

Abstract
Type 2 diabetes (T2D) is associated with vascular dysfunction. We hypothesized that increased nuclear factor-κB (NF-κB) signaling contributes to vascular dysfunction in T2D. We treated type 2 diabetic (db−/db−) and control (db−/db+) mice with two NF-κB inhibitors (6 mg/kg dehydroxymethylepoxyquinomicin twice a week and 500 μg/kg/day IKK-NBD peptide) for 4 weeks. Pressure-induced myogenic tone was significantly potentiated, while endothelium-dependent relaxation (EDR) was impaired in small coronary arterioles and mesenteric resistance artery from diabetic mice compared with controls. Interestingly, diabetic mice treated with NF-κB inhibitors had significantly reduced myogenic tone potentiation and improved EDR. Importantly, vascular function was also rescued in db−/db−p50NF-κB−/− and db−/db−PARP-1−/− double knockout mice compared with db−/db− mice. Additionally, the acute in vitro downregulation of NF-κB–p65 using p65NF-κB short hairpin RNA lentivirus in arteries from db−/db− mice also improved vascular function. The NF-κB inhibition did not affect blood glucose level or body weight. The RNA levels for Sp-1 and eNOS phosphorylation were decreased, while p65NF-κB phosphorylation, cleaved poly(ADP-ribose) polymerase (PARP)-1, and cyclooxygenase (COX)-2 expression were increased in arteries from diabetic mice, which were restored after NF-κB inhibition and in db−/db−p50NF-κB−/− and db−/db−PARP-1−/− mice. In the current study, we provided evidence that enhanced NF-κB activity impairs vascular function by PARP-1–, Sp-1–, and COX-2–dependent mechanisms in male type 2 diabetic mice. Therefore, NF-κB could be a potential target to overcome diabetes-induced vascular dysfunction.
Diabetes-induced vascular dysfunction is a major clinical problem that is responsible for morbidity and predisposes patients to a variety of cardiovascular diseases (1,2). Vascular endothelial and smooth muscle cell dysfunction are early events in diabetes, characterized by impaired nitric oxide (NO) pathway signaling and potentiation of pressure-induced myogenic tone (3–6). The loss of vascular endothelial NO bioavailability in diabetes results in vasospasm, platelet aggregation, leukocyte adhesion, vascular smooth muscle proliferation, and induction and progression of atherosclerosis (7–9) associated with increases in activity of the proinflammatory transcription factor nuclear factor-κB (NF-κB) (7). The activation of the NF-κB pathway regulates gene expression of cytokines and chemotactic and matrix proteins and induces cell proliferation resulting in the induction and progression of vascular disease (10).
It has been shown that hyperglycemia induces cyclooxygenase (COX)-2 expression through NF-κB pathway (11). This concept is supported by previous studies showing that COX-2 induction is primarily mediated through the activation of the NF-κB pathway (12,13). It has been reported that NF-κB subunits interact with poly(ADP-ribose) polymerase (PARP)-1 in the nucleus and then both bind to DNA to modulate gene expression (14). Recently, we have demonstrated that PARP-1 activity is enhanced in the vasculature in type 2 diabetes and is involved in the impairment of vascular function (15). It also has been shown that NF-κB regulates inflammatory cytokines through the transcription factor Sp-1 (16,17). Thus, the role and mechanism of NF-κB in vascular dysfunction in type 2 diabetes are important questions that remain unanswered. Therefore, in this study we determined whether enhanced NF-κB activity impairs vascular function in type 2 diabetes by PARP-1–,Sp-1–, and COX-2–dependent mechanisms and confirmed that the effect of NF-κB is not specific to one vascular bed by including coronary and mesenteric resistance arteries (MRAs).
RESEARCH DESIGN AND METHODS
All experiments were performed according to the American Guidelines for the Ethical Care of Animals and were approved by Tulane University Health Sciences Center Animal Care and Use Committee. Type 2 diabetic male mice (db−/db−) (8- to 10-week-old males) and their homologous controls were purchased from The Jackson Laboratory (Bar Harbor, ME), housed in groups of five mice, and maintained at a temperature of 23°C with 12-h light/dark cycles. Mice were fed on a solid standard diet (Na+ content 0.4%) and water. Mice were divided into six groups: 1) control mice infused with saline (n = 10), 2) control mice that received dehydroxymethylepoxyquinomicin (DHMEQ) (an NF-κB inhibitor, 6 mg/kg i.p. injection twice a week) for 4 weeks (n = 10), 3) control mice that received IKK-NBD peptide (an NF-κB inhibitor, 500 μg/kg/day daily i.p. injection) for 4 weeks (n = 10), 4) db−/db− mice infused with saline (n = 10), 5) db−/db− mice treated with DHMEQ (6 mg/kg i.p. injection twice per week) for 4 weeks (n = 10), and 6) db−/db− mice treated with IKK-NBD peptide (500 μg/kg/day i.p. daily) for 4 weeks (n = 10). The body weight and blood glucose levels were recorded weekly during the experimental period. Blood glucose measurements were obtained from tail blood samples using a blood glucose meter (Prestige Smart System HDI; Home Diagnostic, Fort Lauderdale, FL) in all groups of mice after a 6-h fast as previously described (18). Systolic blood pressure was measured by the tail-cuff machine as previously described (19).
At the end of the treatment period, mice were anesthetized with isoflurane and blood samples were collected from carotid artery into containing heparin tubes. Then, tissues (heart and MRA) were harvested immediately, placed in physiological salt solution (PSS) solution (composition in millimoles per liter: NaCl 118, KCl 4.7, CaCl2 2.5, KH2PO4 1.2, MgSO4x7H2O 1.2, NaHCO3 25, and glucose 11, pH = 7.4), and processed appropriately for further studies.
In another series of experiments, we used 8-week-old double knockout between db−/db− and p-50NF-κB male mice (db−/db−p50NF-κB−/−, n = 5) and between db−/db− and PARP-1 male mice (db−/db−PARP-1−/−, n = 5). The PARP-1 knockout mice were provided by A.B. The p50NF-κB knockout mice were purchased from The Jackson Laboratory. To generate double knockout, we bred heterozygote db−/db+ with PARP-1 or p50NF-κB knockout mice. Before the animals were killed, the body weight and blood glucose levels were measured. Then mice were anesthetized with isoflurane and then coronary arterioles (CAs), and MRAs were immediately harvested, placed in PSS solution, and processed appropriately for further studies.
To study the metabolic characterization of the mice on drugs and crossed with knockouts, we measured the insulin and cholesterol levels using the Insulin ELISA kit (Mercodia, Uppsala, Sweden) and cholesterol kit (Cayman Chemical Company, Ann Arbor, MI), respectively. We also measured markers of inflammation (interleukin-6 and tumor necrosis factor [TNF]-α [both Bioscience, San Diego, CA).
Vascular reactivity
Ex vivo experiments.
Diameter. The left anterior descending CAs and MRAs were isolated, cannulated with glass micropipettes, and perfused with PSS bubbled with a 95% O2 plus 5% CO2 gas mixture. The vessels were pressurized to 50 mmHg using pressure-servo control perfusion systems (Living Systems Instruments, St. Albans, VT) for a 40-min equilibration period. The vessel diameter was monitored by a video image analyzer as previously described (19,20).
Intraluminal pressure was increased from 25 to 100 mmHg in a stepwise manner to measure myogenic tone. At the end of the experiments, vessels were incubated with a calcium-free PSS to determine passive diameter. Myogenic tone is calculated as the percentage between active and passive diameter. For determination of the endothelium-dependent relaxation (EDR), pressurized arteries were preconstricted with thromboxane agonist (U-46619, 10−7 mol/L) and then cumulative concentrations (10−9 to 10−5 mol/L) of acetylcholine (ACh) were assessed. The NO synthase inhibitor NG-nitro-l-arginine methyl ester (L-NAME) (10−4 mol/L) was added to the superfusate for 30 min before the assessment of the vasodilator response to ACh.
Isometric tension recording. MRAs from control and diabetic mice were carefully cleaned of fat and connective tissue and then cut into rings (2 mm in length). MRAs were mounted in a small-vessel dual-chamber myograph for measurement of isometric tension. After a 30-min equilibration period in PSS solution bubbled with carbogen at 37°C and pH = 7.4, arteries were stretched to their optimal lumen diameter for active tension development. After a second 30-min equilibration period, the vessels were exposed to phenylephrine (PE) (10−5 mol/L) and the presence of functional endothelium was assessed by the ability of ACh (10−6 mol/L) to induce relaxation.
For determination of the role of endothelial NO synthase (eNOS) and COX-2 in the impaired EDR in diabetic mice, MRAs were incubated with NS 398 (10 μmol/L), a COX-2–selective inhibitor, for 1 h and then EDR was performed after precontraction with PE. For determination of the role of eNOS and NADPH oxidase in the impaired EDR in diabetic mice, MRAs were incubated with L-NAME (100 μmol/L) and apocynin (100 μmol/L) for 30 min and then EDR was performed after precontraction with PE.
The same protocol was used for double knockout db−/db− and p50 NF-κB (db−/db−p50NF-κB−/−) and db−/db− and PARP-1 (db−/db−PARP-1−/−) male mice. After precontraction with PE (10−5 mol/L) and the steady maximal contraction, cumulative dose-response curves were obtained for ACh (10−8 to 10−5 mol/L) and sodium nitroprusside (SNP) (10−8 to 10−5 mol/L) in the presence or absence of NS-398, L-NAME, and apocynin.
In vitro experiments.
MRA and CAs from diabetic mice were carefully cleaned of fat and connective tissue and then cut into rings (2 mm in length). Arteries from MRA were mounted in a small-vessel dual-chamber myograph for measurement of isometric tension. Arteries were incubated with either p65NF-κB short hairpin RNA (shRNA) lentiviral particle (Santa Cruz Biotechnology, Santa Cruz, CA), to downregulate p65NF-κB expression, or AG1478 (LC Laboratories, Woburn, MA), epidermal growth factor receptor (EGFR) tyrosine kinase (EGFRtk) inhibitor for 4 h. After precontraction with PE (10−5 mol/L) and the steady maximal contraction, cumulative dose-response curves were obtained for ACh (10−8 to 10−5 mol/L) and SNP (10−8 to 10−5 mol/) in the presence or absence of L-NAME as described above.
Western blot analysis.
Freshly isolated hearts MRAs from all groups were immediately frozen in liquid nitrogen and then homogenized in ice-cold lysis buffer as previously described (18–20). Western blot analysis was performed for phosphorylated and total eNOS (1:1,000 dilution; Cell Signaling, Boston, MA), phosphorylated and total p65NF-κB (1:1,000 dilution; Cell Signaling), cleaved and total PARP-1 (1:1,000 dilution; Cell Signaling), and COX-2 (1:500 dilution, Santa Cruz Biotechnology) using specific antibodies. Blots were stripped and then reprobed with either β-actin (1:2,000 dilution, Santa Cruz Biotechnology) or glyceraldehyde-3-phosphate dehydrogenase (1:2,000 dilution, Santa Cruz Biotechnology) antibodies to verify the equal loading among the samples.
Endothelial cell culture, transient transfection, and luciferase assays.
Mouse coronary arterial endothelial cells (ECs) were purchased from Celprogen (San Pedro, CA), and they were cultured according to the manufacturer instructions using complete growth media. Cultures were incubated at 37°C in a humidified 5% CO2 incubator, and the medium was changed every 2 days. Confluent cell monolayers were harvested by a brief incubation with 0.25% trypsin-EDTA solution (GIBCO; Invitrogen, Grand Island, NY) and subcultured to perform further experiments between passages 3 and 7.
ECs were transiently transfected with lipofectamine LTX and Plus transfection reagents (Invitrogen) in 80% confluent 12-well culture dishes, including 1 µg of the reporter plasmid pGL2-eNOSPromoter luciferase containing 1,621-bp fragment of human eNOS promoter and 0.5 µg of the reporter plasmid pRL-TK (Promega, Fitchburg, WI) in the presence or absence of 1 µg of the expression vector pCMV4-p65. The plasmids pGL2-eNOSPromoter and pCMV4-p65 were purchased from Addgene (cat. nos. 19297 and 21966, respectively). After 48 h of incubation with normal glucose (5 mmol/L) or high glucose (25 mmol/L), cells were lysed in lysis buffer (Promega), and luciferase and renilla activities were measured with the dual Luciferase Assay System (Promega) with a luminometer (Fluoroskan Ascent FL; Laboratory Systems, Kilsyth, Victoria, Australia). The renilla activity was used to normalize the transfection efficiency. In another set of experiments, the ECs were only transfected with 1 µg of the expression vector pCMV4-p65 and incubated with normal or high glucose to isolate RNA for further experiments.
Real-time PCR.
Sp1 and eNOS mRNA levels were determined in heart tissues and ECs. Total RNA from heart was obtained using the RNeasy Fibrous Tissue Mini Kit (Quiagen, Valencia, CA) according to the manufacturer’s recommendations or using the TRI Reagent (Sigma-Aldrich, St. Louis, MO) for cells. A total of 1 μg DNase I–treated RNA was reverse transcribed into cDNA using the High Capacity cDNA Archive kit (Applied Biosystems, Foster City, CA) with random hexamers in a 20-μL reaction. PCR was performed in duplicate for each sample using 1 μL cDNA as a template ×1 of TaqMan Universal PCR Master Mix (Applied Biosystems) and ×10 of Taqman Gene Expression Assays (Applied Biosystems) in a 20-μL reaction. Assays-on-Demand (Applied Biosystems) of TaqMan fluorescent real-time PCR primers and probes were used for Sp1 (Mm00489039_m1), Nos3 (Mm00435217_m1), and glyceraldehyde-3-phosphate dehydrogenase (Mm99999915_g1), which was used as endogenous control to normalize results. Quantitative RT-PCR was carried out in an ABI PRISM 7000 Sequence Detection System (Applied Biosystems) using the following conditions: 2 min at 50°C and 10 min at 95°C followed by 40 cycles of 15 s at 95°C and 1 min at 60°C. Relative mRNA levels were determined using the 2-∆∆Ct method. Results are expressed as the relative expression of mRNA in treated mice or transfected cells compared with untreated mice or control cells.
Drugs.
PI hydrochloride, ACh, L-NAME, apocynin, and NS398 were obtained from Sigma-Aldrich. IKK-NBD peptide was purchased from Enzo Lifesciences (Farmingdale, NY). The DHMEQ was synthesized by K.U. (Aichi Medical University, Nagakute, Japan).
Statistical analysis.
Results are expressed as means ± SEM. Concentration response curves were analyzed using the GraphPad Prism 4.0 software (GraphPad, La Jolla, CA). One-way or two-way ANOVA was used to compare each parameter when appropriate. Comparisons between groups were performed with t tests when the ANOVA test was statistically significant. Values of P < 0.05 were considered significant. Differences between specified groups were analyzed using the Student t test (two tailed) for comparing two groups, with P < 0.05 considered statistically significant.
RESULTS
General parameters.
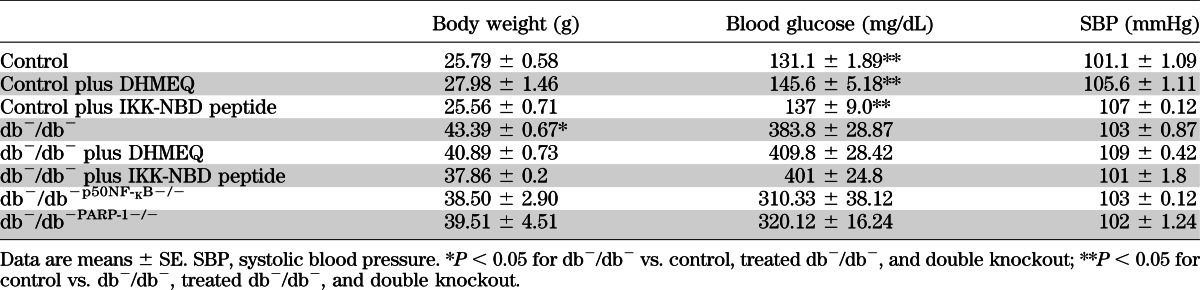
Blood glucose levels and body weight were elevated in diabetic mice (383.1 ± 28.8 mg/dL and 43.3 ± 0.6 g, respectively) compared with control mice (131.1 ± 1.8 mg/dL and 25.7 ± 0.5 g) but were not affected by the NF-κB inhibitors or in double knockout mice (db−/db−p50NF-κB−/− and db−/db−PARP-1−/−), indicating that enhanced NF-κB and PARP-1 activity are involved in type 2 diabetes–induced vascular complication (Table 1). Systolic blood pressure was similar in all groups (Table 1).
TABLE 1.
Blood glucose, body weight, and systolic blood pressure measurements
Insulin levels as well as cholesterol, interleukin-6, and TNF-α were increased in the diabetic mice compared with control mice (Figure Suppl. 1). Treatment with either DHMEQ or peptide had no effect on the control group. Additionally, NF-κB inhibitors reduced interleukin-6 levels and TNF-α in the diabetic group with no effect on insulin and cholesterol. Interestingly, all of these parameters were reduced in double knockout mice (db−/db−p50NF-κB−/− and db−/db−PARP-1−/−) (Supplementary Fig. 1).
NF-κB and coronary and MRA reactivity.
Myogenic tone was significantly increased in the CA and MRA from diabetic mice compared with control mice and was normalized after NF-κB inhibition (Fig. 1A and B). The EDR was also impaired in CA and MRAs from diabetic mice and was rescued after NF-κB inhibition (Fig. 1C and D). To strengthen our data, we examined myogenic tone and the EDR in CAs and MRAs from db−/db− p50NF-κB−/− and db−/db−PARP-1−/− double knockout mice. It was observed that the myogenic tone and EDR in CAs and MRAs were also normalized in these two double knockout mice (Fig. 1E–H).
FIG. 1.
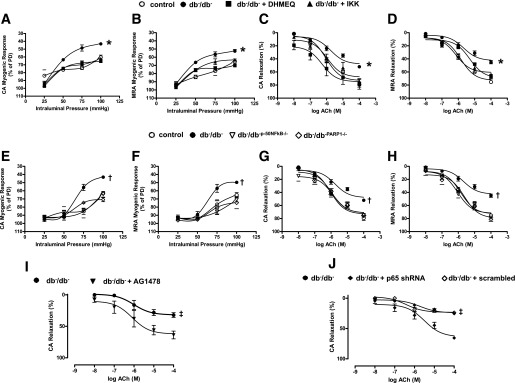
Effect of the NF-kB inhibition on myogenic tone and EDR in coronary and MRAs. Pressure-induced myogenic responses in coronary arteries and MRAs from control and type 2 diabetic (db−/db−) mice treated with or without DHMEQ and IKK-NBD (IKK) peptide (A and B) and db−/db−p50NF-kB−/− and db−/db−PARP-1−/− mice (E and F). *P < 0.05 for db−/db− vs. control, db−/db− treated with DHMEQ or IKK-NBD; †P < 0.05 for db−/db− vs. db−/db−p50NF-kB−/− and db−/db−PARP-1−/− mice. EDR in response to cumulative doses of ACh (10−8 to 10−4 mol/L) in coronary arteries and MRAs from control and db−/db− mice treated with or without DHMEQ and IKK-NBD (C and D) and db−/db−p50NF-kB−/− and db−/db−PARP-1−/− mice (G and H).*P < 0.05 for db−/db− vs. control, db−/db− treated with DHMEQ or IKK-NBD; †P < 0.05 for db−/db− vs. db−/db−p50NF-kB−/− and db−/db−PARP-1−/−. EDR in response to cumulative doses of ACh (10−8 to 10−4 mol/L) in coronary arteries from db−/db− incubated with either AG1478 (EGFRtk inhibitor) (I) or p65NFkB shRNA lentiviral particle (J). ‡P < 0.05 for db−db− vs. db−/db− with AG1478 or db−/db− with p65NF-kB shRNA lentiviral particle. PD, passive diameter.
We did not observe any effect on myogenic tone or EDR in CAs or MRAs from control mice treated with DHMEQ and IKK-NBD peptide (Supplementary Fig. 2A–D). The inhibition of eNOS with L-NAME reduced the EDR in all groups of mice (Supplementary Fig. 2E and F).
Effect of acute EGFRtk inhibition and downregulation of p65NF-κB on CA reactivity.
To determine whether NF-κB is downstream of EGFRtk and that the acute downregulation of NF-κB improves coronary vascular function in diabetes, we incubated CAs from db−/db− mice with the EGFRtk inhibitor (AG1478, 1 μmol/L) for 4 h. Our results show a significant improvement in EDR associated with a reduction in NF-κB activity (Figs. 1I and 2G).
FIG. 2.

Western blot analysis and quantitative data in homogenized CA from control and type 2 diabetic mice (db−/db−) treated with or without DHMEQ (DHM) or IKK-NBD (IKK), showing P-p65 and T-p65 (A), P-eNOS and T-eNOS (B), C-PARP-1 and and T-PARP-1 (C), and glyceraldehyde-3-phosphate dehydrogenase (GAPDH). $P < 0.05 for db−/db− vs. control, control treated with DHMEQ or IKK-NBD, db−/db− treated with DHMEQ or IKK-NBD. #P < 0.05 for db−/db−, db−/db− treated with DHMEQ or IKK-NBD vs. control, control treated with DHMEQ or IKK-NBD, db−/db−, and db−/db−p50NF-kB−/− showing p50 (D), P-eNOS and T-eNOS (E), C-PARP-1 and T-PARP-1 (F), and glyceraldehyde-3-phosphate dehydrogenase. Control, db−/db−, and db−/db−PARP-1−/−, showing P-p-65 and T-p-65 (H), P-eNOS and T-eNOS (I), T-PARP-1 (J), and GAPDH. Control and db−/db− incubated with either AG1478 (EGFRtk inhibitor) or p65NF-kB shRNA lentiviral particles showing P-p65, T-p65, and GAPDH (G and K). P, phosphorylated; T, total; C, active.
Furthermore, in isolated coronary artery from db−/db− mice transfected with p65NF-κB shRNA lentiviral particles for 4 h, there was a significant reduction in p65NF-κB expression and improvement in coronary EDR (Figs. 1J and 2K). As a control experiment, CA from db−/db− mice transfected with scrambled shRNA did not show improved coronary EDR (Fig. 1J).
NF-κB and signaling.
To confirm the effect of NF-κB inhibition, we performed Western blot analysis and found that NF-κB activity was elevated in db−/db− mice compared with db−/db− mice treated with DHMEQ and IKK-NBD peptide and compared with the double knockout db−/db− p50NF-κB−/− and db−/db−PARP-1−/− mice (Fig. 2A, D, and H). The eNOS phosphorylation at Ser 1177 and the expression were significantly reduced in CA from db−/db− mice compared with controls, db−/db− mice treated with DHMEQ and IKK-NBD peptide, and db−/db− p50NF-κB−/− and db−/db−PARP-1−/− mice (Fig. 2B, E, and I).
To study the effect of NF-κB inhibition on PARP-1 signaling, we performed Western blot in CA from all groups. Our data indicate that cleaved PARP-1 levels and total PARP-1 were increased in the diabetic group compared with diabetic mice treated with DHMEQ and IKK-NBD peptide and in double knockout db−/db− p50NF-κB−/− and db−/db−PARP-1−/− mice (Fig. 2C, F, and J).
NF-κB and EDR in MRAs.
Figure 3A shows the key representative curves made in the wire myograph to test the EDR. Our data demonstrated that EDR is impaired in MRAs in db−/db− mice and the inhibition of NF-κB with DHMEQ and IKK-NBD peptide significantly improved the EDR (Fig. 3B and Supplementary Table 1).
FIG. 3.

Effect of the NF-kB inhibition on EDR in MRAs. Key representative traces showing EDR curves to ACh from control and type 2 diabetic mice (db−/db−) treated with or without DHMEQ or IKK-NBD peptide, db−/db−p50NF-kB−/− mice, and db−/db−PARP-1−/−) mice (A). EDR in response to cumulative doses of ACh (10−8 to 10−5 mol/L) in MRAs precontracted with PE (10−5 mol/L) from control and db−/db− treated with or without DHMEQ or IKK-NBD peptide (B) and incubated with or without apocynin (Apo) (NADPH oxidase inhibitor) (C) or NS 398 (COX-2 inhibitor) (D). *P < 0.05 for db−/db− vs. control, db−/db− treated with DHMEQ or IKK-NBD. &P < 0.05 for db−/db− treated with DHMEQ or IKK-NBD vs. control. EDR in MRA from control, db−/db−, db−/db−p50NF-kB−/−, and db-/db-PARP-1−/− (E) and incubated with COX-2 inhibitor, NS 398 (F), and db−/db− incubated with either AG1478 (EGFRtk inhibitor) (G) or P65NF-kB shRNA lentiviral particles (H). *P < 0.05 for db−/db− vs. control, db−/db−p50NF-kB−/−, or db−/db−PARP-1−/−. #P < 0.05 for db−/db−PARP-1−/− vs. control, db−/db−p50NF-kB−/−. $P < 0.05 for db−/db− vs. db−/db− plus AG1478. @P < 0.05 for db−/db−, db−/db− plus scrambled vs. db−/db− plus p65 shRNA.
We did not observe a change in EDR in MRA from control mice treated with DHMEQ and IKK-NBD peptide (Supplementary Fig. 3A). The inhibition of eNOS with L-NAME reduced EDR in all groups of mice (Supplementary Fig. 3B). To further elucidate the mechanism of vascular endothelial dysfunction in diabetic mice related to NF-κB, we studied the role of the COX-2 and oxidative stress pathways. Thus, the inhibition of NADPH oxidases by apocynin showed a significant improvement in MRA (Fig. 3C and Supplementary Table 1). In addition, the COX-2 inhibition by NS398 significantly improved the EDR in MRA (Fig. 3D and Supplementary Table 1). Western blot analysis revealed that in MRA from db−/db− mice, eNOS activity was significantly reduced, while NF-κB and PARP-1 activity and COX-2 expression were significantly augmented (Fig. 4A–D). Interestingly, the treatment of db−/db− mice with NF-κB inhibitors (DHMEQ and IKK-NBD peptide) increased eNOS activity and reduced NF-κB, PARP-1 activity, and COX-2 expression (Fig. 4A–D).
FIG. 4.

Western blot analysis and quantitative data in homogenized MRAs from control and type 2 diabetic mice (db−/db−) treated with or without DHMEQ (DHM) or IKK-NBD (IKK) showing P-eNOS and T-eNOS (A) P-p65, T-p65 (B), c-PARP-1 and T-PARP-1 (C), COX-2 (D), and β-actin. $P < 0.05 for db−/db− vs. control, control treated with DHMEQ or IKK-NBD, db−/db− treated with DHMEQ or IKK-NBD. #P < 0.05 for db−/db−, db−/db− treated with DHMEQ or IKK-NBD vs. control, control treated with DHMEQ or IKK-NBD. Western blot analysis in MRA from control, db−/db−, and db−/db−p50NFkB−/−) showing P-eNOS, T-eNOS (E), p-50 (G), c-PARP-1 and T-PARP-1 (I and J), COX-2 (K and L), and β-actin and db−/db−PARP-1−/− showing P-eNOS, T-eNOS (F), P-p65 and T-p65 (H), T-PARP-1 (J), COX-2 (L), and β-actin. Western blot analysis in MRA from db−/db− incubated with either AG1478 (EGFRtk inhibitor) or p65NF-kB shRNA lentiviral particles, showing P-p65, T-p65 (M and N), and β-actin. P, phosphorylated; T, total; C, active.
To strengthen our studies, we examined the EDR in MRA in double knockout mice (db−/db− p50NF-κB−/−). Western blot analysis confirmed the absence of p50NF-κB in MRA in db−/db− p50NF-κB−/− mice (Fig. 4G). Furthermore, the EDR was significantly improved in MRA isolated from db−/db− p50NF-κB−/− mice associated with increased eNOS activity (Figs. 3E and 4E and Supplementary Table 1). To further determine the mechanism of NF-κB in vascular function, we studied the role of PARP-1 and COX-2 as downstream signaling for NF-κB. Thus, EDR was significantly improved in double knockout db−/db−PARP-1−/− mice and arteries from db−/db− mice treated with COX-2 inhibitor (Fig. 3E and F and Table 1). These data were also associated with reduction in cleaved PARP-1 and COX-2 expression in MRA from db−/db− p50NF-κB−/− mice (Fig. 4I and K). Furthermore, Western blot analysis revealed that in MRA from db−/d− PARP-1−/− mice, PARP-1 was absent and this was associated with reduced p65NF-κB phosphorylation or COX-2 expression or with an increase in eNOS phosphorylation (Fig. 4F, H, J, and L). Endothelium-independent relaxation in response to SNP in MRA was similar in control, db−/db−, and db−/db− p50NF-κB−/− mice (Supplementary Fig. 4A).
Effect of acute EGFRtk inhibition and downregulation of p65NF-κB on MRA reactivity.
Isolated MRAs from db−/db− mice treated with EGFRtk inhibitor significantly improved the EDR (Fig. 3G and H and Supplementary Table 1). Western blot analysis confirmed the reduction in p65NF-κB phosphorylation in MRA (Fig. 4M) indicating that p65NF-κB is downstream of EGFRtk. The acute downregulation of p65NF-κB in isolated MRAs from db−/db− mice transfected with p65NF-κB shRNA lentiviral particles significantly improved the EDR, which is inhibited by L-NAME (Fig. 3H and Supplementary Table 1). Western blot analysis confirmed the downregulation of p65NF-κB in MRA transfected with p65NF-κB shRNA lentiviral particles (Fig. 4N). Endothelium-independent relaxation in response to SNP in MRA showed no difference between controls, db−/db−, and db−/db− transfected with lentivirus shRNA p65NF-κB (Supplementary Fig. 4B).
Effect of high glucose and overexpression of p65NF-κB on eNOS promoter.
To elucidate the mechanism by which eNOS expression and activity were decreased in diabetes, we studied the Sp-1 expression known as downstream signaling of NF-κB. Thus, the mRNA levels of Sp-1 were significantly reduced in coronary artery isolated from db−/db− mice compared with controls (Fig. 5A). Interestingly, db−/db− mice treated with NF-κB inhibitor significantly increased Sp-1 expression in CAs (Fig. 5A). We did not see any change in Sp-1 expression in control mice treated with NF-κB inhibitor (Fig. 5A). In primary ECs isolated from CAs, transfected with a reporter plasmid containing the eNOS promoter, the stimulation of these cells with high glucose and/or overexpression of p65NF-κB significantly reduced Sp-1 expression (Fig. 5B). In addition, eNOS promoter activity was significantly reduced in ECs stimulated with high glucose and/or transfected with p65NF-κB (Fig. 5C). These results were associated with the reduction in eNOS expression (Fig. 5D).
FIG. 5.
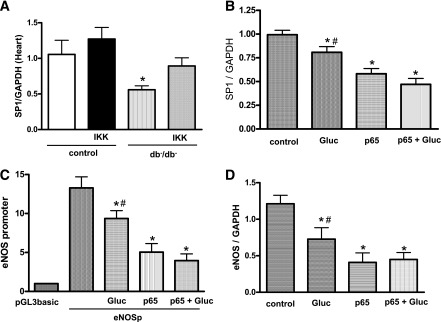
The regulation of eNOS activity. Sp-1 mRNA levels in CAs isolated from control and type 2 diabetic mice (db−/db−) untreated or treated with IKK-NBD (IKK). *P < 0.05 for db−/db− vs. control, control treated with IKK-NBD, db−/db− treated with IKK-NBD (A); Sp-1 and eNOS mRNA levels in ECs from CAs stimulated with high glucose or overexpressing P65NF-kB. *P < 0.05 for high glucose, p65NF-kB or high glucose plus p65NF-kB vs. control, #P < 0.05 for high glucose vs. p65-NF-kB or high glucose plus p65NF-kB (B and D); eNOS promoter activity in transfected primary ECs from CAs stimulated with high glucose or overexpressed with P65NF-kB. *P < 0.05 for high glucose, p65NF-kB, or high glucose plus p65NF-kB vs. control; #P < 0.05 for high glucose vs. p65NF-kB or high glucose plus p65NF-kB (C). P-GL3basic indicates the cells transfected with the reporter plasmid alone without containing the eNOS promoter driving luciferase gene expression, and it is used as control of luciferase basal activity.
DISCUSSION
Our study demonstrates that enhanced NF-κB activity is an important factor involved in macro- and microvascular dysfunction in type 2 diabetes. This evidence came from our ex vivo and in vitro experiments where vascular EDR and myogenic tone were significantly improved in type 2 diabetic mice treated with NF-κB inhibitors and double knockout mice (db−/db−p50NF-κB−/− and db−/db−PARP-1) and with the downregulation of NF-κB using shRNA lentiviral particles. We further demonstrated that in diabetes, NF-κB inhibition improves vascular function by PARP-1–, Sp-1–, and COX-2–dependent mechanisms. These results suggest that the NF-κB pathway could be an important target for a therapeutic approach to reverse diabetes-induced vascular dysfunction.
In diabetes, vascular complications are major causes for morbidity and mortality (21–24). Several mechanisms have been proposed such as hyperglycemia (25), altered COX-2 activity (26), oxidative stress (27), insulin resistance, and inflammation (28). Although previous studies established the relationship between NF-κB, diabetes, and cardiovascular diseases (29,30), the role and mechanism of NF-κB in diabetes-induced vascular dysfunction are still unknown.
Myogenic tone is a characteristic of resistance arteries, which plays an important role in the local regulation of vascular diameter change and tissue blood flow (31). Previous studies have investigated impaired small artery and arteriolar function in diabetic patients and have shown that vasomotor dysfunction affects both endothelial and smooth muscle cell–mediated regulatory mechanisms (32,33). In type 2 diabetic mice, we and other investigators have demonstrated an increase in myogenic tone (15,34,35). Interestingly, in the current study we observed that the augmented myogenic tone in type 2 diabetes is the result of enhanced NF-κB and PARP-1 activity. Thus, NF-κB and PARP-1 pathway inhibition, using pharmacologic and genetic approaches, reduced the potentiated myogenic tone in diabetic mice.
Vascular reactivity is also regulated by EDR mechanism. This mechanism is highly dependent not only on the bioavailability of NO in large arteries but also on the endothelium-dependent hyperpolarizing factor and prostaglandin metabolites in resistance arteries (36). Our results indicate that EDR is impaired in CA and MRA in type 2 diabetes, which is in agreement with previous studies (31,37,38). It has been shown that loss of endothelium-derived NO is associated with increases in NF-κB activity (39). In addition, it has been reported that cultured porcine coronary EC function is impaired with senescence, associated with enhanced NF-κB activity (40). In this study, we demonstrated that type 2 diabetes is associated with increased vascular NF-κB activity and impaired vascular function. Thus, the inhibition of NF-κB activity with different approaches (pharmacologic, double-knockout mouse models and downregulation of protein expression by NF-κB shRNA lentiviral particles) significantly improved EDR in CA and MRA. Importantly, the effect of NF-κB activity inhibition is not specific to one vascular bed indicating the critical importance of the NF-κB pathway in the global regulation of vasculature function in type 2 diabetes. It is also interesting to note that NF-κB inhibition did not reduce blood glucose and insulin levels or body weight, suggesting that enhanced NF-κB is a consequence of type 2 diabetes.
It is well-known that active NF-κB interacts with PARP-1 and translocates to the nucleus to target promoters (41). In addition, we previously reported that inhibition of PARP-1 activity improved vascular function in type 2 diabetes (15). In the current study, we found an increase in PARP-1 activity in arteries from type 2 diabetic mice. The inhibition of PARP-1 activity in db−/db− mice, using genetic deletion, significantly improved vascular function. In addition, the inhibition of the NF-κB pathway reduced PARP-1 activity, indicating that NF-κB regulates vascular function in type 2 diabetes by a PARP-1–dependent mechanism. These results are supported by studies in the literature demonstrating the formation of a complex NF-κB–PARP-1 in the nucleus, which binds to DNA to modulate gene expression (38,42). This activity of this chemical complex is also supported by our experiments showing an improvement in vascular function in double knockout db−/db−PARP-1−/− mice.
To further investigate the downstream mechanism of NF-κB and PARP-1 and because of the importance of the transcription factor Sp-1 in the regulation of eNOS promoter activity, we studied Sp-1 expression. Interestingly, we found that Sp-1 mRNA levels were significantly reduced and were rescued by the inhibition of NF-κB in CAs from type 2 diabetic mice. In addition, in vitro studies show that overexpression of p65NF-κB in transfected primary cultured coronary ECs or incubation with high glucose reduced eNOS promoter activity and eNOS and Sp-1 mRNA levels. Our results are in agreement with studies indicating that the transcription factor Sp-1 binds to the eNOS promoter (43). In addition, it has recently been demonstrated that p65NF-κB subunit interacts with Sp-1 and negatively regulates gene expression (44). Therefore, our results suggest that enhanced NF-κB signaling impairs vascular function by PARP-1– and Sp-1–dependent mechanisms.
To provide information about the mechanism of impaired vascular function caused by enhanced NF-κB activity in diabetes, we observed that vascular COX-2 expression was enhanced in db−/db− mice. These data are consistent with studies showing the induction of COX-2 in type 2 diabetes (45,46).
The NF-κB is a ubiquitous family of transcription factors that also control the expression of genes involved in the inflammatory response, such as COX-2. Our studies showed that inhibition of NF-κB activity in db−/db− mice reduced COX-2 expression indicating that COX-2 is downstream to NF-κB. In addition, the in vitro inhibition of COX-2 improves vascular function in db−/db− mice. These data indicate that inhibition of NF-κB improved vascular function by a COX-2–dependent mechanism.
Although a previous population-based study showed that COX-2 inhibitors might lead to increase the risk of myocardial infarction in the general population (47), it has been shown that a post hoc analysis for different coxibs revealed a significant association with incident atrial fibrillation for etoricoxib but not for celecoxib. These results indicate that the issue is mostly related to the nature of COX-2 inhibitors (48).
In summary, our study demonstrates the importance of NF-κB in the regulation of vascular function in type 2 diabetes by PARP-1–, Sp-1–, and COX-2–dependent mechanisms (Supplementary Fig. 5). Therefore, NF-κB could be a potential target for a novel therapeutic strategy to reverse diabetes-induced vascular dysfunction.
Novelty and significance.
Although there are studies involving NF-κB in diabetes, the proposed study is innovative, in our opinion, because our approach provided direct evidence that enhanced NF-κB activity causes vascular dysfunction in terms of myogenic tone and EDR by PARP-1–, Sp-1– and COX-2–dependent mechanisms in two different vascular beds. There is a paucity of studies that explore the role of the NF-κB pathway and the mechanism (PARP-1, Sp-1, and COX-2) by which type 2 diabetes impairs the regulation of arteries, which leads to heart disease.
Emerging evidence from experimental and clinical research indicates that an NF-κB pathway plays pivotal roles in cardiovascular diseases. Lack of such knowledge is a fundamental problem because without it, endothelial dysfunction that causes coronary artery disease will still present a high risk for myocardial infarction in diabetic patients. Thus, our ex vivo and in vitro data clearly indicate an enhanced NF-κB pathway associated with impaired EDR in three vascular beds. Importantly, the inhibition of NF-κB activity improves vascular EDR. Thus, in our study we demonstrated the role of enhanced NF-κB activity in the dysfunction of vascular EDR by PARP-1–, Sp-1–, and COX-2–dependent mechanism.
Supplementary Material
ACKNOWLEDGMENTS
The authors acknowledge grant support from the National Institutes of Health (1R01HL095566 [principal investigator K.M.] and 5R01HL097111 [principal investigator M.T.]).
No potential conflicts of interest relevant to this article were reported.
M.K., S.-K.C., and M.G. conducted experiments and wrote the manuscript. A.B. generated PARP-1 knockout mice. K.U. generated and provided DHMEQ. M.T. and S.B. participated in discussion and wrote the manuscript. K.M. was the principal investigator, designed the research, and wrote the manuscript. K.M. is the guarantor of this work and, as such, had full access to all the data in the study and takes full responsibility for the integrity of the data and the accuracy of the data analysis.
Footnotes
This article contains Supplementary Data online at http://diabetes.diabetesjournals.org/lookup/suppl/doi:10.2337/db12-1374/-/DC1.
REFERENCES
- 1.Ruderman NB, Williamson JR, Brownlee M. Glucose and diabetic vascular disease. FASEB J 1992;6:2905–2914 [DOI] [PubMed] [Google Scholar]
- 2.Cosentino F, Lüscher TF. Endothelial dysfunction in diabetes mellitus. J Cardiovasc Pharmacol 1998;32(Suppl. 3):S54–S61 [PubMed] [Google Scholar]
- 3.Cai S, Khoo J, Mussa S, Alp NJ, Channon KM. Endothelial nitric oxide synthase dysfunction in diabetic mice: importance of tetrahydrobiopterin in eNOS dimerisation. Diabetologia 2005;48:1933–1940 [DOI] [PubMed] [Google Scholar]
- 4.Hadi HA, Suwaidi JA. Endothelial dysfunction in diabetes mellitus. Vasc Health Risk Manag 2007;3:853–876 [PMC free article] [PubMed] [Google Scholar]
- 5.Ding H, Triggle CR. Endothelial cell dysfunction and the vascular complications associated with type 2 diabetes: assessing the health of the endothelium. Vasc Health Risk Manag 2005;1:55–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yang G, Lucas R, Caldwell R, Yao L, Romero MJ, Caldwell RW. Novel mechanisms of endothelial dysfunction in diabetes. J Cardiovasc Dis Res 2010;1:59–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Creager MA, Lüscher TF, Cosentino F, Beckman JA. Diabetes and vascular disease: pathophysiology, clinical consequences, and medical therapy: Part I. Circulation 2003;108:1527–1532 [DOI] [PubMed] [Google Scholar]
- 8.Harrison DG. Cellular and molecular mechanisms of endothelial cell dysfunction. J Clin Invest 1997;100:2153–2157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yang Z, Ming XF. Recent advances in understanding endothelial dysfunction in atherosclerosis. Clin Med Res 2006;4:53–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Guijarro C, Egido J. Transcription factor-kappa B (NF-kappa B) and renal disease. Kidney Int 2001;59:415–424 [DOI] [PubMed] [Google Scholar]
- 11.Shanmugam N, Gaw Gonzalo IT, Natarajan R. Molecular mechanisms of high glucose-induced cyclooxygenase-2 expression in monocytes. Diabetes 2004;53:795–802 [DOI] [PubMed] [Google Scholar]
- 12.Crofford LJ, Tan B, McCarthy CJ, Hla T. Involvement of nuclear factor kappa B in the regulation of cyclooxygenase-2 expression by interleukin-1 in rheumatoid synoviocytes. Arthritis Rheum 1997;40:226–236 [DOI] [PubMed] [Google Scholar]
- 13.Charalambous MP, Lightfoot T, Speirs V, Horgan K, Gooderham NJ. Expression of COX-2, NF-kappaB-p65, NF-kappaB-p50 and IKKalpha in malignant and adjacent normal human colorectal tissue. Br J Cancer 2009;101:106–115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hassa PO, Haenni SS, Buerki C, et al. Acetylation of poly(ADP-ribose) polymerase-1 by p300/CREB-binding protein regulates coactivation of NF-kappaB-dependent transcription. J Biol Chem 2005;280:40450–40464 [DOI] [PubMed] [Google Scholar]
- 15.Choi SK, Galán M, Kassan M, Partyka M, Trebak M, Matrougui K. Poly(ADP-ribose) polymerase 1 inhibition improves coronary arteriole function in type 2 diabetes mellitus. Hypertension 2012;59:1060–1068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Oliver FJ, Ménissier-de Murcia J, Nacci C, et al. Resistance to endotoxic shock as a consequence of defective NF-kappaB activation in poly (ADP-ribose) polymerase-1 deficient mice. EMBO J 1999;18:4446–4454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hassa PO, Hottiger MO. A role of poly (ADP-ribose) polymerase in NF-kappaB transcriptional activation. Biol Chem 1999;380:953–959 [DOI] [PubMed] [Google Scholar]
- 18.Choi SK, Galán M, Partyka M, Trebak M, Belmadani S, Matrougui K. Chronic inhibition of epidermal growth factor receptor tyrosine kinase and extracellular signal-regulated kinases 1 and 2 (ERK1/2) augments vascular response to limb ischemia in type 2 diabetic mice. Am J Pathol 2012;180:410–418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kassan M, Galan M, Partyka M, Trebak M, Matrougui K. Interleukin-10 released by CD4(+)CD25(+) natural regulatory T cells improves microvascular endothelial function through inhibition of NADPH oxidase activity in hypertensive mice. Arterioscler Thromb Vasc Biol 2011;31:2534–2542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Amin AH, Abd Elmageed ZY, Partyka M, Matrougui K. Mechanisms of myogenic tone of coronary arteriole: Role of down stream signaling of the EGFR tyrosine kinase. Microvasc Res 2011;81:135–142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schalkwijk CG, Stehouwer CD. Vascular complications in diabetes mellitus: the role of endothelial dysfunction. Clin Sci (Lond) 2005;109:143–159 [DOI] [PubMed] [Google Scholar]
- 22.Maser RE, Mitchell BD, Vinik AI, Freeman R. The association between cardiovascular autonomic neuropathy and mortality in individuals with diabetes: a meta-analysis. Diabetes Care 2003;26:1895–1901 [DOI] [PubMed] [Google Scholar]
- 23.Laing SP, Swerdlow AJ, Slater SD, et al. Mortality from heart disease in a cohort of 23,000 patients with insulin-treated diabetes. Diabetologia 2003;46:760–765 [DOI] [PubMed] [Google Scholar]
- 24.Haffner SM, Lehto S, Rönnemaa T, Pyörälä K, Laakso M. Mortality from coronary heart disease in subjects with type 2 diabetes and in nondiabetic subjects with and without prior myocardial infarction. N Engl J Med 1998;339:229–234 [DOI] [PubMed] [Google Scholar]
- 25.Gutterman DD. Vascular dysfunction in hyperglycemia: is protein kinase C the culprit? Circ Res 2002;90:5–7 [PubMed] [Google Scholar]
- 26.Sánchez A, Contreras C, Martínez P, et al. Enhanced cyclooxygenase 2-mediated vasorelaxation in coronary arteries from insulin-resistant obese Zucker rats. Atherosclerosis 2010;213:392–399 [DOI] [PubMed] [Google Scholar]
- 27.Schuhmacher S, Oelze M, Bollmann F, et al. Vascular dysfunction in experimental diabetes is improved by pentaerithrityl tetranitrate but not isosorbide-5-mononitrate therapy. Diabetes 2011;60:2608–2616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Natali A, Toschi E, Baldeweg S, et al. Clustering of insulin resistance with vascular dysfunction and low-grade inflammation in type 2 diabetes. Diabetes 2006;55:1133–1140 [DOI] [PubMed] [Google Scholar]
- 29.Lorenzo O, Picatoste B, Ares-Carrasco S, et al. Potential role of nuclear factor κB in diabetic cardiomyopathy. Mediators Inflamm 2011;2011:652097. [DOI] [PMC free article] [PubMed]
- 30.Rodríguez-Ayala E, Anderstam B, Suliman ME, et al. Enhanced RAGE-mediated NFkappaB stimulation in inflamed hemodialysis patients. Atherosclerosis 2005;180:333–340 [DOI] [PubMed] [Google Scholar]
- 31.Scotland RS, Chauhan S, Vallance PJ, Ahluwalia A. An endothelium-derived hyperpolarizing factor-like factor moderates myogenic constriction of mesenteric resistance arteries in the absence of endothelial nitric oxide synthase-derived nitric oxide. Hypertension 2001;38:833–839 [DOI] [PubMed] [Google Scholar]
- 32.Bagi Z. Mechanisms of coronary microvascular adaptation to obesity. Am J Physiol Regul Integr Comp Physiol 2009;297:R556–R567 [DOI] [PubMed] [Google Scholar]
- 33.Picchi A, Capobianco S, Qiu T, et al. Coronary microvascular dysfunction in diabetes mellitus: A review. World J Cardiol 2010;2:377–390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Belmadani S, Palen DI, Gonzalez-Villalobos RA, Boulares HA, Matrougui K. Elevated epidermal growth factor receptor phosphorylation induces resistance artery dysfunction in diabetic db/db mice. Diabetes 2008;57:1629–1637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lagaud GJ, Masih-Khan E, Kai S, van Breemen C, Dubé GP. Influence of type II diabetes on arterial tone and endothelial function in murine mesenteric resistance arteries. J Vasc Res 2001;38:578–589 [DOI] [PubMed] [Google Scholar]
- 36.Moncada S, Higgs A. The L-arginine-nitric oxide pathway. N Engl J Med 1993;329:2002–2012 [DOI] [PubMed] [Google Scholar]
- 37.Palen DI, Matrougui K. Role of elevated EGFR phosphorylation in the induction of structural remodelling and altered mechanical properties of resistance artery from type 2 diabetic mice. Diabetes Metab Res Rev 2008;24:651–656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Su J, Lucchesi PA, Gonzalez-Villalobos RA, et al. Role of advanced glycation end products with oxidative stress in resistance artery dysfunction in type 2 diabetic mice. Arterioscler Thromb Vasc Biol 2008;28:1432–1438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zeiher AM, Fisslthaler B, Schray-Utz B, Busse R. Nitric oxide modulates the expression of monocyte chemoattractant protein 1 in cultured human endothelial cells. Circ Res 1995;76:980–986 [DOI] [PubMed] [Google Scholar]
- 40.Lee MY, Wang Y, Vanhoutte PM. Senescence of cultured porcine coronary arterial endothelial cells is associated with accelerated oxidative stress and activation of NFkB. J Vasc Res 2010;47:287–298 [DOI] [PubMed] [Google Scholar]
- 41.Zerfaoui M, Errami Y, Naura AS, et al. Poly(ADP-ribose) polymerase-1 is a determining factor in Crm1-mediated nuclear export and retention of p65 NF-kappa B upon TLR4 stimulation. J Immunol 2010;185:1894–1902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ménissier de Murcia J, Ricoul M, Tartier L, et al. Functional interaction between PARP-1 and PARP-2 in chromosome stability and embryonic development in mouse. EMBO J 2003;22:2255–2263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kumar S, Sun X, Wiseman DA, et al. Hydrogen peroxide decreases endothelial nitric oxide synthase promoter activity through the inhibition of Sp1 activity. DNA Cell Biol 2009;28:119–129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Benjamin JT, Carver BJ, Plosa EJ, et al. NF-kappaB activation limits airway branching through inhibition of Sp1-mediated fibroblast growth factor-10 expression. J Immunol 2010;185:4896–4903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bagi Z, Erdei N, Toth A, et al. Type 2 diabetic mice have increased arteriolar tone and blood pressure: enhanced release of COX-2-derived constrictor prostaglandins. Arterioscler Thromb Vasc Biol 2005;25:1610–1616 [DOI] [PubMed] [Google Scholar]
- 46.Zhang J, Lei T, Chen X, et al. Resistin up-regulates COX-2 expression via TAK1-IKK-NF-kappaB signaling pathway. Inflammation 2010;33:25–33 [DOI] [PubMed] [Google Scholar]
- 47.Hippisley-Cox J, Coupland C. Risk of myocardial infarction in patients taking cyclo-oxygenase-2 inhibitors or conventional non-steroidal anti-inflammatory drugs: population based nested case-control analysis. BMJ 2005;330:1366–1372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bäck M, Yin L, Ingelsson E. Cyclooxygenase-2 inhibitors and cardiovascular risk in a nation-wide cohort study after the withdrawal of rofecoxib. Eur Heart J 2012;33:1928–1933 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.