

Abstract
We applied a combined proteomic and metabolomic approach to obtain novel mechanistic insights in PKCε-mediated cardioprotection. Mitochondrial and cytosolic proteins from control and transgenic hearts with constitutively active or dominant negative PKCε were analyzed using difference in-gel electrophoresis (DIGE). Among the differentially expressed proteins were creatine kinase, pyruvate kinase, lactate dehydrogenase, and the cytosolic isoforms of aspartate amino transferase and malate dehydrogenase, the two enzymatic components of the malate aspartate shuttle, which is required for the import of reducing equivalents from glycolysis across the inner mitochondrial membrane. These enzymatic changes appeared to be dependent on PKCε activity, as they were not observed in mice expressing inactive PKCε. High-resolution proton nuclear magnetic resonance (1H-NMR) spectroscopy confirmed a pronounced effect of PKCε activity on cardiac glucose and energy metabolism: normoxic hearts with constitutively active PKCε had significantly lower concentrations of glucose, lactate, glutamine and creatine, but higher levels of choline, glutamate and total adenosine nucleotides. Moreover, the depletion of cardiac energy metabolites was slower during ischemia/reperfusion injury and glucose metabolism recovered faster upon reperfusion in transgenic hearts with active PKCε. Notably, inhibition of PKCε resulted in compensatory phosphorylation and mitochondrial translocation of PKCδ. Taken together, our findings are the first evidence that PKCε activity modulates cardiac glucose metabolism and provide a possible explanation for the synergistic effect of PKCδ and PKCε in cardioprotection.
Keywords: proteomics, metabolism, cardioprotection, protein kinase C
Introduction
Protein kinase C (PKC) is a heterogeneous family of phospholipid-dependent kinases. Notably, the PKCε and the PKCδ isoform have both been implicated in cardioprotection as translocation of activated PKCε and PKCδ to the membrane fraction has been detected in preconditioned hearts [1]. PKCε activation but PKCδ inhibition are thought to be involved in myocardial salvage and combined treatment with PKCδ inhibitor and PKCε activator peptides exerted an additive protective effect on the ischemic heart [2–7]. On the other hand, expression of active PKCδ increased resistance to simulated ischemia in neonatal cardiomyocytes [8]. Thus, although it is now widely accepted that PKC isoforms play a pivotal role in mediating both the early and the late phase of ischemic preconditioning [9–11], the mechanisms responsible for PKC-mediated ischaemic preconditioning are still debated. While numerous studies have addressed potential targets of PKC in cardiac signalling [12–15], the role of these specific PKC isoforms in cardiac metabolism is less clear.
To further advance our understanding of how PKC isoforms alter cardiac metabolism, we have previously identified protein and metabolite changes after ischemic preconditioning [16] and demonstrated that loss of PKCδ altered glucose metabolism resulting in a reduction in the ratio of cardiac glucose to lipid metabolites [17]. We now integrate protein with metabolites changes in cardioprotected mice with transgenic activation of PKCε and unravel metabolic changes occurring during ischemia/reperfusion injury. Our data are the first evidence that the cardioprotective effect of PKCε activation on mitochondrial function may be, in part, an indirect effect of modulating cardiac glucose metabolism providing an explanation for the synergistic effects of PKCε and PKCδ in cardioprotection.
Methods
Detailed methodology is provided in the online data supplement.
Transgenic mice
Animals used in the present study were PKCε transgenic mice (ICR background) generated using a cDNA of active PKCε driven by the α-myosin heavy chain promoter to achieve cardiac-specific expression [12, 18]. AE PKCε transgenic mice express a PKCε molecule that favors the open and active conformation (A to E mutation at A159), whereas DN PKCε mice express a dominant negative form of the same molecule (A to E mutation at A159 and K to R mutation at K436). The hearts of AE PKCε mice exhibit a 6-fold overexpression of PKCε and an inherent resistance to infarction (as compared to wild type, ICR controls) that is not observed in the DN PKCε mice [12]. Transgenic and control mice were sacrificed at the age of 6 months. Only male animals were used in all the experiments. Hearts from 8-week-old PKCδ-deficient mice served as controls for validating the antibody specificity [19].
Subcellular fractionation
Isolation of mitochondria was performed in the cold room at 4°C. Freshly-harvested mouse hearts were minced in 250mM sucrose, 1mM EGTA, 20mM Hepes, pH 7.5 with a glass homogenizer. Nuclei and unbroken cells were pelleted by centrifugation at 1,000g for 10min. The crude mitochondrial and cytosolic fraction was obtained from the supernatant by centrifugation at 4,000g for 30min. The membrane fraction was obtained by centrifugation of the cytosolic fraction at 100,000g for 1hr.
Proteomics
Key techniques involved adaptations of previously published protocols, including those for difference in-gel electrophoresis (DIGE) [20], and nano-liquid chromatography tandem mass spectrometry (nano-LC MS/MS) [21]. Methods are described online. A detailed methodology is also available on our website http://www.vascular-proteomics.com. For proteomic analysis, mitochondrial and cytosolic protein extracts were prepared from 16 murine hearts (n=6 for control and AE mice, n=4 for DN mice). Two samples were pooled for each gel to obtain sufficient material for proteomic analysis. Differences in protein expression were analyzed using the Decyder software (Version 6.5, GE healthcare).
Murine Model of Regional Myocardial Ischemic Injury
Myocardial infarction was induced in the anesthetized mouse using an established protocol of regional ischemic injury[22]. Briefly, male ICR mice were subjected to a 30-min left anterior descending coronary artery occlusion followed by 30min of reperfusion. Sham control group was subjected to open chest surgery without coronary occlusion.
Metabolomics
For metabolomic analysis, metabolite extracts were prepared from 29 murine hearts (n=5 for normoxic control and AE PKCε mice, n=4 for control and n=3 for AE PKCε mice after ischemia, and n=4 for control, AE and DN PKCε mice after 30 min reperfusion, respectively). Water-soluble metabolites were extracted in 6% perchloric acid and proton magnetic resonance spectroscopy (1H-NMR) was performed as described online [23].
Statistical analysis
Statistical analysis was performed using the analysis of variance (ANOVA) and unpaired Student’s t-test. Pairwise comparisons between metabolites were performed using the Bonferroni/Dunn test. Results were given as means±SE. A P value <0.05 was considered significant. Principal component analysis (PCA) was used for the analysis of the multivariate data produced by proteomic and metabolomic technologies, which reduces the data dimensionality. For proteomic datasets, DIGE gels were analysed using the extended data analysis (EDA) module of the Decyder software (GE healthcare). For metabolite profiles, the 1H-NMR spectra were used for generating bucket tables for PCA analysis.
Results
The proteome of PKCε transgenic hearts
To provide insights into potential cellular targets of PKCε, we compared the mitochondrial and cytosolic proteome of control hearts and transgenic hearts with constitutively active (AE) and dominant negative (DN) PKCε by DIGE. The quality of subcellular fractionation was assessed by immunoblotting (Figure 1). The intactness of mitochondria was verified by the calcium swelling assay as described [24]. A representative image of the cardiac mitochondrial and cytosolic fraction as separated by two-dimensional gel electrophoresis (2-DE, pH 3–10 nonlinear) is presented in Figure 2. Differentially expressed spots are numbered and protein identifications as obtained by nano-LC MS/MS analysis are listed in online Table I–III. Only a few mitochondrial proteins in AE transgenic hearts were consistently different from control as well as DN transgenic hearts, including mitofilin (Supplemental Figure 1A), an inner mitochondrial membrane protein and regulator of metabolic flux, and manganese superoxide dismutase, an important mitochondrial antioxidant (Supplemental Table I). The latter was subsequently confirmed by immunoblotting (Supplemental Figure 2). No difference in net expression was observed for mitofillin because post-translational modifications as well as changes in net expression can be visualized on 2-DE gels (Supplemental Figure 1A).
Figure 1. Subcellular fractionation.

Mitochondrial and cytosolic extracts were prepared from murine hearts as described in the Material and Methods section. The purity of the fractions was assessed by Western blotting for VDAC, a mitochondrial outer membrane protein, and myoglobin, an abundant cytosolic protein (A). Panel B illustrates the depletion of nuclear proteins (NuMA) and the enrichment of mitochondrial (prohibitin) compared to other membrane proteins (Na+/K+ ATPase).
Figure 2. 2-DE map of cardiac proteins in the mitochondrial and cytosolic fraction.
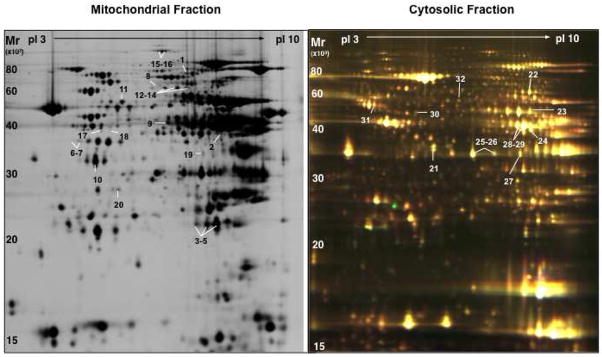
Subcellular protein extracts were pre-labelled with Cy3 and Cy5 using the DIGE approach and co-separated on large format 2-DE gels using pH 3-10NL IPG strips followed by 12% SDS polyacrylamide gels. Images were acquired on a fluorescence scanner and counterstained with silver. A silver-stained image of mitochondrial extracts and a DIGE image of cytosolic extracts is shown in panel A and B, respectively. Analyses using DeCyder® software revealed the spots showing a significant difference in AE or DN hearts. Proteins were numbered and identified by nano-LC MS/MS (Supplemental Table I–III).
Besides mitochondria, several cytosolic enzymes related to glucose and energy metabolism were profoundly influenced by PKCε activation (Supplemental Table III, Supplemental Figure 1B), including pyruvate kinase, enolase, lactate dehydrogenase, creatine kinase as well as cytosolic malate dehydrogenase and cytosolic aspartate aminotransferase. The latter two enzymes constitute the components of the malate-aspartate shuttle, which exchanges cytoplasmic malate for mitochondrial aspartate and allows the transport of glycolytically-derived reducing equivalents across the inner mitochondrial membrane. When cross-validation was performed using principal component analysis (PCA) to investigate the global variation in the proteomic space, the differentially expressed cytosolic proteins allowed a clear separation of control and transgenic hearts (Figure 3A–C).
Figure 3. Differential expression of cytosolic enzymes in PKCε transgenic hearts.

Principal Component Analysis on the set of differentially expressed cytosolic proteins (Anova<0.05) allowed clear discrimination of controls (grey), AE (green) and DN PKCε hearts (red) (A). The differentially expressed enzymes contributing to this discrimination formed a cluster of similar expression profiles (B) and are highlighted on the representative 2-DE gel shown below (C).
Unexpectedly, the proteomic experiments also revealed a pronounced effect of PKCε inhibition on the mitochondrial pyruvate dehydrogenase complex (supplemental Table II). The latter consists of E1 [the pyruvate dehydrogenase (decarboxylating) (alpha)2/(beta)2 heterotetramer], E2 (the lipoyl acetyltransferase) and E3 (the dihydrolipoyl dehydrogenase). Component X is an E3-binding protein and/or an E2 ancillary subunit with E2-like activity. Given that a 1:1 stoichiometry should exist between E1-alpha and E1-beta, the opposing directionality in changes observed in the proteomic experiment, suggests a post-translational modification. This was further substantiated by our finding that pyruvate dehydrogenase kinase 4, an inducible regulatory component, was identified within the protein spot containing E1-alpha. Phosphorylation of E1 by pyruvate dehydrogenase kinase (PDK) inactivates E1 and subsequently the entire pyruvate dehydrogenase complex. No statistically significant differences were found for cytosolic proteins. When the differentially expressed proteins were submitted to Ingenuity Pathway Analysis (Ingenuity System, Mountain View, CA), the computational algorithms built two dominant protein association networks (Supplemental Figure 3) and returned pyruvate metabolism as the top canonical pathway. Thus, our proteomic data provide strong evidence for a previously unrecognized effect of PKCε activity on cardiac glucose metabolism.
Metabolomic analysis of normoxic PKCε transgenic hearts
To prove the functional relevance of the described enzymatic changes, we applied high-resolution 1H-NMR spectroscopy to measure cardiac metabolites. A representative 1H-NMR spectrum of control and AE PKCε transgenic hearts is shown in Figure 4. A principal component (PCA) and a linear discriminant analysis (LDA) were performed to investigate the global variation of the cardiac samples in the 25-dimensional metabolite space. AE PKCε and control hearts were dissimilar enough that principal component 1 (PC1) alone gave good discrimination (Supplemental Figure 4A). 9/10 samples were correctly classified, with 1 control misclassified (LDA, leave one out). The scores on PC1 were −0.0565, −0.0039, −0.0420, −0.0321, 0.0015 for control and 0.0256, 0.0363, 0.0236, 0.0169, 0.0307 for AE PKCε hearts. The difference between the two means (blue) and PC1 (green) is plotted in the bottom panel of Figure 4. The fact that the difference between the 2 means and the direction of largest variance (PC1) is so small suggests that the two groups are well separated, which is confirmed by the cross-validated classification. Metabolites responsible for the discrimination between the two groups have been assigned. The quantitative data are included as Online Table IV. Levels of lactate, glucose, glutamine, leucine, taurine and total creatine were significantly lower in cardioprotected AE PKCε transgenic hearts, while choline, glutamate and adenosine nucleotides were higher compared to controls (Figure 5A).
Figure 4. Nuclear magnetic resonance spectra of murine hearts.

Representative high-resolution 1H-NMR spectra of AE PKCε transgenic and control hearts. Within the aliphatic region of the NMR spectra (−0.05 to 4.2 parts per million), resonances have been assigned to 25 metabolites, including creatine (Cr), glycine (Gly), taurine (Tau), phosphocholine (PC), succinate (Succ), glutamate (Glu), alanine (Ala), and lactate (Lac). Sodium 3-trimethylsilyl-2,2,3,3-tetradeuteropropionate (TSP) was added into the samples for chemical shift calibration. The bottom panel shows the first principal component (PC1) and the differences between the two means of metabolites in hearts with and without PKCε activation.
Figure 5. Comparison of metabolites during normoxia (A) and after ischemia/reperfusion injury (B).

The histogram shows the fold change of metabolites in AE PKCε hearts normalized to controls. Note the inverse pattern of most cardiac metabolites before (A) and after ischemia/reperfusion injury (B), but the consistent elevation of the adenosine nucleotide pool in AE PKCε hearts. * Significant difference p<0.05, ** p<0.01
Metabolomic analysis after ischemia/reperfusion injury
To trace metabolite changes during ischemia/reperfusion injury, we measured cardiac metabolites after 30 min of ischemia and 30 min reperfusion. Strikingly, no significant metabolite changes were detected between control and AE PKCε transgenic hearts immediately after ischemia (Supplemental Table V), but differences reappeared upon reperfusion (Supplemental Table VI, Supplemental Figure 4B): After only 30 min of reperfusion, the adenosine nucleotide pool was significantly higher in AE PKCε transgenic hearts. Similarly, the decline of the total creatine pool during schemia/reperfusion injury was less pronounced. This maintenance of cardiac energy metabolites in AE PKCε transgenic hearts coincided with a faster recovery of glucose metabolites (Figure 5B). A comparison of average metabolite concentrations in control, AE and DN PKCε transgenic hearts provides further evidence that the activity of PKCε correlates with metabolic recovery upon reperfusion (Figure 6).
Figure 6. Metabolic recovery after ischemia/reperfusion.

Average metabolite concentration after 30 min of reperfusion in control, AE and DN transgenic hearts (A). The short reperfusion time was adapted to assess the early recovery of cardiac metabolism upon restoration of blood flow. Note that concentrations of metabolites related to glucose metabolism (arrows) correlate with PKCε activity (B). * Statistically significant difference from DN hearts, ** statistically significant difference from control as well as DN hearts.
Mitochondrial translocation of PKCδ in PKCε transgenic hearts
While cardiac glucose metabolism was clearly affected by PKCε activity, it remained to be determined whether there was a cross-talk between the PKCε and the PKCδ isoform[25]. Figure 7 provides evidence that activation of PKCε did not lead to a compensatory change in PKCδ expression. Inhibition of PKCε, however, markedly increased phosphorylation of PKCδ at Thr505 and promoted translocation of PKCδ to the cardiac mitochondria (Figure 7A). The specificity of the antibodies was confirmed by using PKCδ–deficient hearts (Figure 7B, C). These results provide additional support for the differential expression of the pyruvate dehydrogenase complex in DN PKCε hearts (Supplemental Table II), as PKCδ Thr505 phosphorylation is known to alter substrate specificity[26] and the pyruvate dehydrogenase complex is one of the likely targets of PKCδ on cardiac mitochondria[16, 17, 27]. Thus, the activation state of PKCε influences PKCδ and the pyruvate dehydrogenase complex, the key enzyme linking glycolysis to mitochondrial respiration.
Figure 7. Mitochondrial translocation of PKCδ.

Mitochondrial extracts of control, AE and DN PKCε hearts were analyzed by Western blotting and probed with antibodies for phospho-PKCδ (Thr505) and total PKCδ. Ponceau red staining served as a loading control. Note the increase in phosphorylation and mitochondrial translocation of PKCδ upon inactivation of PKCε. pT denotes phosphothreonine (A). Verification of the antibody specificity by using hearts from PKCδ-deficient mice (B, C). The antibody for PKCδ-pThr505 showed good specificity. The antibody for total PKCδ recognizes three bands, of which the middle one is specific for PKCδ.
Discussion
In previous studies, our laboratory has shown that transgenic activation of PKCε in the heart is sufficient to significantly reduce myocardial infarction after coronary artery occlusion [12, 28]. In the present study, we used a novel combination of gel-based proteomics and high-resolution 1H-NMR spectroscopy-based metabolomics [29], to investigate mechanisms of PKCε-mediated cardioprotection. This comprehensive analysis revealed that PKCε activation has a previously unrecognized effect on cardiac glucose metabolism [30, 31].
PKCε and cardiac metabolism
Despite abundant information on PKCε signalling networks in the heart [12–14, 32], the manner in which the actions of these various signalling molecules integrate into cardiac metabolism is still not well understood. To our knowledge, only two papers have previously addressed the effects of PKCε activation on cardiac metabolism: Cross et al [33] used 31P-NMR spectroscopy to measure phosphorus metabolites during ischemia reperfusion injury in Langendorff-perfused hearts. Despite maintaining a greater level of myocardial ATP during ischemia and reperfusion, the phosphocreatine content in AE PKCε transgenic hearts was similar to controls. McCarthy [34] et al subsequently evaluated this phenotype in isolated mitochondria: they described a modest basal hyperpolarization in mitochondria from AE PKCε transgenic hearts versus controls. While no difference was observed in basal mitochondrial respiratory function by measuring oxygen consumption, respiratory-control index, and the rate of ATP production, PKCε activation augmented mitochondrial respiratory capacity in response to anoxia-reoxygenation. However, many issues of metabolism in AE PKCε hearts remain unresolved, especially with respect to the hearts in vivo.
Proteomics and metabolomics combined
Previous proteomic analyses of cardiac PKCε signaling complexes revealed metabolic enzymes as potential interacting partners of this serine/threonine kinase [13, 32]. We now demonstrate that some of the previously identified PKCε targets, i.e. creatine kinase, enolase, and the mitochondrial malate carrier, are indeed among the differentially expressed proteins in AE PKCε transgenic hearts. Moreover, as no data were available on how these interactions between PKCε and metabolic enzymes influence cardiac metabolism, we quantified cardiac metabolites in control and transgenic hearts under normoxia, after ischemia and after reperfusion injury. Our 1H-NMR data on extracted cardiac metabolites not only confirmed the 31P-NMR data on Langendorff-perfused hearts [33] in that cardiac energy metabolites were better preserved in AE PKCε transgenic hearts, but also provide a mechanistic underpinning for the observed cardioprotective phenotype: activation of PKCε resulted in lower lactate levels and a similar downregulation of cytosolic malate dehydrogenase isoforms as previously observed in preconditioned wildtype hearts[16]. Thus, PKCε activation induces proteomic and metabolic characteristics of a preconditioned cardiac phenotype, which were previously found to be abolished in hearts with targeted disruption of the PKCδ gene [16].
Cross-talk between PKCε and PKCδ
The striking consistency between different genetically engineered mouse models suggests that the metabolic effects of PKCε may, at least partially, be interrelated with PKCδ. Subsequent experiments confirmed a pronounced effect of PKCε activity on PKCδ translocation to cardiac mitochondria. Previously, increased expression and activation of PKCδ was observed in PKCε−/− mice [35, 36]. However, increased PKCδ activity in the complete absence of PKCε did not result in mitochondrial translocation of the PKCδ isoform. In our study, cardiac-specific manipulation of PKCε activity did not alter PKCδ expression, but inhibition of PKCε was associated with increased PKCδ loop phosphorylation, mitochondrial translocation and differential expression of the pyruvate dehydrogenase complex. These data confirm our previous results in PKCδ-deficient mice, where altered activity of the pyruvate dehydrogenase complex was postulated to be a likely explanation for the observed metabolic effects after deletion of the PKCδ gene [16, 17, 37].
Notably, despite increased PKCδ on mitochondria, transgenic hearts with DN PKCε showed no cardioprotective phenotype, which may be consistent with previous reports claiming that translocation of PKCδ is required for mediating cardioprotection after pharmacological[38], but not ischemic preconditioning [39]. On the other hand, a moderate increase in mitochondrial PKCδ was also observed in AE PKCε hearts and similar to mice deficient for PKCε [35], PKCδ knockout mice lost cardioprotection in response to ischemic preconditioning [16]. In fact, cardiac damage was even exaggerated by ischemic preconditioning in the absence of PKCδ [16]. This finding is in line with a recent clinical trial [40], demonstrating that the intra-coronary application of high doses of a PKCδ inhibitor during primary percutaneous coronary intervention were associated with an increase, rather than a decrease, in infarct size [41]. Thus, deficiency for either PKCε or PKCδ was sufficient to abrogate the cardioprotective effects of ischemic preconditioning, indicating that an individual PKC isoform may not be cardioprotective per se, but that cardioprotection may require the interaction of both PKC isoforms and that the cardioprotective effect may be the consequence of their combined orchestration of cardiac glucose and energy metabolism (Figure 8).
Figure 8. Model for PKCε-initiated metabolic changes linked to cardioprotection.
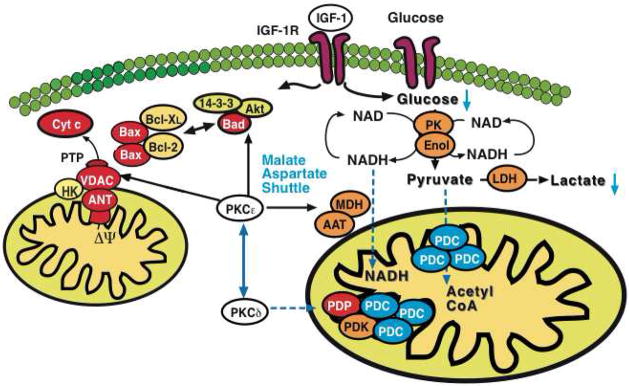
PKCε activation resulted in differential expression of pyruvate kinase (PK), enolase (Enol) and lactate dehydrogenase (LDH) with a corresponding reduction of glucose and lactate in murine hearts. Notably, a pronounced effect was also observed on cytosolic malate dehydrogenase (MDH) and aspartate aminotransferase (AAT), the two enzymatic components of the malate-aspartate shuttle. The latter transfers electrons from the cytoplasm to mitochondria and is important in allowing maximum release of the free energy in glycolysis under aerobic conditions. In contrast, inhibition of PKCε stimulated loop phosphorylation and translocation of PKCδ to cardiac mitochondria, where it targets the pyruvate dehydrogenase complex (PDC), the enzyme responsible for converting the glycolytic endproduct pyruvate to acetyl-CoA. The pyruvate dehydrogenase complex is located at the inner mitochondrial membrane and inhibited when phosphorylated by pyruvate dehydrogenase kinase (PDK) and activated upon dephosphorylation by pyruvate dehydrogenase phosphatase (PDP). Thus, PKCε and PKCδ activities influence key enzymatic reactions bridging aerobic and anaerobic glucose metabolism.
Limitations of the study
Although proteomics and metabolomics offer a suite of tools to interrogate tissue metabolism [29, 37, 42], it is important to acknowledge that both techniques are biased to high-abundant components. Moreover, metabolites were extracted directly from snap-frozen hearts and a reduction of a metabolite as measured by 1H-NMR spectroscopy may be the consequence of increased consumption or decreased production. As enzymatic activity in tissues is reflected in the expression and post-translational modifications of enzymes [43], 2-DE offers a particular advantage by visualizing posttranslational modifications of high abundant enzymes as a shift in isoelectric point or molecular weight [44]. In combination, these emerging technologies contribute to a better understanding of enzymatic and metabolite changes associated with PKCε-mediated cardioprotection.
Conclusion
While activation of PKCε and inhibition of PKCδ were known to be cardioprotective against ischemia/reperfusion injury, the common denominator in cardioprotective signalling between the two PKC isoforms remained unknown. Our studies demonstrate that PKCε and PKCδ influence key steps in glucose metabolism of murine hearts. Thus, these kinases appear to be uniquely positioned to integrate signaling with metabolism, a central determinant of cardiomyocyte life and death. Future studies will have to address whether a similar interplay between the two PKC isoforms is also observed in human hearts and other experimental models of cardioprotection.
Acknowledgments
This work emanates from the European Vascular Genomics Network (http://www.evgn.org), a Network of Excellence supported by the European Community’s sixth Framework Programme for Research Priority 1 “Life sciences, genomics and biotechnology for health” (Contract N°LSHM-CT-2003-503254). This work was supported in part by grants from the British Heart Foundation to M. Mayr and Q. Xu, by the Oak Foundation to Q. Xu, by NIH grants HL-76526, HL63901, HL65431, HL 80691, and HL-80111 to P. Ping, and Laubisch Endowment to P Ping.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Mackay K, Mochly-Rosen D. Localization, anchoring, and functions of protein kinase C isozymes in the heart. J Mol Cell Cardiol. 2001 Jul;33(7):1301–7. doi: 10.1006/jmcc.2001.1400. [DOI] [PubMed] [Google Scholar]
- 2.Hahn HS, Yussman MG, Toyokawa T, Marreez Y, Barrett TJ, Hilty KC, et al. Ischemic protection and myofibrillar cardiomyopathy: dose-dependent effects of in vivo deltaPKC inhibition. Circ Res. 2002 Oct 18;91(8):741–8. doi: 10.1161/01.res.0000037091.64492.69. [DOI] [PubMed] [Google Scholar]
- 3.Chen L, Hahn H, Wu G, Chen CH, Liron T, Schechtman D, et al. Opposing cardioprotective actions and parallel hypertrophic effects of delta PKC and epsilon PKC. Proc Natl Acad Sci U S A. 2001 Sep 25;98(20):11114–9. doi: 10.1073/pnas.191369098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Inagaki K, Hahn HS, Dorn GW, 2nd, Mochly-Rosen D. Additive Protection of the Ischemic Heart Ex Vivo by Combined Treatment With {delta}-Protein Kinase C Inhibitor and {epsilon}-Protein Kinase C Activator. Circulation. 2003 Jul 14; doi: 10.1161/01.CIR.0000081943.93653.73. [DOI] [PubMed] [Google Scholar]
- 5.Murriel CL, Churchill E, Inagaki K, Szweda LI, Mochly-Rosen D. Protein Kinase C{delta} Activation Induces Apoptosis in Response to Cardiac Ischemia and Reperfusion Damage: A MECHANISM INVOLVING BAD AND THE MITOCHONDRIA. J Biol Chem. 2004 Nov 12;279(46):47985–91. doi: 10.1074/jbc.M405071200. [DOI] [PubMed] [Google Scholar]
- 6.Dorn GW, 2nd, Souroujon MC, Liron T, Chen CH, Gray MO, Zhou HZ, et al. Sustained in vivo cardiac protection by a rationally designed peptide that causes epsilon protein kinase C translocation. Proc Natl Acad Sci U S A. 1999 Oct 26;96(22):12798–803. doi: 10.1073/pnas.96.22.12798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liu GS, Cohen MV, Mochly-Rosen D, Downey JM. Protein kinase C-epsilon is responsible for the protection of preconditioning in rabbit cardiomyocytes. J Mol Cell Cardiol. 1999 Oct;31(10):1937–48. doi: 10.1006/jmcc.1999.1026. [DOI] [PubMed] [Google Scholar]
- 8.Zhao J, Renner O, Wightman L, Sugden PH, Stewart L, Miller AD, et al. The expression of constitutively active isotypes of protein kinase C to investigate preconditioning. J Biol Chem. 1998 Sep 4;273(36):23072–9. doi: 10.1074/jbc.273.36.23072. [DOI] [PubMed] [Google Scholar]
- 9.Bolli R. The late phase of preconditioning. Circ Res. 2000;87(11):972–83. doi: 10.1161/01.res.87.11.972. [DOI] [PubMed] [Google Scholar]
- 10.Kloner RA, Jennings RB. Consequences of brief ischemia: stunning, preconditioning, and their clinical implications: part 1. Circulation. 2001 Dec 11;104(24):2981–9. doi: 10.1161/hc4801.100038. [DOI] [PubMed] [Google Scholar]
- 11.Kloner RA, Jennings RB. Consequences of brief ischemia: stunning, preconditioning, and their clinical implications: part 2. Circulation. 2001 Dec 18;104(25):3158–67. doi: 10.1161/hc5001.100039. [DOI] [PubMed] [Google Scholar]
- 12.Ping P, Song C, Zhang J, Guo Y, Cao X, Li RC, et al. Formation of protein kinase C(epsilon)-Lck signaling modules confers cardioprotection. J Clin Invest. 2002 Feb;109(4):499–507. doi: 10.1172/JCI13200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ping P, Zhang J, Pierce WM, Jr, Bolli R. Functional proteomic analysis of protein kinase C epsilon signaling complexes in the normal heart and during cardioprotection. Circ Res. 2001 Jan 19;88(1):59–62. doi: 10.1161/01.res.88.1.59. [DOI] [PubMed] [Google Scholar]
- 14.Baines CP, Zhang J, Wang GW, Zheng YT, Xiu JX, Cardwell EM, et al. Mitochondrial PKCepsilon and MAPK form signaling modules in the murine heart: enhanced mitochondrial PKCepsilon-MAPK interactions and differential MAPK activation in PKCepsilon-induced cardioprotection. Circ Res. 2002 Mar 8;90(4):390–7. doi: 10.1161/01.res.0000012702.90501.8d. [DOI] [PubMed] [Google Scholar]
- 15.Zhang J, Baines CP, Zong C, Cardwell EM, Wang G, Vondriska TM, et al. Functional proteomic analysis of a three-tier PKCepsilon-Akt-eNOS signaling module in cardiac protection. Am J Physiol Heart Circ Physiol. 2005 Feb;288(2):H954–61. doi: 10.1152/ajpheart.00756.2004. [DOI] [PubMed] [Google Scholar]
- 16.Mayr M, Metzler B, Chung YL, McGregor E, Mayr U, Troy H, et al. Ischemic preconditioning exaggerates cardiac damage in PKC-{delta} null mice. Am J Physiol Heart Circ Physiol. 2004 Aug;287(2):H946–H56. doi: 10.1152/ajpheart.00878.2003. [DOI] [PubMed] [Google Scholar]
- 17.Mayr M, Chung YL, Mayr U, McGregor E, Troy H, Baier G, et al. Loss of PKC-{delta} alters cardiac metabolism. Am J Physiol Heart Circ Physiol. 2004 Aug;287(2):H937–H45. doi: 10.1152/ajpheart.00877.2003. [DOI] [PubMed] [Google Scholar]
- 18.Pass JM, Zheng Y, Wead WB, Zhang J, Li RC, Bolli R, et al. PKCepsilon activation induces dichotomous cardiac phenotypes and modulates PKCepsilon-RACK interactions and RACK expression. Am J Physiol Heart Circ Physiol. 2001 Mar;280(3):H946–55. doi: 10.1152/ajpheart.2001.280.3.H946. [DOI] [PubMed] [Google Scholar]
- 19.Leitges M, Mayr M, Braun U, Mayr U, Li C, Pfister G, et al. Exacerbated vein graft arteriosclerosis in protein kinase Cdelta-null mice. J Clin Invest. 2001 Nov;108(10):1505–12. doi: 10.1172/JCI12902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mayr M, Zampetaki A, Sidibe A, Mayr U, Yin X, De Souza AI, et al. Proteomic and metabolomic analysis of smooth muscle cells derived from the arterial media and adventitial progenitors of apolipoprotein E-deficient mice. Circ Res. 2008 May 9;102(9):1046–56. doi: 10.1161/CIRCRESAHA.108.174623. [DOI] [PubMed] [Google Scholar]
- 21.Gomes AV, Zong C, Edmondson RD, Li X, Stefani E, Zhang J, et al. Mapping the murine cardiac 26S proteasome complexes. Circ Res. 2006 Aug 18;99(4):362–71. doi: 10.1161/01.RES.0000237386.98506.f7. [DOI] [PubMed] [Google Scholar]
- 22.Wang G, Liem DA, Vondriska TM, Honda HM, Korge P, Pantaleon DM, et al. Nitric oxide donors protect murine myocardium against infarction via modulation of mitochondrial permeability transition. Am J Physiol Heart Circ Physiol. 2005 Mar;288(3):H1290–5. doi: 10.1152/ajpheart.00796.2004. [DOI] [PubMed] [Google Scholar]
- 23.Mayr M, Yusuf S, Weir G, Chung YL, Mayr U, Yin X, et al. Combined metabolomic and proteomic analysis of human atrial fibrillation. J Am Coll Cardiol. 2008 Feb 5;51(5):585–94. doi: 10.1016/j.jacc.2007.09.055. [DOI] [PubMed] [Google Scholar]
- 24.Baines CP, Song CX, Zheng YT, Wang GW, Zhang J, Wang OL, et al. Protein kinase Cepsilon interacts with and inhibits the permeability transition pore in cardiac mitochondria. Circ Res. 2003 May 2;92(8):873–80. doi: 10.1161/01.RES.0000069215.36389.8D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rybin VO, Guo J, Gertsberg Z, Elouardighi H, Steinberg SF. Protein kinase Cepsilon (PKCepsilon) and Src control PKCdelta activation loop phosphorylation in cardiomyocytes. J Biol Chem. 2007 Aug 10;282(32):23631–8. doi: 10.1074/jbc.M701676200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liu Y, Belkina NV, Graham C, Shaw S. Independence of protein kinase C-delta activity from activation loop phosphorylation: structural basis and altered functions in cells. J Biol Chem. 2006 Apr 28;281(17):12102–11. doi: 10.1074/jbc.M600508200. [DOI] [PubMed] [Google Scholar]
- 27.Churchill EN, Murriel CL, Chen CH, Mochly-Rosen D, Szweda LI. Reperfusion-induced translocation of deltaPKC to cardiac mitochondria prevents pyruvate dehydrogenase reactivation. Circ Res. 2005 Jul 8;97(1):78–85. doi: 10.1161/01.RES.0000173896.32522.6e. [DOI] [PubMed] [Google Scholar]
- 28.Ping P, Zhang J, Qiu Y, Tang XL, Manchikalapudi S, Cao X, et al. Ischemic preconditioning induces selective translocation of protein kinase C isoforms epsilon and eta in the heart of conscious rabbits without subcellular redistribution of total protein kinase C activity. Circ Res. 1997 Sep;81(3):404–14. doi: 10.1161/01.res.81.3.404. [DOI] [PubMed] [Google Scholar]
- 29.Mayr M, Madhu B, Xu Q. Proteomics and metabolomics combined in cardiovascular research. Trends Cardiovasc Med. 2007 Feb;17(2):43–8. doi: 10.1016/j.tcm.2006.11.004. [DOI] [PubMed] [Google Scholar]
- 30.Depre C, Vanoverschelde JL, Taegtmeyer H. Glucose for the heart. Circulation. 1999 Feb 2;99(4):578–88. doi: 10.1161/01.cir.99.4.578. [DOI] [PubMed] [Google Scholar]
- 31.Vogt AM, Poolman M, Ackermann C, Yildiz M, Schoels W, Fell DA, et al. Regulation of glycolytic flux in ischemic preconditioning. A study employing metabolic control analysis. J Biol Chem. 2002 Jul 5;277(27):24411–9. doi: 10.1074/jbc.M201138200. [DOI] [PubMed] [Google Scholar]
- 32.Edmondson RD, Vondriska TM, Biederman KJ, Zhang J, Jones RC, Zheng Y, et al. Protein kinase C epsilon signaling complexes include metabolism- and transcription/translation-related proteins: complimentary separation techniques with LC/MS/MS. Mol Cell Proteomics. 2002 Jun;1(6):421–33. doi: 10.1074/mcp.m100036-mcp200. [DOI] [PubMed] [Google Scholar]
- 33.Cross HR, Murphy E, Bolli R, Ping P, Steenbergen C. Expression of activated PKC epsilon (PKC epsilon) protects the ischemic heart, without attenuating ischemic H(+) production. J Mol Cell Cardiol. 2002 Mar;34(3):361–7. doi: 10.1006/jmcc.2001.1518. [DOI] [PubMed] [Google Scholar]
- 34.McCarthy J, McLeod CJ, Minners J, Essop MF, Ping P, Sack MN. PKCepsilon activation augments cardiac mitochondrial respiratory post-anoxic reserve--a putative mechanism in PKCepsilon cardioprotection. J Mol Cell Cardiol. 2005 Apr;38(4):697–700. doi: 10.1016/j.yjmcc.2005.02.010. [DOI] [PubMed] [Google Scholar]
- 35.Gray MO, Zhou HZ, Schafhalter-Zoppoth I, Zhu P, Mochly-Rosen D, Messing RO. Preservation of base-line hemodynamic function and loss of inducible cardioprotection in adult mice lacking protein kinase C epsilon. J Biol Chem. 2004 Jan 30;279(5):3596–604. doi: 10.1074/jbc.M311459200. [DOI] [PubMed] [Google Scholar]
- 36.Klein G, Schaefer A, Hilfiker-Kleiner D, Oppermann D, Shukla P, Quint A, et al. Increased collagen deposition and diastolic dysfunction but preserved myocardial hypertrophy after pressure overload in mice lacking PKCepsilon. Circ Res. 2005 Apr 15;96(7):748–55. doi: 10.1161/01.RES.0000161999.86198.1e. [DOI] [PubMed] [Google Scholar]
- 37.Mayr M, Siow R, Chung YL, Mayr U, Griffiths JR, Xu Q. Proteomic and metabolomic analysis of vascular smooth muscle cells: role of PKCdelta. Circ Res. 2004 May 28;94(10):e87–96. doi: 10.1161/01.RES.0000131496.49135.1d. [DOI] [PubMed] [Google Scholar]
- 38.Fryer RM, Wang Y, Hsu AK, Gross GJ. Essential activation of PKC-delta in opioid-initiated cardioprotection. Am J Physiol Heart Circ Physiol. 2001 Mar;280(3):H1346–53. doi: 10.1152/ajpheart.2001.280.3.H1346. [DOI] [PubMed] [Google Scholar]
- 39.Fryer RM, Hsu AK, Wang Y, Henry M, Eells J, Gross GJ. PKC-delta inhibition does not block preconditioning-induced preservation in mitochondrial ATP synthesis and infarct size reduction in rats. Basic Res Cardiol. 2002 Jan;97(1):47–54. doi: 10.1007/s395-002-8387-x. [DOI] [PubMed] [Google Scholar]
- 40.Bates E, Bode C, Costa M, Gibson CM, Granger C, Green C, et al. Intracoronary KAI-9803 as an adjunct to primary percutaneous coronary intervention for acute ST-segment elevation myocardial infarction. Circulation. 2008 Feb 19;117(7):886–96. doi: 10.1161/CIRCULATIONAHA.107.759167. [DOI] [PubMed] [Google Scholar]
- 41.Metzler B, Xu Q, Mayr M. Letter by Metzler et al regarding article, “Intracoronary KAI-9803 as an adjunct to primary coronary intervention for acute ST-segment elevation myocardial infarction”. Circulation. 2008 Jul 22;118(4):e80. doi: 10.1161/CIRCULATIONAHA.108.772772. [DOI] [PubMed] [Google Scholar]
- 42.Mayr M, Chung YL, Mayr U, Yin X, Ly L, Troy H, et al. Proteomic and metabolomic analyses of atherosclerotic vessels from apolipoprotein E-deficient mice reveal alterations in inflammation, oxidative stress, and energy metabolism. Arterioscler Thromb Vasc Biol. 2005 Oct;25(10):2135–42. doi: 10.1161/01.ATV.0000183928.25844.f6. [DOI] [PubMed] [Google Scholar]
- 43.Sun J, Morgan M, Shen RF, Steenbergen C, Murphy E. Preconditioning results in S-nitrosylation of proteins involved in regulation of mitochondrial energetics and calcium transport. Circ Res. 2007 Nov 26;101(11):1155–63. doi: 10.1161/CIRCRESAHA.107.155879. [DOI] [PubMed] [Google Scholar]
- 44.Mayr M, Mayr U, Chung YL, Yin X, Griffiths JR, Xu Q. Vascular proteomics: Linking proteomic and metabolomic changes. Proteomics. 2004 Dec;4(12):3751–61. doi: 10.1002/pmic.200400947. [DOI] [PubMed] [Google Scholar]