

Abstract
Autophagy is a critical cellular housekeeping process that is essential for removal of damaged or unwanted organelles and protein aggregates. Under conditions of starvation, it is also a mechanism to break down proteins to generate amino acids for synthesis of new and more urgently needed proteins. In the heart, autophagy is upregulated by starvation, reactive oxygen species, hypoxia, exercise, and ischemic preconditioning, the latter a well-known potent cardioprotective phenomenon. The observation that upregulation of autophagy confers protection against ischemia/reperfusion injury and inhibition of autophagy is associated with a loss of cardioprotection conferred by pharmacological conditioning suggests that the pathway plays a key role in enhancing the heart’s tolerance to ischemia. While many of the antecedent signaling pathways of preconditioning are well-defined, the mechanisms by which preconditioning and autophagy converge to protect the heart are unknown. In this review we discuss mechanisms that potentially underlie the linkage between cardioprotection and autophagy in the heart.
Keywords: Autophagy, Mitochondria, Homeostasis, Lysosomes, Preconditioning, Cardioprotection
Signaling to regulate autophagy
Macroautophagy (autophagy) is a downstream effector pathway that is essential for clearing cellular damage and the long-term homeostasis of cells and tissues [12, 13, 35, 58, 71, 89]. Information obtained using multiple model systems shows that the autophagy/lysosomal pathway responds to a variety of environmental and internal stimuli to mediate the bulk clearance of extraneous or damaged cellular components. The observation that starvation or caloric restriction rapidly induces autophagy in many tissues and is in part regulated by the insulin pathway (via TOR kinase) suggests that suppression of autophagy, due to excessive caloric intake, may significantly contribute to deleterious health effects associated with obesity, metabolic syndrome, type-II diabetes or cellular processes linked to aging [18, 22, 26, 94].
The link between metabolic syndrome and pathological metabolic profiles has brought into focus this condition as a risk factor for poor cardiac outcome, both in terms of cardiovascular disease and progressive heart failure. Our recent work has demonstrated that murine myocytes mount a robust autophagic response, which is protective during periods of acute stress such as simulated ischemia and reperfusion [33, 35, 40]. Fasting is well known to induce autophagy in cardiac myocytes [40, 59, 68]. There is growing evidence that aerobic exercise may mediate its cardioprotective effects by upregulating autophagy [17, 29, 45, 54, 56, 77, 85]. These findings imply that the cardioprotective effects afforded by a robust autophagic response during an acute episode of ischemia may be suppressed in people that have metabolic syndrome, Type II diabetes, or sedentary lifestyles. Interestingly, amino acids (particularly Leu, Tyr, and Gln) and insulin suppress autophagy through independent pathways [43] and Elazar’s group recently showed autophagy is potently induced by removal of a single amino acid if it is arginine, lysine, leucine, or methionine [87]. Further work will be required to establish whether high-protein diets or dietary amino acid supplements can suppress autophagy in mammalian cardiomyocytes.
In recent studies of autophagy gene expression patterns in Drosophila, message levels of the Atg2, Atg8a, Atg18 and bchs genes decline with age while substrates normally cleared by the pathway, like insoluble ubiquitinated proteins, increase dramatically [89]. Changes in ubiquitinated protein profiles are similar to those detected when key autophagy genes from both Drosophila and mouse models are genetically altered, a finding which underscores the highly conserved nature of the pathway [27, 36, 50]. Recently an additional link between autophagy and metabolic disorders has arisen from work done on the autophagic protein, Sequestosome-1 [p62 or Ref(2)P in Drosophila] [39, 69]. The p62 protein is required to specify and facilitate the clearance of protein inclusions or cellular aggregates by autophagy. Interestingly, genetic deletion of murine p62 results in viable adult mice that develop insulin resistance and obesity as early as 4 months of age [83].
Machinery of autophagy
Autophagy is a remarkably flexible and dynamic cellular process that involves the de novo synthesis of cytoplasmic organelles, which are transported to and fuse with lysosomes, where their contents are degraded and recycled. Figure 1 is a schematic representation of the pathway and illustrates the sequestration of individual cargos located in the cytoplasm (i.e. organelles, ubiquitinated protein inclusions) by a growing isolation membrane or phagophore. Early molecular events required for vesicle formation involve the coordinated response of two ubiquitin-like pathways that involve the conjugation of the Atg12 and Atg8 proteins and are illustrated in Fig. 2. In the case of the Atg12 protein, it first undergoes conjugation to the E1-like ligase Atg7 via its C-terminal glycine. Atg12 is then “transferred” to Atg10 (E2-like protein) followed by a final ligation with the Atg5 protein. The Atg12–Atg5 conjugate then goes on to form a tight non-covalent association with Atg16 protein, producing a trimeric complex. Upon formation of a high molecular weight Atg12/Atg5/Atg16 multimer (350KD), the complex localizes to the outer surface of a growing cup-shaped phagophore.
Fig. 1.
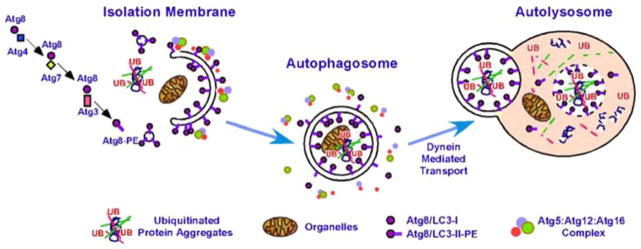
Key cellular components required for autophagosome formation, transport, and fusion. Adaptor proteins recognize targets for engulfment (Bchs/Alfy recognizes ubiquitin-rich protein aggregates) in conjunction with p62/Sequestosome1/Ref(2)P. The forming autophagosome (phagophore) is initially decorated with Atg5:Atg12 conjugates complexed with Atg16, followed by Atg8/LC3, which is covalently linked to phosphatidylethanolamine (PE). Once the autophagosome seals around its target, externally exposed Atg proteins dissociate (internal LC3 is retained) and the vesicle is transported along microtubules to fuse with a lysosome. The vacuolar proton ATPase acidifies the autophagosome before fusion with a lysosome (also acidified). Upon fusion, the contents of the autophagolysosome are degraded, including LC3
Fig. 2.

Autophagy requires the activity of two-ubiquitin-like pathways. Atg8 and Atg12 both resemble ubiquitin. Atg4 is a cysteine protease that removes the terminal cysteine of Atg8 (LC3) to expose the glycine that is used for conjugation via Atg7 (E1-like) and Atg3 (E2-like) enzymes to the acceptor phosphatidylethanolamine. Atg12 is conjugated to Lys130 in Atg5 via Atg7 and Atg10 (E2-like). Following conjugation of Atg12–Atg5, a heteromeric complex forms with Atg16 that binds to the convex face of the phagophore
At the same time, a second ubiquitin-like molecule, Atg8, also known as microtubule-associated light chain 3 (LC3), undergoes C-terminal cleavage (via the Atg4 cysteine protease) to expose a reactive glycine. Atg8/LC3 then undergoes conjugation to Atg7 (E1-like) before being passed on to the Atg3 protein (E2-like enzyme via a thio-ester bond). From there, Atg8/LC3 forms a C-terminal covalent linkage to phosphatidylethanolamine lipid (PE) through an amide bond (Atg8-PE) and becomes an integral component of the membrane located on both external and lumenal sides of the growing phagophore. Once autophagosome formation is complete (closure of the phagophore into a mature vesicle), most of the external “coat” protein complexes (Atg12/5/16) dissociate and the mature autophagosome is ready for transport to and fusion with the lysosome. The Atg8-PE molecules located within the vesicle lumen are retained along with the sequestered cargo and will eventually be degraded by low lysosomal pH and hydrolases, but until its degradation, Atg8/LC3 is a reliable molecular marker of autophagosomes and autophagolysosomes (also called autolysosomes). Other critical elements of autophagy that are the subject of intense research include mechanisms of substrate specification, lipid recruitment to the growing phagophore (i.e. Atg9, Atg11), microtubule transport and vesicle fusion events.
Other components of autophagy/lysosomal pathway are known to have a role in the heart’s response to stress. Both Class I and Class III phosphatidylinositol 3-kinases (PI3K) regulate autophagy by suppressing or activating the pathway through the production of phosphorylated phosphatidylinositol derivatives. The class III PI3K is composed of a trimeric protein complex consisting of Beclin1 (Atg6), Vps15 and hVps34 (Vps14 is also required in yeast) and produces the second messenger molecule phosphatidylinositol 3-phosphate. In contrast, the Class I PI3K is located mainly at the plasma membrane and inhibits autophagy by activating the Akt pathway, with signaling to mTOR. Confusingly, Akt signaling is cardioprotective [88] despite its predicted suppression of autophagy. Enhanced expression of Beclin1 in cultured cardiomyocytes (HL-1 cells) was shown to increase autophagosome formation and pathway flux, following simulated ischemia reperfusion (sI/R) [7, 35]. Conversely, knockdown of Beclin1 (via RNAi) perturbs the formation of autophagosomes in cardiomyocyte as well as the cell’s ability to respond to starvation and sI/R. Similar to the protection afforded by rapamycin, cells transiently transfected with a Beclin1 expression construct show significant protection against sI/R-induced GFP-Bax translocation [35].
Clearance of autophagosomes depends upon their degradation after fusion with the lysosome. The fusion event depends upon acidification, which is accomplished by the vacuolar proton ATPase (VPATPase) [46]. Like autophagy genes Atg5 and Atg7, lysosomal function is critical for the long-term function and maintenance of the heart. Lysosomal cysteine peptidase cathepsin L (CTSL) is the primary protease involved with degradation of intracellular protein. Targeted knockout of the mouse CTSL gene leads to dilated cardiomyopathy phenotypes that are similar to those seen in humans [76]. At the subcellular level CTSL−/− myocytes show an increased number of lysosomes with abnormal characteristics that suggest vesicle fusion defects. Autophagic defects are also associated with mutations to the lysosomal-associated membrane protein 2 (LAMP-2). Mutations in the human LAMP-2 gene are associated with Danon disease [21]. Patients with this disorder show glycogen storage defects and abnormal accumulation of autophagic compartments in the heart and skeletal muscle which leads to acute cardiomyopathy and myopathy. Transgenic mice lacking LAMP-2 also show similar accumulation defects in autophagic vesicles found in heart, muscle, pancreas and liver cells [25]. Together these finding further highlight the importance of the autophagic/lysosomal pathway in maintaining cardiac viability.
Pharmacologic and molecular tools to study autophagy
The study of autophagy was accelerated by the cloning of many of the genes involved in the pathway in yeast [62]. Subsequently it was found that these genes are highly conserved throughout evolution [63, 64]. A second advance was the introduction of GFP–LC3. Overexpression of this fusion protein allowed visualization of autophagosomes in real time in cells and subsequently followed in tissues of the GFP–LC3 transgenic mouse [65]. Recently, we generated transgenic mice expressing mCherry–LC3 driven by the α-myosin heavy chain promoter, which has allowed monitoring of autophagy in the heart [40]. Imaging autophagosome abundance with these fluorescent fusion proteins in cells or transgenic animals has become the “industry standard” for measuring autophagy. A thorough discussion of the advantages and disadvantages of various methods to monitor autophagy was published by Klionsky et al. [49] in 2008 and represents the consensus of over 200 authors.
Autophagosomes can accumulate when fusion with lysosomes is prevented, leading to a mistaken interpretation that autophagy is upregulated in all cases where numerous LC3-labeled puncta are seen; thus it is important to assess flux in order to have an accurate understanding of the process. One strategy for measuring flux is to compare the abundance of autophagosomes at steady state and after lysosomal degradation is inhibited [7]. In the case of ischemia/reperfusion, flux is impaired and the accumulation of autophagosomes is not an indication of increased autophagy [35]. Lysosomal degradation can be blocked with Bafilomycin A1, an inhibitor of the vacuolar proton ATPase that drives acidification of the organelle [101]. This is often used in tandem with lysosomal protease inhibitors such as pepstatin and E64d, but blocking acidification is generally sufficient, as lysosomal proteases such as cathepsin D have a pH optimum below 4.0 and are inactive above pH 5.5 [78]. Activity of cathepsin B, a predominant lysosomal protease, can be assessed using (z-RR)2-MagicRed-Cathepsin B substrate (B-Bridge). Bafilomycin is a natural product derived from a fungus, and as such, nature has evolved a molecule with high specificity; however, this reagent is moderately expensive. A modestly less effective reagent is chloroquine, which is a weak base that accumulates in lysosomes and neutralizes the pH, thereby preventing lysosomal degradation of autophagosomes (and possibly fusion of autophagosomes with lysosomes), resulting in an accumulation of autophagosomes. An advantage of chloroquine is its low cost, making it suitable for use in vivo [40]. However, chronic chloroquine administration is known to induce a myopathy characterized by accumulation of double-membrane LC3-positive vacuoles (autophagosomes) [91], supporting the idea that chloroquine prevents autophagosome-lysosome fusion.
Many investigators have used processing of LC3 to assess autophagy [42]. When autophagy is activated, LC3-I is lipidated, resulting in a form that has faster mobility on SDS-PAGE (LC3-II). The LC3-II/LC3-I ratio is often used as an indication of increased autophagy. However, this fails to take flux into account, as LC3-II will be degraded after the autophagosome fuses with the lysosome [40]. LC3-I turns over rapidly—possibly by the ubiquitin-proteasome system—so levels can rise if the ubiquitin-proteasome system is not functioning well [16]. However, elevated levels of LC3-II after inhibition of lysosomal degradation (e.g., with Bafilomycin A1 or chloroquine) can be taken as an indication of increased autophagy, if appropriate controls are performed.
Studies with LC3–GFP and LysoTracker Red™ (Invitrogen), which labels lysosomes, have found limited co-localization, in part because GFP fluorescence diminishes in the acidic environment of the lysosome. LC3 associated with the outer face of the autophagosomal membrane is removed following lysosomal fusion, while internally exposed LC3 is proteolytically degraded. In contrast, LC3 fused to red fluorescent protein (RFP) or mCherry does not quench in acidic pH, and thus persists for a longer time after lysosomal fusion. A tandem LC3–GFP–RFP construct has been described that allows estimation of flux by comparing the ratio of GFP to RFP. Monodansylcadaverine has been used by some to measure autophagy [66]. This fluorescent diamine co-localizes with LC3–mCherry-labeled structures in the heart [40]. Whether the accumulation is related to pH of the vesicle or its unique lipid architecture has been debated [70], but we have been able to label autophagosomes from frozen and thawed heart tissue; while it is possible that the pH gradient is retained, we consider it more likely that the dye is incorporated due to the double-membrane lipid structure and potentially unique lipid composition. Subsequently we have adopted cadaverine probes that are modified with either Alexa-Fluor 488™ or BODIPY-Texas Red™ (Invitrogen) and have developed a protocol to measure autophagy in frozen biopsy tissue, based on cadaverine binding [75].
An important approach to understanding the functional significance of autophagy is to perturb the system and monitor the effects on cells. Two small-molecule reagents for inhibiting autophagy are available: 3-methyladenine (3MA), and wortmannin. Both agents inhibit the initiation phase of autophagy by inhibiting PI3K, but wortmannin and 3MA inhibit class I PI3K (activator of Akt) as well as class III PI3K Vps34 (activator of autophagy). Since Akt functions in critical survival pathways, inadvertently inhibiting Akt could complicate interpretation of experiments designed to assess the role of autophagy when wortmannin is employed. The 3MA also suffers from limitations, as it has effects on intermediary metabolism [8]. Isoform-selective PI3K inhibitors are in development, but their use for selective inhibition of autophagy remains to be evaluated [98]. Bafilomycin and chloroquine can inhibit the degradation phase of autophagy, but this requires some time for feedback inhibition of the process and so these inhibitors may not be informative. Many agents are known to stimulate autophagy; best-known is rapamycin, the cognate inhibitor of mTOR, which functions as the central control for catabolic and anabolic pathways. Recently, contradictory studies have emerged, in which agents either increase or decrease autophagy, including statins and aminoimidazole carboxamide ribonucleotide (AICAR) [1, 86, 97]. This may be related to the cell type under study, so it will be important to characterize the effects of the agents in the particular system under study. For instance, we have found that the adenosine A1 receptor agonist stimulates autophagy in HL-1 cells [100], while others have reported that it inhibits autophagy in hepatocytes [86].
For these reasons, there has been a move to genetic models, notably Beclin1(+/−) mice, Atg5(−/−) mice, and others. These models are informative, but here again, caution is necessary, and compensatory upregulation of other gene products may occur that confound the interpretation. For instance, ERK is substantially upregulated in Atg5(−/−) embryonic fibroblasts, which could offer protection against some insults unrelated to autophagy [80]. Moreover, both Beclin1 and Atg5 contain a BH3 domain [57, 79]. Cleavage of Atg5 by calpain exposes the pro-apoptotic BH3 domain and triggers apoptosis [102]. We have developed a cell-permeable inhibitor of autophagy, Tat-Atg5K130R, which blocks the induction of autophagy within a few minutes of its introduction, and which therefore allows a more targeted and selective intervention. It is important to point out a caveat in the use of LC3–GFP (or LC3–mCherry) transgenic mice: since LC3 is a rate-limiting protein in the autophagy pathway, constitutive overexpression may upregulate basal levels of autophagy, with resulting effects on cellular and organ physiology. For this reason it is important to select the lowest-expressing line and to characterize the relevant parameters of the system under study. These tools can be used to understand the role of autophagy in clinically relevant contexts such as preconditioning.
Role of autophagy in preconditioning
Despite major advances in the treatment of heart disease, post-ischemic myocardial stunning and infarction remain a major cause of complications and death in patients undergoing heart surgery and percutaneous coronary interventions. For these reasons, considerable efforts are underway to develop new therapeutic strategies to increase the heart’s tolerance to ischemia. One approach has been to elucidate the heart’s endogenous defense mechanisms and to develop pharmacological agents that mimic the cardioprotection conferred by ischemic preconditioning and postconditioning [61, 67].
To date, the results using this approach, however, have been disappointing. This is due, in part, to the fact that most of these efforts have focused primarily on various upstream triggers and signaling pathways rather than defining the precise intracellular mechanisms that are involved or the end effectors. Recently, it was shown that agents known to mimic pharmacological conditioning are also potent inducers of autophagy. This offers new insights into the endogenous mechanisms that regulate protection in the human heart and thereby provides a roadmap for the development of therapeutic agents that target autophagy. A number of studies have implicated autophagy in cardioprotection. In 2005 Yan et al. [99] showed that autophagy was upregulated in chronically ischemic myocardium. They noted that autophagosomes were seen in surviving cells, not apoptotic ones, suggesting that autophagy might play a beneficial role in chronic low-flow ischemia. Subsequently, Dosenko [19, 20] noted a connection between preconditioning and autophagy. More recently, Gurusamy et al. [31, 32] reported that cardio-protection by adaptation to ischemia (preconditioning) was mediated by BAG-1 and upregulation of autophagy. Adenosine is well known as a mediator of preconditioning, yet its connection to autophagy was unknown until recently [11]. The essential role for autophagy is revealed in the study by our group [100], in which we showed that 2-chloro-N(6)-cyclopentyladenosine (CCPA), an adenosine A1 receptor agonist and potent cardioprotective agent, markedly induces autophagy within 10 min of treatment in GFP–LC3 infected HL-1 cells, neonatal, and adult rat cardiomyocytes. The salutary effect of this agent in the setting of simulated ischemia/reperfusion on cell survival was abolished when the cells were treated with a specific inhibitor of autophagy, the dominant negative protein Atg5K130R. It is also possible to infer a role for autophagy from other studies. Rapamycin, a powerful inducer of autophagy, has been reported to reduce infarct size in a Langendorff isolated perfused rat heart model [48]. Statins, which have been shown to be beneficial in the post-MI setting in patients, regardless of effects on cholesterol levels, are also powerful inducers of autophagy. Interestingly, Pravastatin, which showed less benefit in the PROVE IT-TIMI 22 trial, failed to induce autophagy [1, 55]. Exercise is another intervention that has been shown to confer cardioprotection [4, 77] as well as to induce autophagy [56]. Similarly, lipopolysaccharide elicits preconditioning and autophagy [96, 103]. Preconditioning is considered to mediate its beneficial effects during the reperfusion phase [37], and that would suggest that autophagy plays its role in cardioprotection during the same window. However, if autophagy is important, it begs the question, “Why?” The next paragraphs address some possible explanations for the mechanism of cardioprotection by autophagy.
Elimination of protein aggregates
The process of autophagy in the heart is also responsible for the clearance of protein aggregates that occur due to natural progresses of cellular aging [damage from reactive oxygen species (ROS)], pressure overload or genetic mutations that increase the risk of endogenous proteins to misfold and aggregate. Direct analysis of autophagy in mice and Drosophila has shown that basal levels of the pathway are required to remove cytoplasmic protein inclusions or aggregates containing ubiquitin. Conditional knockout alleles in the mouse Atg5 and Atg7 genes and multiple loss-of-function mutations in Drosophila show that endogenous ubiquitinated proteins in the nervous system are not effectively eliminated and accumulate as a result of impaired autophagy [41, 50]. For mice and flies, the increase in protein aggregates is not due to the presence of mutant aggregate-prone peptides but rather the accumulation of oxidatively modified proteins due to failed autophagy [36, 89]. Once the misfolded proteins exceed the capacity of the proteasome for removal, they form insoluble aggregates, which can only be eliminated by macroautophagy. Cardiac-specific deficiency of Atg5 in adult mice also results in cardiac hypertrophy followed by ventricular dilatation and contractile defects accompanied by increased levels of ubiquitinated proteins [51, 68]. Functional loss of Atg5 during embryogenesis does not affect cardiac development or biogenesis, but in adult mice subjected to pressure overload, left ventricular dilatation develops. These results suggest that basal autophagy serves a protective homeostatic function in the heart, contributing to the elimination of damaged proteins that could form aggregates that would further compromise cardiomyocyte function and viability.
While the relationship between aggregate-prone proteins and autophagic clearance is an established hypothesis in neurodegenerative diseases, the pathway’s ability to eliminate misfolded or aggregate-prone cardiac proteins is also receiving attention. As with familial cases of neurodegeneration, gain-of-function mutations in sarcomere or cytoskeletal proteins can increase the tendency of proteins to misfold. This defect can then go on to produce early onset cardiomyopathies or heart failure. These mutations, as well as environmental stresses, can promote cellular protein misfolding and can affect the function and turnover of individual proteins. The best examples are associated with the mutations in the desmin (intermediate filament protein) and its chaperone αB-cystallin (small heat shock-like protein) genes, which result in desmin-related myopathy disorders (DRM) [92]. Defects in either gene lead to altered protein profiles, a build-up of misfolded proteins and aggresomes, and cardiac amyloidosis. Recent studies have shown upregulation of autophagy in cardiac myocyte is an adaptive response to the disorder and suppressing the pathway accelerates heart failure.
Sequestration of damaged organelles (mitophagy), and mitochondrial quality control
Mitochondria undergo continuous remodeling and excess mitochondria are eliminated by autophagy when ATP production requirements are low. Mitochondrial quality control is the process of sorting well-functioning mitochondrial components from damaged and dysfunctional elements through fusion and fission. Highly functional mitochondria, characterized by efficient ATP production, high membrane potential, and low levels of ROS production, few oxidatively modified proteins, and high levels of OPA-1 can continue to participate in fusion and fission events, while damaged mitochondria have low membrane potential, low levels of OPA-1, are excluded from subsequent fusion events, and are removed by autophagy [95]. This culling process is important to maintaining a population of highly functional mitochondria. Recent studies have shown that mitochondria are frequent targets of engulfment by autophagosomes during prolonged starvation [15, 52], and our studies in the heart support this observation. During times of increased energy demand, mitochondrial biogenesis will be stimulated, and this may be enhanced if a cycle of mitophagy preceded it. Since autophagy is upregulated by caloric restriction or exercise [6, 56], one can imagine that increased mitochondrial culling will result in a population of high-functioning, robust mitochondria. This may explain the observation that caloric restriction or exercise results in mitochondria that show a higher threshold for permeability transition pore opening [45], and may also be reflected in an altered protein composition of the mitochondrial population [28].
Mitochondria are targets of autophagy during cellular stress including ischemia/reperfusion or oxidative stress [24]. Since mitochondria can release cytochrome c and other pro-apoptotic factors and can generate substantial amounts of ROS after I/R, it is conceivable that autophagy may protect the cell by sequestering and degrading the mitochondria that are most likely to trigger apoptosis (through cytochrome c release) and necrosis [through catastrophic mitochondrial permeability transition (MPT)]. Removal of ROS-producing mitochondria through autophagy will also decrease the overall oxidative stress experienced by a cell, which may explain in part why caloric restriction is associated with less oxidative damage [77]. Conceivably, any stimulus that results in enhanced mitochondrial quality control through autophagy will have benefits with respect to organ function and aging.
Proteomic analysis of mitochondria from preconditioned hearts reveals changes in post-translational modifications and protein composition [2]. It is unknown whether some of this can be attributed to removal of a subset of effete mitochondria through autophagy, but is certainly a possibility given the critical role of autophagy in preconditioning [31, 32, 74, 100]. What is clear from these studies is that autophagy plays a critical role in cardioprotection. Studies from the Atg5(−/−) animals have already shown an essential role for housekeeping autophagy in maintaining cardiac function. Thus, autophagy appears to play an important role in the heart. Since cardiomyocytes are long-lived cells, it would be important to be able to maintain optimal function of each cell, and the evidence now suggests that autophagy is essential to that process. Oxidative stress leads to damaged mitochondria, which are removed through autophagy in what may a Bnip3-dependent process [38, 53]. Subsequently, mitochondrial biogenesis is stimulated, and may be linked to autophagy [81, 82]. Studies of heart proteins over the circadian cycle has revealed substantial changes in the abundance of many mitochondrial proteins [23]; removal of mitochondria by autophagy during periods of low activity would lower the oxidative stress experienced by the cell, and would facilitate mitochondrial biogenesis, resulting in replacement of deteriorated components. Mitochondria are important targets of autophagy, which serves a beneficial role in mitochondrial quality control. High-quality mitochondria exhibit efficient ATP production and resistance to stimuli that would trigger the MPT. Conditions that suppress mitophagy—such as caloric excess—would be expected to result in the accumulation of damaged, inefficient mitochondria with a low threshold for MPT.
There is growing evidence to support the notion that damaged mitochondria are selectively removed by autophagy. Lemasters and collaborators [24] showed that photodamaged mitochondria were selectively engulfed by autophagosomes, and Shirihai’s group showed that mitochondria with lower membrane potential were removed by autophagy while those with high membrane potential underwent successive cycles of fusion and fission [95]. Since mitochondrial protein import depends upon high membrane potential, only this subset of mitochondria can replace damaged proteins. Inhibition of autophagy prevents removal of the low membrane potential mitochondria, with the result that oxidatively damaged proteins and poorly functioning mitochondria accumulate within the cell, accompanied by inefficient ATP production. This suggests that autophagy is important to maintain high-functioning mitochondria. For this reason, suppressing autophagy will have significant adverse consequences for mitochondrial homeostasis, and may explain, in part, the severe cardiomyopathy seen in several mouse models in which autophagy is impaired (Atg5-null, Atg7-null, LAMP-null, and Danon disease) (see reviews [33, 34]). Furthermore, we suggest that clinical conditions in which autophagy is suppressed will also lead to the accumulation of poor quality mitochondria; such conditions include hyperinsulinemia and caloric excess. Like so much of biology, too much of a good thing can be bad. There are conditions when excessive autophagy (and mitophagy) may lead to depletion of mitochondria beyond a critical level required to support the needs of the cell. This may explain why partial inactivation of autophagy [Beclin1(+/−) mice] do better than their wild type counterparts when subjected to severe aortic banding [84].
Protein breakdown to yield metabolic substrates for ATP production
There is profound nutrient limitation during ischemia. Autophagy represents a compensatory response to nutrient limitation and is regulated by AMP-activated kinase (AMPK), hypoxia, and low ATP levels [59, 60, 73]. However, our studies indicate that the formation of autophagosomes is suppressed in HL-1 cells during simulated ischemia. During reoxygenation or reperfusion, more autophagosomes are present, but we still find that clearance of the autophagosomes is impaired [35]. When autophagy proceeds efficiently, products of protein degradation (short peptides and amino acids) can be transported out of the autophagolysosome. The amino acids can be used as substrates for oxidative phosphorylation; however, they can also be used for non-oxidative ATP production. For instance, glutamine can serve as an important energy source independent of mitochondrial respiration [14]. Interestingly, the peptides can be used for antigen presentation on Class II major histocompatibility complex molecules, revealing another connection between autophagy and immunity. Little work has been done to characterize the amino acid transporters present in the autophagolysosome.
Amino acid transport for glutathione biosynthesis
A proteomic study of hepatic autophagosomes identified numerous enzymes involved in sulfhydryl repair [72]. The purpose of these enzymes for repair of oxidatively damaged proteins in the lumen of the autophagosome seems unlikely; rather, we hypothesize that the autophagolysosomal membrane serves as a scaffold for repair of cytoplasmic proteins. Amino acids liberated by proteolytic degradation of lumenal contents would be transported across the membrane, providing the driving force for glutathione biosynthesis. Multiple other enzymes involved in sulfhydryl repair might assemble in proximity to the site of glutathione biosynthesis, as proposed in Fig. 3. It is appealing to think that autophagy, which is potently induced by ROS, would be part of a cellular homeostatic response enabling repair of oxidatively modified sulfhydryls. Considerable work lies ahead to determine whether this speculation will be shown to be true.
Fig. 3.
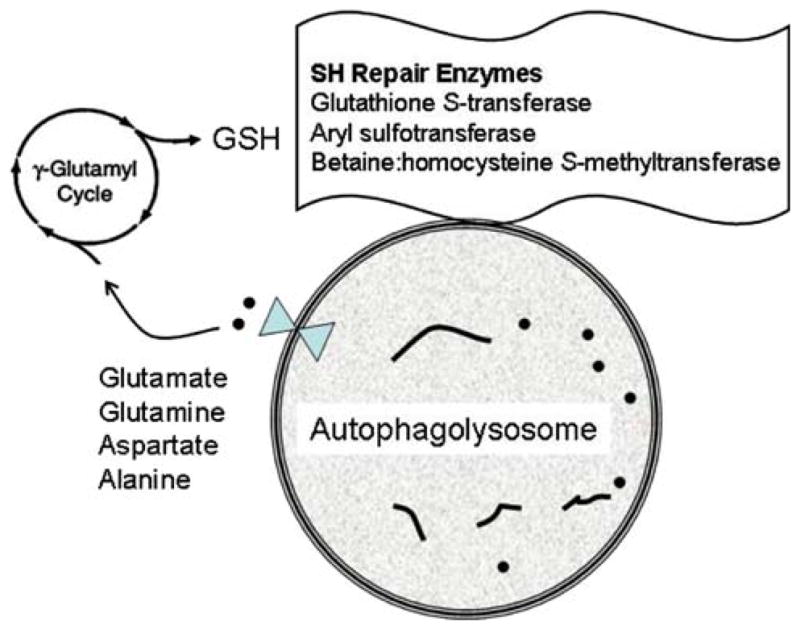
Putative connection between protein thiol repair and autophagy. Lysosomal hydrolases degrade proteins into peptides (short lines) and amino acids (dots), which are transported out of the autophagolysosome, where they may enter the gamma-glutamyl cycle to generate glutathione, which is utilized by thiol repair enzymes associated with the autophagolysosome, including glutathione S-transferase, aryl sulfotransferase and betaine:homocysteine S-methyltransferase
Proton pumping to support ion homeostasis
During ischemia, intracellular pH (pHi) drops rapidly to below 6.6 as lactic acid accumulates and protons are released from ATP hydrolysis [90]. During early reperfusion, the Na+/H+ exchanger operates to eliminate protons, taking advantage of the driving force of high extracellular [Na+], resulting in normalization of pHi (sometimes even an overshoot) and increased intracellular [Na+]. The intracellular Na+ competes with Ca++ for extrusion via the Na+/Ca++ exchanger, resulting in Ca++ overload, which in turn activates calpains, leading to proteolytic activation of pro-apoptotic Bid [9, 10], as well as opening of the mitochondrial permeability transition pore (MPTP). Ischemic preconditioning limits intracellular acidification, thereby preventing Na+ and Ca++ overload. In previous work we showed that this depended upon activity of the vacuolar proton ATPase (VPATPase), which is the proton pump responsible for lysosomal acidification [30, 44]. We now suggest that upregulation of autophagy would necessarily be linked to activation of the VPATPase. In this case, upregulation of autophagy would be predicted to limit Na+ and Ca++ overload (see diagram). Bafilomycin A1 is a potent and specific inhibitor of the VPATPase, which is essential for receptor internalization and endosomal trafficking and could therefore affect adenosine receptor signaling [93]. One could then argue that the loss of protection observed by Yitzhaki et al. is due to inhibition of endosomal trafficking rather than blockade of autophagy. However, Atg5 is not required for endosome trafficking [5].
Stimulation of mitochondrial biogenesis
An intriguing link between autophagy and mitochondrial biogenesis is suggested by studies in which cardiomyocytes exposed to LPS upregulate autophagy and mitochondrial biogenesis [38, 103]. Mitochondria are a common target of autophagy, but a mechanistic link between mitophagy (autophagy of mitochondria) and mitochondrial biogenesis is not yet established. Here again, there is room for speculation that elimination of mitochondria directly signals mitochondrial biogenesis. To draw a lesson from bacteria (with which mitochondria share ancestry), bacterial populations secrete factors that provide information about the relative density of the population [3]. The concentration of these factors serves to regulate rates of bacterial replication and expression of bacterial and host cell genes [47]. By analogy, mitochondria might also secrete factors that coordinate their biogenesis. The simplest example of this is ATP as the ‘secreted factor’ that slows (mitochondrial) replication by suppressing AMPK; low [ATP] activates AMPK which in turn activates the key transcription factor PGC1α to initiate mitochondrial biogenesis. Removal of mitochondria by autophagy would decrease the concentration of the ‘secreted factor’ (whether ATP or another molecule), thereby triggering replacement of the missing mitochondria.
Summary
Autophagy is mechanism to recycle proteins and to remove unwanted or damaged organelles. It is important for the removal of oxidized or aggregated proteins. In dividing cells with rapid turnover, autophagy may not be essential; however, in long-lived cells such as cardiomyocytes and neurons, removal of damaged organelles and protein aggregates is critical to maintaining optimal cellular function. In particular, autophagy appears to be essential to mitochondrial quality control. Matching energy production to cellular energy needs is controlled at multiple levels, including transcription/translation, post-translational control, and allosteric regulation; it appears that autophagy participates in regulation by removing effete mitochondria, thereby permitting biogenesis. Failure of autophagy results in accumulation of poor-functioning mitochondria and aggregated proteins. Autophagy may serve additional functions in the acute setting of stress, including production of metabolic substrates for energy production and thiol repair, and may also contribute to pH and ion homeostasis through proton sequestration. A more detailed understanding of the role of autophagy in cardiac health and disease may offer new therapeutic approaches to the management of ischemia/reperfusion injury, hypertrophy, and failure.
Acknowledgments
This work was supported by NIH grants P01HL85577, R01HL060590, R01AG033283 (to RAG), R21AG030187 (to KDF), and R01HL034579 (to RMM).
Footnotes
Open Access This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Contributor Information
Roberta A. Gottlieb, Email: robbieg@sciences.sdsu.edu, The BioScience Center, San Diego State University, 5500 Campanile Drive, San Diego, CA 92182-4650, USA
Kim D. Finley, Email: kfinley@sciences.sdsu.edu, The BioScience Center, San Diego State University, 5500 Campanile Drive, San Diego, CA 92182-4650, USA
Robert M. Mentzer, Jr, Email: rmentzer@med.wayne.edu, School of Medicine, WSU Cardiovascular Research Institute, Wayne State University, 540 E. Canfield 1241 Scott Hall, Detroit, MI 48201, USA.
References
- 1.Araki M, Motojima K. Hydrophobic statins induce autophagy in cultured human rhabdomyosarcoma cells. Biochem Biophys Res Commun. 2008;367:462–467. doi: 10.1016/j.bbrc.2007.12.166. [DOI] [PubMed] [Google Scholar]
- 2.Arrell DK, Elliott ST, Kane LA, Guo Y, Ko YH, Pedersen PL, Robinson J, Murata M, Murphy AM, Marban E, Van Eyk JE. Proteomic analysis of pharmacological preconditioning: novel protein targets converge to mitochondrial metabolism pathways. Circ Res. 2006;99:706–714. doi: 10.1161/01.RES.0000243995.74395.f8. [DOI] [PubMed] [Google Scholar]
- 3.Asad S, Opal SM. Bench-to-bedside review: quorum sensing and the role of cell-to-cell communication during invasive bacterial infection. Crit Care. 2008;12:236. doi: 10.1186/cc7101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ascensao A, Ferreira R, Magalhaes J. Exercise-induced cardioprotection—biochemical, morphological and functional evidence in whole tissue and isolated mitochondria. Int J Cardiol. 2007;117:16–30. doi: 10.1016/j.ijcard.2006.04.076. [DOI] [PubMed] [Google Scholar]
- 5.Bampton ET, Goemans CG, Niranjan D, Mizushima N, Tolkovsky AM. The dynamics of autophagy visualized in live cells: from autophagosome formation to fusion with endo/lysosomes. Autophagy. 2005;1:23–36. doi: 10.4161/auto.1.1.1495. [DOI] [PubMed] [Google Scholar]
- 6.Bergamini E, Cavallini G, Donati A, Gori Z. The anti-ageing effects of caloric restriction may involve stimulation of macroautophagy and lysosomal degradation, and can be intensified pharmacologically. Biomed Pharmacother. 2003;57:203–208. doi: 10.1016/s0753-3322(03)00048-9. [DOI] [PubMed] [Google Scholar]
- 7.Brady NR, Hamacher-Brady A, Yuan H, Gottlieb RA. The autophagic response to nutrient deprivation in the HL-1 cardiac myocyte is modulated by Bcl-2 and sarco/endoplasmic reticulum calcium stores. FEBS J. 2007;274:3184–3197. doi: 10.1111/j.1742-4658.2007.05849.x. [DOI] [PubMed] [Google Scholar]
- 8.Caro LHP, Plomp PJAM, Wolvetang EJ, Kerkhof C, Meijer AJ. 3-Methyladenine, an inhibitor of autophagy, has multiple effects on metabolism. Eur J Biochem. 1988;175:325–329. doi: 10.1111/j.1432-1033.1988.tb14200.x. [DOI] [PubMed] [Google Scholar]
- 9.Chen M, He H, Zhan S, Krajewski S, Reed JC, Gottlieb RA. Bid is cleaved by calpain to an active fragment in vitro and during myocardial ischemia/reperfusion. J Biol Chem. 2001;276:30724–30728. doi: 10.1074/jbc.M103701200. [DOI] [PubMed] [Google Scholar]
- 10.Chen M, Won DJ, Krajewski S, Gottlieb RA. Calpain and mitochondria in ischemia/reperfusion injury. J Biol Chem. 2002;277:29181–29186. doi: 10.1074/jbc.M204951200. [DOI] [PubMed] [Google Scholar]
- 11.Cohen MV, Downey JM. Adenosine: trigger and mediator of cardioprotection. Basic Res Cardiol. 2008;103:203–215. doi: 10.1007/s00395-007-0687-7. [DOI] [PubMed] [Google Scholar]
- 12.Cuervo AM, Bergamini E, Brunk UT, Droge W, Ffrench M, Terman A. Autophagy and aging: the importance of maintaining “clean” cells. Autophagy. 2005;1:131–140. doi: 10.4161/auto.1.3.2017. [DOI] [PubMed] [Google Scholar]
- 13.Cuervo AM, Dice JF. How do intracellular proteolytic systems change with age? Front Biosci. 1998;3:d25–d43. doi: 10.2741/a264. [DOI] [PubMed] [Google Scholar]
- 14.DeBerardinis RJ, Mancuso A, Daikhin E, Nissim I, Yudkoff M, Wehrli S, Thompson CB. Beyond aerobic glycolysis: Transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc Natl Acad Sci USA. 2007;104:19345–19350. doi: 10.1073/pnas.0709747104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dengjel J, Kristensen AR, Andersen JS. Ordered bulk degradation via autophagy. Autophagy. 2008;4:1057–1059. doi: 10.4161/auto.6824. [DOI] [PubMed] [Google Scholar]
- 16.Ding WX, Ni HM, Gao W, Yoshimori T, Stolz DB, Ron D, Yin XM. Linking of autophagy to ubiquitin-proteasome system is important for the regulation of endoplasmic reticulum stress and cell viability. Am J Pathol. 2007;171:513–524. doi: 10.2353/ajpath.2007.070188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dohm GL, Tapscott EB, Kasperek GJ. Protein degradation during endurance exercise and recovery. Med Sci Sports Exerc. 1987;19:S166–S171. [PubMed] [Google Scholar]
- 18.Donati A. The involvement of macroautophagy in aging and anti-aging interventions. Mol Aspects Med. 2006;27:455–470. doi: 10.1016/j.mam.2006.08.003. [DOI] [PubMed] [Google Scholar]
- 19.Dosenko VE, Nagibin VS, Tumanovska LV, Moibenko AA. Protective effect of autophagy in anoxia-reoxygenation of isolated cardiomyocyte? Autophagy. 2006;2:305–306. doi: 10.4161/auto.2946. [DOI] [PubMed] [Google Scholar]
- 20.Dosenko VE, Nagibin VS, Tumanovskaya LV, Zagoriy VY, Moibenko AA, Vaage J. Proteasomal proteolysis in anoxia-reoxygenation, preconditioning and postconditioning of isolated cardiomyocytes. Pathophysiology. 2006;13:119–125. doi: 10.1016/j.pathophys.2006.01.003. [DOI] [PubMed] [Google Scholar]
- 21.Dougu N, Joho S, Shan L, Shida T, Matsuki A, Uese K, Hirono K, Ichida F, Tanaka K, Nishino I, Inoue H. Novel LAMP-2 mutation in a family with Danon disease presenting with hypertrophic cardiomyopathy. Circulation. 2009;73:376–380. doi: 10.1253/circj.cj-08-0241. [DOI] [PubMed] [Google Scholar]
- 22.Droge W, Schipper HM. Oxidative stress and aberrant signaling in aging and cognitive decline. Aging Cell. 2007;6:361–370. doi: 10.1111/j.1474-9726.2007.00294.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Durgan DJ, Young ME. Linking the cardiomyocyte circadian clock to myocardial metabolism. Cardiovasc Drugs Ther. 2008;22:115–124. doi: 10.1007/s10557-008-6086-y. [DOI] [PubMed] [Google Scholar]
- 24.Elmore SP, Qian T, Grissom SF, Lemasters JJ. The mitochondrial permeability transition initiates autophagy in rat hepatocytes. FASEB J. 2001;15:2286–2287. doi: 10.1096/fj.01-0206fje. [DOI] [PubMed] [Google Scholar]
- 25.Eskelinen EL. Roles of LAMP-1 and LAMP-2 in lysosome biogenesis and autophagy. Mol Aspects Med. 2006;27:495–502. doi: 10.1016/j.mam.2006.08.005. [DOI] [PubMed] [Google Scholar]
- 26.Etgen GJ, Oldham BA, Johnson WT, Broderick CL, Montrose CR, Brozinick JT, Misener EA, Bean JS, Bensch WR, Brooks DA, Shuker AJ, Rito CJ, McCarthy JR, Ardecky RJ, Tyhonas JS, Dana SL, Bilakovics JM, Paterniti JR, Jr, Ogilvie KM, Liu S, Kauffman RF. A tailored therapy for the metabolic syndrome: the dual peroxisome proliferator-activated receptor-alpha/gamma agonist LY465608 ameliorates insulin resistance and diabetic hyperglycemia while improving cardiovascular risk factors in preclinical models. Diabetes. 2002;51:1083–1087. doi: 10.2337/diabetes.51.4.1083. [DOI] [PubMed] [Google Scholar]
- 27.Finley KD, Edeen PT, Cumming RC, Mardahl-Dumesnil MD, Taylor BJ, Rodriguez MH, Hwang CE, Benedetti M, McKeown M. Blue cheese mutations define a novel, conserved gene involved in progressive neural degeneration. J Neurosci. 2003;23:1254–1264. doi: 10.1523/JNEUROSCI.23-04-01254.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Franch HA. Nutrition and muscle catabolism in maintenance hemodialysis: does feeding make muscle cells selective self-eaters? J Ren Nutr. 2009;19:86–90. doi: 10.1053/j.jrn.2008.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.French JP, Hamilton KL, Quindry JC, Lee Y, Upchurch PA, Powers SK. Exercise-induced protection against myocardial apoptosis and necrosis: MnSOD, calcium-handling proteins, and calpain. FASEB J. 2008;22:2862–2871. doi: 10.1096/fj.07-102541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gottlieb RA, Gruol DL, Zhu JY, Engler RL. Preconditioning in rabbit cardiomyocytes: role of pH, vacuolar proton ATPase, and apoptosis. J Clin Invest. 1996;97:2391–2398. doi: 10.1172/JCI118683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gurusamy N, Lekli I, Gherghiceanu M, Popescu LM, Das DK. BAG-1 induces autophagy for cardiac cell survival. Autophagy. 2009;5:120–121. doi: 10.4161/auto.5.1.7303. [DOI] [PubMed] [Google Scholar]
- 32.Gurusamy N, Lekli I, Gorbunov N, Gherghiceanu M, Popescu LM, Das DK. Cardioprotection by adaptation to ischemia augments autophagy in association with BAG-1 protein. J Cell Mol Med. 2008 doi: 10.1111/j.1582-4934.2008.00495.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gustafsson AB, Gottlieb RA. Eat your heart out: role of autophagy in myocardial ischemia/reperfusion. Autophagy. 2008;4:416–421. doi: 10.4161/auto.5655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gustafsson AB, Gottlieb RA. Recycle or die: the role of autophagy in cardioprotection. J Mol Cell Cardiol. 2008;44:654–661. doi: 10.1016/j.yjmcc.2008.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hamacher-Brady A, Brady NR, Gottlieb RA. Enhancing macroautophagy protects against ischemia/reperfusion injury in cardiac myocytes. J Biol Chem. 2006;281:29776–29787. doi: 10.1074/jbc.M603783200. [DOI] [PubMed] [Google Scholar]
- 36.Hara T, Nakamura K, Matsui M, Yamamoto A, Nakahara Y, Suzuki-Migishima R, Yokoyama M, Mishima K, Saito I, Okano H, Mizushima N. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature. 2006;441:885–889. doi: 10.1038/nature04724. [DOI] [PubMed] [Google Scholar]
- 37.Hausenloy DJ, Wynne AM, Yellon DM. Ischemic preconditioning targets the reperfusion phase. Basic Res Cardiol. 2007;102:445–452. doi: 10.1007/s00395-007-0656-1. [DOI] [PubMed] [Google Scholar]
- 38.Hickson-Bick DL, Jones C, Buja LM. Stimulation of mitochondrial biogenesis and autophagy by lipopolysaccharide in the neonatal rat cardiomyocyte protects against programmed cell death. J Mol Cell Cardiol. 2008;44:411–418. doi: 10.1016/j.yjmcc.2007.10.013. [DOI] [PubMed] [Google Scholar]
- 39.Ichimura Y, Kumanomidou T, Sou YS, Mizushima T, Ezaki J, Ueno T, Kominami E, Yamane T, Tanaka K, Komatsu M. Structural basis for sorting mechanism of p62 in selective autophagy. J Biol Chem. 2008;283:22847–22857. doi: 10.1074/jbc.M802182200. [DOI] [PubMed] [Google Scholar]
- 40.Iwai-Kanai E, Yuan H, Huang C, Sayen MR, Perry-Garza CN, Kim L, Gottlieb RA. A method to measure cardiac autophagic flux in vivo. Autophagy. 2008;4:322–329. doi: 10.4161/auto.5603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Juhasz G, Erdi B, Sass M, Neufeld TP. Atg7-dependent autophagy promotes neuronal health, stress tolerance, and longevity but is dispensable for metamorphosis in Drosophila. Genes Dev. 2007;21:3061–3066. doi: 10.1101/gad.1600707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, Kominami E, Ohsumi Y, Yoshimori T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000;19:5720–5728. doi: 10.1093/emboj/19.21.5720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kanazawa T, Taneike I, Akaishi R, Yoshizawa F, Furuya N, Fujimura S, Kadowaki M. Amino acids and insulin control autophagic proteolysis through different signaling pathways in relation to mTOR in isolated rat hepatocytes. J Biol Chem. 2004;279:8452–8459. doi: 10.1074/jbc.M306337200. [DOI] [PubMed] [Google Scholar]
- 44.Karwatowska-Prokopczuk E, Nordberg J, Li HL, Engler RL, Gottlieb RA. Effect of the vacuolar proton ATPase on intracellular pH, calcium, and on apoptosis in neonatal cardiomyocytes during metabolic inhibition and recovery. Circ Res. 1998;82:1139–1144. doi: 10.1161/01.res.82.11.1139. [DOI] [PubMed] [Google Scholar]
- 45.Kavazis AN, McClung JM, Hood DA, Powers SK. Exercise induces a cardiac mitochondrial phenotype that resists apoptotic stimuli. Am J Physiol Heart Circ Physiol. 2008;294:H928–H935. doi: 10.1152/ajpheart.01231.2007. [DOI] [PubMed] [Google Scholar]
- 46.Kawai A, Uchiyama H, Takano S, Nakamura N, Ohkuma S. Autophagosome-lysosome fusion depends on the pH in acidic compartments in CHO cells. Autophagy. 2007;3:154–157. doi: 10.4161/auto.3634. [DOI] [PubMed] [Google Scholar]
- 47.Kendall MM, Sperandio V. Quorum sensing by enteric pathogens. Curr Opin Gastroenterol. 2007;23:10–15. doi: 10.1097/MOG.0b013e3280118289. [DOI] [PubMed] [Google Scholar]
- 48.Khan S, Salloum F, Das A, Xi L, Vetrovec GW, Kukreja RC. Rapamycin confers preconditioning-like protection against ischemia-reperfusion injury in isolated mouse heart and cardiomyocytes. J Mol Cell Cardiol. 2006;41:256–264. doi: 10.1016/j.yjmcc.2006.04.014. [DOI] [PubMed] [Google Scholar]
- 49.Klionsky DJ, Abeliovich H, Agostinis P, Agrawal DK, Aliev G, Askew DS, Baba M, Baehrecke EH, Bahr BA, Ballabio A, Bamber BA, Bassham DC, Bergamini E, Bi X, Biard-Piechaczyk M, Blum JS, Bredesen DE, Brodsky JL, Brumell JH, Brunk UT, Bursch W, Camougrand N, Cebollero E, Cecconi F, Chen Y, Chin LS, Choi A, Chu CT, Chung J, Clarke PG, Clark RS, Clarke SG, Clave C, Cleveland JL, Codogno P, Colombo MI, Coto-Montes A, Cregg JM, Cuervo AM, Debnath J, Demarchi F, Dennis PB, Dennis PA, Deretic V, Devenish RJ, Di Sano F, Dice JF, Difiglia M, Dinesh-Kumar S, Distelhorst CW, Djavaheri-Mergny M, Dorsey FC, Droge W, Dron M, Dunn WA, Jr, Duszenko M, Eissa NT, Elazar Z, Esclatine A, Eskelinen EL, Fesus L, Finley KD, Fuentes JM, Fueyo J, Fujisaki K, Galliot B, Gao FB, Gewirtz DA, Gibson SB, Gohla A, Goldberg AL, Gonzalez R, Gonzalez-Estevez C, Gorski S, Gottlieb RA, Haussinger D, He YW, Heidenreich K, Hill JA, Hoyer-Hansen M, Hu X, Huang WP, Iwasaki A, Jaattela M, Jackson WT, Jiang X, Jin S, Johansen T, Jung JU, Kadowaki M, Kang C, Kelekar A, Kessel DH, Kiel JA, Kim HP, Kimchi A, Kinsella TJ, Kiselyov K, Kitamoto K, Knecht E, et al. Guidelines for the use and interpretation of assays for monitoring autophagy in higher eukaryotes. Autophagy. 2008;4:151–175. doi: 10.4161/auto.5338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Komatsu M, Waguri S, Chiba T, Murata S, Iwata J, Tanida I, Ueno T, Koike M, Uchiyama Y, Kominami E, Tanaka K. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature. 2006;441:880–884. doi: 10.1038/nature04723. [DOI] [PubMed] [Google Scholar]
- 51.Komatsu M, Waguri S, Ueno T, Iwata J, Murata S, Tanida I, Ezaki J, Mizushima N, Ohsumi Y, Uchiyama Y, Kominami E, Tanaka K, Chiba T. Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. J Cell Biol. 2005;169:425–434. doi: 10.1083/jcb.200412022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kristensen AR, Schandorff S, Hoyer-Hansen M, Nielsen MO, Jaattela M, Dengjel J, Andersen JS. Ordered organelle degradation during starvation-induced autophagy. Mol Cell Proteomics. 2008;7:2419–2428. doi: 10.1074/mcp.M800184-MCP200. [DOI] [PubMed] [Google Scholar]
- 53.Kubli DA, Quinsay MN, Huang C, Lee Y, Gustafsson AB. Bnip3 functions as a mitochondrial sensor of oxidative stress during myocardial ischemia and reperfusion. Am J Physiol Heart Circ Physiol. 2008;295:H2025–H2031. doi: 10.1152/ajpheart.00552.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.LePage C, Noirez P, Courty J, Riou B, Swynghedauw B, Besse S. Exercise training improves functional post-ischemic recovery in senescent heart. Exp Gerontol. 2008 doi: 10.1016/j.exger.2008.10.003. [DOI] [PubMed] [Google Scholar]
- 55.Lotfi A, Schweiger MJ, Giugliano GR, Murphy SA, Cannon CP. High-dose atorvastatin does not negatively influence clinical outcomes among clopidogrel treated acute coronary syndrome patients—a pravastatin or atorvastatin evaluation and infection therapy-thrombolysis in myocardial infarction 22 (PROVE IT-TIMI 22) analysis. Am Heart J. 2008;155:954–958. doi: 10.1016/j.ahj.2007.12.009. [DOI] [PubMed] [Google Scholar]
- 56.Mackenzie MG, Hamilton DL, Murray JT, Baar K. mVps34 is activated by an acute bout of resistance exercise. Biochem Soc Trans. 2007;35:1314–1316. doi: 10.1042/BST0351314. [DOI] [PubMed] [Google Scholar]
- 57.Maiuri MC, Le Toumelin G, Criollo A, Rain JC, Gautier F, Juin P, Tasdemir E, Pierron G, Troulinaki K, Tavernarakis N, Hickman JA, Geneste O, Kroemer G. Functional and physical interaction between Bcl-X(L) and a BH3-like domain in Beclin-1. EMBO J. 2007;26:2527–2539. doi: 10.1038/sj.emboj.7601689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Martinez-Vicente M, Cuervo AM. Autophagy and neurodegeneration: when the cleaning crew goes on strike. Lancet Neurol. 2007;6:352–361. doi: 10.1016/S1474-4422(07)70076-5. [DOI] [PubMed] [Google Scholar]
- 59.Matsui Y, Takagi H, Qu X, Abdellatif M, Sakoda H, Asano T, Levine B, Sadoshima J. Distinct roles of autophagy in the heart during ischemia and reperfusion: roles of AMP-activated protein kinase and Beclin 1 in mediating autophagy. Circ Res. 2007;100:914–922. doi: 10.1161/01.RES.0000261924.76669.36. [DOI] [PubMed] [Google Scholar]
- 60.Meley D, Bauvy C, Houben-Weerts JH, Dubbelhuis PF, Helmond MT, Codogno P, Meijer AJ. AMP-activated protein kinase and the regulation of autophagic proteolysis. J Biol Chem. 2006;281:34870–34879. doi: 10.1074/jbc.M605488200. [DOI] [PubMed] [Google Scholar]
- 61.Miura T, Miki T. Limitation of myocardial infarct size in the clinical setting: current status and challenges in translating animal experiments into clinical therapy. Basic Res Cardiol. 2008;103:501–513. doi: 10.1007/s00395-008-0743-y. [DOI] [PubMed] [Google Scholar]
- 62.Mizushima N, Noda T, Yoshimori T, Tanaka Y, Ishii T, George MD, Klionsky DJ, Ohsumi M, Ohsumi Y. A protein conjugation system essential for autophagy. Nature. 1998;395:395–398. doi: 10.1038/26506. [DOI] [PubMed] [Google Scholar]
- 63.Mizushima N, Ohsumi Y, Yoshimori T. Autophagosome formation in mammalian cells. Cell Struct Funct. 2002;27:421–429. doi: 10.1247/csf.27.421. [DOI] [PubMed] [Google Scholar]
- 64.Mizushima N, Sugita H, Yoshimori T, Ohsumi Y. A new protein conjugation system in human. The counterpart of the yeast Apg12p conjugation system essential for autophagy. J Biol Chem. 1998;273:33889–33892. doi: 10.1074/jbc.273.51.33889. [DOI] [PubMed] [Google Scholar]
- 65.Mizushima N, Yamamoto A, Matsui M, Yoshimori T, Ohsumi Y. In vivo analysis of autophagy in response to nutrient starvation using transgenic mice expressing a fluorescent autophagosome marker. Mol Biol Cell. 2004;15:1101–1111. doi: 10.1091/mbc.E03-09-0704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Munafo DB, Colombo MI. A novel assay to study autophagy: regulation of autophagosome vacuole size by amino acid deprivation. J Cell Sci. 2001;114:3619–3629. doi: 10.1242/jcs.114.20.3619. [DOI] [PubMed] [Google Scholar]
- 67.Mykytenko J, Reeves JG, Kin H, Wang NP, Zatta AJ, Jiang R, Guyton RA, Vinten-Johansen J, Zhao ZQ. Persistent beneficial effect of postconditioning against infarct size: role of mitochondrial K(ATP) channels during reperfusion. Basic Res Cardiol. 2008;103:472–484. doi: 10.1007/s00395-008-0731-2. [DOI] [PubMed] [Google Scholar]
- 68.Nakai A, Yamaguchi O, Takeda T, Higuchi Y, Hikoso S, Taniike M, Omiya S, Mizote I, Matsumura Y, Asahi M, Nishida K, Hori M, Mizushima N, Otsu K. The role of autophagy in cardiomyocytes in the basal state and in response to hemodynamic stress. Nat Med. 2007;13:619–624. doi: 10.1038/nm1574. [DOI] [PubMed] [Google Scholar]
- 69.Nezis IP, Simonsen A, Sagona AP, Finley K, Gaumer S, Contamine D, Rusten TE, Stenmark H, Brech A. Ref(2)P, the Drosophila melanogaster homologue of mammalian p62, is required for the formation of protein aggregates in adult brain. J Cell Biol. 2008;180:1065–1071. doi: 10.1083/jcb.200711108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Niemann A, Takatsuki A, Elsasser HP. The lysosomo-tropic agent monodansylcadaverine also acts as a solvent polarity probe. J Histochem Cytochem. 2000;48:251–258. doi: 10.1177/002215540004800210. [DOI] [PubMed] [Google Scholar]
- 71.Nixon RA. Autophagy in neurodegenerative disease: friend, foe or turncoat? Trends Neurosci. 2006;29:528–535. doi: 10.1016/j.tins.2006.07.003. [DOI] [PubMed] [Google Scholar]
- 72.Overbye A, Fengsrud M, Seglen PO. Proteomic analysis of membrane-associated proteins from rat liver autophagosomes. Autophagy. 2007;3:300–322. doi: 10.4161/auto.3910. [DOI] [PubMed] [Google Scholar]
- 73.Papandreou I, Lim AL, Laderoute K, Denko NC. Hypoxia signals autophagy in tumor cells via AMPK activity, independent of HIF-1, BNIP3, and BNIP3L. Cell Death Differ. 2008;15:1572–1581. doi: 10.1038/cdd.2008.84. [DOI] [PubMed] [Google Scholar]
- 74.Park HK, Chu K, Jung KH, Lee ST, Bahn JJ, Kim M, Lee SK, Roh JK. Autophagy is involved in the ischemic preconditioning. Neurosci Lett. 2009;451:16–19. doi: 10.1016/j.neulet.2008.12.019. [DOI] [PubMed] [Google Scholar]
- 75.Perry C, Gottlieb R. Novel Methods for measuring cardiac autophagy in vivo. Methods Enzymol. 2009 doi: 10.1016/S0076-6879(08)04016-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Petermann I, Mayer C, Stypmann J, Biniossek ML, Tobin DJ, Engelen MA, Dandekar T, Grune T, Schild L, Peters C, Reinheckel T. Lysosomal, cytoskeletal, and metabolic alterations in cardiomyopathy of cathepsin L knockout mice. FASEB J. 2006;20:1266–1268. doi: 10.1096/fj.05-5517fje. [DOI] [PubMed] [Google Scholar]
- 77.Powers SK, Quindry JC, Kavazis AN. Exercise-induced cardioprotection against myocardial ischemia-reperfusion injury. Free Radic Biol Med. 2008;44:193–201. doi: 10.1016/j.freeradbiomed.2007.02.006. [DOI] [PubMed] [Google Scholar]
- 78.Puizdar V, Turk V. Cathepsinogen D: characterization and activation to cathepsin D and inhibitory peptides. FEBS Lett. 1981;132:299–304. doi: 10.1016/0014-5793(81)81184-2. [DOI] [PubMed] [Google Scholar]
- 79.Pyo J-O, Jang M-H, Kwon Y-K, Lee H-J, Jun J-IL, Woo H-N, Cho D-H, Choi B, Lee H, Kim J-H, Mizushima N, Oshumi Y, Jung Y-K. Essential Roles of Atg5 and FADD in autophagic cell death: dissection of autophagic cell death into vacuole formation and cell death. J Biol Chem. 2005;280:20722–20729. doi: 10.1074/jbc.M413934200. [DOI] [PubMed] [Google Scholar]
- 80.Pyo JO, Nah J, Kim HJ, Lee HJ, Heo J, Lee H, Jung YK. Compensatory activation of ERK1/2 in Atg5-deficient mouse embryo fibroblasts suppresses oxidative stress-induced cell death. Autophagy. 2008;4:315–321. doi: 10.4161/auto.5525. [DOI] [PubMed] [Google Scholar]
- 81.Rasbach KA, Schnellmann RG. PGC-1alpha over-expression promotes recovery from mitochondrial dysfunction and cell injury. Biochem Biophys Res Commun. 2007;355:734–739. doi: 10.1016/j.bbrc.2007.02.023. [DOI] [PubMed] [Google Scholar]
- 82.Rasbach KA, Schnellmann RG. Signaling of mitochondrial biogenesis following oxidant injury. J Biol Chem. 2007;282:2355–2362. doi: 10.1074/jbc.M608009200. [DOI] [PubMed] [Google Scholar]
- 83.Rodriguez A, Duran A, Selloum M, Champy MF, Diez-Guerra FJ, Flores JM, Serrano M, Auwerx J, Diaz-Meco MT, Moscat J. Mature-onset obesity and insulin resistance in mice deficient in the signaling adapter p62. Cell Metab. 2006;3:211–222. doi: 10.1016/j.cmet.2006.01.011. [DOI] [PubMed] [Google Scholar]
- 84.Rothermel BA, Hill JA. Autophagy in load-induced heart disease. Circ Res. 2008;103:1363–1369. doi: 10.1161/CIRCRESAHA.108.186551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Salminen A, Vihko V. Autophagic response to strenuous exercise in mouse skeletal muscle fibers. Virchows Arch B Cell Pathol Incl Mol Pathol. 1984;45:97–106. doi: 10.1007/BF02889856. [DOI] [PubMed] [Google Scholar]
- 86.Samari HR, Seglen PO. Inhibition of hepatocytic autophagy by adenosine, aminoimidazole-4-carboxamide riboside, and N6-mercaptopurine riboside. Evidence for involvement of AMP-activated protein kinase. J Biol Chem. 1998;273:23758–23763. doi: 10.1074/jbc.273.37.23758. [DOI] [PubMed] [Google Scholar]
- 87.Shvets E, Fass E, Elazar Z. Utilizing flow cytometry to monitor autophagy in living mammalian cells. Autophagy. 2008;4:621–628. doi: 10.4161/auto.5939. [DOI] [PubMed] [Google Scholar]
- 88.Siddall HK, Warrell CE, Yellon DM, Mocanu MM. Ischemia-reperfusion injury and cardioprotection: investigating PTEN, the phosphatase that negatively regulates PI3K, using a congenital model of PTEN haploinsufficiency. Basic Res Cardiol. 2008;103:560–568. doi: 10.1007/s00395-008-0735-y. [DOI] [PubMed] [Google Scholar]
- 89.Simonsen A, Cumming RC, Brech A, Isakson P, Schubert DR, Finley KD. Promoting basal levels of autophagy in the nervous system enhances longevity and oxidant resistance in adult Drosophila. Autophagy. 2008;4:176–184. doi: 10.4161/auto.5269. [DOI] [PubMed] [Google Scholar]
- 90.Steenbergen C, Perlman ME, London RE, Murphy E. Mechanism of preconditioning: Ionic alterations. Circ Res. 1993;72:112–125. doi: 10.1161/01.res.72.1.112. [DOI] [PubMed] [Google Scholar]
- 91.Suzuki T, Nakagawa M, Yoshikawa A, Sasagawa N, Yoshimori T, Ohsumi Y, Nishino I, Ishiura S, Nonaka I. The first molecular evidence that autophagy relates rimmed vacuole formation in chloroquine myopathy. J Biochem. 2002;131:647–651. doi: 10.1093/oxfordjournals.jbchem.a003147. [DOI] [PubMed] [Google Scholar]
- 92.Tannous P, Zhu H, Nemchenko A, Berry JM, Johnstone JL, Shelton JM, Miller FJ, Jr, Rothermel BA, Hill JA. Intra-cellular protein aggregation is a proximal trigger of cardiomyocyte autophagy. Circulation. 2008;117:3070–3078. doi: 10.1161/CIRCULATIONAHA.107.763870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Tong H, Rockman HA, Koch WJ, Steenbergen C, Murphy E. G protein-coupled receptor internalization signaling is required for cardioprotection in ischemic preconditioning. Circ Res. 2004;94:1133–1141. doi: 10.1161/01.RES.0000126048.32383.6B. [DOI] [PubMed] [Google Scholar]
- 94.Toth ML, Sigmond T, Borsos E, Barna J, Erdelyi P, Takacs-Vellai K, Orosz L, Kovacs AL, Csikos G, Sass M, Vellai T. Longevity pathways converge on autophagy genes to regulate life span in Caenorhabditis elegans. Autophagy. 2008;4:330–338. doi: 10.4161/auto.5618. [DOI] [PubMed] [Google Scholar]
- 95.Twig G, Elorza A, Molina AJ, Mohamed H, Wikstrom JD, Walzer G, Stiles L, Haigh SE, Katz S, Las G, Alroy J, Wu M, Py BF, Yuan J, Deeney JT, Corkey BE, Shirihai OS. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 2008;27:433–446. doi: 10.1038/sj.emboj.7601963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Valeur HS, Valen G. Innate immunity and myocardial adaptation to ischemia. Basic Res Cardiol. 2009;104:22–32. doi: 10.1007/s00395-008-0756-6. [DOI] [PubMed] [Google Scholar]
- 97.Viana R, Aguado C, Esteban I, Moreno D, Viollet B, Knecht E, Sanz P. Role of AMP-activated protein kinase in autophagy and proteasome function. Biochem Biophys Res Commun. 2008;369:964–968. doi: 10.1016/j.bbrc.2008.02.126. [DOI] [PubMed] [Google Scholar]
- 98.Ward SG, Finan P. Isoform-specific phosphoinositide 3-kinase inhibitors as therapeutic agents. Curr Opin Pharmacol. 2003;3:426–434. doi: 10.1016/s1471-4892(03)00078-x. [DOI] [PubMed] [Google Scholar]
- 99.Yan L, Vatner DE, Kim SJ, Ge H, Masurekar M, Massover WH, Yang G, Matsui Y, Sadoshima J, Vatner SF. Autophagy in chronically ischemic myocardium. Proc Natl Acad Sci USA. 2005;102:13807–13812. doi: 10.1073/pnas.0506843102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Yitzhaki S, Huang C, Liu W, Gustafsson AB, Mentzer RM, Gottlieb RA. Autophagy is required for preconditioning by the adenosine A1 receptor-selective agonist CCPA. Basic Res Cardiol. 2009 doi: 10.1007/s00395-009-0006-6. (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Yoshimori T, Yamamoto A, Moriyama Y, Futai M, Tashiro Y. Bafilomycin A1, a specific inhibitor of vacuolar-type H(+)-ATPase, inhibits acidification and protein degradation in lysosomes of cultured cells. J Biol Chem. 1991;266:17707–17712. [PubMed] [Google Scholar]
- 102.Yousefi S, Perozzo R, Schmid I, Ziemiecki A, Schaffner T, Scapozza L, Brunner T, Simon H-U. Calpain-mediated cleavage of Atg5 switches autophagy to apoptosis. Nat Cell Biol. 2006;8:1124–1132. doi: 10.1038/ncb1482. [DOI] [PubMed] [Google Scholar]
- 103.Yuan H, Perry CN, Huang C, Iwai-Kanai E, Carreira RS, Glembotski CC, Gottlieb RA. LPS-Induced Autophagy Is Mediated by Oxidative Signaling in Cardiomyocytes and is Associated with Cytoprotection. Am J Physiol Heart Circ Physiol. 2008 doi: 10.1152/ajpheart.01051.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]