

Abstract
Activation of AMP-activated protein kinase (AMPK) signaling reduces hepatic steatosis and hepatic insulin resistance; however, its regulatory mechanisms are not fully understood. In this study, we sought to determine whether vasodilator-stimulated phosphoprotein (VASP) signaling improves lipid metabolism in the liver and, if so, whether VASP’s effects are mediated by AMPK. We show that disruption of VASP results in significant hepatic steatosis as a result of significant impairment of fatty acid oxidation, VLDL-triglyceride (TG) secretion, and AMPK signaling. Overexpression of VASP in hepatocytes increased AMPK phosphorylation and fatty acid oxidation and reduced hepatocyte TG accumulation; however, these responses were suppressed in the presence of an AMPK inhibitor. Restoration of AMPK phosphorylation by administration of 5-aminoimidazole-4-carboxamide riboside in Vasp−/− mice reduced hepatic steatosis and normalized fatty acid oxidation and VLDL-TG secretion. Activation of VASP by the phosphodiesterase-5 inhibitor, sildenafil, in db/db mice reduced hepatic steatosis and increased phosphorylated (p-)AMPK and p-acetyl CoA carboxylase. In Vasp−/− mice, however, sildendafil treatment did not increase p-AMPK or reduce hepatic TG content. These studies identify a role of VASP to enhance hepatic fatty acid oxidation by activating AMPK and to promote VLDL-TG secretion from the liver.
Obesity is often associated with hepatic steatosis in a large proportion of obese patients, which leads to nonalcoholic fatty liver disease (NAFLD) and, in some cases, to nonalcoholic steatohepatitis (1–3). Hepatic lipid accumulation results from an imbalance between lipid availability (from circulating lipid uptake or de novo lipogenesis) and lipid disposal (via fatty acid oxidation or triglyceride [TG]-rich lipoprotein secretion).
In the liver, most fatty acids are metabolized through β-oxidation, which occurs mainly in mitochondria but also in peroxisomes (2). During fasting, hepatic fatty acid oxidation and secretion of VLDL-TG are enhanced to respond to energy demand, while in the fed state these responses are suppressed by insulin. Fasting increases free fatty acid (FFA) oxidation by inducing the phosphorylation of AMPK, a critical energy sensor, which inactivates acetyl CoA carboxylase (ACC) and activates carnitine palmitoyltransferase 1a (4)—all important steps in the transport of long-chain fatty acids to the mitochondria. Peroxisome proliferator–activated receptor (PPAR)α participates in the regulation of fatty acid oxidation through altered transcription of several key enzymes such as acyl-CoA oxidase 1 (Acox1), UCP2, nuclear respiratory factor, and mitochondrial transcription factor (Tfam) (2).
Microsomal TG transfer protein, an endoplasmic reticulum resident protein that acts as both a lipid transfer protein (5) and a facilitator of apolipoprotein B folding and translocation, is a critical factor for hepatic VLDL production (6,7). Recently, insulin-dependent suppression of microsomal TG transfer protein was shown to require forkhead transcription factor 1 (8), yet molecular mechanisms regulating fatty acid oxidation and VLDL-TG secretion are not fully understood.
Vasodilator-stimulated phosphoprotein (VASP) belongs to the ENA/VASP family of adaptor proteins linking the cytoskeletal systems to signal transduction pathways and functions in cytoskeletal organization, fibroblast migration, platelet activation, and axon guidance (9,10). We have shown previously that endothelial nitric oxide (NO)/cGMP/VASP signaling attenuates high-fat–mediated insulin resistance and inflammatory activation in hepatic tissue (11), whereas the absence of VASP increases nuclear factor (NF)-κB signaling in the liver, impairs insulin signaling, and increases hepatic TG content (11). Hepatic inflammation and insulin resistance are commonly associated with an elevated hepatic TG content. NO/cGMP is implicated with mitochondrial biogenesis (12), which influences fatty acid oxidation capacity (13). Therefore, in this study we sought to investigate whether VASP, a downstream target of NO/cGMP, reduces hepatic TG levels by enhancing fatty acid oxidation in the liver.
RESEARCH DESIGN AND METHODS
Vasp+/− mice on a C57BL/6 background have previously been described (14). C57BL/6 wild-type (WT) mice were purchased from The Jackson Laboratory. Vasp+/− mice were crossed to obtain Vasp−/− and WT littermate control mice. Male db/db and lean controls, db/+m mice, were purchased from The Jackson Laboratory. For evaluation of VLDL secretion, Triton WR 1339 (500 mg/kg i.p.) was injected after a 16-h fast and blood was sampled from the tail vein. 5-Aminoimidazole-4-carboxamide riboside (AICAR) (200 mg/kg) was injected (subcutaneously) daily for 5 days. Twelve-week-old db/db and db/+m (control) mice received daily oral administration of either vehicle or the phosphodiesterase-5 (PDE)-5 inhibitor, sildenafil (30 mg/kg/day), for 4 weeks. For the obesity-induced hepatic steatosis study, WT and Vasp−/− mice were maintained on a high-fat (60% saturated fat, D12450B; Research Diet, New Brunswick, NJ) diet for 8 weeks, and for the last 2 weeks of the diet study mice received 30 mg/kg/day oral sildenafil or vehicle. In all experiments, male, age-matched groups were maintained in a temperature-controlled facility with a 12-h light-dark cycle. All procedures were approved by the University of Washington Institutional Animal Care and Use Committee.
Quantitative RT-PCR analyses.
RNA was extracted using RNAase kit (Quiagen; Valencia, CA). For gene expression analysis, real-time RT-PCR reactions were conducted as described previously (15) using TaqMan Gene Expression Analysis (Applied Biosystems; Foster City, CA).
Western blotting.
Cell lysis and tissue extraction were performed as previously described (16). All Western blots used equal amounts of total protein for each condition from individual experiments and were performed as previously described (17).
Materials.
Anti-VASP, anti-phosphorylated (p-)VASP (Ser239), anti–p-AMPK-α (Thr172), anti–AMPK-α, anti–p-ACC (Ser79), and anti-ACC were obtained from Cell Signaling (Denvers, MA). Anti–glyceraldehyde-3-phosphate dehydrogenase rabbit polyclonal antibody was obtained from Santa Cruz Biotechnology (Santa Cruz, CA). Triton WR 1339 and STO-609 were purchased from Sigma (St Louis, MO). AICAR and compound c were purchased from Calbiochem (Darmstadt, Germany). Sildenafil was purchased from Pfizer (New York, NY). Oleic acid was purchased from NU-CHEK PREP (Elysian, MN).
Measurement of metabolic parameter and hepatic TG content.
Plasma insulin was measured by a mouse insulin ELISA kit (Crystal Chem, Downers Grove, IL). Plasma TG and FFA were measured enzymatically (L-Type TG M, Wako, and NEFA-HR, Wako, respectively; Richmond, VA). Alanine aminotransferase (ALT) and serum albumin were measured using an autoanalyzer through the Nutrition Obesity Research Center at the University of Washington. Determinations of body lean and fat mass were made in conscious mice using quantitative magnetic resonance (EchoMRI 3-in-1 body composition analyzer; Echo Medical Systems, Houston, TX) (Nutrition Obesity Research Center). Hepatic TG content was enzymatically measured (L-Type TG M; Wako) in liver lysates as previously described (18).
Cell culture.
Alpha mouse liver 12 (AML12) hepatocytes were purchased from American Type Culture Collection (Manassas, VA) and cultured in Dulbecco’s modified Eagle’s medium/F-12 50/50 (10-090-CV; Cellgro) with 3.151 mg/mL d-glucose, 0.005 mg/mL insulin, 0.005 mg/mL transferrin, 5 ng/mL selenium, 40 ng/mL dexamethasone, and 10% FBS as previously described (11). AML12 hepatocytes were treated with oleic acid (1.3 mmol/L BSA) or BSA (1.3 mmol/L) alone for 24 h, followed by homogenization with 2-propanol. The supernatant was concentrated (Savant SpeedVac), and TG content was measured enzymatically (L-Type TG M; Wako). A retroviral construct containing human wild-type VASP in the retroviral vector LXSN (14) was used for overexpression studies. AML12 cells were infected with LXSN or LXSN-WT-VASP virus. Multiple clones were selected, propagated, and maintained in the presence of G418 (0.6 mg/mL; Cellgro, Manassas, VA). Primary hepatocyte cell culture was performed as previously described (11). In brief, after hepatic perfusion through the portal vein with liver digest medium (GIBCO) containing 0.05 mg/mL collagenase type 4 (Worthington), the liver was minced and transferred into a 50-mL conical tube through a 70-μm cell strainer. After centrifugation (5 min at 50g), primary hepatocytes (the cell pellet) were collected and cultured in the media used for AML12 cells.
Knockdown of AMPK-α1 and -α2 by small interfering RNA in AML12 cells.
AML12 hepatocytes were transfected with either scramble small interfering RNA (siRNA) (4390843; Ambion, Austin, TX) or siRNA for AMPK-α1 (Prkaa1, s98535; Ambion) and AMPK-α2 (Prkaa2, s99117; Ambion) using siPORT NeoFX Transfection Agent (Ambion) according to the manufacturer’s protocol. Sequences of siRNA are as follows: Prkaa1, sense 5′ CGGGAUCCAUCAGCAACUATT 3′, antisense 5′ UAGUUGCUGAUGGAUCCCGAT 3′; Prkaa2, sense 5′ GGAUUUGCCCAGCUACCUATT 3′, antisense 5′ UAGGUAGCUGGGCAAAUCCTG 3′.
Fatty acid oxidation.
The production of acid soluble metabolites was measured as previously described (19) with minor modification and was used as an index of the β-oxidation of fatty acids. Briefly, AML12 cells were incubated in Dulbecco’s modified Eagle’s medium plus 0.5% fatty acid–free BSA with 0.05 mmol/L carnitine and 0.25 μCi [1-14C] palmitate (GE Healthcare Life Sciences; Pittsburgh, PA) per well in six-well plates. After 24 h, 800 μL medium was harvested on ice, and 200 μL ice-cold 70% perchloric acid was added in order to precipitate BSA–fatty acid complexes. The samples were centrifuged for 10 min at 14,000g, and the radioactivity of the supernatant was determined by liquid scintillation (19).
Statistical analysis.
In all experiments, densitometry measurements were normalized to controls incubated with vehicle and fold increase above the control condition was calculated. Analysis of the results was performed using the Graphpad statistical package. Data are expressed as means ± SEM, and values of P < 0.05 were considered statistically significant. A two-tailed t test was used to compare mean values in two-group comparisons. For four-group comparison, two-way ANOVA, and Bonferroni post hoc comparison tests were used to compare mean values between groups.
RESULTS
Mice lacking VASP develop hepatic steatosis on chow diet.
Since hepatic insulin resistance is associated with impaired hepatic lipid metabolism, we first asked whether an absence of VASP is associated with increased hepatic lipid content. Chow-fed Vasp−/− and littermate control mice were killed at 12 weeks of age either in a fasting condition (overnight) or in a fed condition. VASP deficiency was not associated with significant differences in body weight or adiposity (Table 1); however, in Vasp−/− mice, enzymatical measurements of hepatic TG content demonstrated a significant increase in hepatic lipid content compared with littermate controls (Fig. 1A). No significant differences in changes in liver function tests (albumin and ALT) (Table 1) were noted. These results suggest that absence of VASP is associated with hepatic steatosis without obvious changes in synthetic liver function.
TABLE 1.
Metabolic parameters of WT and Vasp−/− mice fed a chow diet
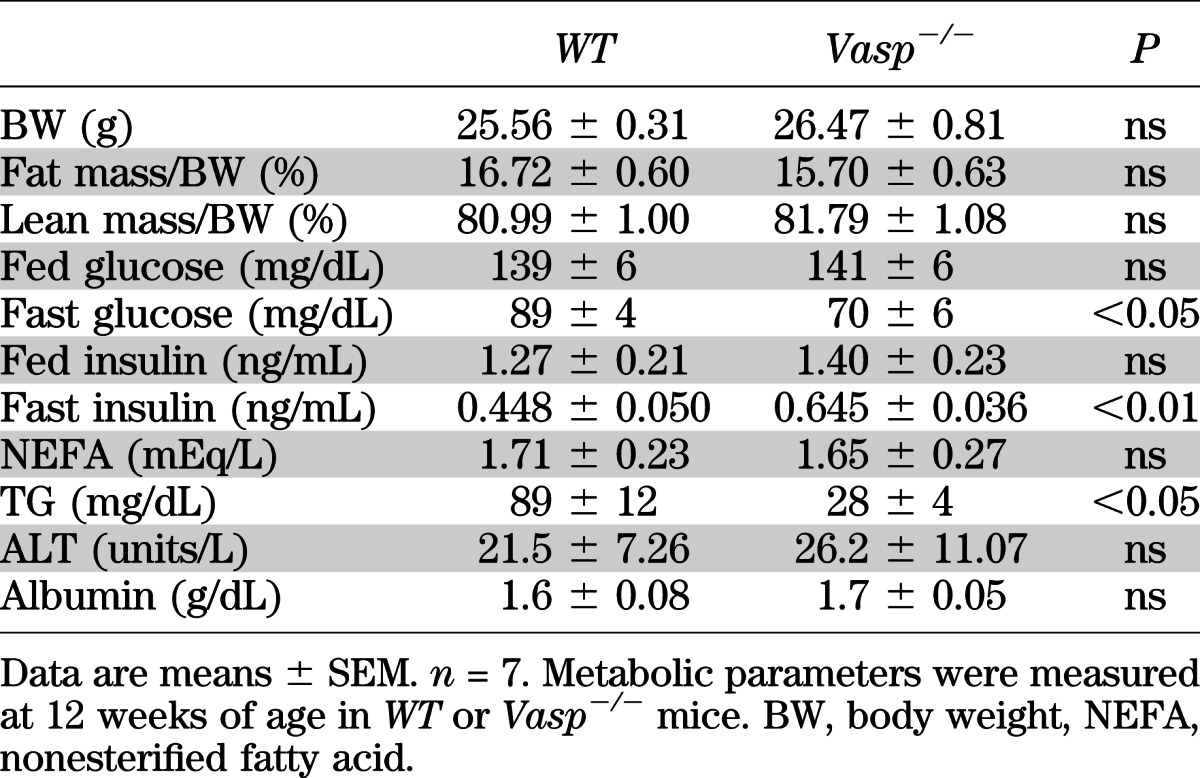
FIG. 1.

Hepatic steatosis in Vasp−/− mice fed on a chow diet. A: Enzymatic measurement of hepatic TG content collected after 16-h fast (n = 7). B: RT-PCR analysis of lipolysis-related genes in the epididymal adipose tissue collected after 16-h fast (n = 6). C: RT-PCR analysis of lipid metabolism–related genes in the liver collected either in fed mice or after 16-h fast. Expression of Gapdh as shown by the ratio of threshold cycle (CT) value (n = 6). D: Administration of Triton WR1339 (500 mg/kg i.p.), followed by measurement of plasma TG level (n = 5). *P < 0.05.
VASP deficiency is associated with an impairment of fatty acid oxidation and VLDL-related TG secretion.
Hepatic TG content is determined by a balance between circulating FFA, incorporation of FFA into the liver, lipogenesis, liver fatty acid oxidation, and VLDL-related TG secretion. An imbalance of these lipid metabolism factors is implicated in the development of hepatic steatosis (2,3). Therefore, we sought to determine whether VASP deficiency alters all or a combination of these factors leading to increased hepatic steatosis. No differences were noted in plasma FFA levels (Table 1), mRNA expression of hormone-sensitive lipase (Hsl) or adipose tissue TG lipase (Atgl), enzymes that regulate lipolysis in epididymal white adipose tissue (Fig. 1B), hepatic incorporation of FFA as measured by mRNA expressions of Caveolin-1, Cd36, or fatty acid transport protein 5 (Fatp5) expression (2) or in gene expression of factors in the lipogenesis pathway [sterol regulatory element binding protein 1c (Srebp1c), fatty acid synthase (Fas), stearoyl-CoA desaturase 1 (Scd1), and diacylglycerol acyltransferase 1 (Dgat1)] (Fig. 1C).
Fasting increases fatty acid oxidation at the level of mitochondrial and peroxisomal β-oxidation, and this was observed in littermate wild-type control mice, whereas in the Vasp−/− mice, fasting failed to increase fatty acid oxidation at the level of Acox1, Pparα, Cpt1a, Ucp2, PPARγ coactivator 1α (Pgc1α), Nrf-1, and Tfam (2,13,20) (Fig. 1C). In addition, mRNA of Mtp (involved in VLDL secretion) (Fig. 1C) and fasting TG levels were significantly lower in Vasp−/− compared with littermate control mice (Table 1), suggesting an impairment of VLDL secretion from the liver. To investigate this further, we treated fasted WT and Vasp−/− mice with Triton WR1339, an agent known to inhibit lipoprotein lipase–mediated TG removal from plasma. This results in a progressive increase in plasma TG levels, which provides a measure of the rate of VLDL secretion. Triton WR1339–dependent increase in plasma TG was inhibited in Vasp−/− mice compared with WT mice, suggesting an impairment of VLDL secretion from the liver (Fig. 1D). Collectively, these results suggest that development of hepatic steatosis in Vasp−/− mice may be attributed to alterations in fatty acid oxidation and VLDL secretion.
Overexpression of VASP in AML12 cells increases fatty acid oxidation.
Since the absence of VASP signaling in mice reduces fatty acid oxidation and VLDL release, we next asked whether VASP signaling is sufficient to increase fatty acid oxidation directly in hepatocytes. Human VASP or empty vector control was overexpressed in AML12 hepatocytes by retroviral gene transfer, and VASP overexpression was confirmed by Western blot (Fig. 2A). VASP overexpression was associated with increased fatty acid oxidation and Mtp gene expression compared with control hepatocytes (Fig. 2B) in the absence of significant changes in fatty acid uptake or lipogenic gene expression.
FIG. 2.
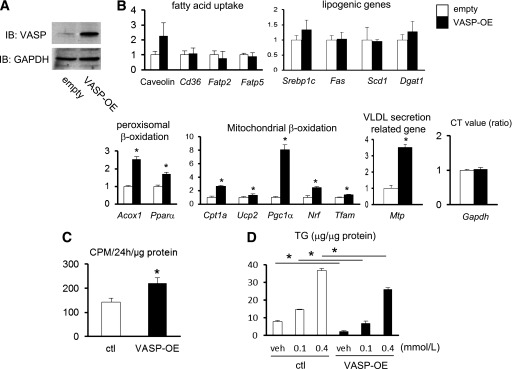
The effect of VASP on lipid metabolism in AML12 cells. A: AML12 cells were transduced with VASP (VASP-OE) or control (ctl) (empty) vector. B: RT-PCR analysis of lipid metabolism–related genes in AML12 cells. Expression of Gapdh as shown by the ratio of CT value (n = 3). C: Rate of [1-14C]palmitate incorporation into acid-soluble metabolites (n = 4). D: Oleic acid (either 0.1 or 0.4 mmol/L)-induced accumulation of TG in AML12 cells. BSA (1.3 mmol/L) was used as a control (n = 3). *P < 0.05. CT, threshold cycle. GAPDH, glyceraldehyde-3-phosphate dehydrogenase; IB, immunoblot.
To determine the effect of VASP on fatty acid oxidation directly, we next assessed the rate of [1-14C]palmitate incorporation into acid-soluble metabolites. Compared with the controls, VASP-overexpressing AML12 cells demonstrated a higher rate of fatty acid oxidation (Fig. 2C), consistent with fatty acid oxidation gene expression data.
To model the effect of hepatic steatosis in vitro, we treated AML12 hepatocytes with increasing doses of oleic acid, which leads to increased TG accumulation (21). VASP overexpression in hepatoctyes decreased oleic acid–dependent TG accumulation compared with empty vector control hepatocytes (Fig. 2D). These data collectively suggest a direct role of VASP in liver cells to enhance fatty acid oxidation and Mtp.
VASP regulates hepatic AMPK signaling during fasting.
During fasting, activation of AMPK (4) activates PPARα and cofactor PGC1α (22–26), both of which are key upstream transcription factors regulating fatty acid oxidation gene expression (Acox1, Ucp2, Nrf-1, and Tfam) (22,27–31). Since VASP similarly regulates these responses (Fig. 1C and B), we hypothesized that VASP-dependent effects on lipid metabolism are mediated by AMPK. To test this hypothesis, we analyzed AMPK signaling in AML12 hepatocytes and from hepatic tissues of WT and Vasp−/− mice. Overexpression of VASP in AML12 hepatocytes was associated with increased p-AMPK and p-ACC (Fig. 3A) compared with empty vector–transduced hepatocytes, whereas primary hepatocytes isolated from Vasp−/− mice exhibit reduced AMPK signaling and rate of fatty acid oxidation (Fig. 3B). In vivo, overnight fasting increased hepatic levels of p-AMPK and p-ACC. However, this expected response was attenuated in Vasp−/− mice (Fig. 3C). Collectively, these data suggest roles of VASP in AMPK signaling and fatty acid oxidation in hepatocytes.
FIG. 3.

AMPK signaling is regulated by VASP in the liver. A: Phosphorylation of AMPK (Thr172) and ACC (Ser79) in AML12 cells with VASP overexpression. AICAR was used as a positive control. Western blot from one of three independent experiments is shown. B: Primary hepatocytes were isolated from WT or Vasp−/− mice and cultured. p-AMPK (Thr172) and p-ACC (Ser79) as measured by Western blot. Representative blots are shown. Rate of [1-14C]palmitate incorporation into acid-soluble metabolites (n = 3). C: p-AMPK and p-ACC in the liver collected after an overnight fast (n = 6). Representative blots are shown. *P < 0.05. GAPDH, glyceraldehyde-3-phosphate dehydrogenase; IB, immunoblot.
AMPK is necessary for the protective effect of VASP on hepatic lipid metabolism.
To determine whether AMPK is necessary for the effect of VASP on hepatic FFA oxidation, we used an RNAi-mediated knockdown of AMPK-α1 and AMPK-α2, catalytic subunits of AMPK, both of which are important for hepatic fatty acid oxidation (4,23) (Fig. 4A). As expected, overexpression of VASP in AML12 hepatocytes increased peroxisomal and mitochondrial β-oxidation and VLDL-TG secretion; however, in the presence of siRNA for AMPKα, these responses were significantly attenuated (Fig. 4B). Furthermore, even though VASP overexpression reduced oleic acid–induced hepatic TG accumulation, inhibition of AMPK attenuated VASP’s protective responses (Fig. 4C). Similar results were obtained using a pharmacological inhibitor of AMPK, compound c (Supplementary Fig. 1A and B). These data collectively suggest that intact AMPK signaling is required for VASP to attenuate TG accumulation by enhancing fatty acid oxidation and upregulating Mtp gene expression in the liver.
FIG. 4.

Involvement of AMPK signaling in the effect of VASP. A: AMPK-α protein levels after siRNA for AMPK-α1 (Prkaa1, 5 nmol/L, 48 h) and AMPK-α2 (Prkaa2, 5 nmol/L, 48 h) in AML12 hepatocytes (n = 2). B: AML12 hepatocytes were transduced with VASP (VASP-OE) or control (ctl) (empty) vector. RT-PCR analysis of fatty acid oxidation genes or Mtp gene in AML12 cells with siRNAs for AMPK-α1 (Prkaa1, 5 nmol/L, 48 h) and -α2 (Prkaa2, 5 nmol/L, 48 h) (n = 4). C: Oleic acid (0.1 mmol/L) was used to treat AML12 cells for 24 h with or without siRNAs for AMPK-α1 (Prkaa1, 5 nmol/L, 48 h) and -α2 (Prkaa2, 5 nmol/L, 48 h) (n = 4). D: Either AICAR (200 mg/kg) or PBS was injected (subcutaneously) daily for 5 days in WT and Vasp−/− mice, after which mice were fasted overnight and then killed. Relative mRNA expression in the liver as measured by RT-PCR is shown (n = 5). E: Hepatic TG content as measured enzymatically (n = 5). F: At the end of the 5-day treatment of AICAR protocol, Triton WR1339 (500 mg/kg) was administered intraperitoneally after an overnight fast, followed by a measurement of plasma TG level (n = 5). *P < 0.05. CT, threshold cycle; ctl, control; IB, immunoblot.
Restoration of AMPK signaling in Vasp−/− mice attenuates fatty liver through a normalization of impaired fatty acid oxidation and VLDL secretion.
Since reduced AMPK signaling is associated with the development of hepatic steatosis in Vasp−/− mice, we next determined whether restoration of AMPK signaling would restore fatty acid oxidation in Vasp−/− mice. Administration of an AMPK activator, AICAR, daily for 5–7 days has been shown to enhance fatty acid oxidation and ameliorate hepatic steatosis not only in rodent models but also in human subjects (31–33). Vasp−/− mice received daily AICAR injections (200 mg/kg) for 5 days and were then killed after an overnight fast. Restoration of p-AMPK and p-ACC (Supplementary Fig. 2) by the administration of AICAR did not change body weight (Supplementary Table 1); however, AICAR reduced hepatic TG content and increased peroxisomal β-oxidation gene (Acox1 and Pparα), mitochondrial β-oxidation gene (Cpt1a, Ucp2, and Pgc1α), (Fig. 4D and E) and Mtp gene (Fig. 4D) expression in Vasp−/− mice. Finally, triton-induced VLDL-TG secretion in Vasp−/− mice was normalized in response to AICAR treatment (Fig. 4F). These data suggest that restoration of AMPK signaling in Vasp−/− mice restores hepatic fatty acid oxidation and VLDL-TG secretion.
Activation of VASP by sildenafil reduces hepatic steatosis in db/db mice and during diet-induced obesity.
We next determined whether activation of VASP signaling reduces hepatic steatosis in a genetic model of obesity. Since activation of VASP (phosphorylation of VASP at Ser239) is governed by cGMP-dependent protein kinase (34), we first tested whether 8Br-cGMP, an analog of cGMP, could directly regulate AMPK activation and fatty acid oxidation in hepatocytes. We found that cGMP enhances hepatic fatty acid oxidation, and this was mediated by AMPK (Supplementary Fig. 3A–C).
In order to investigate this further in vivo, we used the PDE-5 inhibitor, sildenafil, which is known to increase intracellular cGMP levels, activate cGMP-dependent protein kinase, and phosphorylate (serine 239) VASP (11,34,35). db/db and control db/+m mice received daily doses of vehicle or sildenafil (30 mg/kg) for 4 weeks. At the end of the study, liver lysates were analyzed for VASP, AMPK, and ACC phosphorylation by Western blot. As expected, db/db mice exhibited reduced levels of p-VASP, p-AMPK, and p-ACC in liver lysates compared with db/+m control mice, whereas sildenafil treatment in db/db mice restored p-VASP (Ser239), p-AMPK, and p-ACC levels comparable with control db/+m mice (Fig. 5A). These observations were accompanied by a reduction of hepatic steatosis in the db/db mice (Fig. 5B). Collectively, pharmacological activation of VASP by sildenafil reduces hepatic steatosis in db/db mice through increased AMPK signaling and fatty acid oxidation in the liver.
FIG. 5.

Pharmacological activation of VASP ameliorates hepatic steatosis during the fasting state in db/db mice and mice with high-fat diet–induced obesity. Twelve-week-old db/db and db/+m mice fed chow diet received daily oral administration of either vehicle or the PDE-5 inhibitor sildenafil (30 mg/kg/day) for 4 weeks. For the obesity-induced hepatic steatosis study, WT and Vasp−/− mice were maintained on a high-fat diet for 8 weeks, and for the last 2 weeks of the diet study mice received 30 mg/kg/day oral sildenafil or vehicle. A: p-VASP (Ser239), p-AMPK (Thr172), and p-ACC (Ser79) as measured by Western blot in the liver samples collected after an overnight fast. Representative Western blots are shown. B: Hepatic TG content as measured enzymatically. *P < 0.05 (db/+m, n = 3; db/db, n = 6). C: p-AMPK (Thr172) as measured by Western blot in the liver sample collected after an overnight fast. Representative Western blots are shown. D: Hepatic TG content as measured enzymatically. *P < 0.05 (n = 5). GAPDH, glyceraldehyde-3-phosphate dehydrogenase; IB, immunoblot.
Sildenafil does not ameliorate hepatic steatosis in Vasp−/− mice.
Finally, we sought to test whether VASP is required for sildenafil to enhance AMPK signaling and ameliorate hepatic steatosis. Previously, we demonstrated that sildenafil reduces hepatic steatosis induced during high-fat feeding (measured in the fed state) (11); however, the effect of sildenafil on hepatic steatosis during fasting is unknown. WT and Vasp−/− mice were maintained on a high-fat diet for 8 weeks, and for the last 2 weeks of the diet study mice received 30 mg/kg/day oral sildenafil or vehicle. While sildenafil increased p-AMPK in WT mice after an overnight fast, this was not evident in Vasp−/− mice (Fig. 5C). Similarly, sildenafil reduced hepatic TG content in WT mice but not in Vasp−/− mice (Fig. 5D). These data suggest that the effect of sildenafil to reduce hepatic steatosis requires VASP and its downstream target, AMPK.
DISCUSSION
Hepatic steatosis is a commonly observed hepatic manifestation of the metabolic syndrome in diabetes and obesity and is thought to be the initial abnormality in NAFLD (2). NAFLD encompasses a spectrum of liver disease ranging from benign fatty liver to the more severe nonalcoholic steatohepatitis—a condition that may progress to cirrhosis in up to 25% of patients (2).
By using both loss-of-function and gain-of-function studies, we demonstrate that VASP enhances fatty acid oxidation by activating AMPK signaling in the liver. Conversely, the absence of VASP reduces AMPK activation and reduces hepatic fatty acid oxidation, resulting in increased hepatic steatosis. These results suggest a novel role for VASP in attenuating the development of hepatic steatosis during obesity and, furthermore, may identify VASP as a potential therapeutic target.
AMPK is a serine/threonine protein kinase, which acts as a sensor of cellular energy status and regulates a wide variety of gene regulatory and metabolic pathways, including fatty acid oxidation in liver (4). Activation of AMPK occurs by phosphorylation at Thr172 catalyzed by liver kinase B1 in response to an increase in the AMP-to-ATP ratio and by calmodulin-dependent protein kinase kinase β (CaMKKβ) in response to elevated Ca2+ levels (4). NO has recently been reported as an endogenous activator of AMPK through a CaMKK-dependent mechanism in endothelial cells (36). Therefore, it is reasonable to hypothesize that in hepatocytes, CaMKK signaling may link VASP to AMPK activation. We tested this hypothesis using STO-609, a pharmacological inhibitor of CaMKK, and found that in the presence of STO-609, AMPK signaling (Supplementary Fig. 4A) and fatty acid oxidation (Supplementary Fig. 4B) are reduced. Additional studies are necessary, since STO-609 may have off-target effects (37). Other possible mechanisms may involve liver kinase B1 or protein phosphatase-2C, a phosphatase that dephosphorylates and inactivates AMPK (4).
Since AMPK plays an important role in the regulation of gluconeogenesis and lipid metabolism, therapeutic strategies targeting AMPK signaling are promising therapies to reverse both glucose and lipid abnormalities associated with type 2 diabetes (38,39). Our current studies suggest that targeting VASP (e.g., by the PDE-5 inhibitor, sildenafil) could lead to an improvement of hepatic steatosis by activating AMPK signaling.
Vasp−/− mice display fasting hypoglycemia (Table 1), despite the presence of hepatic steatosis (Fig. 1A) and insulin resistance at the level of Akt/IRS2 signaling (11). Hepatic steatosis and fasting hypoglycemia are also observed in Pparα−/− mice, a mice model of impaired mitochondrial fatty acid oxidation (40,41). Hypoglycemia occurring in the context of defective mitochondrial fatty acid oxidation is likely due to a combination of glycogen depletion and a blunted gluconeogenic response (41). Since Pparα was regulated by VASP (Figs. 1C and 2B), mechanisms of steatosis and fasting hypoglycemia in Vasp−/− mice might be similar to those of Pparα−/− mice.
In Vasp−/− mice, fasting plasma TG level was reduced (Table 1), and this was associated with decreased Mtp gene expression (Fig. 1C) and impaired VLDL-TG secretion (Fig. 1D). Mechanistically, since transcription of Mtp is positively regulated by PPARα (29) and since VASP activates AMPK and PPARα (Figs. 2B and 3A), VASP may regulate Mtp thorough PPARα.
AMPK not only enhances fatty acid oxidation but also suppresses NF-κB signaling (42). In Vasp−/− mice, reduction of fatty acid oxidation (Fig. 1C) was coincident with elevation of inflammation (11). These alterations might be explained by reduced AMPK signaling (Fig. 3B and C). Anti-inflammatory effect of VASP (11) might be mediated by AMPK. Indeed, restoration of AMPK by AICAR attenuated hepatic inflammation in Vasp−/− mice (Supplementary Fig. 5).
Another study provides different evidence linking inflammation to AMPK, in that they show that inflammatory pathway downregulates AMPK phosphorylation and activity (43). Since Vasp−/− mice demonstrated increased inflammation not only in liver (11) but also in adipose tissue (44), it is possible that reduced AMPK signaling in Vasp−/− mice liver could be attributed to the increased inflammation. However, alterations of AMPK signaling by VASP in vitro (Fig. 3A and B) indicate a direct effect of VASP in hepatocytes. Furthermore, we demonstrated that this was CaMKK dependent (Supplementary Fig. 4A and B).
NO/cGMP, regulators of VASP, are implicated with mitochondrial biogenesis in adipocytes (12), which influences fatty acid oxidation capacity (13). This effect can also be exerted in the liver. NO/cGMP has been reported to enhance fatty acid oxidation in rat hepatocytes (45) and indeed, enos−/− mice, in which this signaling is downregulated, develop hepatic steatosis (11,46). We consider that VASP, a downstream target, has similar roles. Since NO/cGMP/VASP signaling was downregulated during diet-induced obesity (11), reduced NO/cGMP/VASP signaling might cause impaired mitochondrial biogenesis, followed by susceptibility to develop steatosis.
VASP protein expression is found in hepatocytes, Kuppfer cells, and hepatic sinusoidal endothelial cells, and the cell-specific contribution of VASP to the development of hepatic steatosis remains an important unanswered question. We previously demonstrated that the absence of VASP is associated with increased hepatocyte as well as Kupffer cell NF-κB activation during high-fat feeding (11). Since M1 (proinflammatory) activated Kuppfer cells have been reported to contribute to the development of hepatic steatosis (47), additional studies are needed to clarify the role of VASP signaling in M1 activated Kupffer cells (11) and in the development of hepatic steatosis in Vasp−/− mice.
Since hepatic expression of Fatp2 was increased in Vasp−/− mice (Fig. 1C), elevated incorporation of FFA into the liver other than reduced fatty acid oxidation and VLDL-TG secretion might also contribute the development of steatosis. However, Fatp2 was not decreased by the overexpression of VASP (Fig. 2B). Thus, whether VASP is associated with an uptake of fatty acid into the liver has not been fully revealed yet.
Recently, activation of NO and cGMP signaling (34) has been reported to activate AMPK signaling in various type of cells, including endothelial cells (36) and human muscle cells (48). Our current study, which indicates that VASP enhances fatty acid oxidation by activating AMPK, is the first evidence that links VASP, a downstream molecule of cGMP, to AMPK signaling.
In summary, we have demonstrated that in liver, VASP has a role to enhance fatty acid oxidation by activating AMPK. We also identified a role of VASP to promote VLDL-TG secretion through alteration of Mtp gene expression. Improvement of hepatic steatosis in db/db mice by pharmacological enhancement of VASP signaling additionally suggests that direct activation of VASP could be a promising target for the treatment of hepatic steatosis.
Supplementary Material
ACKNOWLEDGMENTS
This study was supported by National Institutes of Health grants DK-073878 (to F.K.), HL-94352 and HL-92969 (to A.C.), and HL-062887, HL-92969, and HL-097365 (to K.E.B.); Cardiovascular Postdoctoral Training Grant T32 HL-07828 (to A.M.C.); Uehara Memorial Foundation (to S.T.); and grants from the John L. Locke, Jr., Charitable Trust and from the Kenneth H. Cooper Endowed Professorship in Preventive Cardiology (to F.K.). Nutrition Obesity Research Center at the University of Washington is supported by the National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases Grant P30 DK-035816.
No potential conflicts of interest relevant to this article were reported.
S.T. designed and performed experiments, provided data analysis, contributed to discussion, and wrote, reviewed, and edited the manuscript. N.R.-D.L. designed and performed experiments. A.M.C., K.E.B., and A.C. designed experiments and contributed to discussion. P.H. performed experiments and contributed to discussion. V.M.-S., K.O., and J.E.K. performed experiments. G.D. and A.W.C. reviewed and edited the manuscript. F.K. interpreted data and wrote, reviewed, and edited the manuscript. S.T. is the guarantor of this work and, as such, had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Footnotes
This article contains Supplementary Data online at http://diabetes.diabetesjournals.org/lookup/suppl/doi:10.2337/db12-0325/-/DC1.
REFERENCES
- 1.Unger RH. Minireview: weapons of lean body mass destruction: the role of ectopic lipids in the metabolic syndrome. Endocrinology 2003;144:5159–5165 [DOI] [PubMed] [Google Scholar]
- 2.Musso G, Gambino R, Cassader M. Recent insights into hepatic lipid metabolism in non-alcoholic fatty liver disease (NAFLD). Prog Lipid Res 2009;48:1–26 [DOI] [PubMed] [Google Scholar]
- 3.Postic C, Girard J. Contribution of de novo fatty acid synthesis to hepatic steatosis and insulin resistance: lessons from genetically engineered mice. J Clin Invest 2008;118:829–838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Viollet B, Lantier L, Devin-Leclerc J, et al. Targeting the AMPK pathway for the treatment of Type 2 diabetes. Front Biosci 2009;14:3380–3400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wetterau JR, Combs KA, Spinner SN, Joiner BJ. Protein disulfide isomerase is a component of the microsomal triglyceride transfer protein complex. J Biol Chem 1990;265:9800–9807 [PubMed] [Google Scholar]
- 6.Davis RA, Hui TY. 2000 George Lyman Duff Memorial Lecture: atherosclerosis is a liver disease of the heart. Arterioscler Thromb Vasc Biol 2001;21:887–898 [DOI] [PubMed] [Google Scholar]
- 7.Fisher EA, Ginsberg HN. Complexity in the secretory pathway: the assembly and secretion of apolipoprotein B-containing lipoproteins. J Biol Chem 2002;277:17377–17380 [DOI] [PubMed] [Google Scholar]
- 8.Kamagate A, Qu S, Perdomo G, et al. FoxO1 mediates insulin-dependent regulation of hepatic VLDL production in mice. J Clin Invest 2008;118:2347–2364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ball LJ, Kühne R, Hoffmann B, et al. Dual epitope recognition by the VASP EVH1 domain modulates polyproline ligand specificity and binding affinity. EMBO J 2000;19:4903–4914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Machesky LM. Putting on the brakes: a negative regulatory function for Ena/VASP proteins in cell migration. Cell 2000;101:685–688 [DOI] [PubMed] [Google Scholar]
- 11.Tateya S, Rizzo NO, Handa P, et al. Endothelial NO/cGMP/VASP signaling attenuates Kupffer cell activation and hepatic insulin resistance induced by high-fat feeding. Diabetes 2011;60:2792–2801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nisoli E, Clementi E, Paolucci C, et al. Mitochondrial biogenesis in mammals: the role of endogenous nitric oxide. Science 2003;299:896–899 [DOI] [PubMed] [Google Scholar]
- 13.Adam T, Opie LH, Essop MF. AMPK activation represses the human gene promoter of the cardiac isoform of acetyl-CoA carboxylase: Role of nuclear respiratory factor-1. Biochem Biophys Res Commun 2010;398:495–499 [DOI] [PubMed] [Google Scholar]
- 14.Chen L, Daum G, Chitaley K, et al. Vasodilator-stimulated phosphoprotein regulates proliferation and growth inhibition by nitric oxide in vascular smooth muscle cells. Arterioscler Thromb Vasc Biol 2004;24:1403–1408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kim F, Pham M, Maloney E, et al. Vascular inflammation, insulin resistance, and reduced nitric oxide production precede the onset of peripheral insulin resistance. Arterioscler Thromb Vasc Biol 2008;28:1982–1988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kim F, Pham M, Luttrell I, et al. Toll-like receptor-4 mediates vascular inflammation and insulin resistance in diet-induced obesity. Circ Res 2007;100:1589–1596 [DOI] [PubMed] [Google Scholar]
- 17.Kim F, Tysseling KA, Rice J, et al. Free fatty acid impairment of nitric oxide production in endothelial cells is mediated by IKKbeta. Arterioscler Thromb Vasc Biol 2005;25:989–994 [DOI] [PubMed] [Google Scholar]
- 18.Kanda H, Tateya S, Tamori Y, et al. MCP-1 contributes to macrophage infiltration into adipose tissue, insulin resistance, and hepatic steatosis in obesity. J Clin Invest 2006;116:1494–1505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Golej DL, Askari B, Kramer F, et al. Long-chain acyl-CoA synthetase 4 modulates prostaglandin E2 release from human arterial smooth muscle cells. J Lipid Res 2011;52:782–793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Scarpulla RC. Nuclear activators and coactivators in mammalian mitochondrial biogenesis. Biochim Biophys Acta 2002;1576:1–14 [DOI] [PubMed] [Google Scholar]
- 21.Lee JY, Cho HK, Kwon YH. Palmitate induces insulin resistance without significant intracellular triglyceride accumulation in HepG2 cells. Metabolism 2010;59:927–934 [DOI] [PubMed] [Google Scholar]
- 22.Scarpulla RC. Transcriptional paradigms in mammalian mitochondrial biogenesis and function. Physiol Rev 2008;88:611–638 [DOI] [PubMed] [Google Scholar]
- 23.Cantó C, Auwerx J. AMP-activated protein kinase and its downstream transcriptional pathways. Cell Mol Life Sci 2010;67:3407–3423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lee WJ, Kim M, Park HS, et al. AMPK activation increases fatty acid oxidation in skeletal muscle by activating PPARalpha and PGC-1. Biochem Biophys Res Commun 2006;340:291–295 [DOI] [PubMed] [Google Scholar]
- 25.Bronner M, Hertz R, Bar-Tana J. Kinase-independent transcriptional co-activation of peroxisome proliferator-activated receptor alpha by AMP-activated protein kinase. Biochem J 2004;384:295–305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Longuet C, Sinclair EM, Maida A, et al. The glucagon receptor is required for the adaptive metabolic response to fasting. Cell Metab 2008;8:359–371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Varanasi U, Chu R, Huang Q, Castellon R, Yeldandi AV, Reddy JK. Identification of a peroxisome proliferator-responsive element upstream of the human peroxisomal fatty acyl coenzyme A oxidase gene. J Biol Chem 1996;271:2147–2155 [DOI] [PubMed] [Google Scholar]
- 28.Yoon M. The role of PPARalpha in lipid metabolism and obesity: focusing on the effects of estrogen on PPARalpha actions. Pharmacol Res 2009;60:151–159 [DOI] [PubMed] [Google Scholar]
- 29.Koves TR, Li P, An J, et al. Peroxisome proliferator-activated receptor-gamma co-activator 1alpha-mediated metabolic remodeling of skeletal myocytes mimics exercise training and reverses lipid-induced mitochondrial inefficiency. J Biol Chem 2005;280:33588–33598 [DOI] [PubMed] [Google Scholar]
- 30.Spann NJ, Kang S, Li AC, et al. Coordinate transcriptional repression of liver fatty acid-binding protein and microsomal triglyceride transfer protein blocks hepatic very low density lipoprotein secretion without hepatosteatosis. J Biol Chem 2006;281:33066–33077 [DOI] [PubMed] [Google Scholar]
- 31.Villarroya F, Iglesias R, Giralt M. PPARs in the control of uncoupling proteins gene expression. PPAR Res 2007;2007:74364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cool B, Zinker B, Chiou W, et al. Identification and characterization of a small molecule AMPK activator that treats key components of type 2 diabetes and the metabolic syndrome. Cell Metab 2006;3:403–416 [DOI] [PubMed] [Google Scholar]
- 33.Boon H, Bosselaar M, Praet SF, et al. Intravenous AICAR administration reduces hepatic glucose output and inhibits whole body lipolysis in type 2 diabetic patients. Diabetologia 2008;51:1893–1900 [DOI] [PubMed] [Google Scholar]
- 34.Oelze M, Mollnau H, Hoffmann N, et al. Vasodilator-stimulated phosphoprotein serine 239 phosphorylation as a sensitive monitor of defective nitric oxide/cGMP signaling and endothelial dysfunction. Circ Res 2000;87:999–1005 [DOI] [PubMed] [Google Scholar]
- 35.Ayala JE, Bracy DP, Julien BM, Rottman JN, Fueger PT, Wasserman DH. Chronic treatment with sildenafil improves energy balance and insulin action in high fat-fed conscious mice. Diabetes 2007;56:1025–1033 [DOI] [PubMed] [Google Scholar]
- 36.Zhang J, Xie Z, Dong Y, Wang S, Liu C, Zou MH. Identification of nitric oxide as an endogenous activator of the AMP-activated protein kinase in vascular endothelial cells. J Biol Chem 2008;283:27452–27461 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 37.Bain J, Plater L, Elliott M, et al. The selectivity of protein kinase inhibitors: a further update. Biochem J 2007;408:297–315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Winder WW. Can patients with type 2 diabetes be treated with 5′-AMP-activated protein kinase activators? Diabetologia 2008;51:1761–1764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang BB, Zhou G, Li C. AMPK: an emerging drug target for diabetes and the metabolic syndrome. Cell Metab 2009;9:407–416 [DOI] [PubMed] [Google Scholar]
- 40.Kersten S, Seydoux J, Peters JM, Gonzalez FJ, Desvergne B, Wahli W. Peroxisome proliferator-activated receptor alpha mediates the adaptive response to fasting. J Clin Invest 1999;103:1489–1498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Leone TC, Weinheimer CJ, Kelly DP. A critical role for the peroxisome proliferator-activated receptor alpha (PPARalpha) in the cellular fasting response: the PPARalpha-null mouse as a model of fatty acid oxidation disorders. Proc Natl Acad Sci USA 1999;96:7473–7478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Salminen A, Hyttinen JM, Kaarniranta K. AMP-activated protein kinase inhibits NF-κB signaling and inflammation: impact on healthspan and lifespan. J Mol Med (Berl) 2011;89:667–676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yang Z, Kahn BB, Shi H, Xue BZ. Macrophage alpha1 AMP-activated protein kinase (alpha1AMPK) antagonizes fatty acid-induced inflammation through SIRT1. J Biol Chem 2010;285:19051–19059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Handa P, Tateya S, Rizzo NO, et al. Reduced vascular nitric oxide-cGMP signaling contributes to adipose tissue inflammation during high-fat feeding. Arterioscler Thromb Vasc Biol 2011;31:2827–2835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.García-Villafranca J, Guillén A, Castro J. Involvement of nitric oxide/cyclic GMP signaling pathway in the regulation of fatty acid metabolism in rat hepatocytes. Biochem Pharmacol 2003;65:807–812 [DOI] [PubMed] [Google Scholar]
- 46.Schild L, Dombrowski F, Lendeckel U, Schulz C, Gardemann A, Keilhoff G. Impairment of endothelial nitric oxide synthase causes abnormal fat and glycogen deposition in liver. Biochim Biophys Acta 2008;1782:180–187 [DOI] [PubMed] [Google Scholar]
- 47.Huang W, Metlakunta A, Dedousis N, et al. Depletion of liver Kupffer cells prevents the development of diet-induced hepatic steatosis and insulin resistance. Diabetes 2010;59:347–357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Deshmukh AS, Long YC, de Castro Barbosa T, et al. Nitric oxide increases cyclic GMP levels, AMP-activated protein kinase (AMPK)alpha1-specific activity and glucose transport in human skeletal muscle. Diabetologia 2010;53:1142–1150 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.