

Cardiovascular complications are more prevalent in individuals with conditions associated with insulin resistance. Knowledge is evolving concerning the mechanistic link between insulin resistance and vascular disease, but complete clarity does not exist. This is a high-priority area of investigation because individual and societal costs associated with diabetic vascular complications are increasing, and the need to elucidate effective therapeutic targets and intervention strategies with minimal off-target side effects is urgent.
Insulin-resistant conditions such as type 2 diabetes and diet-induced obesity are associated with an altered endothelial cell phenotype, i.e., endothelial dysfunction. A crucial component of endothelial cell dysfunction is reduced nitric oxide (NO) bioavailability. Reduced NO bioavailability contributes importantly to pathologies specific to large (atherosclerosis, cardiomyopathy) and small (retinopathy, nephropathy, neuropathy) blood vessels (1). Oxidative stress, hyperglycemia, lipotoxicity, activation of the renin-angiotensin system, and increased proinflammatory cytokines are systemic disturbances in patients with conditions associated with insulin resistance. Each of these factors contributes independently and synergistically to decreasing NO bioavailability by impairing its synthesis and/or by enhancing its degradation (2). In this issue of Diabetes, Sukumar et al. (3) demonstrate that endothelial cell–specific insulin resistance increases NADPH oxidase (Nox) isoform 2 (Nox2) expression to an extent that elevates superoxide anion (O2·−) production and precipitates endothelial dysfunction.
Reactive oxygen species (ROS) are highly reactive metabolites of oxygen that participate in oxidation-reduction reactions (4). A traditional definition of oxidative stress is when ROS accumulation overwhelms the cellular antioxidant defense mechanisms and the redox state of the biological compartment is shifted toward one that is more oxidizing (5,6). Cellular sources of ROS include NADPH-dependent oxidases, xanthine oxidase, lipoxygenases, mitochondrial oxidases, and NO synthases (7). Nox is a major source of O2·− in insulin-resistant humans (8,9) and mice (10,11). Seven isoforms of Nox have been described in mammals (Fig. 1). Each isoform comprises a core catalytic subunit, i.e., Nox1–Nox5 and dual oxidase (DUOX) 1 and DUOX 2 (4). Each Nox catalytic isoform contains up to five regulatory subunits that determine 1) maturation and expression of Nox and DUOX subunits in biological membranes (e.g., p22phox), 2) enzyme activation (e.g., p67phox, RAC1/RAC2), and 3) spatial organization (e.g., p47phox). Endothelial cells express Nox1, Nox2 (gp91phox), Nox4, and Nox5; vascular smooth muscle cells express Nox1, Nox4, and Nox5; and adventitial fibroblasts express Nox2 and Nox4 (5).
FIG. 1.
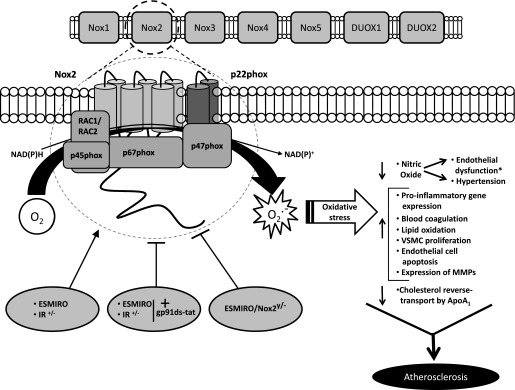
Hypothesized contributions from Nox2-generated oxidative stress to the process of atherosclerosis. When NADPH donates electrons to molecular oxygen via Nox2, O2·− generation occurs. Oxidative stress occurs when O2·− production exceeds the antioxidant capacity of the immediate environment. Oxidative stress can contribute to atherosclerosis progression by various mechanisms (7). ↑, increase; ↓, decrease. Mice with endothelial cell–specific insulin resistance (ESMIRO and IR+/− mice) exhibit increased Nox2-mediated O2·− generation to an extent that evokes endothelial dysfunction. Treatment of ESMIRO and IR+/− mice with the Nox inhibitor gp91ds-tat (+gp91ds-tat) or endothelial cell–specific knockout of Nox2 in ESMIRO mice (ESMIRO / Nox2y/−) reduces O2·− production and restores endothelial function. *Endothelial function was assessed in the current study (3), but the influence of endothelial cell–specific Nox2 deletion on other potential contributors to atherosclerosis depicted in the figure has not been investigated. VSMC, vascular smooth muscle cell; MMPs, matrix metalloproteinases.
Earlier work from this laboratory group indicates that mice with germ-line haploinsufficiency of the insulin receptor (IR+/− mice) display hypertension and an age-dependent attenuation of arterial vasorelaxation that is associated with elevated Nox-mediated O2·− production (12,13). Importantly, impaired endothelial function in IR+/− mice has physiological relevance in models of endothelial injury (14) and atherosclerotic plaque development (15). A valid concern when interpreting these results is that metabolic disruptions known to exist in IR+/− and apoE mice might contribute to impaired endothelial function. To address this, mice with endothelium-targeted overexpression of a dominant-negative mutant human insulin receptor (endothelium-specific mutant insulin receptor overexpressing mice [ESMIRO]) were studied (10). Compared with wild-type littermates, ESMIRO mice have preserved glucose homeostasis and are normotensive. However, endothelium-dependent vasorelaxation and endothelial NO synthase (eNOS) phosphorylation are blunted in aortae from ESMIRO versus wild-type mice, despite similar eNOS protein expression. Because mRNA expression of Nox2 and Nox4 was increased in endothelial cells and aortae of ESMIRO mice, and endothelium-dependent dysfunction was normalized when aortae from ESMIRO mice were incubated with a superoxide dismutase mimetic, the authors concluded that Nox-mediated O2·− generation contributed importantly to dysfunction observed in arteries from mutant mice (10).
Sukumar et al. investigated a relatively new approach to the challenge of preventing arterial dysfunction in the context of insulin resistance, i.e., to suppress O2·− generation locally by disrupting an NADPH enzyme complex located in the endothelial cell. First, the authors confirmed the preserved metabolic phenotype and disrupted vascular phenotype that exists in IR+/− and ESMIRO mice, and demonstrated that exaggerated O2·− production and endothelial dysfunction exhibited in aortae from both groups could be attenuated by acute or chronic Nox inhibition using gp91ds-tat (16). Because Nox2 mRNA expression was elevated in endothelial cells from IR+/− and ESMIRO mice and Nox2 siRNA suppressed O2·− generation in endothelial cells from ESMIRO mice, the authors sought to determine whether Nox2–mediated O2·− production is sufficient to contribute to arterial dysfunction in the context of insulin resistance. To explore this, mice with endothelial cell–specific insulin resistance and deletion of Nox2 were generated i.e., ESMIRO / Nox2y/− mice. As hypothesized, increased O2·− generation and vascular dysfunction were prevented in endothelial cells and/or aortae from ESMIRO/Nox2y/− versus ESMIRO mice. Importantly, systemic glucose homeostasis and blood pressure were similar between groups. These findings provide strong evidence to consider the Nox2 isoform as a therapeutic target for the treatment or prevention of vascular disease in the context of insulin resistance.
Strengths of this particular study include 1) the use of double transgenic mice wherein insulin resistance and Nox2 deletion is created specifically in the endothelial cell; 2) demonstrating that systemic glucose homeostasis and arterial pressure was not altered in double transgenic versus ESMIRO mice, 3) using redundant methodologies to confirm O2·− generation; and 4) verifying that Nox2 expression and O2·− production in the context of insulin resistance was negated in denuded arteries. Further exploration of 1) the systemic metabolic environment (with particular focus on factors known to alter Nox2 expression, e.g., angiotensin II), 2) arterial function (e.g., acetylcholine-evoked vasorelaxation and receptor/nonreceptor-mediated contraction ± NOS inhibition ± endothelial denudation), 3) the impact of Nox2 inhibition/deletion on the function of other tissues known to express high levels of Nox2 (e.g., pancreatic β-cells) (17), and 4) indices of vascular signal transduction (e.g., in vivo and/or in vitro insulin-mediated arterial eNOS phosphorylation and NO production) in ESMIRO/Nox2y/− and ESMIRO mice could yield interesting findings to provide even greater insight. Future research questions arising from this important contribution to the field include 1) Does Nox2 deletion in endothelial cells influence the expression/function of other Nox isoforms throughout the arterial wall and/or unmask beneficial effects of vasoprotective (?) isoforms such as Nox4 (5,18,19)? 2) Will endothelial cell–specific deletion of Nox2 mitigate the endothelial dysfunction and hypertension that exist in the context of obesity in high-fat diet–fed mice (11,20) and/or atherosclerosis in ApoE mice (15)? 3) Because females have lower Nox expression and activity than males (21), do sex-specific differences concerning Nox2-mediated O2·− generation exist?
The prevalence of diagnosed and undiagnosed cases of type 2 diabetes—a condition characterized by insulin resistance and endothelial dysfunction—is estimated to increase to 33% by 2050 (22). Sukumar et al. have provided novel mechanistic insight into defining a molecular target that should be pursued further in an effort to delay the onset and/or lessen the severity of vascular disease that exists in patients with conditions associated with insulin resistance.
ACKNOWLEDGMENTS
J.D.S. is supported by National Institutes of Health grant 2R15HL091493, American Diabetes Association Research grants 1-12-BS-208 and 7-08-RA-164, and the University of Utah College of Health and School of Medicine.
No potential conflicts of interest relevant to this article were reported.
The author is grateful to Anthony J. Donato (Department of Internal Medicine, University of Utah) and Sihem Boudina (Department of Molecular Medicine, University of Utah) for their expert advice and critical evaluation of the manuscript. The author also thanks Paula Fernandes D’Elia and Matthew Walker (University of Utah) for their help with the figure.
Footnotes
See accompanying brief report, p. 2130.
REFERENCES
- 1.Triggle CR, Ding H. A review of endothelial dysfunction in diabetes: a focus on the contribution of a dysfunctional eNOS. J Am Soc Hypertens 2010;4:102–115 [DOI] [PubMed] [Google Scholar]
- 2.Symons JD, Abel ED. Lipotoxicity contributes to endothelial dysfunction: A focus on the contribution from ceramide. Rev Endocr Metab Disord 2013;14:59–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sukumar P, Viswambharan H, Imrie H, et al. Nox2 NADPH oxidase has a critical role in insulin resistance–related endothelial cell dysfunction. Diabetes 2013;62:2130–2134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bedard K, Krause KH. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol Rev 2007;87:245–313 [DOI] [PubMed] [Google Scholar]
- 5.Drummond GR, Selemidis S, Griendling KK, Sobey CG. Combating oxidative stress in vascular disease: NADPH oxidases as therapeutic targets. Nat Rev Drug Discov 2011;10:453–471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cai H, Harrison DG. Endothelial dysfunction in cardiovascular diseases: the role of oxidant stress. Circ Res 2000;87:840–844 [DOI] [PubMed] [Google Scholar]
- 7.Guzik TJ, Harrison DG. Vascular NADPH oxidases as drug targets for novel antioxidant strategies. Drug Discov Today 2006;11:524–533 [DOI] [PubMed] [Google Scholar]
- 8.Silver AE, Beske SD, Christou DD, et al. Overweight and obese humans demonstrate increased vascular endothelial NAD(P)H oxidase-p47(phox) expression and evidence of endothelial oxidative stress. Circulation 2007;115:627–637 [DOI] [PubMed] [Google Scholar]
- 9.Guzik TJ, Mussa S, Gastaldi D, et al. Mechanisms of increased vascular superoxide production in human diabetes mellitus: role of NAD(P)H oxidase and endothelial nitric oxide synthase. Circulation 2002;105:1656–1662 [DOI] [PubMed] [Google Scholar]
- 10.Duncan ER, Crossey PA, Walker S, et al. Effect of endothelium-specific insulin resistance on endothelial function in vivo. Diabetes 2008;57:3307–3314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Symons JD, McMillin SL, Riehle C, et al. Contribution of insulin and Akt1 signaling to endothelial nitric oxide synthase in the regulation of endothelial function and blood pressure. Circ Res 2009;104:1085–1094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wheatcroft SB, Shah AM, Li JM, et al. Preserved glucoregulation but attenuation of the vascular actions of insulin in mice heterozygous for knockout of the insulin receptor. Diabetes 2004;53:2645–2652 [DOI] [PubMed] [Google Scholar]
- 13.Duncan ER, Walker SJ, Ezzat VA, et al. Accelerated endothelial dysfunction in mild prediabetic insulin resistance: the early role of reactive oxygen species. Am J Physiol Endocrinol Metab 2007;293:E1311–E1319 [DOI] [PubMed] [Google Scholar]
- 14.Kahn MB, Yuldasheva NY, Cubbon RM, et al. Insulin resistance impairs circulating angiogenic progenitor cell function and delays endothelial regeneration. Diabetes 2011;60:1295–1303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rask-Madsen C, Li Q, Freund B, et al. Loss of insulin signaling in vascular endothelial cells accelerates atherosclerosis in apolipoprotein E null mice. Cell Metab 2010;11:379–389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rey FE, Cifuentes ME, Kiarash A, Quinn MT, Pagano PJ. Novel competitive inhibitor of NAD(P)H oxidase assembly attenuates vascular O(2)(-) and systolic blood pressure in mice. Circ Res 2001;89:408–414 [DOI] [PubMed] [Google Scholar]
- 17.Li N, Li B, Brun T, et al. NADPH oxidase NOX2 defines a new antagonistic role for reactive oxygen species and cAMP/PKA in the regulation of insulin secretion. Diabetes 2012;61:2842–2850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gavazzi G, Deffert C, Trocme C, Schäppi M, Herrmann FR, Krause KH. NOX1 deficiency protects from aortic dissection in response to angiotensin II. Hypertension 2007;50:189–196 [DOI] [PubMed] [Google Scholar]
- 19.Ray R, Murdoch CE, Wang M, et al. Endothelial Nox4 NADPH oxidase enhances vasodilatation and reduces blood pressure in vivo. Arterioscler Thromb Vasc Biol 2011;31:1368–1376 [DOI] [PubMed] [Google Scholar]
- 20.Zhang QJ, Holland WL, Wilson L, et al. Ceramide mediates vascular dysfunction in diet-induced obesity by PP2A-mediated dephosphorylation of the eNOS-Akt complex. Diabetes 2012;61:1848–1859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Miller AA, De Silva TM, Jackman KA, Sobey CG. Effect of gender and sex hormones on vascular oxidative stress. Clin Exp Pharmacol Physiol 2007;34:1037–1043 [DOI] [PubMed] [Google Scholar]
- 22.Boyle JP, Thompson TJ, Gregg EW, Barker LE, Williamson DF. Projection of the year 2050 burden of diabetes in the US adult population: dynamic modeling of incidence, mortality, and prediabetes prevalence. Popul Health Metr 2010;8:29. [DOI] [PMC free article] [PubMed] [Google Scholar]