

Abstract
The precise role of AMP-activated protein kinase (AMPK), a target of metformin, in pancreatic β cells remains controversial, even though metformin was recently shown to enhance the expression of incretin receptors (GLP-1 and GIP receptors) in pancreatic β cells. In this study, we investigated the effect of AMPK in the regulation of incretin receptors expression in pancreatic islets. The phosphorylation of AMPK in the mouse islets was decreased by increasing glucose concentrations. We showed the expression of incretin receptors in bell-shaped response to glucose; Expression of the incretin receptors in the isolated islets showed higher levels under a medium glucose concentration (11.1 mM) than that under a low glucose concentration (2.8 mM), but was suppressed under a high glucose concentration (22.2 mM). Both treatment with an AMPK inhibitor and DN-AMPK expression produced a significant increase of the incretin receptors expression under a low glucose concentration. By contrast, in hyperglycemic db/db islets, the enhancing effect of the AMPK inhibitor on the expression of incretin receptors was diminished under a low glucose concentration. Taken together, AMPK is involved in the regulation of incretin receptors expression in pancreatic islets under a low glucose concentration.
Introduction
Type 2 diabetes is characterized by impaired β cell function and peripheral insulin resistance [1], [2]. In regard to β cell function, the incretin effect, i.e., the increased insulin response to glucose, is markedly reduced in patients with type 2 diabetes [3]. While the incretin effect accounts for more than 50% of the meal-related insulin secretion in healthy individuals [4], its contribution to the overall insulin response after oral glucose ingestion may amount to <20% in patients with type 2 diabetes [3]. The two main incretins are glucagon-like peptide 1 (GLP-1) [5] and glucose-dependent insulinotropic peptide (GIP) [6].
The mechanisms underlying the downregulation of the incretin effect in patients with type 2 diabetes are still incompletely understood. Impaired insulinotropic activity of the incretins may be responsible for the downregulation of the incretin receptors expression. The idea of a reduced expression of GIP receptors in patients with type 2 diabetes has already expounded in 1997 [7]. The results of previous animal studies lend support to this contention [8]–[10]. Moreover, in a recent study, exposure of β cells to high blood glucose concentrations led to downregulation of the GLP-1 receptor expression and impaired insulin secretion in response to GLP-1, both in vivo and in vitro [8]. These studies provided the rationale for the design of new therapies, based on increment of the incretin receptor expressions.
Recently, GLP-1 receptor agonists [11] and dipeptidyl-peptidase-4 inhibitors [11] have come to be widely used as treatment targeting β cell dysfunction in patients with type 2 diabetes [12]. In addition, metformin has been increasingly used in combination with incretin-based compounds [13]. According to previous reports, metformin not only increases the plasma GLP-1 levels in rodents [14]–[16], and humans [17]–[19], but also enhances the expressions of GLP-1 and GIP receptor in INS-1 β cells and mouse islets [14], [20]. However, the molecular target of metformin is still elusive. The main effect of metformin is to decrease hepatic glucose production and metformin has been shown to inhibit gluconeogenesis through the AMPK-dependent and –independent mechanism [21]. The effects of metformin on the expression of the incretin receptors in β cells are reported to be mediated by a PPAR-α-dependent, AMPK-independent mechanism [14]. On the contrary, Pan et al. reported that 5-aminoimidazole-4-carboxamide-1-β-d-riboruranosid (AICAR) mimicked the actions of metformin on the expression of the incretin receptors [20]. The interactions between metformin and the incretin axis also remain to be clearly elucidated.
AMPK is a heterotrimeric serine/threonine protein kinase that acts as a metabolic sensor of the cellular energy status [22]. AMPK activity is activated by a wide array of metabolic stresses, including hypoxia, ischemia, and oxidative stresses [23]. In general, activation of AMPK triggers glucose uptake and lipid oxidation to produce energy, while turning off energy-consuming processes including glucose and lipid production to restore energy balance. AMPK activity is regulated both allosterically by AMP, and through reversible phosphorylation at Thr-172 of the α-subunit by an upstream kinase. AMPK controls whole-body glucose homeostasis by regulating metabolism in multiple peripheral tissues, such as skeletal muscle, liver, adipose tissue, and pancreatic β cells-key tissues in pathogenesis of type 2 diabetes. While AMPK activation in the muscle and liver improves the insulin sensitivity and is now established as a strategy for glucose lowering [23], the role of AMPK in the pancreatic β cells is controversial [24]. It has been reported that elevated glucose concentrations suppress AMPK phosphorylation and activation, and that the AMPK activity is inversely correlated with insulin release [25]–[28]. However, other studies have shown that insulin release is normal in isolated islets from mice lacking either the α1 or α2 catalytic subunit of AMPK [29]–[31]. Despite its profound effect on insulin release, the role of AMPK in β cell is unclear and downstream targets of AMPK that mediate these physiological processes remain to be identified. Thus, it also remains to be established whether AMPK plays a role in the regulation of incretin receptors expression. In the present study, we examined the effects of AMPK on the incretin receptors expression in mouse pancreatic islets.
Materials and Methods
Ethics statement
This study was approved by the Animal Care Committee of the Yokohama City University (Permit Number: F11-28, F11-29). All the animal procedures were performed in accordance with the institutional animal care guidelines and the guidelines of the Animal Care Committee of the Yokohama City University. Special care was taken to reduce animal suffering and to use the minimum number of animals for all studies.
Animals
C57BL/6J male mice aged 8–9 weeks were purchased from CLEA Japan (Tokyo, Japan). Littermate db/db and db/+ mice aged 7 weeks were purchased from Charles River Japan (Yokohama, Japan). All mice were fed standard chow (MF, Oriental Yeast, Tokyo, Japan) and were allowed free access to water and the diets. The animal housing rooms were maintained at a constant room temperature (25°C) under a 12-h light (7:00 A.M.)/dark (7:00 P.M.) cycle.
Treatment of the islets with chemicals
The islets were isolated with collagenase (Sigma-Aldrich), as described elsewhere [32]. The AMPK activator, 5-aminoimidazole-4-carboxamide-1-β-d-riboruranosid (AICAR), and the AMPK inhibitor, compound C, were purchased from Sigma-Aldrich (St. Louis, MO). The glucokinase activator (GKA), compound A, was purchased from Calbiochem (Darmstadt, Germany). Isolated islets were incubated for 24 hours in RPMI1640 medium containing 2.8 or 11.1 mM glucose and 30 µM compound A, 1 mM AICAR, 40 µM compound C, or vehicle.
Glucose-stimulated insulin secretion by islets
After culture for 24 hours in RPMI 1640 medium at 2.8 mM glucose supplemented with 10% fetal bovine serum and vehicle or 40 µM compound C, 10 islets were incubated at 37°C for 1 h in Krebs-Ringer bicarbonate (KRB) buffer containing 2.8 mM glucose. The buffer was then replaced with fresh KRB buffer containing 2.8 mM glucose with or without 10 nM GLP-1 and 10 nM GIP (Sigma-Aldrich) for 1.5 h, followed by replacement with KRB buffer containing 11.1 mM glucose with or without 10 nM GLP-1 and 10 nM GIP for an additional 1.5 h. Compound C (40 µM) was added to the KRB/glucose for throughout the study.
Western blot analysis
The isolated islets (100 islets per mouse) were incubated in each well of 12-well plate. After culture for 24 hours, the isolated islets (100 islets per sample) were lysed, as described elsewhere [32]. After centrifugation, the extracts were subjected to immunoblotting with antibodies to AMPKα, p-AMPKα (Cell signaling Technology), anti-GIPR antibody (Santa Cruz Biotechnology, CA, USA), anti-GLP-1R antibody, GAPDH (Abcam, MA, USA). Densitometry was performed using the Multi gauge V3.0 software (Fuji Film Life Science, Tokyo, Japan).
Real-time PCR
Total RNA extraction, cDNA synthesis, and real-time quantitative PCR were performed as described previously [33]. cDNA was prepared using the High Capacity cDNA Reverse Transcription Kits (Applied Biosystems, CA, USA) and quantitative PCR was performed by 7900 real-time PCR system (Applied Biosystems). All the probes were purchased from Applied Biosystems. The data were normalized to the mRNA expression level of β actin and to the control samples.
Suppression of AMPK phosphorylation by adenovirus-mediated expression of a mutant form of AMPK
AdCMV-LacZ and Ad-AMPK-α2-K45R, a dominant-inhibitory AMPK, were kindly provided by Dr. Birnbaum (Howard Hughes Medical Institute and Department of Medicine, University of Pennsylvania school of Medicine, Philadelphia, Pennsylvania, USA). Trypsinized islet cells were cultured in monolayer, and infected with AdCMV-LacZ or Ad-AMPK-α2-K45R viruses at 250 m.o.i. for 24 h in 1.5 ml of RPMI 1640 medium containing 2.8 mM glucose.
Statistical analyses
Results are expressed as means ± S.E. (n). Differences between two groups were analyzed for statistical significance by Student's t test. Individual comparisons among more than 2 groups were assessed with the post-hoc Fisher's PLSD test. P<0.05 was considered statistically significant.
Results
The effect of glucose signal on the phosphorylation level of AMPK and the expression of the incretin receptors in mouse islets
We first confirmed that the phosphorylation of endogenous AMPKα on Thr172 in the mouse islets was decreased by increasing glucose concentrations (Figure 1A). We next evaluated whether the glucose signal regulated the expression of the incretin receptors (GLP-1 and GIP receptors). Expression of the incretin receptors at 11.1 mM glucose showed higher levels than that at 2.8 or 5.6 mM glucose, but was suppressed under high glucose concentration (22.2 mM) (Figure 1B). Moreover, when the islets were incubated at 2.8, 11.1 or 22.2 mM glucose for up to 48 h, the expression of the incretin receptors at 11.1 mM glucose was higher than those at 2.8 mM glucose, however, the expression was suppressed at 22.2 mM glucose at any of the indicated times (Figure 1C).
Figure 1. The effect of glucose signal on the phosphorylation level of AMPK and the expressions of the incretin receptors in mouse islets.

A), B) Isolated islets were incubated for 24 h in the presence of 2.8, 5.6, 11.1 or 22.2 mM glucose. A) Total cell extracts from the isolated islets were subjected to immunoblotting for p-AMPKα (Thr172) and AMPKα. Data shown are representative of three independent experiments. B) The GLP-1 and GIP receptor expressions in the islets were determined by real-time quantitative RT-PCR and normalized to the expression level of β actin mRNA and to the samples at 2.8 mM glucose (n = 4). The samples are from independent experiments. *P<0.05 and ** P<0.01. C) Isolated islets were incubated for the indicated times in the presence of 2.8, 11.1 or 22.2 mM glucose. The GLP-1 and GIP receptor expressions were determined as shown in B. (n = 4). The samples are from independent experiments. * P<0.05 and ** P<0.01 vs. 2.8 mM glucose. † P<0.05 and †† P<0.01 vs. 22.2 mM glucose.
The effect of glucokinase activator on the expression of the incretin receptors under a low glucose concentration
It has shown that glucokinase, the main enzyme involved in the phosphorylation of glucose in the β cells [34], plays a crucial role in the glucose-induced dephosphorylation of AMPK [35]. Glucokinase activator (GKA) is an allosteric activator of glucokinase in the β cells and liver [36]. To assess whether the glucose signal might be involved in the expression of the incretin receptors, we treated isolated islets with GKA under low glucose concentration (2.8 mM). Treatment with GKA produced dephosphorylation of AMPK in the islets at 2.8 mM glucose (Figure 2A). The expression of the incretin receptors was significantly increased by treatment with GKA at 2.8 mM glucose (Figure 2B).
Figure 2. The effect of glucokinase activator on the expression of the incretin receptors under a low glucose concentration.
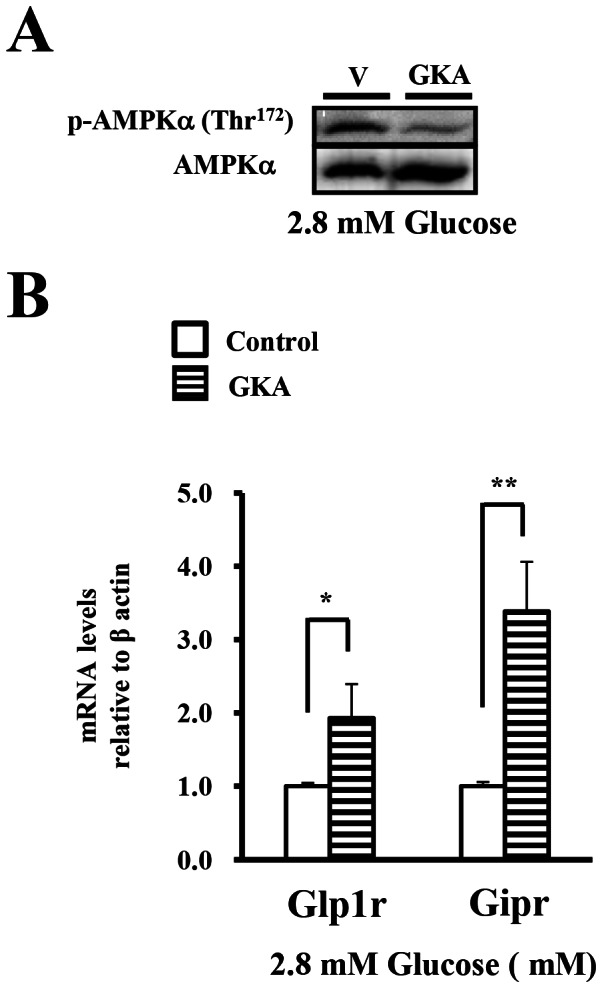
A), B). Isolated islets were incubated with DMSO (ctl) or GKA (G: 30 µM compound A) in the presence of 2.8 mM glucose for 24 hours. Total cell extracts from the isolated islets were subjected to immunoblotting for p-AMPKα (Thr172) and AMPKα. Data shown are representative of three independent experiments. B) The GLP-1 and GIP receptor expressions were determined by real-time quantitative RT-PCR and normalized to the β actin mRNA level and to the control samples (n = 4). *P<0.05, ** P<0.01.
The effect of pharmacologic modulation of AMPK phosphorylation on the expression of the incretin receptors under a low glucose concentration
To assess whether the phosphorylation state of AMPK regulated the expression of the incretin receptors in the islets under lower glucose concentrations (2.8–11.1 mM), we treated isolated islets with an AMPK inhibitor under a low glucose concentration (2.8 mM) to activate the endogenous AMPK. The AMPK inhibitor decreased the phosphorylation level of AMPK to 30% (Figure 3A). Treatment with the AMPK inhibitor resulted in a significant increase in the expression of the incretin receptors as compared with the levels in the control at 2.8 mM glucose (Figure 3B). These results suggested that the expression of the incretin receptors was dependent, at least in part, on the phosphorylation state of AMPK under low glucose concentrations.
Figure 3. The effect of pharmacologic modulation of AMPK phosphorylation on the expressions of the incretin receptors.

A)–C) Isolated islets were treated for 24 h with vehicle (Veh; DMSO) or the AMPK inhibitor (AMPKi; 40 µM compound C) in the presence of 2.8 mM glucose. A) Total islet extracts from the isolated islets were subjected to immunoblotting for p-AMPKα (Thr172) and AMPKα (n = 3). Data shown are representative of three independent experiments. B) The GLP-1 and GIP receptor expressions were determined by real-time quantitative RT-PCR and normalized to the expression level of β actin mRNA and normalized to the vehicle-treated samples (n = 4–7). C) Insulin secretion by the islets treated with the AMPK inhibitor or vehicle (ctl) in the presence of 2.8 mM glucose for 24 h with or without addition of 10 nM GLP-1 or 10 nM GIP. The results are expressed in ng of insulin/10 islets/90 min (n = 3). *P<0.05 and ** P<0.01.
To investigate the effect of AMPK inhibitor on the sensitivity of the islets to GLP-1 and GIP-stimulated insulin secretion, we examined insulin response to incretins at 2.8 and 11.1 mM glucose in isolated islets treated with vehicle or AMPK inhibitor at 2.8 mM glucose for 24 h. AMPK inhibitor increased insulin secretion from islet at 2.8 and 11.1 mM glucose, although the difference was not statistically significant (Figure 3C). The relative effects of GLP-1 and GIP on insulin secretion were not significantly increased by AMPK inhibitor treatment (Figure 3C).
The impact of manipulation of AMPK phosphorylation by dominant-negative AMPK expression on the expression of the incretin receptors under a low glucose concentration
To confirm whether the phosphorylation state of AMPK regulated the expression of the incretin receptors in the islets, we manipulated AMPK phosphorylation in the islet cells by inducing the expression of a dominant-negative form of the AMPKα subunit (DN-AMPK) carrying a point mutation (K45R). Overexpression of DN-AMPK with adenoviral vector in isolated islets decreased the ratio of phosphorylated to total AMPK to 60% under a low glucose concentration (2.8 mM) to activate the endogenous AMPK (Figure 4A). The expression of the incretin receptors was significantly decreased in isolated islets treated with overexpression of AdCMV-LacZ compared with in control islets (Figure 4B), which was due to the direct effect of adenoviral infection. We confirmed that the morphology of trypsinized islet cells was unaffected by adenoviral infection (Figure. S1). In addition, we next evaluated the apoptosis in islets infected with the viruses using TUNEL staining. The results showed that the ratio of TUNEL-positive β cells were equally around 10% across the three groups (Mock control islet cells, islet cells infected with AdCMV-LacZ, and islet cells infected with Ad-AMPK-α2-K45R). These results indicated that the apoptosis was not induced by adenoviral infection. (Figure. S1). AMPK dephosphorylation by DN-AMPK expression led to significant increases in the expression of the incretin receptors as compared with islets treated with overexpression of AdCMV-LacZ at 2.8 mM glucose (Figure 4B). These results lend support to our hypothesis that the expression of the incretin receptors was dependent, at least in part, on the phosphorylation state of AMPK under a low glucose concentration.
Figure 4. The impact of manipulation of AMPK phosphorylation by DN-AMPK expression on the expressions of the incretin receptors.

A), B) Isolated islet cells in monolayer were infected with the Ad-AMPK-α2-K45R or AdCMV-LacZ virus in the presence of 2.8 mM glucose for 24 h. A) Total islet extracts from the uninfected cells and virus-infected cells were subjected to immunoblotting for p-AMPKα (Thr172) and AMPKα. Data shown are representative of three independent experiments. B) The GLP-1 and GIP receptor expressions were determined by real-time quantitative RT-PCR and normalized to the β actin mRNA level and to the control samples (n = 3). The samples are from independent experiments. *P<0.05 and ** P<0.01.
The effect of glucokinase activator on the expression of the incretin receptors under a medium glucose concentration
To assess whether the glucose signal might be involved in the expression of the incretin receptors, we treated isolated islets with GKA under a medium glucose concentration (11.1 mM). Treatment with GKA produced dephosphorylation of AMPK in the islets at 11.1 mM glucose (Figure 5A). The GLP-1 receptor expression was decreased by treatment with GKA at 11.1 mM glucose, whereas no significant decrease of the GIP receptor was observed (Figure 5B). The result suggested that an excessively glucose signal suppressed the expression of GLP-1 receptor.
Figure 5. The effect of glucokinase activator on the expression of the incretin receptors under a medium glucose concentration.
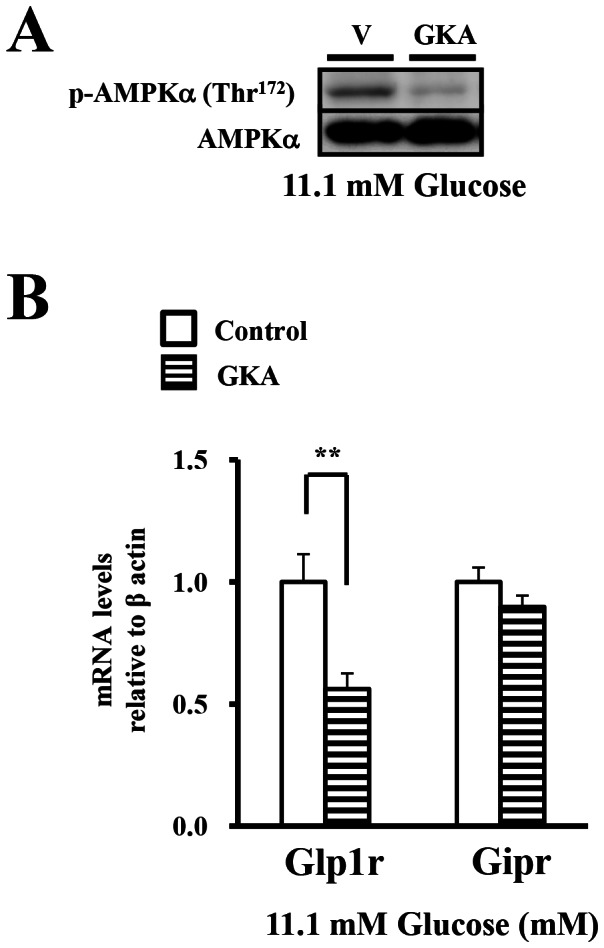
A), B). Isolated islets were incubated with DMSO (ctl) or GKA (G: 30 µM compound A) in the presence of 11.1 mM glucose for 24 hours. Total cell extracts from the isolated islets were subjected to immunoblotting for p-AMPKα (Thr172) and AMPKα. Data shown are representative of three independent experiments. B) The GLP-1 and GIP receptor expressions were determined by real-time quantitative RT-PCR and normalized to the β actin mRNA level and to the control samples (n = 4). *P<0.05, ** P<0.01.
The effect of pharmacologic modulation of AMPK phosphorylation on the expression of the incretin receptors under a medium glucose concentration
To assess whether the phosphorylation state of AMPK regulated the expression of the incretin receptors in the islets under lower glucose concentrations (2.8–11.1 mM), we treated isolated islets with AICAR under a medium glucose concentration (11.1 mM) to suppress the endogenous enzyme to some extent. AICAR increased the phosphorylation levels of AMPK at 11.1 mM glucose, as compared to that in the control (Figure 6A). The expression of the incretin receptors in the islets was not significantly decreased by treatment with AICAR at 11.1 mM glucose (Figure 6B). We next assess the expression of incretin receptors in islets co-treated with AICAR and the AMPK inhibitor. Co-treatment with the AMPK inhibitor significantly increased the expression of GIP receptor in islets treated with AICAR, but not GLP-1 receptor (Figure 6C).
Figure 6. The effect of pharmacologic modulation of AMPK phosphorylation on the expression of the incretin receptors under a medium glucose concentration.

A), B) Isolated islets were treated for 24 h with vehicle (V, water) or AICAR (A, 1 mM) in the presence of 11.1 mM glucose. A) Total islet extracts from the isolated islets were subjected to immunoblotting for p-AMPKα (Thr172) and AMPKα.(n = 4) B) The GLP-1 and GIP receptor expressions were determined by real-time quantitative RT-PCR and normalized to the β actin mRNA level and to the control samples (n = 4). C) Isolated islets were pre-treated for 15 min with vehicle (Veh; DMSO) or the AMPK inhibitor (AMPKi; 40 µM compound C) in the presence of 11.1 mM glucose, followed by the addition of vehicle (V; water) or AICAR (A, 1 mM) for an additional 24 h. The GLP-1 and GIP receptor expressions were determined as shown in B) (n = 3). White bars, vehicle; Black bars; AMPKi. *P<0.05 and ** P<0.01.
The phosphorylation levels of AMPK and the expression of the incretin receptors in db/db mice
We next evaluated the phosphorylation levels of AMPK and the expression of the incretin receptors in the islets of the db/db mice, a model of chronic hyperglycemia. In the db/db islets, the ratio of phosphorylated-AMPK/total-AMPK at 2.8 mM glucose was decreased as compared to that in the euglycemic db/+ islets (Figure 7A). In the db/db islets, expression of the incretin receptors was decreased at 11.1 mM glucose as compared with that in the euglycemic db/+ islets, although the difference was not statistically significant (Figure 7B). In addition, we also evaluated the effect of the AMPK inhibitor on the expression of incretin receptors in the db/db islets under a low glucose concentration (2.8 mM) to activate the endogenous AMPK. The AMPK inhibitor decreased the ratio of phosphorylated-AMPK/total-AMPK to 64% in the db/+ islets and to 89% in the db/db islets (Figure 7A). The enhancing effect of the AMPK inhibitor on the GIP receptor expression in the db/db islets was reduced to one third compared to the effect in the db/+ islets. No significant increase of the GLP-1 receptor was observed in the db/db islets following AMPK inhibitor treatment at 2.8 mM glucose (Figure 7B). Taken together, the enhancing effect of the AMPK inhibitor on the expression of incretin receptors was diminished under a low glucose concentration in the db/db islets.
Figure 7. The impact of pharmacologic modulation of AMPK phosphorylation on the expressions of the incretin receptors in db/db mice.

A), B) Isolated islets from 7-week-old db/+ and db/db mice were pre-treated for 15 min with vehicle (Veh; DMSO) or an AMPK inhibitor (AMPKi; 40 µM compound C) in the presence of 2.8 mM glucose, or with vehicle (Veh; DMSO) in the presence of 11.1 mM glucose, followed by the addition of vehicle (Veh; water) for an additional 24 h. A) Total islet extracts from the isolated islets were subjected to immunoblotting for p-AMPKα (Thr172) and AMPKα (n = 2–4). B) The GLP-1 and GIP receptor expressions were determined by real-time quantitative RT-PCR and normalized to the β actin mRNA expression level and to the vehicle-treated control samples at 2.8 mM glucose (n = 2–3). *P<0.05 and ** P<0.01.
The impact of treatment with metformin on the phosphorylation level of AMPK and the expression of the incretin receptors in islets in vitro and in vivo
We next assessed the effects of metformin on the expression of the incretin receptors in primary mouse islets in vitro and in vivo. Treatment with metformin significantly reduced the expression of the incretin receptors in vitro under a medium glucose concentration (11.1 mM) (Figure. S2A). To investigate the effect of metformin on the sensitivity of the islets to GLP-1 and GIP-stimulated insulin secretion, we examined insulin response to incretins at 2.8 and 11.1 mM glucose in isolated islets treated with vehicle or metformin at 11.1 mM glucose for 24 h. GLP-1 significantly increased GSIS in response to 11.1 mM glucose in the control islets, but not in the metformin-treated islets (Figure. S2B). We confirmed that the islet morphology and the insulin content in islets were unaffected by metformin treatment in vitro (Figure. S2C, D). The phosphorylation levels of AMPK in the control islets at 11.1 mM glucose were significantly decreased as compared with that at 2.8 mM glucose. As expected, treatment with metformin increased the phosphorylation levels of AMPK in the islets in vitro at 11.1 mM glucose (Figure. S2E).
We next evaluated the effect of administration of metformin on the expression of the incretin receptors in the islets in vivo. In contrast to the findings in vitro, treatment with metformin for 2 days significantly increased the expression of the incretin receptors in the islets in vivo (Figure. S3A). Pparα expression was also significantly increased by metformin treatment in the islets in vivo (Figure. S3A). We examined the effect of GLP-1 and GIP on GSIS in response to 2.8 or 11.1 mM glucose after treatment with metformin or vehicle control in vivo (Figure. S3B). GSIS in response to 11.1 mM glucose was significantly increased by GLP-1 and GIP in the islets of the vehicle- and metformin-treated mice, although the relative insulin response to incretins was comparable between the two groups (Figure. S3B). We next evaluated the phosphorylation levels of AMPK in the islets of the metformin-treated mice. Amazingly, the islets from the metformin-treated mice showed decreased phosphorylation level of AMPK (Figure. S3C). We also examined the phosphorylation levels of AMPK in the livers and skeletal muscles of the vehicle- and metformin-treated mice. Unlike in the islets, the phosphorylation levels of AMPK in the livers and skeletal muscles of the metformin-treated mice were significantly increased as compared to those in the untreated controls (Figure. S3C).
Immunoblot analysis with the commercial antibody did not demonstrate the significant changes in the protein expression levels of incretin receptors
To assess the protein expression levels of GLP-1 receptor and GIP receptor in islets, we performed immunoblot analysis by the commercial available antibody. However, recent reports have shown that almost commercially available GLP-1 receptor antibodies do not detect authentic GLP-1 receptor protein, even using optimally enhanced methods [37], [38]. Our immunoblotting also showed that the anticipated bands of incretin receptors detected by the antibody were not affected by glucose concentration (Figure. S4), treatment with GKA (Figure. S5, S6), AMPK inhibitor (Figure. S7), AICAR (Figure. S8), db/db genotype (Figure. S9), or metformin (Figure. S2F, S3D).
Discussion
In this study, we investigated the regulation of incretin receptor expressions in the pancreatic islets, and described that AMPK partly controlled them in a glucose concentration-dependent manner.
Although the expression of the incretin receptors in the islets was suppressed under chronic high concentration of glucose (22.2 mM), the glucose signal increased them at lower glucose concentrations (2.8–11.1 mM). A previous report also showed the expression of GLP-1 receptor in bell-shaped response to glucose [20]. This finding was supported by our results that GKA-induced glucose phosphorylation increased the expression of the expression of GLP-1 receptor in the islets under a low concentration of glucose (2.8 mM), while it decreased the expression under a medium concentration of glucose (11.1 mM). In addition, suppression of the incretin receptors expression under a high glucose concentration could be accounted for by the decreased expression of incretin receptors in the islets of db/db mice, a model of hyperglycemia, as shown in a previous report [8]. By what mechanism did glucose signal increase the expression of the incretin receptors under lower glucose concentrations and suppress the expression under a high glucose concentration? A previous report showed that GIP receptor expression was glucose-dependently increased in isolated rat islets incubated for 48 hours, and the result suggested that the rapid increase in GIP receptor could contribute to the maintenance of insulin output during the elevated metabolic demand induced by glucose elevation [8]. In contrast, decrease in the expression of the incretin receptors after exposure to high glucose concentrations has also been reported from previous studies [8], [20], [39]. Xu et al indicated that PKCα may be involved in the downregulation of the GLP-1 receptor expression at 20 mM glucose [8]. Cheong et al. showed that a high glucose concentration (30 mM glucose) reduced the GLP-1 receptor expression through endoplasmic reticulum (ER) stress [39]. Rajan et al. recently showed that downregulation of GLP-1 receptor signaling at a high glucose concentration was due to the reduction of cell surface trafficking of GLP-1 receptor by small ubiquitin-related modifier protein (SUMO) [40]. Persistence of hyperglycemia may interfere with incretin receptor signaling by an additive effect of the transcriptional attenuation and the posttranslational modification of receptors. The change in expression of GIP receptor was somewhat different as compared with those found with the GLP-1 receptor expression by treatment with GKA at 11.1 mM glucose. Xu et al showed that increases in GIP receptor expression were found in the short-term glucose infusion model and cultured islets with exposure to high glucose, although reduction in GLP-1 receptor was observed in the models [8]. These differences in the expression of GIP and GLP-1 receptor may be due to timing and we need to elucidate the differences in future.
The finding in our study, demonstrating that the phosphorylation of AMPK was decreased by increasing glucose concentrations, although the expression of the incretin receptors was in bell-shaped response to glucose, suggested that the glucose signal induced-regulation of incretin receptors expression appeared to be an AMPK-independent pathway. However, under a high glucose concentration, above-mentioned factors largely seem to affect the regulation of the expression of the incretin receptors. Therefore, our hypothesis was that AMPK was partly involved in the regulation of the incretin receptors expression under lower glucose concentrations (2.8–11.1 mM). By contrast, we also demonstrated that the enhancing effect of AMPK inhibitor on the expression of the incretin receptors was diminished in db/db islets under chronic hyperglycemic conditions. The data in db/db islets also suggested that AMPK is not involved in the regulation of the incretin receptors expression under a high glucose concentration. It has been shown that not only hyperglycemia, but also ER stress [41] and oxidative stress [42], [43] may contribute to β cell dysfunction in the db/db mice. Therefore, it is possible that AMPK activity fails to contribute to the regulation of the expression of the incretin receptors under ER or oxidative stress conditions. Since the disruption of leptin receptor expression in the pancreas directly affects β cell function [44], the impact of the leptin receptor-mediated mechanisms on insulin secretion in islets may possibly reflect the diminished effects of AMPK regulation on the incretin receptors expression. Collectively, these results suggested that AMPK was involved in the regulation in the expression of incretin receptors under low and medium glucose concentrations, but not under a high glucose concentration.
The fact that AICAR failed to significantly decrease the expression of the incretin receptors, suggests that the regulation in the expression appear to be an AMPK-independent mechanism. In this study, we treated isolated islets with AICAR under a medium glucose concentration (11.1 mM) to suppress the endogenous enzyme to some extent. However, phosphorylation of AMPK in islet was not fully suppressed under a medium glucose concentration (Figure 1A). Therefore, it may be difficult to assess the direct effect of AICAR on the expression of the incretin receptors in islet under the condition. We need to assess the impact of AICAR on the expression of the incretin receptors in islets under other conditions to suppress the phosphorylation of AMPK to a large extent.
The results in our study also demonstrated that inhibition of AMPK increased insulin secretion, as shown in a previous report [28]. The previous finding showed that inhibition of AMPK with DN-AMPK led to the activation of insulin secretion via Ca2+-independent mechanism [28]. It is possible that AMPK dephosphorylation-induced insulin secretion may make it difficult to evaluate the insulin response to incretins in islets treated with AMPK inhibitor in our current models. Moreover, intact islets contain not only β cells but α or δ cells. Although, a previous report showed that the expression of GLP-1 receptor in alpha cells was <0.2% of that in β cells [45], AMPK is regulated by glucose in pancreatic alpha cells, and increases in AMPK activity are sufficient and necessary for the stimulation of glucagon release [46]. Therefore, we need to assess the effect of AMPK regulation in alpha cells on the incretin receptors expression in islets in the future.
The correlation between the expression of the incretin receptors and the phosphorylation level of AMPK in vitro and in vivo suggests that metformin may partly regulate the expression in islets via AMPK-dependent pathway. However, the inhibitory effect of metformin on mitochondrial electron transport chain also led to AMPK activation secondarily [47], therefore the secondary effect of metformin on AMPK activation may partially regulate the incretin receptors expression in islets in this study. Moreover, the time for isolating islets in vivo and the differences in the concentrations of metformin between in vitro and in vivo may affect the discrepant results. By contrast, the finding that metformin and AICAR increased AMPK phosphorylation, but only metformin significantly decreased the expression of the incretin receptors, suggests that an AMPK-independent mechanism may be involved in the regulation of the expression by metformin. We should further assess what mechanism underlying the regulation of the expression of the incretin receptors by metformin using other experimental models.
In this study, we showed that there were no significant changes in the protein expression of the incretin receptors in islets between control group and experimental group in each experiment. There is a possibility that the commercially available antibody of GLP-1 receptor failed to detect authentic GLP-1 receptor protein. It is well-known that the GLP-1 receptor and other GPCRs are membrane-spanning molecules. However, it has been shown that pancreatic islets are known to frequently give rise to false-positive staining in immunohistochemistry studies [48]. More recently, Panjwani et al. have shown that the commercial antibodies could not detect the native GLP-1R protein in conventional Western blot analysis, and the antibodies did detect several immunoreactive bands ranging in size from approximately 54–65 kDa even in lung extracts from both Glp1r+/+ and Glp1r−/− mice [37]. Pyke et al. have also shown that the commercial antibodies recommended for IHC reacted with equal intensity with both GLP-1R-transfected and untransfected cells and, when further diluted to a concentration where there was negligible staining of untransfected cells, did not react with GLP-1R-transfected cells [38]. Based upon the fact, it is possible that the commercial antibody used in this study detected non-specific bands, but not authentic GLP-1 receptor protein. Therefore, we would very much like to assess the reliable GLP-1 receptor protein expression data using other technical challenge. In previous studies, many groups reported that a lack of specificity of commercial antibodies against GPCRs (G protein-coupled protein receptors) was shown to be the rule rather than the exception [49]–[54]. The lack of specificity of GPCRs antibodies may also apply to the GIP receptor. However, we also should assess the protein expression of GIP receptor as with that of GLP-1 receptor using other technical procedure.
In summary, we show here that AMPK is involved in the regulation in the expression of incretin receptors under a low glucose concentration. The results of the current study have important implications in relation to the design of new therapies targeting the expression of the incretin receptors, however, further research is needed to clarify the underlying mechanisms.
Supporting Information
The effect of adenoviral infection on the apoptosis in islets. Trypsinized islet cells of indicated groups were subjected to a TUNEL assay. Insulin is stained green and TUNEL-positive nuclei are stained red. DAPI (blue) and differential interference contrast (DIC) images were also merged. TUNEL staining was performed using the ApopTag In Situ Detection Kit (Chemicon). For TUNEL staining, at least 40 islets per trypsinized islets group attached to poly-L-lysine coated coverslips (Falcon) were analyzed using the FLUOVIEW FV 1000-D confocal laser scanning microscope (OLYMPUS) to assess the proportion of immunostained nuclei among the insulin-positive cells.
(TIF)
The impact of treatment with metformin on the phosphorylation level of AMPK and the expressions of the incretin receptors in islets in vitro . A), B) Isolated islets were incubated with or without 1 mM metformin (Dainippon Sumitomo Pharma, Osaka, Japan) for 24 h in the presence of 2.8 or 11.1 mM glucose. A) The Glp1, Gip receptor and Pparα expressions in the islets were determined by real-time quantitative RT-PCR and normalized to the expression level of β actin mRNA and to the vehicle-treated control samples at 2.8 mM glucose (n = 7–8). *P<0.05. B) Insulin secretion by the islets treated with metformin or vehicle (ctl) in the presence of 11.1 mM glucose for 24 h with or without addition of 10 nM GLP-1 or 10 nM GIP. The results are expressed in ng of insulin/10 islets/90 min (n = 4). * P<0.05 and ** P<0.01. C), D) Isolated islets were incubated with or without 1 mM metformin for 24 h in the presence of 11.1 mM glucose. C) Islet morphology of the vehicle- and metformin-treated islets. D) The insulin content in the islets was determined after acid ethanol extraction (n = 3). White bars, vehicle; Black bars; metformin. E) Total islet extracts from the isolated islets were subjected to immunoblotting for p-AMPKα (Thr172) and AMPKα. The intensities of the signals were quantified by densitometry and normalized to the vehicle-treated control samples at 2.8 mM glucose (n = 3). The results shown are the means of three independent experiments. F) Total cell extracts from the isolated islets were subjected to immunoblotting for p-AMPKα (Thr172), AMPKα, anti-GLP1R antibody, and β actin.
(TIF)
The impact of treatment with metformin on the phosphorylation level of AMPK and the expressions of the incretin receptors in islets in vivo . Mice were given metformin in drinking water at 300 mg/kg daily or standard drinking water for 48 hours. Four hours prior to the islet isolation, the food was removed from the cages and the mice were orally administrated metformin (75 mg/kg) or vehicle (water) [14]. A) The Glp1, Gip receptor and Pparα expressions in the islets were determined by real-time quantitative RT-PCR and normalized to the expression level of β actin mRNA and to the samples in vehicle-treated mice (n = 6–7). Experiments were performed on mice treated with metformin or vehicle (ctl). *P<0.05 and ** P<0.01. B) Insulin secretion from the islets of mice treated with metformin or vehicle (ctl) in the presence of 2.8 mM or 11.1 mM glucose, with or without addition of 10 nM GLP-1 or 10 nM GIP. The results are expressed as ng of insulin/10 islets/90 min (n = 4). * P<0.05 and ** P<0.01. C) Freshly isolated total islets, the extracted liver and skeletal muscle tissues from the vehicle or metformin-treated mice were subjected to immunoblotting for p-AMPKα (Thr172) and AMPKα. The intensities of the signals were quantified by densitometry and normalized to the samples in vehicle-treated mice (n = 3). The results shown are the means of three independent experiments. D) Total cell extracts from the isolated islets were subjected to immunoblotting for p-AMPKα (Thr172), AMPKα, anti-GLP1R antibody, and β actin (n = 3).
(TIF)
The effect of glucose signal on the protein expression of the incretin receptors in islets. Isolated islets were incubated for 24 h in the presence of 2.8, 5.6, 11.1 or 22.2 mM glucose. Total cell extracts from the isolated islets were subjected to immunoblotting for p-AMPKα (Thr172), AMPKα, anti-GLP1R antibody, anti-GIPR antibody, and GAPDH.
(TIF)
The effect of glucokinase activator on the protein expression of the incretin receptors under a low glucose concentration. Isolated islets were incubated with DMSO (ctl) or GKA (G: 30 µM compound A) in the presence of 2.8 mM glucose for 24 hours. Total cell extracts from the isolated islets were subjected to immunoblotting for p-AMPKα (Thr172), AMPKα, anti-GLP1R antibody, anti-GIPR antibody, and GAPDH (n = 3).
(TIF)
The effect of glucokinase activator on the protein expression of the incretin receptors under a medium glucose concentration. Isolated islets were incubated with DMSO (ctl) or GKA (G: 30 µM compound A) in the presence of 11.1 mM glucose for 24 hours. Total cell extracts from the isolated islets were subjected to immunoblotting for p-AMPKα (Thr172), AMPKα, anti-GLP1R antibody, anti-GIPR antibody, and GAPDH (n = 3).
(TIF)
The effect of pharmacologic modulation of AMPK phosphorylation on the protein expressions of the incretin receptors under a low glucose concentration. Isolated islets were treated for 24 h with vehicle (Veh; DMSO) or the AMPK inhibitor (AMPKi; 40 µM compound C) in the presence of 2.8 mM glucose. Total cell extracts from the isolated islets were subjected to immunoblotting for p-AMPKα (Thr172), AMPKα, anti-GLP1R antibody, anti-GIPR antibody, and GAPDH (n = 3).
(TIF)
The effect of pharmacologic modulation of AMPK phosphorylation on the protein expression of the incretin receptors under a medium glucose concentration. Isolated islets were treated for 24 h with vehicle (V, water) or AICAR (A, 1 mM) in the presence of 11.1 mM glucose. Total cell extracts from the isolated islets were subjected to immunoblotting for p-AMPKα (Thr172), AMPKα, anti-GLP1R antibody, anti-GIPR antibody, and GAPDH (n = 4)
(TIF)
The impact of pharmacologic modulation of AMPK phosphorylation on the protein expressions of the incretin receptors in db/db mice. Isolated islets from 7-week-old db/+ and db/db mice were pre-treated for 15 min with vehicle (Veh; DMSO) or an AMPK inhibitor (AMPKi; 40 µM compound C) in the presence of 2.8 mM glucose, or with vehicle (Veh; DMSO) in the presence of 11.1 mM glucose, followed by the addition of vehicle (Veh; water) for an additional 24 h. Total cell extracts from the isolated islets were subjected to immunoblotting for p-AMPKα (Thr172), AMPKα, anti-GLP1R antibody, anti-GIPR antibody, and GAPDH.
(TIF)
Acknowledgments
The authors thank Dr. Birnbaum (Howard Hughes Medical Institute and Department of Medicine, University of Pennsylvania school of Medicine, Philadelphia, Pennsylvania, USA) for providing the AdCMV-LacZ, and Ad-AMPK-α2-K45R virus, and Ms. Misa Katayama (Yokohama City University) for her secretarial assistance. The authors also wish to thank Dainippon Sumitomo Pharma (Osaka, Japan) for providing the metformin for this study.
Funding Statement
This work was supported in part by Grants-in-Aid for Scientific Research (B) 19390251, (B) 21390282 and (B) 24390235 from the Ministry of Education, Culture, Sports, Science and Technology (MEXT) of Japan, a Grant for the Strategic Japanese-Danish Cooperative Program on Molecular Diabetology from the Japan Science and Technology Agency, a Medical Award from the Japan Medical Association, a Grant-in-Aid from the Japan Diabetes Foundation, a Grant-in-Aid from the Suzuken Memorial Foundation, a Grant-in-Aid from the Naito Foundation, a Grant-in-Aid from the Uehara Memorial Foundation (to Y.T.), and a Grant-in-Aid for JSPS Fellows, a Grant-in-Aid from Yokohama General Promotion Foundation, a Grant-in-Aid from the Novo Nordisk Insulin Research Foundation, a Grant-in-Aid from Japan Foundation for Applied Enzymology (to J.S.). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Butler AE, Janson J, Bonner-Weir S, Ritzel R, Rizza RA, et al. (2003) Beta-cell deficit and increased beta-cell apoptosis in humans with type 2 diabetes. Diabetes 52: 102–110. [DOI] [PubMed] [Google Scholar]
- 2. Polonsky KS, Sturis J, Bell GI (1996) Seminars in Medicine of the Beth Israel Hospital, Boston. Non-insulin-dependent diabetes mellitus - a genetically programmed failure of the beta cell to compensate for insulin resistance. N Engl J Med 334: 777–783. [DOI] [PubMed] [Google Scholar]
- 3. Nauck M, Stockmann F, Ebert R, Creutzfeldt W (1986) Reduced incretin effect in type 2 (non-insulin-dependent) diabetes. Diabetologia 29: 46–52. [DOI] [PubMed] [Google Scholar]
- 4. Nauck MA, Vardarli I, Deacon CF, Holst JJ, Meier JJ (2011) Secretion of glucagon-like peptide-1 (GLP-1) in type 2 diabetes: what is up, what is down? Diabetologia 54: 10–18. [DOI] [PubMed] [Google Scholar]
- 5. Mojsov S, Weir GC, Habener JF (1987) Insulinotropin: glucagon-like peptide I (7-37) co-encoded in the glucagon gene is a potent stimulator of insulin release in the perfused rat pancreas. J Clin Invest 79: 616–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Holst JJ, Gromada J (2004) Role of incretin hormones in the regulation of insulin secretion in diabetic and nondiabetic humans. Am J Physiol Endocrinol Metab 287: E199–206. [DOI] [PubMed] [Google Scholar]
- 7. Holst JJ, Gromada J, Nauck MA (1997) The pathogenesis of NIDDM involves a defective expression of the GIP receptor. Diabetologia 40: 984–986. [DOI] [PubMed] [Google Scholar]
- 8. Xu G, Kaneto H, Laybutt DR, Duvivier-Kali VF, Trivedi N, et al. (2007) Downregulation of GLP-1 and GIP receptor expression by hyperglycemia: possible contribution to impaired incretin effects in diabetes. Diabetes 56: 1551–1558. [DOI] [PubMed] [Google Scholar]
- 9. Lynn FC, Pamir N, Ng EH, McIntosh CH, Kieffer TJ, et al. (2001) Defective glucose-dependent insulinotropic polypeptide receptor expression in diabetic fatty Zucker rats. Diabetes 50: 1004–1011. [DOI] [PubMed] [Google Scholar]
- 10. Lynn FC, Thompson SA, Pospisilik JA, Ehses JA, Hinke SA, et al. (2003) A novel pathway for regulation of glucose-dependent insulinotropic polypeptide (GIP) receptor expression in beta cells. FASEB J 17: 91–93. [DOI] [PubMed] [Google Scholar]
- 11. Tahrani AA, Piya MK, Kennedy A, Barnett AH (2010) Glycaemic control in type 2 diabetes: targets and new therapies. Pharmacol Ther 125: 328–361. [DOI] [PubMed] [Google Scholar]
- 12. Tahrani AA, Bailey CJ, Del Prato S, Barnett AH (2011) Management of type 2 diabetes: new and future developments in treatment. Lancet 378: 182–197. [DOI] [PubMed] [Google Scholar]
- 13. Cho YM, Kieffer TJ (2011) New aspects of an old drug: metformin as a glucagon-like peptide 1 (GLP-1) enhancer and sensitiser. Diabetologia 54: 219–222. [DOI] [PubMed] [Google Scholar]
- 14. Maida A, Lamont BJ, Cao X, Drucker DJ (2011) Metformin regulates the incretin receptor axis via a pathway dependent on peroxisome proliferator-activated receptor-alpha in mice. Diabetologia 54: 339–349. [DOI] [PubMed] [Google Scholar]
- 15. Green BD, Irwin N, Duffy NA, Gault VA, O'Harte FP, et al. (2006) Inhibition of dipeptidyl peptidase-IV activity by metformin enhances the antidiabetic effects of glucagon-like peptide-1. Eur J Pharmacol 547: 192–199. [DOI] [PubMed] [Google Scholar]
- 16. Yasuda N, Inoue T, Nagakura T, Yamazaki K, Kira K, et al. (2002) Enhanced secretion of glucagon-like peptide 1 by biguanide compounds. Biochem Biophys Res Commun 298: 779–784. [DOI] [PubMed] [Google Scholar]
- 17. Migoya EM, Bergeron R, Miller JL, Snyder RN, Tanen M, et al. (2010) Dipeptidyl peptidase-4 inhibitors administered in combination with metformin result in an additive increase in the plasma concentration of active GLP-1. Clin Pharmacol Ther 88: 801–808. [DOI] [PubMed] [Google Scholar]
- 18. Mannucci E, Ognibene A, Cremasco F, Bardini G, Mencucci A, et al. (2001) Effect of metformin on glucagon-like peptide 1 (GLP-1) and leptin levels in obese nondiabetic subjects. Diabetes Care 24: 489–494. [DOI] [PubMed] [Google Scholar]
- 19. Cuthbertson J, Patterson S, O'Harte FP, Bell PM (2009) Investigation of the effect of oral metformin on dipeptidylpeptidase-4 (DPP-4) activity in Type 2 diabetes. Diabet Med 26: 649–654. [DOI] [PubMed] [Google Scholar]
- 20. Pan QR, Li WH, Wang H, Sun Q, Xiao XH, et al. (2009) Glucose, metformin, and AICAR regulate the expression of G protein-coupled receptor members in INS-1 beta cell. Horm Metab Res 41: 799–804. [DOI] [PubMed] [Google Scholar]
- 21. Viollet B, Guigas B, Sanz Garcia N, Leclerc J, Foretz M, et al. (2012) Cellular and molecular mechanisms of metformin: an overview. Clin Sci (Lond) 122: 253–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kahn BB, Alquier T, Carling D, Hardie DG (2005) AMP-activated protein kinase: ancient energy gauge provides clues to modern understanding of metabolism. Cell Metab 1: 15–25. [DOI] [PubMed] [Google Scholar]
- 23. Long YC, Zierath JR (2006) AMP-activated protein kinase signaling in metabolic regulation. J Clin Invest 116: 1776–1783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Rutter GA, Leclerc I (2009) The AMP-regulated kinase family: enigmatic targets for diabetes therapy. Mol Cell Endocrinol 297: 41–49. [DOI] [PubMed] [Google Scholar]
- 25. Salt IP, Johnson G, Ashcroft SJ, Hardie DG (1998) AMP-activated protein kinase is activated by low glucose in cell lines derived from pancreatic beta cells, and may regulate insulin release. Biochem J 335 ((Pt 3)) 533–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. da Silva Xavier G, Leclerc I, Salt IP, Doiron B, Hardie DG, et al. (2000) Role of AMP-activated protein kinase in the regulation by glucose of islet beta cell gene expression. Proc Natl Acad Sci U S A 97: 4023–4028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sun G, Tarasov AI, McGinty J, McDonald A, da Silva Xavier G, et al. (2010) Ablation of AMP-activated protein kinase alpha1 and alpha2 from mouse pancreatic beta cells and RIP2.Cre neurons suppresses insulin release in vivo. Diabetologia 53: 924–936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. da Silva Xavier G, Leclerc I, Varadi A, Tsuboi T, Moule SK, et al. (2003) Role for AMP-activated protein kinase in glucose-stimulated insulin secretion and preproinsulin gene expression. Biochem J 371: 761–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Viollet B, Andreelli F, Jorgensen SB, Perrin C, Flamez D, et al. (2003) Physiological role of AMP-activated protein kinase (AMPK): insights from knockout mouse models. Biochem Soc Trans 31: 216–219. [DOI] [PubMed] [Google Scholar]
- 30. Viollet B, Andreelli F, Jorgensen SB, Perrin C, Geloen A, et al. (2003) The AMP-activated protein kinase alpha2 catalytic subunit controls whole-body insulin sensitivity. J Clin Invest 111: 91–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Gleason CE, Lu D, Witters LA, Newgard CB, Birnbaum MJ (2007) The role of AMPK and mTOR in nutrient sensing in pancreatic beta-cells. J Biol Chem 282: 10341–10351. [DOI] [PubMed] [Google Scholar]
- 32. Shirakawa J, Amo K, Ohminami H, Orime K, Togashi Y, et al. (2011) Protective effects of dipeptidyl peptidase-4 (DPP-4) inhibitor against increased beta cell apoptosis induced by dietary sucrose and linoleic acid in mice with diabetes. J Biol Chem 286: 25467–25476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Shirakawa J, Fujii H, Ohnuma K, Sato K, Ito Y, et al. (2011) Diet-induced adipose tissue inflammation and liver steatosis are prevented by DPP-4 inhibition in diabetic mice. Diabetes 60: 1246–1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Matschinsky FM (2009) Assessing the potential of glucokinase activators in diabetes therapy. Nat Rev Drug Discov 8: 399–416. [DOI] [PubMed] [Google Scholar]
- 35. Garcia-Haro L, Garcia-Gimeno MA, Neumann D, Beullens M, Bollen M, et al. (2010) The PP1-R6 protein phosphatase holoenzyme is involved in the glucose-induced dephosphorylation and inactivation of AMP-activated protein kinase, a key regulator of insulin secretion, in MIN6 beta cells. FASEB J 24: 5080–5091. [DOI] [PubMed] [Google Scholar]
- 36. Porat S, Weinberg-Corem N, Tornovsky-Babaey S, Schyr-Ben-Haroush R, Hija A, et al. (2011) Control of pancreatic beta cell regeneration by glucose metabolism. Cell Metab 13: 440–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Panjwani N, Mulvihill EE, Longuet C, Yusta B, Campbell JE, et al. (2013) GLP-1 Receptor Activation Indirectly Reduces Hepatic Lipid Accumulation But Does Not Attenuate Development of Atherosclerosis in Diabetic Male ApoE−/− Mice. Endocrinology 154: 127–139. [DOI] [PubMed] [Google Scholar]
- 38. Pyke C, Knudsen LB (2013) The Glucagon-Like Peptide-1 Receptor–or Not? Endocrinology 154: 4–8. [DOI] [PubMed] [Google Scholar]
- 39. Cheong YH, Kim MK, Son MH, Kaang BK (2011) Glucose exposure pattern determines glucagon-like peptide 1 receptor expression and signaling through endoplasmic reticulum stress in rat insulinoma cells. Biochem Biophys Res Commun 414: 220–225. [DOI] [PubMed] [Google Scholar]
- 40. Rajan S, Torres J, Thompson MS, Philipson LH (2012) SUMO downregulates GLP-1-stimulated cAMP generation and insulin secretion. Am J Physiol Endocrinol Metab 302: E714–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Laybutt DR, Preston AM, Akerfeldt MC, Kench JG, Busch AK, et al. (2007) Endoplasmic reticulum stress contributes to beta cell apoptosis in type 2 diabetes. Diabetologia 50: 752–763. [DOI] [PubMed] [Google Scholar]
- 42. Ishida H, Takizawa M, Ozawa S, Nakamichi Y, Yamaguchi S, et al. (2004) Pioglitazone improves insulin secretory capacity and prevents the loss of beta-cell mass in obese diabetic db/db mice: Possible protection of beta cells from oxidative stress. Metabolism 53: 488–494. [DOI] [PubMed] [Google Scholar]
- 43. Yamamoto M, Yamato E, Toyoda S, Tashiro F, Ikegami H, et al. (2008) Transgenic expression of antioxidant protein thioredoxin in pancreatic beta cells prevents progression of type 2 diabetes mellitus. Antioxid Redox Signal 10: 43–49. [DOI] [PubMed] [Google Scholar]
- 44. Morioka T, Asilmaz E, Hu J, Dishinger JF, Kurpad AJ, et al. (2007) Disruption of leptin receptor expression in the pancreas directly affects beta cell growth and function in mice. J Clin Invest 117: 2860–2868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. De Marinis YZ, Salehi A, Ward CE, Zhang Q, Abdulkader F, et al. (2010) GLP-1 inhibits and adrenaline stimulates glucagon release by differential modulation of N- and L-type Ca2+ channel-dependent exocytosis. Cell Metab 11: 543–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Leclerc I, Sun G, Morris C, Fernandez-Millan E, Nyirenda M, et al. (2011) AMP-activated protein kinase regulates glucagon secretion from mouse pancreatic alpha cells. Diabetologia 54: 125–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Hinke SA, Martens GA, Cai Y, Finsi J, Heimberg H, et al. (2007) Methyl succinate antagonises biguanide-induced AMPK-activation and death of pancreatic beta-cells through restoration of mitochondrial electron transfer. Br J Pharmacol 150: 1031–1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Dichmann DS, Rescan C, Frandsen U, Serup P (2003) Unspecific labeling of pancreatic islets by antisera against fibroblast growth factors and their receptors. J Histochem Cytochem 51: 397–400. [DOI] [PubMed] [Google Scholar]
- 49. Jositsch G, Papadakis T, Haberberger RV, Wolff M, Wess J, et al. (2009) Suitability of muscarinic acetylcholine receptor antibodies for immunohistochemistry evaluated on tissue sections of receptor gene-deficient mice. Naunyn Schmiedebergs Arch Pharmacol 379: 389–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Pradidarcheep W, Stallen J, Labruyere WT, Dabhoiwala NF, Michel MC, et al. (2009) Lack of specificity of commercially available antisera against muscarinergic and adrenergic receptors. Naunyn Schmiedebergs Arch Pharmacol 379: 397–402. [DOI] [PubMed] [Google Scholar]
- 51. Bodei S, Arrighi N, Spano P, Sigala S (2009) Should we be cautious on the use of commercially available antibodies to dopamine receptors? Naunyn Schmiedebergs Arch Pharmacol 379: 413–415. [DOI] [PubMed] [Google Scholar]
- 52. Lu X, Bartfai T (2009) Analyzing the validity of GalR1 and GalR2 antibodies using knockout mice. Naunyn Schmiedebergs Arch Pharmacol 379: 417–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Jensen BC, Swigart PM, Simpson PC (2009) Ten commercial antibodies for alpha-1-adrenergic receptor subtypes are nonspecific. Naunyn Schmiedebergs Arch Pharmacol 379: 409–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Hamdani N, van der Velden J (2009) Lack of specificity of antibodies directed against human beta-adrenergic receptors. Naunyn Schmiedebergs Arch Pharmacol 379: 403–407. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The effect of adenoviral infection on the apoptosis in islets. Trypsinized islet cells of indicated groups were subjected to a TUNEL assay. Insulin is stained green and TUNEL-positive nuclei are stained red. DAPI (blue) and differential interference contrast (DIC) images were also merged. TUNEL staining was performed using the ApopTag In Situ Detection Kit (Chemicon). For TUNEL staining, at least 40 islets per trypsinized islets group attached to poly-L-lysine coated coverslips (Falcon) were analyzed using the FLUOVIEW FV 1000-D confocal laser scanning microscope (OLYMPUS) to assess the proportion of immunostained nuclei among the insulin-positive cells.
(TIF)
The impact of treatment with metformin on the phosphorylation level of AMPK and the expressions of the incretin receptors in islets in vitro . A), B) Isolated islets were incubated with or without 1 mM metformin (Dainippon Sumitomo Pharma, Osaka, Japan) for 24 h in the presence of 2.8 or 11.1 mM glucose. A) The Glp1, Gip receptor and Pparα expressions in the islets were determined by real-time quantitative RT-PCR and normalized to the expression level of β actin mRNA and to the vehicle-treated control samples at 2.8 mM glucose (n = 7–8). *P<0.05. B) Insulin secretion by the islets treated with metformin or vehicle (ctl) in the presence of 11.1 mM glucose for 24 h with or without addition of 10 nM GLP-1 or 10 nM GIP. The results are expressed in ng of insulin/10 islets/90 min (n = 4). * P<0.05 and ** P<0.01. C), D) Isolated islets were incubated with or without 1 mM metformin for 24 h in the presence of 11.1 mM glucose. C) Islet morphology of the vehicle- and metformin-treated islets. D) The insulin content in the islets was determined after acid ethanol extraction (n = 3). White bars, vehicle; Black bars; metformin. E) Total islet extracts from the isolated islets were subjected to immunoblotting for p-AMPKα (Thr172) and AMPKα. The intensities of the signals were quantified by densitometry and normalized to the vehicle-treated control samples at 2.8 mM glucose (n = 3). The results shown are the means of three independent experiments. F) Total cell extracts from the isolated islets were subjected to immunoblotting for p-AMPKα (Thr172), AMPKα, anti-GLP1R antibody, and β actin.
(TIF)
The impact of treatment with metformin on the phosphorylation level of AMPK and the expressions of the incretin receptors in islets in vivo . Mice were given metformin in drinking water at 300 mg/kg daily or standard drinking water for 48 hours. Four hours prior to the islet isolation, the food was removed from the cages and the mice were orally administrated metformin (75 mg/kg) or vehicle (water) [14]. A) The Glp1, Gip receptor and Pparα expressions in the islets were determined by real-time quantitative RT-PCR and normalized to the expression level of β actin mRNA and to the samples in vehicle-treated mice (n = 6–7). Experiments were performed on mice treated with metformin or vehicle (ctl). *P<0.05 and ** P<0.01. B) Insulin secretion from the islets of mice treated with metformin or vehicle (ctl) in the presence of 2.8 mM or 11.1 mM glucose, with or without addition of 10 nM GLP-1 or 10 nM GIP. The results are expressed as ng of insulin/10 islets/90 min (n = 4). * P<0.05 and ** P<0.01. C) Freshly isolated total islets, the extracted liver and skeletal muscle tissues from the vehicle or metformin-treated mice were subjected to immunoblotting for p-AMPKα (Thr172) and AMPKα. The intensities of the signals were quantified by densitometry and normalized to the samples in vehicle-treated mice (n = 3). The results shown are the means of three independent experiments. D) Total cell extracts from the isolated islets were subjected to immunoblotting for p-AMPKα (Thr172), AMPKα, anti-GLP1R antibody, and β actin (n = 3).
(TIF)
The effect of glucose signal on the protein expression of the incretin receptors in islets. Isolated islets were incubated for 24 h in the presence of 2.8, 5.6, 11.1 or 22.2 mM glucose. Total cell extracts from the isolated islets were subjected to immunoblotting for p-AMPKα (Thr172), AMPKα, anti-GLP1R antibody, anti-GIPR antibody, and GAPDH.
(TIF)
The effect of glucokinase activator on the protein expression of the incretin receptors under a low glucose concentration. Isolated islets were incubated with DMSO (ctl) or GKA (G: 30 µM compound A) in the presence of 2.8 mM glucose for 24 hours. Total cell extracts from the isolated islets were subjected to immunoblotting for p-AMPKα (Thr172), AMPKα, anti-GLP1R antibody, anti-GIPR antibody, and GAPDH (n = 3).
(TIF)
The effect of glucokinase activator on the protein expression of the incretin receptors under a medium glucose concentration. Isolated islets were incubated with DMSO (ctl) or GKA (G: 30 µM compound A) in the presence of 11.1 mM glucose for 24 hours. Total cell extracts from the isolated islets were subjected to immunoblotting for p-AMPKα (Thr172), AMPKα, anti-GLP1R antibody, anti-GIPR antibody, and GAPDH (n = 3).
(TIF)
The effect of pharmacologic modulation of AMPK phosphorylation on the protein expressions of the incretin receptors under a low glucose concentration. Isolated islets were treated for 24 h with vehicle (Veh; DMSO) or the AMPK inhibitor (AMPKi; 40 µM compound C) in the presence of 2.8 mM glucose. Total cell extracts from the isolated islets were subjected to immunoblotting for p-AMPKα (Thr172), AMPKα, anti-GLP1R antibody, anti-GIPR antibody, and GAPDH (n = 3).
(TIF)
The effect of pharmacologic modulation of AMPK phosphorylation on the protein expression of the incretin receptors under a medium glucose concentration. Isolated islets were treated for 24 h with vehicle (V, water) or AICAR (A, 1 mM) in the presence of 11.1 mM glucose. Total cell extracts from the isolated islets were subjected to immunoblotting for p-AMPKα (Thr172), AMPKα, anti-GLP1R antibody, anti-GIPR antibody, and GAPDH (n = 4)
(TIF)
The impact of pharmacologic modulation of AMPK phosphorylation on the protein expressions of the incretin receptors in db/db mice. Isolated islets from 7-week-old db/+ and db/db mice were pre-treated for 15 min with vehicle (Veh; DMSO) or an AMPK inhibitor (AMPKi; 40 µM compound C) in the presence of 2.8 mM glucose, or with vehicle (Veh; DMSO) in the presence of 11.1 mM glucose, followed by the addition of vehicle (Veh; water) for an additional 24 h. Total cell extracts from the isolated islets were subjected to immunoblotting for p-AMPKα (Thr172), AMPKα, anti-GLP1R antibody, anti-GIPR antibody, and GAPDH.
(TIF)