide I signal in the C=O stretching frequency range was reported.154 The feasibility of using the near total-internal-reflection geometry to monitor SFG amide I signals contributed from interfacial proteins was demonstrated.154
SFG has been used to investigate fibrinogen adsorption at polymer/protein solution interfaces in both the C-H and C=O stretching frequency ranges. We will discuss these studies in later section 5.2 as examples for time-dependent SFG investigations. The above studies show that SFG is a powerful tool to study protein adsorption at various interfaces in situ.
3.2.2 SFG studies on model cell membranes
Cell membrane is an important biological interface in nature. The interaction of biomolecules and other small molecules with lipid membranes is of general interest in biology, biophysics and medical research. For example, antimicrobial peptides (AMPs) interact with certain types of membrane lipid components in a specific way. Molecular level understanding of such interactions in situ is crucial to reveal general peptide-membrane interaction mechanisms. SFG has proved to be powerful in monitoring such interactions. More specifically, SFG has been applied to investigate the molecular behavior of a novel membrane-active antimicrobial arylamide oligomer 1,155 and peptides such as magainin 2,156 melittin,55 MSI594,157 tachyplesin I,100 pexiganan (MSI-78),144 cell penetrating peptide,145 alamethicin,158 and amyloid polypeptide159, 160 in model cell membranes. Supported lipid bilayers have been extensively used as model cell membranes in these studies, which can be prepared by the Langmuir-Blodgett-Schaeffer method.161 The detailed results reported in these publications will be discussed in the following sections as examples of orientation analysis and time dependent SFG investigations. Here we will just briefly discuss the peak assignments of amide I signal for different secondary structures in SFG experiments.
Combined with IR and Raman studies, it was shown that in SFG spectra, the α-helical signal is centered at ~1650 cm−1, whereas the β-sheet structures have featured peaks centered at ~1635 and ~1685 cm−1. Such assignments were proved using two model peptides with well characterized structures: MSI594, which adopts an α-helical structure at the interface, and tachyplesin I, which has a rigid antiparallel β-sheet structure both in solution and at interfaces.157 Recent studies on the peptide alamethicin indicate that when an α-helix connects to a 310-helix, it may contribute to a peak centered at 1670 cm−1. The 310-helix itself may also contribute to a 1635 cm−1 peak.56, 158
It is also possible to use SFG to analyze the interaction of larger molecules such as proteins with lipids. The interaction of proteins with artificial lipid membranes will be discussed in section 4.2 as examples for SFG molecular orientation study. Here we will just use one protein as an example for the interfacial presence study.
Human islet amyloid polypeptide (hIAPP) has been studied using polarized SFG at the air/water interface.159 hIAPP consists of 37 amino acids and is intrinsically disordered. Upon interaction with cell membranes, hIAPP can misfold into β-sheet aggregate structure. This change is considered to be associated with human type II diabetes. However, rat islet amyloid polypeptide (rIAPP), which differs from hIAPP by six amino acids, does not misfold into β-sheet structures at cell membranes. A fundamental study of interactions between cell membrane and hIAPP at the molecular level can help to better reveal the molecular pathology of type II diabetes. SFG spectra in the amide I region of hIAPP and rIAPP were collected in the presence and absence of 1,2-dipalmitoyl-sn-glycero-3-[phospho-rac-(1-glycerol)] (DPPG) lipid at the air/water and air/D2O interfaces under ssp and psp polarizations. In the psp polarization, which is more sensitive to chiral structures, SFG spectra allowed the assignment of ~1620 cm−1 and ~1660 cm−1 bands to asymmetric and symmetric stretches of parallel β-sheet, respectively. Further, β-sheet psp signal could not be detected for hIAPP unless DPPG was present, and could not be detected for rIAPP either in the presence or absence of DPPG (Figure 4). For ssp spectrum, which is more sensitive for achiral structures, the only case with spectral change in shape and peak position over time was when both hIAPP and DPPG were present. These results suggest that the interaction between membrane lipids and hIAPP leads to misfolding of hIAPP to ordered parallel β-sheet structures.159 Another SFG study on hIAPPs combined chiral SFG and ab initio quantum chemistry calculations to analyze its orientation at the lipid/aqueous interface.160
Figure 4.

The psp spectra of hIAPP without DPPG (t=0 and 10 h) and after addition of DPPG (t=10h) at the (a) air/D2O and (b) air/H2O interfaces. The psp spectra of rIAPP without DPPG (t=0 and 10 h) and after addition of DPPG (t=10h) at the (a) air/D2O and (b) air/H2O interfaces. Reprinted with permission from ref. 159. (2010 American Chemical Society)
3.2.3 SFG studies on DNAs
DNA is another type of important biological molecule besides peptides and proteins and has been extensively studied. However, many of the fundamental principles and behaviors of DNA such as oligonucleotide hybridization, adsorption orientation, etc. are not clearly understood due to the lack of appropriate analytical techniques. The uniqueness of SFG in the study of DNA lies in the ability to provide in situ structural information at biological interfaces. C-H oscillators such as methyl and methylene groups, together with carbonyl and amine groups in DNA offer a wide vibrational spectral window for SFG studies on DNA. Studies have shown that SFG can provide important structural information of DNA strands at interfaces.
DNA can form complexes with cationic lipids and can enter cells through endocytosis. The study of molecular level interactions of DNA/lipid association has the potential to improve gene therapy. In an SFG study of DNA/lipid complex by Wurpel et al., the electric screening of a 1,2-dipalmitoyl-3-trimethylammonium propane (DPTAP) monolayer by polyanionic DNA was observed indirectly by monitoring the water signal decrease.162 Water at the charged lipid interface is more ordered than in the bulk through the alignment of water dipoles by the interfacial electric field. The addition of ions such as NaCl or λ-phage DNA resulted in a screening of the positively charged lipid and changed the interfacial water ordering. As the concentration of NaCl or λ-phage increased, SFG water signals decreased. Further, it was shown that as the water reoriented, less than one monolayer of water remained between the DNA and DPTAP monolayers.162 A schematic representation is shown in Figure 5. Further studies focused on DNA interaction with different lipids including DPTAP, diC14-amidine and a zwitterionic lipid 1,2-dipalmitoyl-sn-glycero-3-phosphocholine (DPPC) in the presence and absence of calcium ion.163 Again, the DNA/lipid complexation was monitored indirectly through the O-D stretching signal of D2O. It was shown that D2O structure differs between DNA adsorbed to cationic lipid monolayers and to zwitterionic lipid monolayers. The interaction of DNA and diC14-amidine could be monitored in the C-H stretching frequency range directly, and it suggested that the adsorption of DNA to the lipid increases the ordering of lipid tails.163
Figure 5.
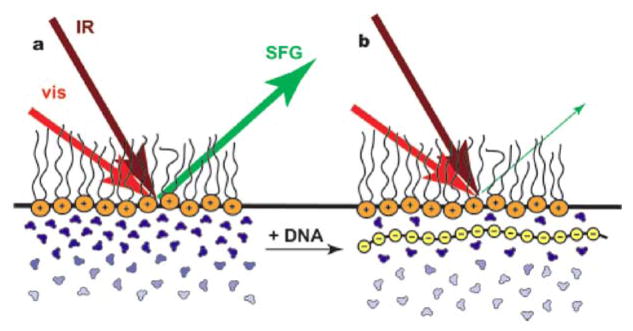
(a) The presence of a cationic lipid monolayer at the air-water interface aligns the first few water layers, generating strong vibrational SFG signal. (b) The binding of DNA and cationic lipids screens the electric charges, disorders the water molecules, leads to a sharp decrease of the water signal. Reprinted with permission from ref. 162. (2007 American Chemical Society)
The impact of metal monovalent and divalent cations on 20-mer single-stranded DNA (ssDNA) and double-stranded DNA (dsDNA) covalently bound to silicon (111) surface has recently been investigated using SFG.164 The molecular structural change of DNA was indirectly monitored through the change in a silane linker layer. The SFG intensity ratio of asymmetric methyl and methylene C-H stretches (r−/d−) provided a good measurement of the molecular order of the silane self-assembled monolayer (SAM) linker. The presence of monovalent cations such as Na+ and K+ increased ordering along the ssDNA strands, whereas the divalent cations such as Mg2+ and Ca2+ disordered the strands. Divalent cations have stronger affinities with DNA, so they tend to distort the ssDNA strands at the interface more than monovalent cations. It was also reported that upon hybridization, the formerly discussed trend was reversed. Moreover, the distortion of dsDNA in the presence of divalent cations is much less than that of ssDNA, which may be attributed to the better rigidity of the duplex compared to single strands. A hypothetical representation of cation-induced structural changes of a DNA/linker monolayer on a silicon surface is shown in Figure 6.
Figure 6.
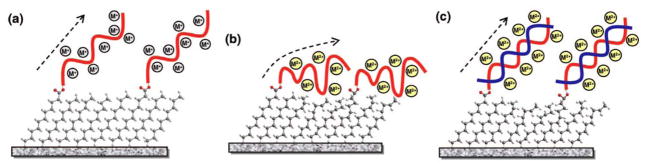
(a) Monovalent cations do not affect the geometry of ssDNA. The underlying linker monolayer keeps its ordered conformation. (b) Divalent cations induce ssDNA deformation and the deformation perturbs the linker monolayer. (c) Hybridization in the presence of divalent cations does not introduce further disruption to the linker monolayer structure. Reprinted with permission from ref. 164. (2008 American Chemical Society)
Gold has also been used as a DNA immobilization substrate. Howell et al. chemically immobilized ssDNA on gold substrate via the thiol groups and studied the air/solid, phosphate buffer saline solution (PBS)/solid, and D2O/solid interfaces.165 Adenine and thymine homo-oligonucleotides showed significant structure differences in the C-H stretching frequency region in different environments such as air and D2O. The signal in the amide I vibrational region indicated that in air, non-thiolated DNA tended to lie down on the gold surface whereas thiol-linked DNA tended to arrange in a brush-like structure. Thiol-linked DNA also showed significant structural differences in air and D2O.165 Other SFG studies of DNA,166, 167 including on platinum substrates have also been reported.168
To summarize, SFG studies on DNA can help to develop molecular level understanding of DNA structures at different biological surfaces or interfaces in situ.
3.3 Other interfaces
In this paper we only focus on SFG studies of certain polymer and biological interfaces. Here we showed that SFG is a unique interfacial selective technique that can probe interfacial molecular presence in situ with high sensitivity. The in situ detection capability ensures that the interfaces do not need to be broken to expose to different environments. SFG can probe interfaces that are accessible to IR and visible laser beams, providing vibrational measurements similar to IR spectroscopy for ordered molecular functional groups, and giving interfacial molecular presence information with high sensitivity. In addition to the interfaces discussed above, recent research has also demonstrated this unique advantage of SFG through studies on polymer/metal interfaces,169, 170 polymer/polymer interfaces,171 water/oil interfaces,172, 173 water/vapor interfaces,88, 174 liquid/gas interfaces,175, 176 solid/vapor interfaces,177 etc. It is difficult to summarize all such published SFG research. Other review papers in the field may help to gain further understanding of the technique and its applications on many other different systems.40, 45, 53, 73, 75–94
4. Molecular orientations at interfaces can be derived using polarized SFG
In situ molecular level detection of the interfacial presence of functional groups is just one of the unique advantages of SFG. Here we will briefly introduce the theory of using SFG to measure interfacial molecular orientations of certain functional groups, which is another important advantage that further demonstrates the uniqueness of SFG as a quantitative interfacial analysis technique.
4.1 C-H functional group orientation analysis
The orientation analysis of functional groups at interfaces using SFG lies in the relation between interfacial second order nonlinear optical susceptibility and molecular hyperpolarizability demonstrated in equation (8). Three angles, azimuthal angle φ, twist angle ψ, and tilt angle θ are used in Euclidean space to describe the orientation of a rigid body. Therefore, the orientation of a functional group, such as a methyl group, pointing in any direction, can be described using these three angles in the lab frame. The relationship between and as described in equation (8) has been deduced systematically.95 It has also been shown that the relationship can be greatly simplified based on certain assumptions or prior knowledge. An example is a functional group of a molecule on an isotropic surface with a free rotation symmetry vs. the principle axis, where azimuthal angle φ and twist angle ψ can be averaged from 0 to 2π.95 To deduce the orientation angles, sometimes certain components of the molecular hyperpolarizability (or ) need to be known. In such cases the bond additivity method is usually needed to obtain from measured or calculated Raman polarizability component αij and dipole transition moment component μk of the vibration mode under study.111
What we can quantitatively obtain from an SFG spectrum after fitting the signal using the Lorentzian equation (2) is the effective second order nonlinear optical susceptibility . Using the relations described in equations (3)~(6), can be correlated to through Fresnel coefficients, which can be calculated at certain input angles for certain species with known refractive indices. Moreover, can be further correlated to molecular hyperpolarizability through surface number density N of the functional group and the three orientation angles as described in equation (8)~(10). The value of N may be estimated, but for many cases it is not required for orientation determination. “N” can be eliminated by measuring orientation using the ratio of certain components of the nonlinear susceptibility, which can be measured using different polarizations of the input and output laser beams in the SFG experiment, or using the SFG signals detected in the same polarization but belonging to different vibrational modes. To further explain these approaches, we will use PMMA as an example to show how to derive the surface molecular orientation of an ester methyl group in air.
Wang et al. have shown that ester methyl groups dominate the PMMA surface in air.51 Methyl groups can be treated as holding C3v symmetry in most cases. Molecular hyperpolarizability can be simplified based on the molecular symmetry.111 In SFG, such hyperpolarizability can be further simplified due to the isotropic surface and rotational symmetry for the C3v symmetry. For example, in C3v, the non-vanishing hyperpolarizability components for the C-H symmetric stretches are and , while for asymmetric stretch, the only non-zero component is .51, 95 For the second-order nonlinear optical susceptibility of C-H stretching vibrational modes of molecules with C3v symmetry, there are only four non-vanishing components: .49 The relation between different second order nonlinear optical susceptibility components in the lab frame and molecular hyperpolarizability components in the molecular coordinate system can be simplified and expressed as:38, 51, 95
For the symmetric C-H stretch:
| (11) |
| (12) |
| (13) |
For the asymmetric stretch:
| (14) |
| (15) |
| (16) |
Here ‘s’ and ‘as’ indicate symmetric and asymmetric C-H stretching modes, respectively. For symmetric C-H stretching modes, . In these equations, it is assumed that the polymer film is isotropic, and the ester methyl group can rotate freely around the principle axis. Accordingly, the azimuthal angle φ and twist angle ψ are averaged from 0 to 2π. If we assume the distribution of θ is a δ-function, then 〈cosθ〉 and 〈cos3θ〉 can be substituted by cosθ and cos3θ. Sometimes, the orientation angle distribution can be modeled by a Gaussian function: f(θ)= C·exp[−(θ − θ0)2/(2σ2)], where θ0 is the center of distribution, σ is the root-mean-square distribution width.178, 179 Here the asymmetric stretching signal is considered to be contributed by the two degenerate modes.180 Although some reports showed that the two modes may actually not be degenerate,181 for most cases we believe that it is reasonable to use the above equations for methyl group orientation studies by assuming that the asymmetric stretching modes are degenerate.51, 182
An important relation for the above equations is that and always have the opposite sign as and . This indicates under above assumptions, symmetric and asymmetric modes of a methyl group always negatively interfere with each other in an ssp spectrum. It is difficult to accurately measure the number density of surface molecules probed in the SFG experiment. Therefore, it is usually necessary to eliminate the number density N when using the above equations for orientation calculations. By taking the ratio of, for example, , the number density N and molecular hyperpolarizability βccc can be eliminated. Thus the ratio of symmetric C-H stretch can be expressed as a function of molecular tilt angle θ and r. The value of r can be obtained by calculation.111 However, sometimes it is difficult to obtain an accurate value of r. Instead, a range of r values were reported in the literature.38 To avoid the use of r, one can choose to use the hyperpolarizability component ratio of the asymmetric C-H stretch such as , which is only a function of θ. To deduce the tilt angle θ, can be plotted as a function of θ (equation (14) and (15)). In SFG experiments, (e.g., and ) can be measured in different polarizations (e.g. ssp and sps polarizations), and the value can be obtained through spectral fitting using equation (2). Further, value can be associated to value through equations (3)–(6). Finally, according to the value deduced from measurement and plotted as the function of θ (obtained from equation (14) and (15)), the tilt angle θ can be deduced.
Using the asymmetric stretching mode of PMMA and the method mentioned above, the PMMA ester methyl group surface orientation can be derived to be between 33° vs. surface normal with a δ-angle distribution and 0° vs. surface normal with a Gaussian angle distribution of 31°.51 Interfacial structures of poly(n-butyl methacrylate) (PBMA)/air and PBMA/water have also been investigated by Wang, et al.182 It was found that side chain methyl groups of PBMA dominated the surfaces in both air and water. As shown in Figure 7, the peak at 2960 cm−1 is assigned to the side chain methyl asymmetric stretch. It can be clearly resolved in the sps spectra in both air and water, and in the ssp spectrum in water. In ssp spectrum collected in air, this peak shows as a shoulder of the peak at 2940 cm−1, which was assigned to a methyl Fermi resonance. Side chain methyl group orientation can be measured using obtained from fitting the ssp and sps spectra. The tilt angle of the side chain methyl group of PBMA in air assuming a δ-distribution is 37° vs. the surface normal, while in water, the angle is measured to be 57° (Figure 8). This indicates that side chain methyl group tends to stand up in air, while tilts more toward the surface in water. Further analysis showed that PBMA side chain methyl group orientation distribution was narrower in water than in air, which suggested that the PBMA surface became more ordered in water.182
Figure 7.

SFG spectra of PBMA in air and water for (a) ssp, (b) sps polarization combinations. Reprinted with permission from ref. 182. (2002 American Chemical Society)
Figure 8.

Calculated |A yyz,as/Ayzy,as| of methyl group as a function of orientation angle θ0 and angle distribution σ. Here the asymmetric vibrational mode of side chain methyl has the same peak width under different polarization measurements, thus . Reprinted with permission from ref. 182. (2002 American Chemical Society)
Similar orientation analysis method has also been developed for methylene group, which has C2v symmetry. For a system with no azimuthal angle and twist angle dependence, the non-vanishing susceptibilities for methylene group are:183
For symmetric stretch:
| (17) |
| (18) |
| (19) |
For asymmetric stretch:
| (20) |
| (21) |
| (22) |
From these equations we can find that the methylene asymmetric stretch bears the same expressions as those in methyl group, except the difference of hyperpolarizability and . These equations can be further simplified through the relations of elements.111 For example, the relation eliminates all terms with ,111,183 and all symmetric susceptibilities only have a 〈cosθ〉 term or vanish. The relation further associates asymmetric stretches with symmetric stretches.111, 183 The fact that ‘opposite signs need to be applied to symmetric and asymmetric stretches’ also holds here for methylene group as for methyl group.
Orientation analysis methods have been developed for studying more complicated methyl groups. For example, a (CH3)2X moiety with two neighboring methyl groups has a fixed bond angle of ~112° on average. In this case the vibrational hyperpolarizabilities of the two individual methyl groups need to be combined in the molecular frame. The transformation from lab frame to molecular frame can be achieved by considering each methyl group as a unit atom. Therefore, each methyl group still has a C3v symmetry, but the two methyl groups connected to the same Si atom are considered to adopt a C2v symmetry. A vector bisecting the angle between two methyl groups is then defined as the molecular orientation axis for twist and tilt angle (Figure 9). Such SFG data analysis results have been reported.98 Using such a method, a mixture of 2-propanol and water was studied at the liquid/vapor interface.98 At low 2-propanol mole fractions (xiso≤0.025), the two methyl groups tend to lie down on the liquid/vapor interface. As the 2-propanol fraction was increased in the mixture (0.025≤xiso≤0.68), one methyl group started to orientate towards the vapor, while the other one oriented nearly parallel with the interface but pointed down into the mixture slightly. At higher 2-propanol fractions in the mixture (xiso≥0.68), one methyl group was nearly parallel with surface normal to the vapor, whereas the other one was slightly orientated downward to the mixture (Figure 10). A recent study used a similar method to determine PDMS methyl group orientation at buried silica/PDMS and PET/PDMS interfaces considering both tilt angle and twist angle.184 It was found that Si(CH3)2 groups tend to have large tilt angles with small twist angles at the PET/PDMS interface. At the silica/PDMS interface, Si(CH3)2 groups tend to adopt a broad distribution of tilt angles with large twist angles. Furthermore, the absolute orientations of PDMS Si(CH3)2 groups have also been determined at various interfaces. It was shown that PDMS Si(CH3)2 groups orientate towards the PDMS bulk rather than towards the substrates at both interfaces.184
Figure 9.
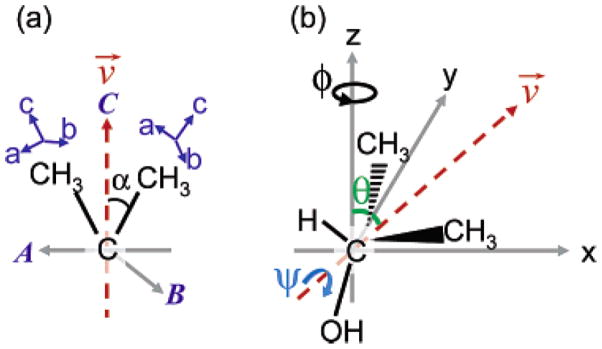
(a) Molecular frame coordinates: the (a,b,c) axis is for methyl group fixed coordinates and the (A,B,C) axis is for isopropyl group fixed coordinates. The C axis bisects the two methyl groups (vector v), and the A axis is set in the plane of the two methyl groups. (b) The geometry of 2-propanol in laboratory frame coordinates. θ, ψ and φ are the tilt angle, the twist angle about v and the azimuthal angle about z-axis for isopropyl group. Reprinted with permission from ref. 98. (2006 American Chemical Society)
Figure 10.
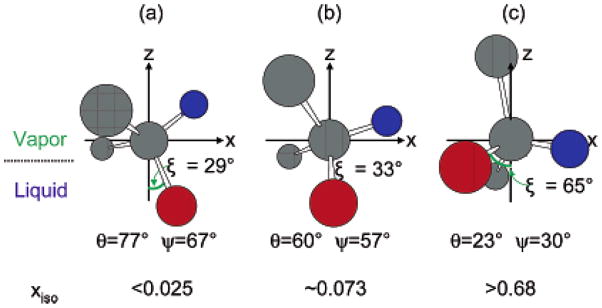
Molecular orientation of 2-propanol at the liquid/vapor interface; gray, red, and blue spheres denote carbon, oxygen, and hydrogen atoms, respectively. (a) xiso<0.025, (b) xiso=0.073, (c) xiso>0.68. Reprinted with permission from ref. 98. (2006 American Chemical Society)
It is also possible to achieve orientation analysis of (CH3)2X using a single methyl orientation method plus some basic assumptions that simplify the system. For example, in order to calculate the orientation angle of PDMS which has two CH3 groups connected to a Si atom, at the surface of water with low PDMS coverage, the polymer backbone can be considered to lie parallel onto the surface. This means that the (CH3)-Si-(CH3) plane is perpendicular to the water surface. Therefore, the two methyl groups tilt angle 〈θ1〉 and 〈θ2〉 satisfy 〈θ1〉 = 110° − 〈θ2〉. Under this assumption, it is possible to use a single methyl group orientation equation to obtain the PDMS methyl group orientation on the water surface.128 PDMS chain conformation at the air/water interface has been derived based on this model, and the results have been discussed in 3.1.2.128
In addition to methyl and methylene group orientations, SFG orientation analysis for other molecular functional groups has been performed. For example, the orientation analysis of phenyl ring has been developed and many compounds with aromatic groups have been studied using SFG.113–117 Phenyl group tilt orientation can be derived utilizing the signal strength ratio of type II (v7b, v20b) and type I (v20a, v2, v7a) vibrational modes in ssp SFG spectra (assuming isotropic surface and free rotation along principle axis):114
| (23) |
where . The value of r and (for example 116) can be obtained using the bond additivity method.116 By fitting SFG spectra, can be obtained and can be associated to through equations (3)–(6). Together with the above equation, the phenyl tilt angle can be derived. It was shown that phenyl rings in PS are oriented more towards the surface normal at the PS/air interface, while they tend to lie down at the PS/sapphire interface.114 Another research on surface structures of epoxy and phenolic resin showed that adsorbed water molecules on the phenolic resin surface can form hydrogen bonds with the surface phenol groups, changing the phenol orientation from the ‘lie down’ to ‘stand up’, as shown in Figure 11.116 SFG has also been used to characterize the interfacial molecular structures and orientations of phenyl groups at polystyrene/comb-polymer interfaces115 and organic counterions bound to a charged surfactant monolayer.113 It has also been shown that for certain aromatic groups on the surface, it is necessary to consider both the tilt and the twist angles.117
Figure 11.
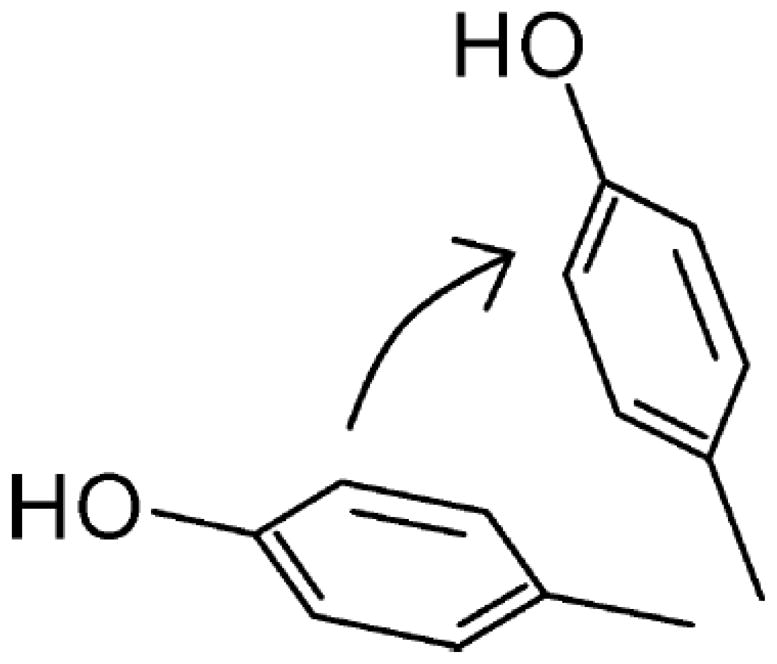
Schematic representation of phenol group stands up more after exposure to humid air. Reprinted with permission from ref. 116. (2009 American Chemical Society)
In this section we summarized the SFG orientation analysis methods of C-H functional groups including methyl, methylene, and aromatic groups. We show that using polarized SFG, it is possible to obtain molecular orientation information of such functional groups at surfaces or interfaces.
4.2 Amide I group orientation analysis
The molecular orientations of biomolecules at different interfaces are of great importance in understanding biomolecule functions and interaction dynamics at such interfaces. In situ molecular level study on biomolecules at interfaces is still challenging due to the lack of appropriated techniques. Recently, SFG was proved to be unique in examining such systems. We can certainly apply the above discussed SFG molecular orientation measurement methods on different C-H functional groups to study biomolecules. However, such studies usually only provide structural information on biomolecule side chains. To determine orientations of biomolecules such as peptides and proteins at interfaces, it would be better to directly study their backbones. As demonstrated in section 3.2.1, SFG can be used to collect peptide/protein amide I signals, which were mainly contributed from the backbone C=O stretches. The methods for interfacial orientations analysis of different secondary structures such as α-helices and β-sheets using SFG amide I signals have been systemically developed.
Here we will first discuss the orientation determination of helices, which are the most distributed secondary structures in proteins. Many antimicrobial peptides adopt α-helical structures in the cell membrane and therefore it is important to study their membrane orientations to understand their antimicrobial activity and selectivity. Similar to the orientation analysis on C-H groups, by combining group theory, coordinate transformation and polarized SFG, it is possible to derive molecular orientation information of an α-helix from SFG experiment.55 For amide I modes of α-helices, both the A and E1 modes have been shown to be SFG active.99 Using a near total reflection SFG experimental geometry, ssp and ppp SFG spectra of amide I signal can be obtained, which correspond to and respectively. For the near total reflection geometry, is only related to in equation (6) because Lxx ≈ 0. Assuming isotropic surface in x-y plane and random twist angle distribution, the relationship between and molecular hyperpolarizability components can be written as:55
For A mode:
| (24) |
| (25) |
For E1 mode:
| (26) |
| (27) |
where . Since the peak centers of the signals contributed from the A and E1 modes are very close, which cannot be distinguished by SFG, the total second order nonlinear optical susceptibility for the amide I signal for α-helices is usually considered to be the sum of the A and E1 modes.99 We therefore have . Utilizing the bond additivity method, it can be shown that and for α-helix.99, 185 By plotting the ratio of as a function of θ, together with the measured value obtained in the polarized SFG spectra (from ssp and ppp spectra), the tilt angle of an α-helix can be derived. According to the plots using equations (24) to (27), it is possible to deduce the ensemble molecular orientation of 〈cosθ〉 and 〈cos3θ〉. If we assume all the helices adopt the same orientation, i.e. a δ-distribution, we can replace 〈cosθ〉 and 〈cos3θ〉 with cosθ and cos3θ.
If α-helical peptides at the interface adopt multiple orientations, e.g., adopt two different orientation angles θ1 and θ2, it would be difficult to use SFG measurements only to deduce the orientation distribution. However, SFG can be combined with other spectroscopic measurements such as ATR-FTIR to determine more complicated orientations. ATR-FTIR can provide an independent orientation measurement on 〈cos2θ〉.186, 187 If molecules have two orientations with two different tilt angles, assuming the fraction of total molecules adopting tilt angle θ1 being M, then the fraction for the molecules adopting θ2 is 1-N.
Thus we have:
| (28) |
After obtaining three independent measurements of 〈cosθ〉, 〈cos2θ〉 and 〈cos3θ〉 from SFG and FTIR, it is possible to derive 〈cosθ1〉, 〈cosθ2〉 and M for interfacial α-helices with two orientation distribution preferences. If the α-helical peptides adopt more than two different orientations, additional independent measurements are needed. For example, 〈cos4θ〉 can be measured using four-wave mixing spectroscopy.187
By applying the orientation analysis method described above, orientations of helical structures at different interfaces can be measured experimentally. For example, SFG was combined with ATR-FTIR spectroscopy to study molecular interactions between melittin and a solid substrate supported DPPG bilayer.55 Several trial distribution functions were used to deduce the orientation distribution of melittin inside a bilayer. It was found that the melittin orientation distribution could not be described using a simple distribution function such as δ-function or single Gaussian distribution. A maximum entropy function has been used to fit the experimental results. The results suggest that melittin α-helices tend to exist in two main populations in the DPPG bilayer at the experimental concentration: about one-fourth of melittin molecules orient nearly perpendicular to the surface, whereas the rest of the molecules orient parallel to the bilayer surface, as shown in Figure 12.55 This study clearly shows that the combination of linear and nonlinear spectroscopic techniques can provide additional molecular orientation information than either technique alone.
Figure 12.
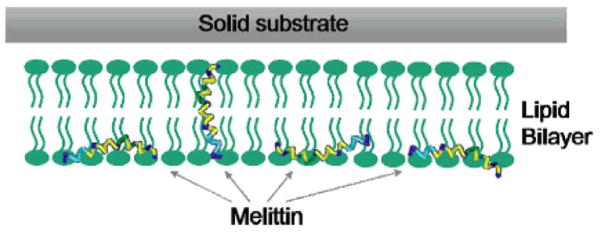
Schematic demonstration of the two orientations of melittin inside a lipid bilayer. Reprinted with permission from ref. 55. (2007 American Chemical Society)
SFG has been applied to investigate how AMPs such as MSI-78 disrupt certain lipid bilayers.144 It is believed that the orientations of AMPs in lipid bilayers are related to their antimicrobial activity and selectivity. An SFG study of MSI-78 compared its interactions with a model bacteria membrane (DPPG or 1-palmitoyl-2-oleoyl-sn-glycero-3-[phospho-rac-(1-glycerol)] (POPG) bilayers) and a model mammalian cell membrane (DPPC or 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) bilayers). It was shown that at the DPPG/d-DPPG bilayer at low peptide concentration of 400 nM, MSI-78 helix oriented with a tilt angle ~70° vs. the surface normal. At a higher peptide concentration of 600 nM, MSI-78 helix inserted into the bilayer with a tilt angle ~25° vs. the surface normal. At even higher concentration, multiple orientations of MSI-78 were observed, suggesting the formation of toroidal-type pores in the lipid bilayer. For POPG/POPG bilayer, MSI-78 can disrupt the bilayer at 500 nM via toroidal pores. However, MSI-78 did not interact with the DPPC bilayer even at a much higher concentration (~12 000nM). For the POPC/POPC bilayer, there was no interaction between MSI-78 and the bilayer below peptide concentration of 800 nM. The SFG measurement results in this research indicated that MSI-78 interacted with bacterial membranes strongly, while interaction with mammalian cell membranes was not as pronounced.144
SFG orientation studies have also been applied to cell penetrating peptides (CPP).145 CPP Pep-1 adopted an orientation nearly perpendicular to a bilayer at a low concentration (0.28 to 1.4 μM), whereas a broad orientation distribution was adopted at a higher concentration (7.0 μM).
An α-helix unit has 18 amino acid residues with five turns. That is to say, in an α-helix every turn has 3.6 peptide units. For a 310 helix, every turn has three peptide units. SFG orientation analysis has been performed on a 310 helix.99 SFG and ATR-FTIR studies have been performed on a 20-residue hydrophobic antibiotic peptide: alamethicin.56 It was shown that alamethicin adopted a mixed α-helical and 310-helical structure in fluid-phase lipid bilayers. In a1,2-dimyristoyl-d54-sn-glycero-3-phosphocholine-1,1,2,2-d4-N,N,N-trimethyl-d9(d-DMPC)/1,2-dimyrist oyl-sn-glycero-3-phosphocholine (DMPC) bilayer, the main α-helix at the N-terminus of alamethicin adopted a tilt angle of about 63° versus the surface normal, while the 310-helix at the C-terminus oriented with a tilt angle of about 43° versus the surface normal. Further study investigated the alamethicin orientation in POPC bilayers in contact with peptide solutions with different pH values.158 At pH=6.7, the α-helix at the N-terminus and the 310-helix at the C-terminus tilted about 72° and 50° versus the surface normal, respectively. At pH=11.9, the tilt angles of the α-helix and 310-helix decreased to about 57° and 45°, respectively. The increase in pH changes the membrane potential and induces the decrease in both bent and tilt angles of helices in alamethicin (Figure 13).
Figure 13.

(A) The definition of tilt angle (θ1 and θ2) and bend angle φ=(θ1−θ2) of alamethicin in POPC/POPC bilayer. (B) Schematic demonstration of pH dependent channel gating action of alamethicin. Reprinted with permission from ref. 158. (2012 American Chemical Society)
Besides helices, β-sheet is also an important secondary protein structure. Similar to α-helices, the orientation analysis method for anti-parallel β-sheets with D2 symmetry has also been developed.100, 109 According to group theory, β-sheets with D2 symmetry have four vibrational modes: A, B1, B2, and B3. The A mode is only Raman active, while all the three B modes are both IR and Raman active, i.e. SFG active. The detailed orientation analysis methods for anti-parallel β-sheets can be found in the literatures.100, 109 The methodology has been successfully applied to derive the orientation of tachyplesin I, a 17 amino acid peptide with an anti-parallel β-sheet structure in aqueous environments, adsorbed to PS surfaces and interacting with a DPPG/d-DPPG bilayer. Both tilt angle and twist angle of the β-sheet at the interfaces were determined.100 Further, it was shown that even with a random twist angle, polarized SFG can be used to detect interfacial chiral signal by detecting B2 and B3 modes.109 Strong chiral SFG vibrational spectra from tachyplesin I peptide at the interface can be detected directly.109 Chiral SFG detection has been reported in a series of publications.188–192 Chiral molecules in bulk materials can contribute chiral SFG signal due to molecular chirality. However, the chiral SFG signal from tachyplesin I at interfaces is contributed by interfacial molecules due to molecular arrangement.109
By monitoring the secondary structures of larger biomolecules using polarized SFG, orientation information of proteins at different interfacial environments can be obtained. An example to demonstrate the feasibility of SFG studies on membrane proteins is to observe the spontaneous membrane insertion process of cytochrome b5 (Cyt-b5) and its mutants.193 A wild-type Cyt-b5 is a tail-anchored membrane protein with molecular weight of ~16 kDa. The protein is composed of three distinct domains with different dynamics: a membrane-spanning anchor, a heme-containing soluble domain, and a linker region connecting the former two domains. Even though Cyt-b5 has been studied extensively, the membrane insertion properties of Cyt-b5 remained unclear. In the SFG study, Cyt-b5 mutants (m-Cyt-b5), in which eight amino acids in the linker region were removed or partially removed, were studied as comparisons to the wild-type Cyt-b5. By measuring the amide I signal in ssp and ppp polarized SFG spectra, the tilt angle for membrane anchoring helix in wild-type Cyt-b5 was determined to be 15° with respect to the dDMPC/dDMPC interface normal at 25 °C, indicating that wild-type Cyt-b5 can insert into a dDMPC/dDMPC bilayer. However, m-Cyt-b5 adopted a different tilt angle compared to the wild-type Cyt-b5 on bilayers. It was deduced that the shorter the linker, the larger the tilt angle vs. the surface normal. For m-Cyt-b5 with eight amino acids deleted in the linker region, the tilt angle of the membrane anchor helix was determined to be ~70° vs. the surface normal using SFG, showing that this mutant could not insert into the lipid bilayer. These experimental results clearly indicated that the length of the linker in Cyt-b5 can influence membrane insertion properties of Cyt-b5. This study also included the effects of temperature and lipid content on Cyt-b5 insertion. It was shown that the insertion of m-Cyt-b5 required higher temperature and was dependent on the lipid phase.193 This research demonstrated the feasibility of determining molecular orientation of a membrane protein in lipid bilayers in situ using SFG.
Orientations of protein complexes have also been examined using SFG, using heterotrimeric guanine nucleotide-binding proteins (G proteins) as examples.194 G proteins are membrane associated proteins that are widely involved in many biological processes such as signal transduction. It is still unclear how G proteins and their subunits interact and order at cell membrane surfaces. By monitoring the SFG signal in the amide I frequency region, the orientation analysis on α-helices shows that Gβγ, a dimer formed by G protein subunits Gβ and Gγ, has a tilt angle −35° against the POPC/POPC bilayer interface normal (Figure 14).194 This study assumed the twist angle of Gβγ is fixed on the bilayer surface. However, this assumption may not be valid. Recently, a computer program was used to deduce the orientation of complicated proteins with many helices.102 The program can read arbitrary PDB protein structures and calculate the susceptibility ratio corresponding to different SFG polarizations as a function of protein orientation (involving both twist and title angles). At the same time, SFG experiments were carried out to observe Gβγ-G protein coupled receptor (GPCR) kinase 2 (GRK2) complex formation at the lipid bilayer surface in situ. GRK2 is a kinase enzyme responsible for cell signaling. The orientation of GRK2 in complex with Gβγ at the lipid bilayer interface was unknown. The membrane orientation of the Gβγ-GRK2 complex obtained from SFG study was different from predictions.102 Gβγ appeared to change its orientation after binding to GRK2. These studies showed that SFG has great potential in deducing orientations of large protein complexes at various interfaces.
Figure 14.
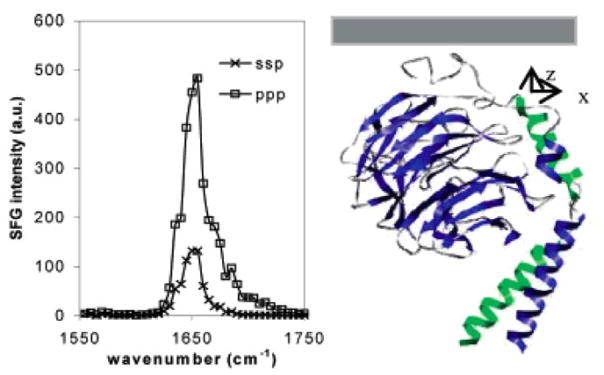
(Left) SFG amide I spectra of interfacial Gβγ (25 μg/mL) adsorbed onto a POPC/POPC bilayer; (Right) Gβγ orientation deduced based on SFG intensity ratio obtain from left spectra. It tilts −35° against the surface normal from the “zero” position defined earlier in ref. 159. Reprinted with permission from ref. 194. (2007 American Chemical Society)
In summary, this section summarizes some of the recent SFG progress in determining molecular orientations at polymer and biomolecule related interfaces which highlight the uniqueness of SFG in interfacial molecular orientation study. Utilizing polarized SFG spectra, it is feasible to obtain interfacial orientations of various functional groups such as methyl, methylene, and phenyl groups, as well as protein secondary structures such as helices and β-sheets.
5. Time dependent SFG studies on molecular structural changes at interfaces
The third important advantage of SFG studies on interfaces is to monitor the real time molecular structural change. In a typical time-dependent SFG experiment, the input laser is tuned to the vibrational resonant frequency that is of interest, and SFG intensity is monitored as a function of time. The repetition rate of the pico-second laser used in our SFG experiments is 20 Hz. To get a reliable data point, usually SFG signal should be collected with at least 20 shots or 1 second. Therefore, the time-dependent change in seconds should be able to be monitored using pico-second SFG system. For a femto-second broadband SFG system, an SFG spectrum usually can be collected in hundreds of millisecond and therefore the time-dependent SFG spectra can be obtained with better time resolution. In this paper, we will focus on the time-dependent processes studied using pico-second SFG systems.
5.1 Time dependent SFG studies on polymer related interfaces
SFG spectroscopy has been applied to study the molecular structure of a moving polymer/liquid interface.135 In the study, SFG was applied to monitor AATMS diffusing into a polymer matrix including PMMA and PS. At initial contact of AATMS with d-PMMA, the SFG signal of silane methoxy groups at 2840 and 2945 cm−1 could be detected (Figure 15). As time passed, the SFG spectral intensity decreased and finally disappeared. Additional experiments utilizing a d-PMMA film on top of a PS film in contact with AATMS proved that silane molecules can diffuse into the polymer matrix, resulting in intensity changes of the silane signal. AATMS diffused into PS much faster than into PMMA. Time dependent SFG intensity was monitored at the AATMS/d-PMMA interface with varying d-PMMA film thickness. Silane required longer time to diffuse through thicker polymer films. The diffusion coefficient of AATMS was calculated to be in the order of 10−13 cm2/s by fitting the time-duration dependence of silane diffusion through the PMMA films with different thicknesses using a Fickian model (Figure 16).135 Compared to typical small molecules such as O2 and CO2, the diffusion of AATMS into polymer films was quite slow.
Figure 15.

SFG spectra (ssp) collected from the d-PMMA/AATM interface as a function of time. The thickness of the d-PMMA film used here is 150 nm. Reprinted with permission from ref. 135. (2004 American Chemical Society)
Figure 16.

(Left) Time-dependent SFG signal intensities at 2840 and 2945 cm−1 as AATM molecules diffuse into d-PMMA films of different thicknesses: (a) 20, (b) 70, (c) 150, (d) 210, and (e) 269 nm. (Right) Diffusion kinetics fitted by the Fickian model. Reprinted with permission from ref. 135. (2004 American Chemical Society)
In another study by Loch et al., SFG was applied to monitor interfacial diffusion of another silane γ-GPS and a silane adhesion promoting mixture SAPM, in contact with polymers.136 γ-GPS and SAPM were found to diffuse through d-PMMA, but dissolved d-PS. However, their diffusion speed and dissolution speed were different. PS/d-PMMA bilayer experiments have also been used to support the diffusion interpretation. Such results were quite different from γ-GPS and SAPM behavior at PET interfaces, which showed no diffusion but segregation of methoxy group at interfaces with a net orientation order.138
As discussed above, SFG is capable of measuring diffusion coefficients of very slow diffusion processes because of its excellent surface selectivity. Only very thin films (tens of nanometers) need to be used in the study. Using a traditional technique, e.g., ATR-FTIR, much thicker films are needed.195–197 The diffusion measurement for slow diffusion would take significantly longer.
5.2 Time dependent SFG studies on biomolecules at interfaces
Recently, time dependent SFG has been applied to study time-dependent behaviors on various biomolecules such as peptides and lipids at interfaces. For example, SFG was used in in situ studies of peptide and protein immobilization on surfaces.7, 198, 199 Chemical immobilization of peptides and proteins on surfaces has found many applications such as biosensors and implant devices. The structure such as orientation of immobilized biomolecules generally determines their function. Therefore, characterizing orientations of immobilized molecules is important. A quantitative SFG study of an α-helical peptide cecropin P1 (CP1) showed that when it was chemically immobilized on a substrate, it adopted a more ordered orientation compared to when it physically adsorbed to the surface.198 In the study, a maleimide-functionalized PS (PS-MA) surface was used as solid substrate for chemical immobilization. CP1 was modified on the c-terminus by a cysteine residue so that it could be chemically bound to the maleimide group of PS-MA. PS film was used as a solid support for physical adsorption of CP1. In order to form a better understanding of environmental effects on the CP1 immobilization process, the study was continued by varying CP1 concentration, assembly state (monomer or dimer), and solvent composition.199 Monitoring the time dependent SFG signal of amide I at 1650 cm−1 showed that with higher peptide concentration, CP1 immobilization reaches equilibrium faster. The time-dependent SFG study also showed that the immobilized CP1 changed orientation as a function of time.199
Time dependent SFG has also been applied to study protein adsorption such as fibrinogen. Fibrinogen (~340K) is a blood protein that is considered to be related to thrombosis.200 It has three hydrophobic domains (one center E domain and two D domains) connected by two α-helical coiled-coils, with each D domain connected to an αC domain by an αC chain (shown in Figure 17). ATR-FTIR and QCM were combined with SFG to study fibrinogen adsorption to different polymers.200 If the adsorbed fibrinogen adopts a linear structure, it nearly has an inversion symmetry, leading to no or weak SFG amide I signal. However, very strong amide I signal centered at ~1650 cm−1 was detected at the PS/protein solution interface. This indicates that fibrinogen adopted a bent structure at the interface. SFG has also been used to study fibrinogen adsorption on different polymer surfaces. Figure 18 shows the time-dependent SFG spectra changes in the amide I frequency range of fibrinogen adsorbed to three biomedical polymers with different hydrophobicities including poly(ether urethane) (PEU), silicone-poly(carbonate urethane) (SPCU), and perfluorinated polymer (PFP). The amide I signal decreased as a function of time for fibrinogen adsorbed on PEU, while it slightly increased for those on SPCU and PFP. It was demonstrated that fibrinogen interacted with hydrophilic surfaces (PEU) via αC domain, whereas it interacted with more hydrophobic surfaces (SPCU or PFP) via the D domain as shown in Figure 19. Further study using SFG supplemented by ATR-FTIR showed that there was no significant secondary structural conversion after fibrinogen adsorbed to PS. However, the side chains of fibrinogen immediately changed after adsorption, whereas the main bent structure of fibrinogen slowly changed into a linear structure that was oriented parallel to the PS surfaces.201
Figure 17.
Structure of a fibrinogen molecule. Reprinted with permission from ref. 200. (2005 American Chemical Society)
Figure 18.
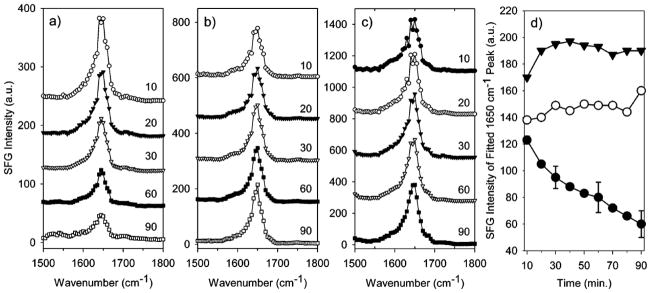
SFG spectra collected in the amide I range of fibrinogen adsorbed to (a) PEU, (b) SPCU, and (c) PFP in PBS buffer at different time (in min). Time dependent SFG signal of α-Helix (d) from fitting SFG spectra for fibrinogen adsorbed to PEU (closed circles), SPCU (open circules), and PFP (closed triangles). Representative error is shown for the fibrinogen/PEU sample. Reprinted with permission from ref. 200. (2005 American Chemical Society)
Figure 19.
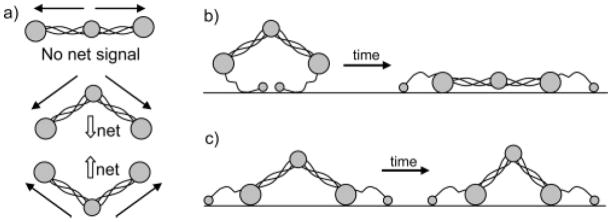
(a) A few possible fibrinogen configurations at the interface. The α-helix SFG signal from each set of coiled coils is shown by solid arrows, and the net α-helix SFG signal is shown by white arrows. Here αC chains are not shown. (b) Schematic of fibrinogen structural changes with time after adsorption on PEU. (c) Schematic of fibrinogen structural changes with time after adsorption on SPCU or PFP. Reprinted with permission from ref. 200. (2005 American Chemical Society)
Time-dependent SFG is an ideal technique to study lipid flip-flop in a membrane bilayer. Lipid flip-flop is also known as lipid translocation, which is the transbilayer phospholipid movement in bilayer membranes. By labeling certain lipid species, studies have shown that lipid flip-flop in a bilayer membrane is possible in the absence of a protein-mediated process, although the flip-flop speed is usually slow, on the order of hours.202, 203 SFG has been applied to monitor the kinetics of flip-flop process by preparing asymmetric supported lipid bilayers with different compositions (deuterated and hydrogenated lipids) in the inner and outer leaflets. For lipid bilayers with identical lipid compositions for both leaflets, the destructive interference of fatty acid methyl stretches in SFG show little signal in the spectrum. Differently, in an asymmetric bilayer, where no destructive interference can occur, both deuterated and hydrogenated vibrational stretches of methyl group can be observed from two different layers. Monitoring such vibrational modes using time dependent SFG, changes in membrane lipid composition due to the exchange between leaflets can be followed by signal decay.204 Using this method, the flip-flop kinetics of an 1,2-distearoyl-sn-glycero-3-phosphocholine (DSPC)/d83-DSPC bilayer at various temperatures was studied using SFG.204 Using an exponential function to fit the decay signal at different temperatures, the rates of DSPC transbilayer movement (k) were determined (as shown in Figure 20). It was found that at 25 C°, the lipid flip-flop was extremely slow (population inversion time is ~18 days). However, at 51.3 C°, just several degrees below the phase-transition temperature of DSPC, the time required for population inversion decreased to 25.9 min. Above the phase-transition temperature of DSPC, the rate of flip-flop was too fast to measure using SFG. A following study focused on the measurement of intrinsic flip-flop rate and activation energy of DMPC, DPPC, DSPC and a labeled lipid 1,2-dipalmitoyl-sn-glycero-3-phosphoethanolamine-n,n-Dimethyl-n-(2′,2′,6′,6′-tetramethyl-4′-piperidy l) (TEMPODPPC).205 It was found that the flip-flop rates for DMPC, DPPC, and DSPC at 5 C° below their main phase transition temperature of the lipids ranged from 196×10−5 s−1, 42.2×10−5 s−1 to 15.2×10−5 s−1. The labeled TEMPODPPC had a significantly lower flip-flop rate compared to DPPC, which indicated that chemical structural modifications of lipids can greatly affect lipid flip-flop kinetics and thermodynamics in model membrane systems.205
Figure 20.

CH3 symmetric SFG intensity decay for DSPC/DSPC-d83 bilayer at various temperatures; the blue line was recorded at 41.7 °C, green at 45.7°C, and red at 50.3°C. The dashed lines are fitted data using equation (3) in ref. 169. Reprinted with permission from ref. 204. (2004 American Chemical Society)
Further studies showed that lipid flip-flop can be affected by transmembrane peptides such as gramicidin A,206 WALP23 and melittin,207 membrane lateral pressure,208 as well as cholesterol.209 These studies provide new insight into transbilayer migrations of phospholipids in different biological conditions and the effects of different membrane factors in membrane dynamics. They suggest that SFG is an ideal analytical technique for membrane lipid dynamics study.
Time-dependent peptide interactions with cell membranes have been widely studied. SFG has been used to study interactions between melittin and a DPPG bilayer.210 Melittin, which has 26 amino acid residues, is among the best studied membrane-active peptides. In the study, a DPPG/d-DPPG or d-DPPG/DPPG (inner leaflet/outer leaflet) bilayer was prepared on a fused silica or CaF2 substrate.210 The asymmetric bilayer generated strong SFG signals from both C-H (in CH3) and C-D (in CD3) vibrational stretches at 2875 and 2070 cm−1. When melittin disrupted the bilayer, the C-H or C-D signal intensities tend to decrease. Time dependent SFG signals at 2875 and 2070 cm−1 indicated that melittin interaction with the DPPG bilayer was concentration dependent. At a high concentration (15.6 μM), melittin disrupted both leaflets immediately after the injection of the peptide to the subphase in contact with the lipid bilayer. At an intermediate concentration (3.9 μM), melittin tended to interact strongly with the outer leaflet first, and then the inner leaflet. As the concentration decreased further (0.78 and 0.156 μM), the interaction of melittin with bilayer becomes weaker. Here for most cases, melittin disrupted the outer leaflet first, then the inner leaflet. However, for tachyplesin I, it was shown that the peptide disrupted both leaflets simultaneously.83 These research shows time dependent SFG can be applied to study many biological interactions involving peptides/proteins and membrane lipid bilayers.
In this section, we used some examples of polymer and biomolecule interfacial studies to show that SFG is a unique analytical tool to monitor and analyze molecular kinetics and dynamics at interfaces in real time and in situ.
6. Summary and outlook
In this review, we focused on three unique features of a nonlinear optical analytical technique SFG. Using various examples in polymer and biomolecule related interfaces, it was shown that SFG has great advantages over other surface sensitive techniques including in situ interfacial presence detection, interfacial orientation analysis, and time dependent interfacial kinetics and dynamics study, all of which at the molecular level. (1) For the interfacial presence detection, we discussed polymer related interfaces including poly(methacrylate)s and PDMS surfaces in water, polymer/silane interfaces, polymer/PDMS interfaces, and polymer/epoxy interfaces. It was shown that SFG can provide molecular level understanding on polymer materials at different interfaces, fouling release mechanism of PDMS, and adhesion mechanism, etc. Biological related interfaces were also discussed. Protein adsorption at interfaces was studied to show different adsorption mechanisms for various substrates and different proteins. The interactions of biological molecules and model cell membranes have been shown as examples for understanding membrane related interactions. DNA behaviors at different interfaces have also been discussed to show the uniqueness of SFG for in situ detection. These studies show that SFG is a very sensitive surface and interface analytical technique which can provide detailed molecular presence information in situ. (2) For the molecular orientation analysis, we summarized the methodologies for studying orientations of methyl, methylene, aromatic functional groups using SFG spectra collected in the C-H stretching frequency region, and for determining orientation of protein secondary structures such as helices, β-sheet using SFG amide I signals. Polymer materials such as PMMA, PBMA, PDMS, PS and phenolic resin were used as examples for polymer orientation analysis. The orientation determination of helices and β-sheet in the amide I frequency region can provide detailed orientation information of the backbone of large biomolecules such as proteins. Using polarized SFG, molecular orientations at interfaces can be determined in situ even with very low interfacial coverage. By combining SFG with other spectroscopic technique such as polarized ATR-FTIR, more detailed molecular orientation information, such as multi-orientation distributions can also be obtained. (3) For time dependent SFG, it was shown that silane diffusion into polymer materials can be detected and measured. SFG can be used to measure systems with very slow diffusion speed which are difficult to measure using other spectroscopic techniques. Time dependent SFG studies on biomolecules interacting with different substrates and model cell membranes have also been shown. It was demonstrated that time dependent SFG study is a powerful analytical tool to study chemical immobilization, protein adsorption, lipid flip-flop in a bilayer, and peptide-lipid bilayer interaction. Time dependent SFG is a powerful spectroscopic technique to study interfacial kinetics and dynamics in situ in real time.
SFG has been applied and is being applied to many different systems to bring new insight or complementary support for scientists. Further research will continue to boost SFG applications in polymer and biological related interfaces. More complicated interfaces will be examined, and other research tools, including various spectroscopic and microscopic techniques will be combined with SFG in future research. SFG data analysis methods will be further developed. With the progress in laser technology and nonlinear optics, SFG systems will be more versatile and powerful in many applications. SFG will also become more compact and affordable for more research groups. Many new research frontiers have been and will be developed utilizing SFG and its combination with other state-of-the-art techniques.
Acknowledgments
The authors want to thank the financial support from Army Research Office (W911NF-11-1-0251), Defense Threat Reduction Agency (HDTRA1-11-1-0019), National Science Foundation (CHE-1111000), National Institutes of Health (GM081655), Office of Naval Research (N00014-12-1-0452), and Semiconductor Research Corporation (P13696).
Biographies

Chi Zhang is currently a PhD candidate in Chemistry at the University of Michigan, advised by Prof. Zhan Chen. He earned his BS and MS in Opto-electronics at Tianjin University in 2006 and 2009. His work now focuses on molecular level understanding of interfaces involving polymers, and developing nonlinear spectroscopic and microscopic techniques for material and biological study.

John Myers received a B.S. degree in Biochemistry from Bradley University in 2009 and a M.S. degree in Analytical Chemistry from Miami University in 2011. He is currently a graduate student in the Department of Chemistry at the University of Michigan advised by Prof. Zhan Chen. His current research focuses on nonlinear optical spectroscopic studies of polymer surfaces and buried interfaces.

Zhan Chen received his B.S. degree in chemistry from Peking University in 1988. He then received his M.S. degree in physics from the Institute of Physics, Chinese Academy of Sciences in 1991. After he worked in the Institute of Physics for 2 years as a research scientist, he went to the Department of Chemistry at the University of California at Berkeley in 1993. He received his Ph.D. degree in 1998, advised by Prof. Herbert Strauss. He then worked as a postdoctoral research fellow in Prof. Gabor Somorjai’s group at Berkeley. He started his own research group in 2000 at the University of Michigan. Currently, he is a professor of Chemistry there. His research is to understand the molecular structures of polymers and biological molecules at interfaces.
References
- 1.Ratner BD, Castner DG. Surface Analysis—The Principal Techniques. John Wiley & Sons; 1997. [Google Scholar]
- 2.McArthur SL. Surf Interface Anal. 2006;38:1380–1385. [Google Scholar]
- 3.Roach P, Parker T, Gadegaard N, Alexander M. Surf Sci Rep. 2010;65:145–173. [Google Scholar]
- 4.Benninghoven A. Surf Sci Rep. 1975;53:596–625. [Google Scholar]
- 5.Henry M, Dupont-Gillain C, Bertrand P. Langmuir. 2003;19:6271–6276. doi: 10.1021/la701850p. [DOI] [PubMed] [Google Scholar]
- 6.Bernsmann F, Lawrence N, Hannig M, Ziegler C, Gnaser H. Anal Bioanal Chem. 2008;391:545–554. doi: 10.1007/s00216-008-2014-z. [DOI] [PubMed] [Google Scholar]
- 7.Baugh L, Weidner T, Baio J, Nguyen PCT, Gamble LJ, Stayton PS, Castner DG. Langmuir. 2010;26:16434–16441. doi: 10.1021/la1007389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Binnig G, Quate CF, Gerber C. Phys Rev Lett. 1986;56:930–933. doi: 10.1103/PhysRevLett.56.930. [DOI] [PubMed] [Google Scholar]
- 9.Giessibl FJ. Rev Mod Phys. 2003;75:949–983. [Google Scholar]
- 10.Hörber J, Miles M. Science. 2003;302:1002–1005. doi: 10.1126/science.1067410. [DOI] [PubMed] [Google Scholar]
- 11.Carnally S, Barrow K, Alexander MR, Hayes CJ, Stolnik S, Tendler SJB, Williams PM, Roberts CJ. Langmuir. 2007;23:3906–3911. doi: 10.1021/la0626205. [DOI] [PubMed] [Google Scholar]
- 12.Liedberg B, Nylander C, Lunström I. Sens Actuators. 1983;4:299–304. [Google Scholar]
- 13.Liedberg B, Nylander C, Lundström I. Biosens Bioelectron. 1995;10:i–ix. doi: 10.1016/0956-5663(95)96965-2. [DOI] [PubMed] [Google Scholar]
- 14.Homola J, Yee SS, Gauglitz G. Sensor Actuat B-Chem. 1999;54:3–15. [Google Scholar]
- 15.Green RJ, Frazier RA, Shakesheff KM, Davies MC, Roberts CJ, Tendler SJB. Biomaterials. 2000;21:1823–1835. doi: 10.1016/s0142-9612(00)00077-6. [DOI] [PubMed] [Google Scholar]
- 16.Eustis S, El-Sayed MA. Chem Soc Rev. 2006;35:209–217. doi: 10.1039/b514191e. [DOI] [PubMed] [Google Scholar]
- 17.De Feijter JA, Benjamins J, Veer FA. Biopolymers. 1978;17:1759–1772. [Google Scholar]
- 18.Azzam RMA, Bashara NM. Ellipsometry and polarized light. North Holland; 1987. [Google Scholar]
- 19.Fujiwara H. Spectroscopic ellipsometry: Principles and Applications. John Wiley&Sons; 2007. [Google Scholar]
- 20.Lekner J. Pure Appl Opt. 1996;5:417–443. [Google Scholar]
- 21.Simpson GJ, Dailey CA, Plocinik RM, Moad AJ, Polizzi MA, Everly RM. Anal Chem. 2005;77:215–224. doi: 10.1021/ac049203m. [DOI] [PubMed] [Google Scholar]
- 22.Harrik N. Internal reflection spectroscopy. Interscience; New York: 1967. [Google Scholar]
- 23.Mirabella FM., Jr Appl Spectrosc Rev. 1985;21:45–178. [Google Scholar]
- 24.Axelsen PH, Citra MJ. Prog Biophys Mol Biol. 1996;66:227–253. doi: 10.1016/s0079-6107(97)00007-2. [DOI] [PubMed] [Google Scholar]
- 25.Le Coutre J, Kaback HR, Patel CKN, Heginbotham L, Miller C. Proc Natl Acad Sci USA. 1998;95:6114–6117. doi: 10.1073/pnas.95.11.6114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vinchurkar MS, Chen KHC, Yu SSF, Kuo SJ, Chiu HC, Chien SH, Chan SI. Biochemistry. 2004;43:13283–13292. doi: 10.1021/bi049016i. [DOI] [PubMed] [Google Scholar]
- 27.Moskovits M, Suh J. J Phys Chem. 1984;88:5526–5530. [Google Scholar]
- 28.Moskovits M. Rev Mod Phys. 1985;57:783–826. [Google Scholar]
- 29.Moskovits M. J Raman Spectrosc. 2005;36:485–496. [Google Scholar]
- 30.Orendorff CJ, Gole A, Sau TK, Murphy CJ. Anal Chem. 2005;77:3261–3266. doi: 10.1021/ac048176x. [DOI] [PubMed] [Google Scholar]
- 31.Nie S, Emory SR. Science. 1997;275:1102–1106. doi: 10.1126/science.275.5303.1102. [DOI] [PubMed] [Google Scholar]
- 32.Michaels AM, Nirmal M, Brus L. J Am Chem Soc. 1999;121:9932–9939. [Google Scholar]
- 33.Xu H, Bjerneld EJ, Käll M, Börjesson L. Phys Rev Lett. 1999;83:4357–4360. [Google Scholar]
- 34.Zhu X, Suhr H, Shen Y. Phys Rev B. 1987;35:3047–3050. doi: 10.1103/physrevb.35.3047. [DOI] [PubMed] [Google Scholar]
- 35.Shen Y. Nature. 1989;337:519–525. [Google Scholar]
- 36.Cremer PS, Su X, Shen YR, Somorjai GA. J Am Chem Soc. 1996;118:2942–2949. [Google Scholar]
- 37.Richter LJ, Petralli-Mallow TP, Stephenson JC. Opt Lett. 1998;23:1594–1596. doi: 10.1364/ol.23.001594. [DOI] [PubMed] [Google Scholar]
- 38.Zhuang X, Miranda P, Kim D, Shen Y. Phys Rev B. 1999;59:12632–12640. [Google Scholar]
- 39.Kim J, Cremer PS. Chem Phys Chem. 2001;2:543–546. doi: 10.1002/1439-7641(20010917)2:8/9<543::AID-CPHC543>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 40.Chen Z, Shen Y, Somorjai GA. Annu Rev Phys Chem. 2002;53:437–465. doi: 10.1146/annurev.physchem.53.091801.115126. [DOI] [PubMed] [Google Scholar]
- 41.Kim J, Somorjai GA. J Am Chem Soc. 2003;125:3150–3158. doi: 10.1021/ja028987n. [DOI] [PubMed] [Google Scholar]
- 42.Ye S, Morita S, Li GF, Noda H, Tanaka M, Uosaki K, Osawa M. Macromolecules. 2003;36:5694–5703. [Google Scholar]
- 43.Hunt J, Guyot-Sionnest P, Shen Y. Chem Phys Lett. 1987;133:189–192. [Google Scholar]
- 44.Bain CD, Davies PB, Ong TH, Ward RN, Brown MA. Langmuir. 1991;7:1563–1566. [Google Scholar]
- 45.Eisenthal KB. Annu Rev Phys Chem. 1992;43:627–661. [Google Scholar]
- 46.Ye S, Nihonyanagi S, Uosaki K. Phys Chem Chem Phys. 2001;3:3463–3469. [Google Scholar]
- 47.Moad AJ, Simpson GJ. J Phys Chem B. 2004;108:3548–3562. [Google Scholar]
- 48.Ma G, Allen HC. Langmuir. 2006;22:5341–5349. doi: 10.1021/la0535227. [DOI] [PubMed] [Google Scholar]
- 49.Guyot-Sionnest P, Hunt J, Shen Y. Phys Rev Lett. 1987;59:1597–1600. doi: 10.1103/PhysRevLett.59.1597. [DOI] [PubMed] [Google Scholar]
- 50.Hirose C, Yamamoto H, Akamatsu N, Domen K. J Phys Chem. 1993;97:10064–10069. [Google Scholar]
- 51.Wang J, Chen C, Buck SM, Chen Z. J Phys Chem B. 2001;105:12118–12125. [Google Scholar]
- 52.Baldelli S. J Phys Chem B. 2003;107:6148–6152. [Google Scholar]
- 53.Chen X, Clarke ML, Wang J, Chen Z. Int J Mod Phys B. 2005;19:691–713. [Google Scholar]
- 54.Zhang D, Shen Y, Somorjai GA. Chem Phys Lett. 1997;281:394–400. [Google Scholar]
- 55.Chen X, Wang J, Boughton AP, Kristalyn CB, Chen Z. J Am Chem Soc. 2007;129:1420–1427. doi: 10.1021/ja067446l. [DOI] [PubMed] [Google Scholar]
- 56.Ye S, Nguyen KT, Chen Z. J Phys Chem B. 2010;114:3334–3340. doi: 10.1021/jp911174d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ma G, Allen HC. J Phys Chem B. 2003;107:6343–6349. [Google Scholar]
- 58.Gopalakrishnan S, Jungwirth P, Tobias DJ, Allen HC. J Phys Chem B. 2005;109:8861–8872. doi: 10.1021/jp0500236. [DOI] [PubMed] [Google Scholar]
- 59.Morkel M, Kaichev VV, Rupprechter G, Freund HJ, Prosvirin IP, Bukhtiyarov VI. J Phys Chem B. 2004;108:12955–12961. [Google Scholar]
- 60.Keszthelyi T, Pászti Z, Rigó T, Hakkel O, Telegdi J, Guczi L. J Phys Chem B. 2006;110:8701–8714. doi: 10.1021/jp057180p. [DOI] [PubMed] [Google Scholar]
- 61.Hankett JM, Zhang C, Chen Z. Langmuir. 2012;28:4654–4662. doi: 10.1021/la2045527. [DOI] [PubMed] [Google Scholar]
- 62.Humbert C, Busson B, Abid JP, Six C, Girault H, Tadjeddine A. Electrochim Acta. 2005;50:3101–3110. [Google Scholar]
- 63.Weidner T, Breen NF, Li K, Drobny GP, Castner DG. Proc Natl Acad Sci USA. 2010;107:13288–13293. doi: 10.1073/pnas.1003832107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zhang D, Gracias D, Ward R, Gauckler M, Tian Y, Shen Y, Somorjai G. J Phys Chem B. 1998;102:6225–6230. [Google Scholar]
- 65.Gracias D, Zhang D, Lianos L, Ibach W, Shen Y, Somorjai G. Chem Phys. 1999;245:277–284. [Google Scholar]
- 66.Chen C, Wang J, Mark A, Chen Z. Macromolecules. 2002;35:8093–8097. [Google Scholar]
- 67.Kim SH, Opdahl A, Marmo C, Somorjai GA. Biomaterials. 2002;23:1657–1666. doi: 10.1016/s0142-9612(01)00292-7. [DOI] [PubMed] [Google Scholar]
- 68.Cimatu K, Baldelli S. J Am Chem Soc. 2006;128:16016–16017. doi: 10.1021/ja067063n. [DOI] [PubMed] [Google Scholar]
- 69.Cimatu K, Moore HJ, Lee TR, Baldelli S. J Phys Chem C. 2007;111:11751–11755. [Google Scholar]
- 70.Uosaki K, Yano T, Nihonyanagi S. J Phys Chem B. 2004;108:19086–19088. [Google Scholar]
- 71.Mermut O, Phillips DC, York RL, McCrea KR, Ward RS, Somorjai GA. J Am Chem Soc. 2006;128:3598–3607. doi: 10.1021/ja056031h. [DOI] [PubMed] [Google Scholar]
- 72.Evans-Nguyen KM, Fuierer RR, Fitchett BD, Tolles LR, Conboy JC, Schoenfisch MH. Langmuir. 2006;22:5115–5121. doi: 10.1021/la053070y. [DOI] [PubMed] [Google Scholar]
- 73.Somorjai GA, York RL, Butcher D, Park JY. Phys Chem Chem Phys. 2007;9:3500–3513. doi: 10.1039/b618805b. [DOI] [PubMed] [Google Scholar]
- 74.Onorato RM, Yoon AP, Lin JT, Somorjai GA. J Phys Chem C. 2012;116:9947–9954. [Google Scholar]
- 75.Bain CD. J Chem Soc, Faraday Trans. 1995;91:1281–1296. [Google Scholar]
- 76.McGilp J. J Phys D: Appl Phys. 1996;29:1812–1821. [Google Scholar]
- 77.Miranda P, Shen Y. J Phys Chem B. 1999;103:3292–3307. [Google Scholar]
- 78.Shultz MJ, Schnitzer C, Simonelli D, Baldelli S. Int Rev Phys Chem. 2000;19:123–153. [Google Scholar]
- 79.Shen YR. Pure Appl Chem. 2001;73:1589–1598. [Google Scholar]
- 80.Lambert AG, Davies PB, Neivandt DJ. Appl Spectrosc Rev. 2005;40:103–145. [Google Scholar]
- 81.Wang J, Clarke ML, Chen X, Even MA, Johnson WC, Chen Z. Surf Sci. 2005;587:1–11. [Google Scholar]
- 82.Wang HF, Gan W, Lu R, Rao Y, Wu BH. Int Rev Phys Chem. 2005;24:191–256. [Google Scholar]
- 83.Chen X, Chen Z. BBA-Biomembranes. 2006;1758:1257–1273. doi: 10.1016/j.bbamem.2006.01.017. [DOI] [PubMed] [Google Scholar]
- 84.Chen Z. Polym Int. 2007;56:577–587. [Google Scholar]
- 85.Somorjai GA, Park JY. Chem Soc Rev. 2008;37:2155–2162. doi: 10.1039/b719148k. [DOI] [PubMed] [Google Scholar]
- 86.Ye S, Nguyen KT, Clair SVL, Chen Z. J Struct Biol. 2009;168:61–77. doi: 10.1016/j.jsb.2009.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Chen Z. Prog Polym Sci. 2010;35:1376–1402. doi: 10.1016/j.progpolymsci.2010.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Jubb AM, Hua W, Allen HC. Annu Rev Phys Chem. 2011;63:107–130. doi: 10.1146/annurev-physchem-032511-143811. [DOI] [PubMed] [Google Scholar]
- 89.Fu L, Wang ZG, Yan ECY. Int J Mol Sci. 2011;12:9404–9425. doi: 10.3390/ijms12129404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Chen Z. Acta Phys-Chim Sin. 2012;28:504–521. [Google Scholar]
- 91.Liu YW, Jasensky J, Chen Z. Langmuir. 2012;28:2113–2121. doi: 10.1021/la203823t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Elliott ABS, Horvath R, Gordon KC. Chem Soc Rev. 2012;41:1929–1946. doi: 10.1039/c1cs15208d. [DOI] [PubMed] [Google Scholar]
- 93.Ahn D, Dhinojwala A. Silicone Surface Science. 2012:23–58. [Google Scholar]
- 94.Hankett JM, Liu Y, Zhang X, Zhang C, Chen Z. J Polym Sci, Part B: Polym Phys. 2012 [Google Scholar]
- 95.Hirose C, Akamatsu N, Domen K. Appl Spectrosc. 1992;46:1051–1072. [Google Scholar]
- 96.Yeh YL, Zhang C, Held H, Mebel A, Wei X, Lin S, Shen Y. J Chem Phys. 2001;114:1837–1843. [Google Scholar]
- 97.Wang J, Clarke ML, Chen Z. Anal Chem. 2004;76:2159–2167. doi: 10.1021/ac049887y. [DOI] [PubMed] [Google Scholar]
- 98.Kataoka S, Cremer PS. J Am Chem Soc. 2006;128:5516–5522. doi: 10.1021/ja060156k. [DOI] [PubMed] [Google Scholar]
- 99.Nguyen KT, Le Clair SV, Ye S, Chen Z. J Phys Chem B. 2009;113:12169–12180. doi: 10.1021/jp904153z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Nguyen KT, King JT, Chen Z. J Phys Chem B. 2010;114:8291–8300. doi: 10.1021/jp102343h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Curtis AD, Asplund MC, Patterson JE. J Phys Chem C. 2011;115:19303–19310. [Google Scholar]
- 102.Boughton AP, Yang P, Tesmer VM, Ding B, Tesmer JJG, Chen Z. Proc Natl Acad Sci USA. 2011;108:E667–E673. doi: 10.1073/pnas.1108236108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Raschke M, Hayashi M, Lin S, Shen Y. Chem Phys Lett. 2002;359:367–372. [Google Scholar]
- 104.Chou K, Westerberg S, Shen Y, Ross P, Somorjai G. Phys Rev B. 2004;69:153413. [Google Scholar]
- 105.Li Q, Hua R, Chou KC. J Phys Chem B. 2008;112:2315–2318. doi: 10.1021/jp0745135. [DOI] [PubMed] [Google Scholar]
- 106.Zhang C, Wang J, Khmaladze A, Liu Y, Ding B, Jasensky J, Chen Z. Opt Lett. 2011;36:2272–2274. doi: 10.1364/OL.36.002272. [DOI] [PubMed] [Google Scholar]
- 107.Shen YR. The principles of nonlinear optics. Wiley; New York: 1984. [Google Scholar]
- 108.Boyd RW. Nonlinear optics. Academic Pr; 2003. [Google Scholar]
- 109.Wang J, Chen XY, Clarke ML, Chen Z. Proc Natl Acad Sci USA. 2005;102:4978–4983. doi: 10.1073/pnas.0501206102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Fu L, Liu J, Yan ECY. J Am Chem Soc. 2011;133:8094–8097. doi: 10.1021/ja201575e. [DOI] [PubMed] [Google Scholar]
- 111.Hirose C, Akamatsu N, Domen K. J Chem Phys. 1992;96:997–1004. [Google Scholar]
- 112.Gan W, Zhang Z, Feng R, Wang H. J Phys Chem C. 2007;111:8726–8738. [Google Scholar]
- 113.Duffy DC, Davies PB, Bain CD. J Phys Chem. 1995;99:15241–15246. [Google Scholar]
- 114.Gautam K, Schwab A, Dhinojwala A, Zhang D, Dougal S, Yeganeh M. Phys Rev Lett. 2000;85:3854–3857. doi: 10.1103/PhysRevLett.85.3854. [DOI] [PubMed] [Google Scholar]
- 115.Harp GP, Rangwalla H, Yeganeh MS, Dhinojwala A. J Am Chem Soc. 2003;125:11283–11290. doi: 10.1021/ja035513v. [DOI] [PubMed] [Google Scholar]
- 116.Lu X, Han J, Shephard N, Rhodes S, Martin AD, Li D, Xue G, Chen Z. J Phys Chem B. 2009;113:12944–12951. doi: 10.1021/jp9058092. [DOI] [PubMed] [Google Scholar]
- 117.Lu X, Spanninga SA, Kristalyn CB, Chen Z. Langmuir. 2010;26:14231–14235. doi: 10.1021/la101866v. [DOI] [PubMed] [Google Scholar]
- 118.Park JB, Lakes RS. Biomaterials: an introduction. Plenum Pub Corp; 1992. [Google Scholar]
- 119.Yebra DM, Kiil S, Dam-Johansen K. Prog Org Coat. 2004;50:75–104. [Google Scholar]
- 120.Chambers LD, Stokes KR, Walsh FC, Wood RJK. Surf Coat Technol. 2006;201:3642–3652. [Google Scholar]
- 121.Krishnan S, Weinman CJ, Ober CK. J Mater Chem. 2008;18:3405–3413. [Google Scholar]
- 122.Joshi RG, Goel A, Mannari VM, Finlay JA, Callow ME, Callow JA. J Appl Polym Sci. 2009;114:3693–3703. [Google Scholar]
- 123.Zhang D, Ward RS, Shen YR, Somorjai GA. J Phys Chem B. 1997;101:9060–9064. [Google Scholar]
- 124.Chen Q, Zhang D, Somorjai G, Bertozzi CR. J Am Chem Soc. 1999;121:446–447. [Google Scholar]
- 125.Wang J, Woodcock SE, Buck SM, Chen CY, Chen Z. J Am Chem Soc. 2001;123:9470–9471. doi: 10.1021/ja0164071. [DOI] [PubMed] [Google Scholar]
- 126.Yoda R. J Biomater Sci, Polym Ed. 1998;9:561–626. doi: 10.1163/156856298x00046. [DOI] [PubMed] [Google Scholar]
- 127.Chen C, Wang J, Chen Z. Langmuir. 2004;20:10186–10193. doi: 10.1021/la049327u. [DOI] [PubMed] [Google Scholar]
- 128.Kim C, Gurau MC, Cremer PS, Yu H. Langmuir. 2008;24:10155–10160. doi: 10.1021/la800349q. [DOI] [PubMed] [Google Scholar]
- 129.Ye S, McClelland A, Majumdar P, Stafslien SJ, Daniels J, Chisholm B, Chen Z. Langmuir. 2008;24:9686–9694. doi: 10.1021/la800769z. [DOI] [PubMed] [Google Scholar]
- 130.Ye S, Majumdar P, Chisholm B, Stafslien S, Chen Z. Langmuir. 2010;26:16455–16462. doi: 10.1021/la1001539. [DOI] [PubMed] [Google Scholar]
- 131.Shi Q, Ye S, Spanninga SA, Su Y, Jiang Z, Chen Z. Soft Matter. 2009;5:3487–3494. [Google Scholar]
- 132.Ye H, Gu Z, Gracias DH. Langmuir. 2006;22:1863–1868. doi: 10.1021/la052030r. [DOI] [PubMed] [Google Scholar]
- 133.Chen C, Loch CL, Wang J, Chen Z. J Phys Chem B. 2003;107:10440–10445. [Google Scholar]
- 134.Loch CL, Ahn D, Chen C, Wang J, Chen Z. Langmuir. 2004;20:5467–5473. doi: 10.1021/la0494526. [DOI] [PubMed] [Google Scholar]
- 135.Chen C, Wang J, Loch CL, Ahn D, Chen Z. J Am Chem Soc. 2004;126:1174–1179. doi: 10.1021/ja0390911. [DOI] [PubMed] [Google Scholar]
- 136.Loch CL, Ahn D, Vázquez AV, Chen Z. J Colloid Interface Sci. 2007;308:170–175. doi: 10.1016/j.jcis.2006.12.029. [DOI] [PubMed] [Google Scholar]
- 137.Mine K, Nishio M, Sumimura S. 4,033,924. US Patent. 1977 Jul 5;
- 138.Loch CL, Ahn D, Chen Z. J Phys Chem B. 2006;110:914–918. doi: 10.1021/jp055377b. [DOI] [PubMed] [Google Scholar]
- 139.Vázquez AV, Shephard NE, Steinecker CL, Ahn D, Spanninga S, Chen Z. J Colloid Interface Sci. 2009;331:408–416. doi: 10.1016/j.jcis.2008.11.065. [DOI] [PubMed] [Google Scholar]
- 140.Zhang C, Shephard NE, Rhodes SM, Chen Z. Langmuir. 2012;28:6052–6059. doi: 10.1021/la300004x. [DOI] [PubMed] [Google Scholar]
- 141.Zhang C, Chen Z. Langmuir. 2012;29:610–619. doi: 10.1021/la3041727. [DOI] [PubMed] [Google Scholar]
- 142.Zhang C, Hankett J, Chen Z. ACS Appl Mater Interfaces. 2012;4:3730–3737. doi: 10.1021/am300854g. [DOI] [PubMed] [Google Scholar]
- 143.Vogel V. Curr Opin Colloid Interface Sci. 1996;1:257–263. [Google Scholar]
- 144.Yang P, Ramamoorthy A, Chen Z. Langmuir. 2011;27:7760–7767. doi: 10.1021/la201048t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Ding B, Chen Z. J Phys Chem B. 2012;116:2545–2552. doi: 10.1021/jp209604m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Wang J, Buck SM, Mark A, Chen Z. J Am Chem Soc. 2002;124:13302–13305. doi: 10.1021/ja026881m. [DOI] [PubMed] [Google Scholar]
- 147.Wang J, Buck SM, Chen Z. J Phys Chem B. 2002;106:11666–11672. [Google Scholar]
- 148.Wang J, Paszti Z, Mark A, Chen Z. J Phys Chem B. 2004;108:3625–3632. [Google Scholar]
- 149.Wang J, Buck SM, Chen Z. Analyst. 2003;128:773–778. doi: 10.1039/b212551j. [DOI] [PubMed] [Google Scholar]
- 150.Wang J, Clarke ML, Zhang Y, Chen X, Chen Z. Langmuir. 2003;19:7862–7866. [Google Scholar]
- 151.Roger L, Mermut O, Phillips DC, McCrea KR, Ward RS, Somorjai GA. J Phys Chem C. 2007;111:8866–8871. [Google Scholar]
- 152.Phillips DC, York RL, Mermut O, McCrea KR, Ward RS, Somorjai GA. J Phys Chem C. 2007;111:255–261. [Google Scholar]
- 153.York RL, Holinga GJ, Somorjai GA. Langmuir. 2009;25:9369–9374. doi: 10.1021/la900654m. [DOI] [PubMed] [Google Scholar]
- 154.Wang J, Mark A, Chen X, Schmaier AH, Waite JH, Chen Z. J Am Chem Soc. 2003;125:9914–9915. doi: 10.1021/ja036373s. [DOI] [PubMed] [Google Scholar]
- 155.Chen X, Tang H, Mark A, Wang J, Tew GN, Chen Z. J Am Chem Soc. 2006;128:2711–2714. doi: 10.1021/ja057029t. [DOI] [PubMed] [Google Scholar]
- 156.Nguyen KT, Le Clair SV, Ye S, Chen Z. J Phys Chem B. 2009;113:12358–12363. doi: 10.1021/jp904154w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Chen X, Wang J, Sniadecki JJ, Mark A, Chen Z. Langmuir. 2005;21:2662–2664. doi: 10.1021/la050048w. [DOI] [PubMed] [Google Scholar]
- 158.Ye S, Li H, Wei F, Jasensky J, Boughton AP, Yang P, Chen Z. J Am Chem Soc. 2012;134:6237–6243. doi: 10.1021/ja2110784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Fu L, Ma G, Yan ECY. J Am Chem Soc. 2010;132:5405–5412. doi: 10.1021/ja909546b. [DOI] [PubMed] [Google Scholar]
- 160.Xiao D, Fu L, Liu J, Batista VS, Yan ECY. J Mol Biol. 2011;421:537–547. doi: 10.1016/j.jmb.2011.12.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Tamm LK, McConnell HM. Biophys J. 1985;47:105–113. doi: 10.1016/S0006-3495(85)83882-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Wurpel GWH, Sovago M, Bonn M. J Am Chem Soc. 2007;129:8420–8421. doi: 10.1021/ja072552o. [DOI] [PubMed] [Google Scholar]
- 163.Campen RK, Ngo TTM, Sovago M, Ruysschaert JM, Bonn M. J Am Chem Soc. 2010;132:8037–8047. doi: 10.1021/ja100838q. [DOI] [PubMed] [Google Scholar]
- 164.Asanuma H, Noguchi H, Uosaki K, Yu HZ. J Am Chem Soc. 2008;130:8016–8022. doi: 10.1021/ja801023r. [DOI] [PubMed] [Google Scholar]
- 165.Howell C, Schmidt R, Kurz V, Koelsch P. Biointerphases. 2008;3:FC47–FC51. doi: 10.1116/1.3064107. [DOI] [PubMed] [Google Scholar]
- 166.Stokes GY, Gibbs-Davis JM, Boman FC, Stepp BR, Condie AG, Nguyen SBT, Geiger FM. J Am Chem Soc. 2007;129:7492–7493. doi: 10.1021/ja071848r. [DOI] [PubMed] [Google Scholar]
- 167.Walter SR, Geiger FM. J Phys Chem Lett. 2009;1:9–15. [Google Scholar]
- 168.Sartenaer Y, Tourillon G, Dreesen L, Lis D, Mani AA, Thiry PA, Peremans A. Biosens Bioelectron. 2007;22:2179–2183. doi: 10.1016/j.bios.2006.10.005. [DOI] [PubMed] [Google Scholar]
- 169.Lu XL, Shephard N, Han JL, Xue G, Chen Z. Macromolecules. 2008;41:8770–8777. [Google Scholar]
- 170.Lu XL, Li DW, Kristalyn CB, Han JL, Shephard N, Rhodes S, Xue G, Chen Z. Macromolecules. 2009;42:9052–9057. [Google Scholar]
- 171.Harp GP, Gautam KS, Dhinojwala A. J Am Chem Soc. 2002;124:7908–7909. doi: 10.1021/ja026528g. [DOI] [PubMed] [Google Scholar]
- 172.Beaman DK, Robertson EJ, Richmond GL. Langmuir. 2011;27:2104–2106. doi: 10.1021/la104390u. [DOI] [PubMed] [Google Scholar]
- 173.Beaman DK, Robertson EJ, Richmond GL. Proc Natl Acad Sci USA. 2012;109:3226–3231. doi: 10.1073/pnas.1200244109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Eisenthal K. Chem Rev. 1996;96:1343–1360. doi: 10.1021/cr9502211. [DOI] [PubMed] [Google Scholar]
- 175.Johnson CM, Tyrode E, Baldelli S, Rutland MW, Leygraf C. J Phys Chem B. 2005;109:321–328. doi: 10.1021/jp047338q. [DOI] [PubMed] [Google Scholar]
- 176.Tyrode E, Johnson CM, Baldelli S, Leygraf C, Rutland MW. J Phys Chem B. 2005;109:329–341. doi: 10.1021/jp047337y. [DOI] [PubMed] [Google Scholar]
- 177.Opdahl A, Somorjai GA. Langmuir. 2002;18:9409–9412. [Google Scholar]
- 178.Wei X, Zhuang X, Hong SC, Goto T, Shen Y. Phys Rev Lett. 1999;82:4256–4259. [Google Scholar]
- 179.Simpson GJ, Rowlen KL. J Am Chem Soc. 1999;121:2635–2636. [Google Scholar]
- 180.Cecchet F, Lis D, Caudano Y, Mani A, Peremans A, Champagne B, Guthmuller J. J Phys: Condens Matter. 2012;24:124110. doi: 10.1088/0953-8984/24/12/124110. [DOI] [PubMed] [Google Scholar]
- 181.Himmelhaus M, Eisert F, Buck M, Grunze M. J Phys Chem B. 2000;104:576–584. [Google Scholar]
- 182.Wang J, Paszti Z, Even MA, Chen Z. J Am Chem Soc. 2002;124:7016–7023. doi: 10.1021/ja012387r. [DOI] [PubMed] [Google Scholar]
- 183.Lu R, Gan W, Wu B, Chen H, Wang H. J Phys Chem B. 2004;108:7297–7306. [Google Scholar]
- 184.Zhang C, Chen Z. J Phys Chem C. 2013 doi: 10.1021/jp307472j. [DOI] [Google Scholar]
- 185.Rintoul L, Carter E, Stewart S, Fredericks P. Biopolymers. 2000;57:19–28. doi: 10.1002/(SICI)1097-0282(2000)57:1<19::AID-BIP4>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- 186.Umemura J, Kamata T, Kawai T, Takenaka T. J Phys Chem. 1990;94:62–67. [Google Scholar]
- 187.Wang J, Paszti Z, Clarke ML, Chen X, Chen Z. J Phys Chem B. 2007;111:6088–6095. doi: 10.1021/jp070383o. [DOI] [PubMed] [Google Scholar]
- 188.Belkin M, Kulakov T, Ernst KH, Yan L, Shen Y. Phys Rev Lett. 2000;85:4474–4477. doi: 10.1103/PhysRevLett.85.4474. [DOI] [PubMed] [Google Scholar]
- 189.Belkin M, Han S, Wei X, Shen Y. Phys Rev Lett. 2001;87:113001. doi: 10.1103/PhysRevLett.87.113001. [DOI] [PubMed] [Google Scholar]
- 190.Belkin M, Shen Y. Phys Rev Lett. 2003;91:213907. doi: 10.1103/PhysRevLett.91.213907. [DOI] [PubMed] [Google Scholar]
- 191.Busson B, Tadjeddine A. J Phys Chem C. 2008;112:11813–11821. [Google Scholar]
- 192.Nagahara T, Kisoda K, Harima H, Aida M, Ishibashi T. J Phys Chem B. 2009;113:5098–5103. doi: 10.1021/jp8105138. [DOI] [PubMed] [Google Scholar]
- 193.Nguyen KT, Soong R, lm SC, Waskell L, Ramamoorthy A, Chen Z. J Am Chem Soc. 2010;132:15112–15115. doi: 10.1021/ja106508f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Chen X, Boughton AP, Tesmer JJG, Chen Z. J Am Chem Soc. 2007;129:12658–12659. doi: 10.1021/ja075542w. [DOI] [PubMed] [Google Scholar]
- 195.Balik C, Simendinger W. Polymer. 1998;39:4723–4728. [Google Scholar]
- 196.Hong S, Barbari T, Sloan J. J Polym Sci, Part B: Polym Phys. 1998;36:337–344. [Google Scholar]
- 197.Sammon C, Yarwood J, Everall N. Polymer. 2000;41:2521–2534. [Google Scholar]
- 198.Ye S, Nguyen KT, Boughton AP, Mello CM, Chen Z. Langmuir. 2009;26:6471–6477. doi: 10.1021/la903932w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Han X, Soblosky L, Slutsky M, Mello CM, Chen Z. Langmuir. 2011;27:7042–7051. doi: 10.1021/la200388y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Clarke ML, Wang J, Chen Z. J Phys Chem B. 2005;109:22027–22035. doi: 10.1021/jp054456k. [DOI] [PubMed] [Google Scholar]
- 201.Wang J, Chen X, Clarke ML, Chen Z. J Phys Chem B. 2006;110:5017–5024. doi: 10.1021/jp0534683. [DOI] [PubMed] [Google Scholar]
- 202.John K, Schreiber S, Kubelt J, Herrmann A, Müller P. Biophys J. 2002;83:3315–3323. doi: 10.1016/S0006-3495(02)75332-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Kol MA, de Kroon AIPM, Killian JA, de Kruijff B. Biochemistry. 2004;43:2673–2681. doi: 10.1021/bi036200f. [DOI] [PubMed] [Google Scholar]
- 204.Liu J, Conboy JC. J Am Chem Soc. 2004;126:8376–8377. doi: 10.1021/ja048245p. [DOI] [PubMed] [Google Scholar]
- 205.Liu J, Conboy JC. Biophys J. 2005;89:2522–2532. doi: 10.1529/biophysj.105.065672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Anglin TC, Liu J, Conboy JC. Biophys J. 2007;92:L01–L03. doi: 10.1529/biophysj.106.096057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Anglin TC, Brown KL, Conboy JC. J Struct Biol. 2009;168:37–52. doi: 10.1016/j.jsb.2009.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Anglin TC, Conboy JC. Biophys J. 2008;95:186–193. doi: 10.1529/biophysj.107.118976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Liu J, Brown KL, Conboy J. Faraday Discuss. 2013;161:45–61. doi: 10.1039/c2fd20083j. [DOI] [PubMed] [Google Scholar]
- 210.Chen X, Wang J, Kristalyn CB, Chen Z. Biophys J. 2007;93:866–875. doi: 10.1529/biophysj.106.099739. [DOI] [PMC free article] [PubMed] [Google Scholar]