

Abstract
While it is clear that astrocytes and microglia cluster around dense-core amyloid plaques in Alzheimer disease (AD), whether they are primarily attracted to amyloid deposits or are just reacting to plaque-associated neuritic damage remains elusive. We postulate that astrocytes and microglia may differentially respond to fibrillar amyloid β (Aβ). Therefore, we quantified the size distribution of dense-core Thioflavin-S (ThioS)-positive plaques in the temporal neocortex of 40 AD patients and the microglial and astrocyte responses in their vicinity (≤50 μm), and performed correlations between both measures. As expected, both astrocytes and microglia were clearly spatially associated with ThioS-positive plaques (p = 0.0001, ≤50 μm vs. >50 μm from their edge), but their relationship to ThioS-positive plaque size differed; larger ThioS-positive plaques were associated with more surrounding activated microglia (p = 0.0026), but this effect was not observed with reactive astrocytes. Microglial response to dense-core plaques appears to be proportional to their size, which we postulate reflects a chemotactic effect of Aβ. By contrast, plaque-associated astrocytic response does not correlate with plaque size and seems to parallel the behavior of plaque-associated neuritic damage.
Keywords: Alzheimer disease, Amyloid plaques, Apolipoprotein E, Astrocytes, Microglia
INTRODUCTION
Dense-core amyloid plaques are one of the pathological hallmarks present in the brain of patients with Alzheimer disease (AD). They are defined by the presence of a compact core of fibrillar amyloid that can be stained with dyes specific for protein aggregates rich in β-pleated sheet structure, such as Thioflavin-S (ThioS) and Congo red. The microenvironment within and around these dense-core plaques is characterized by the presence of both neuritic dystrophies and astrocytic and microglial responses (1-12).
Plaque-associated neuritic changes include distorted dendrites and axons as well as bulbous varicosities or swellings that are generally thought to form as a result of the toxic effect of plaques on the surrounding neuropil (1-4). We have previously reported that, besides these obvious neuritic dystrophies, a more subtle expression of neuritic change consisting of an increased curvature of the trajectory of otherwise normal-looking neurites occurs in the plaque microenvironment (i.e. within 50 μm) (13-15). Importantly, this increased curvature ratio of neurites surrounding dense-core plaques is independent of plaque size, indicating that 1) it is not explained by a “mass effect” of plaques on surrounding neuritis, and 2) there is not proportionality between the extent of amyloid fibrils deposited and this local neurotoxic feature of dense-core plaques (15). Similarly, synaptic loss radiates out from the surface of dense-core plaques and is another feature of this 50-μm halo of neuronal damage that exists around them (16, 17).
The roles of plaque-associated glial responses in AD remain controversial (5-11, 18-20). Reactive astrocytes and activated microglial cells may be primarily attracted to plaques by amyloid β (Aβ) species and/or pro-inflammatory cytokines; however, an alternative and equally plausible explanation is that these glial cells cluster around dense-core plaques because they react to the plaque-induced neuritic damage, perhaps exerting neuroprotective effects (19).
We have previously shown that plaque-associated neuritic curvature is largely independent of plaque size (15); herein, we investigated the relationship between reactive glia and plaque size. We reasoned that if plaque-associated glial responses were chemotactically driven by Aβ deposition, then their magnitude would correlate with plaque size, whereas if glial cells were responding primarily to plaque-associated neuritic changes their numbers would not be influenced by plaque size. We performed stereology-based quantitative measures of reactive astrocytes and activated microglia and correlated them with the size distributions of dense-core (ThioS-positive) amyloid plaques from the temporal neocortex of a large group of AD patients. Our results suggest that astrocytes and microglia differentially respond to fibrillar Aβ deposits. The proportionality between plaque-associated microglial response and plaque size favors the idea of a direct chemotactic effect of Aβ on microglia, whereas the independence of plaque-associated astrocytic response from plaque size resembles the behavior of plaque-associated neuritic changes.
MATERIALS AND METHODS
Subjects
Forty AD patients were selected from the Massachusetts Alzheimer Disease Research Center Brain Bank. Demographic and clinical characteristics of this sample have been reported before (21, 22) (Table 1). All the study subjects or their next-of-kin gave informed consent for the brain donation and the Massachusetts General Hospital Institutional Review Board approved the study protocol. All the subjects fulfilled the NINCDS-ADRDA criteria for probable AD (23), and the NIA-Reagan criteria for high likelihood of AD (24). Because cerebrovascular disease is a major cause of focal gliosis, cases with cerebrovascular disease considered severe enough to contribute to a dementia syndrome were excluded. Cases with Lewy body pathology were also excluded.
Table 1.
Demographic Characteristics of the 40 Subjects with Alzheimer Disease Included in this Quantitative Neuropathological Study
| Sex | n = 26 female (65.0%) |
| Age at death | 77.6 ± 8.6 years |
| Age of onset | 66.9 ± 10.2 years |
| Disease duration | 9.9 years (5.7–15 years) |
| APOE genotype: | |
| APOEε4 carriers | n = 21 (52.5%) |
| APOEε4 alleles | n = 25 (31.2%) |
| Postmortem interval | 14.1 ± 6.2 hours |
Brain Specimens and Immunohistochemical Studies
Eight-μm-thick, formalin-fixed, paraffin-embedded sections from the temporal association isocortex (BA38) were deparaffinized for immunohistochemistry by standard methods. This region was selected because it is a multimodal association area that frequently has moderate numbers of dense-core plaques in AD sufficient to allow the sampling of 100 of these plaques per section (25, 26). Primary and fluorescently labeled secondary antibodies are listed in Table 2. Sections were microwaved for 20 minutes at 95°C in boiling citrate buffer (0.01 M, pH 6.0, 0.05% Tween-20) for antigen retrieval. Glial fibrillary acidic protein (GFAP) was used as a marker for reactive astrocytes (27). Ionized calcium-binding adaptor molecule 1 (Iba1) and CD68 were used as markers for activated microglia. Iba1 is a cytosolic protein upregulated in activated microglia (28), whereas CD68 is a widely used marker for cells with a phagocytic phenotype including macrophages and microglia that mainly locates in the lysosomal membrane (29). After immunostaining, the sections were counterstained with ThioS (Sigma-Aldrich, St. Louis, MO) 0.05% in 50% ethanol for 8 minutes, and cover-slipped with Vectashield mounting media with 4’,6-diamidino-2-phenylindole (DAPI) (Vector Laboratories, Burlingame, CA).
Table 2.
Antibodies Used in the Immunohistochemical Studies
| Primary Antigen | Vendor/Catalog# | Host | Dilution | Secondary Antibody |
|---|---|---|---|---|
| GFAP | Sigma (St. Louis, MO) Cat it#G9269 |
Rabbit | 1:1000 | Gt-Cy3 anti-Rb 1:200 |
| Iba1 | Wako (Osaka, Japan) Cat #019-19741 |
Rabbit | 1:250 | Gt-Cy3 anti-Rb 1:200 |
| CD68 | Dako (Carpinteria, CA) Cat#M0814 |
Mouse | 1:100 | Gt-Cy3 anti-Ms 1:200 |
Abbreviations: Cat, catalogue; GFAP, glial fibrillary acidic protein; Gt, goat; Iba1, ionized calcium-binding adaptor molecule 1. All secondary antibodies were purchased from Jackson ImmunoResearch Labs, West Grove, PA.
Dense-Core Plaque Size Distribution
The size distribution of dense-core plaques in the 40 AD cases was obtained by manually outlining the perimeter of the ThioS-positive plaques photographed for the analysis of glial responses close to plaques with the appropriate tool of the public domain software ImageJ (http://rsbweb.nih.gov/ij/). The fields photographed in each section (142,606 μm2) were randomly selected under the 20× objective of a BX51 Olympus epifluorescence microscope equipped with a DP70 CCD camera and a motorized stage controlled by the CAST stereology software (Olympus, Tokyo, Japan). To reach the end-point of 100 plaques while ensuring an adequate representation of all 6 cortical layers and a sufficient representation of the entire cortical ribbon of the specimens, the meander sampling was set at 1%. Cross-sectional areas of plaques from anti-GFAP, -CD68 and -Iba1-immunostained sections of the same case were pooled because the low meander sampling used made the double measurement of the same plaque in nearly-adjacent sections very unlikely (n = 300).
Stereology-Based Quantitation of Reactive Glial Cells Close to Dense-Core Plaques
Sections from the 40 AD patients were fluorescently stained for reactive astrocytes (GFAP) or activated microglia (both CD68 and Iba1) and dense-core plaques (ThioS-positive). The reactive glial cells surrounding dense-core plaques were quantified using 2 different approaches with the CAST-Olympus system.
First, in a glial-centered approach, 100 GFAP-positive astrocytes or Iba1-positive microglial cells per section were randomly selected under the 20× or the 40× objective, respectively, and their distance with respect to the closest dense-core plaque was measured with the appropriate tool of the CAST software, as previously described (15, 21, 30) (Fig. 1). Briefly, the entire cortical ribbon of the specimen was outlined as the region of interest. To reach the end-point of 100 cells while ensuring that all 6 cortical layers in the entire cortical ribbon were evenly covered, the meander sampling was tailored to each specimen and typically set between 1% and 4%. The size of the optical dissector was set at 10% (3565.2 μm2 for Iba1-positive microglia and 14260.2 μm2 for GFAP-positive astrocytes). Only glial cells with a visible nucleus in the DAPI staining were counted, and only if their cell soma fell within the counting frame or contacting either of its 2 green sides (Fig. 1). Astrocytes and microglial cells were classified as “close to plaques” if located within 50 μm from the edge of a plaque, and “far from plaques” if located beyond this boundary. We selected this 50 μm boundary based on our own and others’ previous data (11, 13-15, 21). Although we cannot exclude the possibility that reactive glial cells located beyond this boundary were actually within 50 μm from the edge of a plaque located above of or below the 8-μm-thick paraffin section, this seems unlikely because reactive astrocytes usually embrace plaques and penetrate them with their process and activated microglial cells are typically embedded within the plaque itself (5, 6). Densities of reactive astrocytes and activated microglial cells close to plaques were then calculated by dividing the number of cells counted by the total area of the dissectors analyzed.
Figure 1.

Glia-centered stereology-based study. (a) Diagram represents a typical dissector map resulting from the random sampling of a paraffin section of temporal cortex using the parameters described in the Materials and Methods section. (b) Only glial cells (in this example, glial fibrillary acidic protein [GFAP]-positive astrocytes, numbered, 1, 2, 3) with a DAPI-visible nucleus and whose soma fell within the counting frame and/or touched one of the 2 green sides of the counting frame (but none of the red sides) were considered. (c, d) Next, the presence of a Thioflavin-S-positive dense-core plaque was evaluated in the blue-green channel (c), and, if present, the distance between the glial cells and the closest plaque was measured with the appropriate tool of the CAST stereology software (d). Glial cells were considered close to plaques if their soma was located within 50 μm from the nearest plaque edge and far from them if located beyond this boundary.
Second, in a plaque-centered approach, 100 ThioS-positive plaques per specimen were randomly selected using the 20x objective and photographed as described above; the GFAP-positive astrocytes, CD68-positive microglia and Iba1-positive microglia close to their edge (within 50 μm) were then counted, measuring the distances with the appropriate tool of ImageJ (Fig. 2). Glial cells located close to 2 or more dense-core plaques according to this criterion were “split” among those plaques (i.e. 0.5 astrocytes or microglial cells for those close to 2 plaques, 0.33 for those close to 3 plaques, 0.25 for those close to 4 plaques, etc.). This conservative criterion was implemented to avoid possible bias of the results due to double counting of glial cells close to dense-core plaques in fields with a high density of plaques. These counts were then normalized to the size of the corresponding plaques (including the 50 μm boundary) to avoid differential sampling of glial cells within the neuropil due to differential plaque sizes. Thus, these results are expressed as number of glial cells per mm2 of area including the plaque and a 50 μm perimeter around the plaque. Again, only glial cells with a visible nucleus in the DAPI staining were considered.
Figure 2.
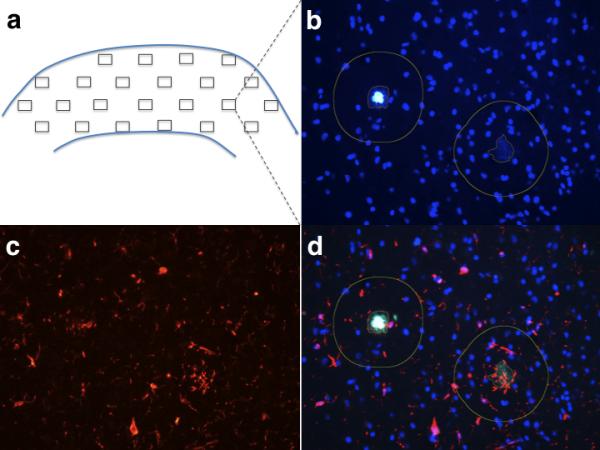
Plaque-centered stereology-based study. (a) Diagram represents a typical map of fields resulting from the random sampling of a paraffin section of temporal cortex using the parameters described in the Methods section. (b, c) Fields with Thioflavin-S (ThioS)-positive plaques present in the blue-green channel also exhibiting DAPI nuclear staining (b) were photographed and the corresponding images of glial cells in the red channel (c) were also taken. (d) Finally, the pairs of images from both channels corresponding to the same fields were merged in the ImageJ software, the sizes of the plaques were measured with the appropriate tool of the software, and numbers of glial cells with a DAPI-visible nucleus located within 50 μm from the edge of each plaque were manually counted.
APOE Genotyping
APOE genotype was determined in all the study subjects by restriction fragment length polymorphism analysis as described previously (31).
Statistics
Normality of datasets was assessed with the D'Agostino-Pearson omnibus test. Plaque size distributions are not Gaussian as they typically exhibit a positive skew (22, 32, 33). To correlate the stereology-based quantitative measures of glial responses with the plaque size distributions, we fit mixed-effects regression models using all the plaques measures from all AD subjects, and allowing for correlation within subject. We tested whether the slopes of the regression lines were different from zero at a 2-sided significance level of p < 0.05. We also compared the distributions of glial cells per dense-core plaque (number of cells within 50 μm of the nearest plaque edge) of AD APOEε4 carriers and non-carriers, using the clustered Wilcoxon rank-sum test at a 1-sided significance level of p < 0.05 (34). Lastly, to compare the density of glial cells in the proximity versus far away from dense-core plaques in each APOE group we performed a Wilcoxon sign-rank t test for paired values, and to compare these densities between both APOE groups we used the Wilcoxon unpaired t test. Statistical analyses were run in S.A.S. (version 9.2, Cary, NC) and GraphPad Prism for Mac (version 4.0, GraphPad Software Inc., La Jolla, CA). Graphs were performed with GraphPad Prism for Mac (version 4.0).
RESULTS
Relationship Between Reactive Glia and Dense-Core Plaque Size
To address the relationship between reactive glia and plaque size in the microenvironment around plaques, we quantified the density of reactive astrocytes and activated microglial cells close to dense-core plaques (≤50 μm) in sections doubly stained for GFAP or Iba1 and ThioS, and then correlated these densities with the size measures of dense-core plaques. First, we observed a significantly increased density of GFAP-positive astrocytes and Iba1-positive microglia in the vicinity of dense-core plaques, as compared to further away (>50 μm) from them (p = 0.0001 for both, Fig. 3), thereby confirming the expected clustering of reactive glia around dense-core plaques. The size of dense-core plaques correlated positively with the density of surrounding Iba1-positive activated microglia (p = 0.0026), although not with the density of GFAP-positive reactive astrocytes (p = 0.1564) (Table 3). These results suggest that larger dense-core plaques are associated with more activated microglial cells in their vicinity than smaller dense-core plaques.
Figure 3.

Association between reactive glia and dense-core plaques. (a, b) Note the intimate interaction between dense-core plaques (stained green with Thioflavin-S) and glial fibrillary acidic protein (GFAP)-positive astrocytes (a) and Iba1-positive microglia (b), in red. (c, d) The densities of GFAP-positive astrocytes or Iba1-positive microglial cells with respect to the nearest dense-core plaque were determined using stereology-based methods in a glia-centered fashion. GFAP-positive astrocytes (c) and Iba1-positive microglia (d) are more abundant in the vicinity (<50 μm) of dense-core plaques than far away from them (>50 μm) (*** p = 0.0001).
Table 3.
Summary of the Results of this Study
| Model 1 | Model 2 | Model 3 | ||||
|---|---|---|---|---|---|---|
| Glia-centered analyses | Estimate (SD) | p value | Estimate (SD) | p value | Estimate (SD) | p value |
| Dense-core plaque size vs. GFAP-positive astrocytes close | 0.8721 (0.6152) | p = 0.1564 | 0.8723 (0.6237) | p = 0.1620 | 0.6382 (0.6690) | p = 0.3402 |
| Dense-core plaque size vs. Iba1-positive microglia close | 1.8182 (0.6030) | p = 0.0026 | 1.8581 (0.6166) | p = 0.0026 | 1.7207 (0.6745) | p = 0.0108 |
| Plaque-centered analyses | Estimate (SD) | p value | Estimate (SD) | p value | Estimate (SD) | p value |
|---|---|---|---|---|---|---|
| Dense-core plaque size vs. GFAP-positive astrocytes close | 0.1642 (0.1313) | p = 0.2111 | 0.1637 (0.1314) | p = 0.2128 | 0.0482 (0.1405) | p = 0.7316 |
| Dense-core plaque size vs. Iba1-positive microglia close | 0.6946 (0.1018) | p < 0.0001 | 0.6950 (0.1019) | p < 0.0001 | 0.6432 (0.1050) | p < 0.0001 |
| Dense-core plaque size vs. CD68-positive microglia close | 0.7484 (0.0904) | p < 0.0001 | 0.7485 (0.0905) | p < 0.0001 | 0.7038 (0.0942) | p < 0.0001 |
Model 1 indicates the results of the bivariate regression analyses depicted in the first column. Model 2 is similar to Model 1 but controlling for the presence of at least one APOEε4 allele (i.e. APOEε4 carriers vs. non-carriers). Model 3 is similar to Model 1 but controlling for the number of APOEε4 alleles (i.e. none vs. 1 vs. 2 alleles). Values represent the estimates (SD) of the slopes and the p values of the corresponding regression models.
GFAP, glial fibrillary acidic protein; Iba1, ionized calcium-binding adaptor molecule 1.
To evaluate the relationship between glia and the size of fibrillar amyloid deposits further, we repeated the quantitative analyses in a plaque-centered fashion. We counted the number of GFAP-positive astrocytes and Iba1-positive microglial cells in the vicinity of randomly selected dense-core plaques (≤50 μm from their edge) and correlated these counts with the size of those individual dense-core plaques. We confirmed the above significant positive correlation between the size of dense-core plaques and the number of surrounding Iba1-positive microglial cells (p < 0.0001). Similar analyses for CD68-positive (phagocytic) microglia yielded the same results (p < 0.0001). By contrast, again GFAP-positive astrocytes did not correlate with plaque size (p = 0.6124) (Table 3; Fig. 4).
Figure 4.

Correlation between dense-core plaque size and number of surrounding activated microglial cells but not of reactive astrocytes. (a-c) Results of the plaque-centered approach in which 100 dense-core plaques per Alzheimer disease case were randomly sampled, and their sizes and the numbers of surrounding Iba1-positive microglial cells, CD68-positive microglial cells, and glial fibrillary acidic protein (GFAP)-positive astrocytes were determined. The numbers of glial cells within 50 μm from the plaque edge (“close to plaques”) are normalized to the plaque size and expressed as a density per mm2. For clarity, plaque size is presented in 500-μm2 intervals. Symbols represent the median values and bars represent the interquartile range. Note the positive correlation between plaque size and both Iba1-positive (a) and CD68-positive (b) activated microglia, but not with GFAP-positive reactive astrocytes (c).
The APOEε4 Genotype Does Not Impact Glial Responses to Plaques
Prior postmortem quantitative studies have established that the APOEε4 allele is associated with a higher cortical amyloid plaque burden (33, 35-38). More recent studies have shown that apoE is a cofactor for the clearance of Aβ by astrocytes and microglia (39, 40), and that the efficiency of this clearance might be isoform-dependent, with apoE4 being less effective than apoE3 (41, 42). In addition, APOEε4 knock-in mice exhibit enhanced astrocytic and microglial activation upon treatment with lipopolysaccharide as compared to APOEε3 knock-in mice (43), and apoE has been involved in the migration of microglia towards common chemotactic stimuli such as the complement factor C5a and ATP in an isoform-differential fashion (apoE3 > apoE4) (44).
We therefore investigated whether astrocytic and microglial responses to dense-core plaques are similar in APOEε4 carriers vs. non-carriers. Both glia- and plaque-centered analyses revealed a quantitatively similar association of reactive astrocytes and activated microglial cells to dense-core plaques in APOEε4 carriers and non-carriers (Fig. 5). Moreover, controlling for the presence of the APOEε4 allele or for the number of APOEε4 alleles had no significant influence in the relationship between plaque size and associated astrocytic or microglial responses (Table 3).
Figure 5.

Lack of differential effect of APOE genotype on the association between reactive glia and dense-core plaques. (a, b) Glia-centered stereology-based study. Glial fibrillary acidic protein (GFAP)-positive astrocytes are more abundant in the vicinity (≤50 μm) of dense-core plaques than far away from them (>50 μm) in both APOEε4 carriers (*** p = 0.0002) and non-carriers (* p = 0.0289). The magnitude of the astroglial response does not differ between both APOE groups in either the proximity to plaques (p = 1.0000) or far from them (p = 0.2334) (a). Similarly, Iba1-positive microglial cells are more abundant in the vicinity of dense-core plaques than far away from them in both APOEε4 carriers (# p < 0.0001) and non-carriers (* p = 0.0401), but no significant difference is observed between both genotypes (close: p = 0.3571; far: p = 0.4011) (b). (c-e) Plaque-centered stereology-based study. Numbers of GFAP-positive astrocytes (c), CD68-positive microglia (e), and Iba1-positive microglia (d) in the vicinity (μ50 μm) of 100 randomly selected dense-core plaques were counted and normalized to the size of those plaques. The distributions of values obtained are remarkably similar in APOEε4 carriers and non-carriers (p = 0.55, p = 0.96, and p = 0.96, respectively). Graphs c–e show the median values for each AD case, but the entire distributions were used in the statistical analyses.
DISCUSSION
The nature of the interaction between amyloid plaques and reactive glia has been debated since the first descriptions of clusters of glial cells within and around senile plaques (5-7). We approached this question by studying the relationship between plaque-associated (within 50 μm from the nearest plaque edge) reactive glia and plaque size. Although postmortem studies cannot establish causal relationships, the results of our present neuropathological quantitative study indicate that these 2 glial cell types may have differential responses to ThioS-positive fibrillar dense-core deposits.
Microglial Response to Amyloid Plaques
Our observation that microglial clustering in the vicinity of dense-core plaques is proportional to their size can be interpreted as evidence favoring a model in which a chemotactic signal engages microglia. Although whether such a signal is Aβ or another plaque-associated component remains unclear (44-48), substantial in vitro data suggest that the Aβ peptide is a strong candidate (49-52). It should be noted that while compact plaques are defined by the presence of a dense core of insoluble Aβ fibrils, they also exhibit a peripheral halo of more soluble and toxic oligomeric species of Aβ (16, 17, 53, 54); hence, our results do not distinguish the effects of fibrillar Aβ from that of soluble oligomeric Aβ.
Our observation is also consistent with a relatively ineffective role of microglia at clearing amyloid fibrils (55-59), or at least argues against a major role of microglia in plaque removal in the AD brain (60, 61). On the other hand, microglial responses to plaques may contribute to stabilizing plaque sizes, which remain relatively stable throughout the clinical course of the disease (22, 33), perhaps by effectively phagocytosing and degrading the smaller and more soluble Aβ species in the halo around dense-core plaques (59).
Astrocytic Response to Amyloid Plaques
In sharp contrast to the microglial response, the magnitude of astrocytic reaction did not correlate with plaque size. This observation resembles our prior finding of the absence of correlation between plaque size and neurite curvature ratio – an expression of plaque-induced neuritic damage subtler than plaque-embedded neuritic dystrophies (15) – and suggests that plaque-associated reactive astrocytes are preferentially reacting to plaque-induce neuritic damage rather than to Aβ itself. Because peri-plaque Aβ oligomers can directly induce the morphological neurodegenerative triad of spine loss, dendritic simplification and neuritic dystrophies (62), it is difficult to dissect the primary trigger of astrocytic reaction. However, our interpretation is supported by a recent study in APP/PS1 mice with a GFAP and vimentin null background. Deletion of both glial filament proteins prevented the contact of astrocytes with plaques and resulted in a marked increase in the number of dystrophic neurites per plaque, suggesting that plaque-associated reactive astrocytes actually protect neurons surrounding the plaques (19). Further studies in experimental systems will be necessary to evaluate the primary cause of plaque-associated astrocytic responses.
Role of APOE Genotype in Glial Responses to Amyloid Plaques
Importantly, carrying the APOEε4 allele had no significant impact on the above results; this argues against a differential effect of the apoE isoforms on glial cell activation and migration towards plaques and extends our prior observation of a similar magnitude of glial responses at postmortem examination between APOEε4 carriers and non-carriers (21, 63). However, because the apolipoprotein E is mainly secreted by astrocytes and, to a lesser extent, microglia, the APOE genotype may still influence other aspects or features of the astrocytic and microglial phenotypic change that occurs in AD by as yet unknown autocrine or paracrine mechanisms (64).
In summary, we observed that activated microglia and reactive astrocytes differentially interact with ThioS-positive fibrillar amyloid deposits. Microglial responses to dense-core plaques appear to be proportional to plaque size, whereas plaque-associated astrocytic responses do not correlate with plaque size. Aβ itself may be the primary chemotactic signal for activated microglia, whereas astrocytes might primarily respond to plaque-associated neuritic damage.
ACKNOWLEDGMENT
The authors want to thank the patients and caregivers involved in research at Massachusetts General Hospital.
This work was funded by the National Institutes of Health (grants P50AG05134 and AG08487). Dr. Alberto Serrano-Pozo was funded by a Research Fellowship from Fundación Alfonso Martín Escudero (Madrid, Spain).
Footnotes
The authors declare that they have no conflict of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Masliah E, Mallory M, Hansen L, et al. Synaptic and neuritic alterations during the progression of Alzheimer's disease. Neurosci Lett. 1994;174:67–72. doi: 10.1016/0304-3940(94)90121-x. [DOI] [PubMed] [Google Scholar]
- 2.Su JH, Cummings BJ, Cotman CW. Plaque biogenesis in brain aging and Alzheimer's disease. I. Progressive changes in phosphorylation states of paired helical filaments and neurofilaments. Brain Res. 1996;739:79–87. doi: 10.1016/s0006-8993(96)00811-6. [DOI] [PubMed] [Google Scholar]
- 3.Su JH, Cummings BJ, Cotman CW. Plaque biogenesis in brain aging and Alzheimer's disease. II. Progressive transformation and developmental sequence of dystrophic neurites. Acta Neuropathol. 1998;96:463–71. doi: 10.1007/s004010050920. [DOI] [PubMed] [Google Scholar]
- 4.Dickson TC, Vickers JC. The morphological phenotype of amyloid-beta deposits and associated neuritic change in Alzheimer's disease. Neuroscience. 2001;105:99–107. doi: 10.1016/s0306-4522(01)00169-5. [DOI] [PubMed] [Google Scholar]
- 5.Itagaki S, McGeer PL, Akiyama H, et al. Relationship of microglia and astrocytes to amyloid deposits of Alzheimer disease. J Neuroimmunol. 1989;24:173–82. doi: 10.1016/0165-5728(89)90115-x. [DOI] [PubMed] [Google Scholar]
- 6.Wisniewski HM, Wegiel J. Spatial relationships between astrocytes and classical plaque components. Neurobiol Aging. 1991;12:593–600. doi: 10.1016/0197-4580(91)90091-w. [DOI] [PubMed] [Google Scholar]
- 7.Ohgami T, Kitamoto T, Shin R-W, et al. Increased senile plaques without microglia in Alzheimer's disease. Acta Neuropathol. 1991;81:242–7. doi: 10.1007/BF00305864. [DOI] [PubMed] [Google Scholar]
- 8.Pike CJ, Cummings BJ, Cotman CW. Early association of reactive astrocytes with senile plaques in Alzheimer's disease. Exp Neurol. 1995;132:172–9. doi: 10.1016/0014-4886(95)90022-5. [DOI] [PubMed] [Google Scholar]
- 9.Fukumoto H, Asami-Odaka A, Suzuki N, et al. Association of Aβ40-positive senile plaques with microglial cells in the brains of patients with Alzheimer's disease and in non-demented aged individuals. Neurodegeneration. 1996;5:13–7. doi: 10.1006/neur.1996.0002. [DOI] [PubMed] [Google Scholar]
- 10.Sheng JG, Mrak RE, Griffin WST. Neuritic plaque evolution in Alzheimer's disease is accompanied by transition of activated microglia from primed to enlarged to phagocytic forms. Acta Neuropathol. 1997;94:1–5. doi: 10.1007/s004010050664. [DOI] [PubMed] [Google Scholar]
- 11.Shah P, Lal N, Leung E, et al. Neuronal and axonal loss are selectively linked to fibrillar amyloid-β within plaques of the aged primate cerebral cortex. Am J Pathol. 2010;177:325–33. doi: 10.2353/ajpath.2010.090937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Serrano-Pozo A, Frosch MP, Masliah E, et al. Neuropathological alterations in Alzheimer disease. Cold Spring Harb Perspect Med. 2011;1:a006189. doi: 10.1101/cshperspect.a006189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Knowles RB, Wyart C, Buldyrev SV, et al. Plaque-induced neurite abnormalities: implications for disruption of neural networks in Alzheimer's disease. Proc Natl Acad Sci USA. 1999;96:5274–9. doi: 10.1073/pnas.96.9.5274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.D'Amore JD, Kajdasz ST, McLellan ME, et al. In vivo multiphoton imaging of a transgenic mouse model of Alzheimer disease reveals marked thioflavin-S-associated alterations in neurite trajectories. J Neuropathol Exp Neurol. 2003;62:137–45. doi: 10.1093/jnen/62.2.137. [DOI] [PubMed] [Google Scholar]
- 15.Serrano-Pozo A, William CM, Ferrer I, et al. Beneficial effect of human anti-amyloid-β active immunization on neurite morphology and tau pathology. Brain. 2010;133:1312–27. doi: 10.1093/brain/awq056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Koffie RM, Meyer-Luehmann M, Hashimoto T, et al. Oligomeric amyloid beta associates with postsynaptic densities and correlates with excitatory synapse loss near senile plaques. Proc Natl Acad Sci. 2009;106:4012–7. doi: 10.1073/pnas.0811698106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Koffie RM, Hashimoto T, Tai HC, et al. Apolipoprotein E4 effects in Alzheimer's disease are mediated by synaptotoxic oligomeric amyloid-β. Brain. 2012;135:2155–68. doi: 10.1093/brain/aws127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vehmas AK, Kawas CH, Stewart WF, et al. Immune reactive cells in senile plaques and cognitive decline in Alzheimer's disease. Neurobiol Aging. 2003;24:321–31. doi: 10.1016/s0197-4580(02)00090-8. [DOI] [PubMed] [Google Scholar]
- 19.Kraft AW, Hu X, Yoon H, et al. Attenuating astrocyte activation accelerates plaque pathogenesis in APP/PS1 mice. FASEB J. 2013;27:1–12. doi: 10.1096/fj.12-208660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wyss-Coray T. Inflammation in Alzheimer disease: driving force, bystander or beneficial response? Nat Med. 2006;12:1005–15. doi: 10.1038/nm1484. [DOI] [PubMed] [Google Scholar]
- 21.Serrano-Pozo A, Mielke ML, Gómez-Isla T, et al. Reactive glia not only associates with plaques but also parallels tangles in Alzheimer disease. Am J Pathol. 2011;179:1373–84. doi: 10.1016/j.ajpath.2011.05.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Serrano-Pozo A, Mielke ML, Muzitansky A, et al. Stable size distribution of amyloid plaques over the course of Alzheimer disease. J Neuropath Exp Neurol. 2012;71:694–701. doi: 10.1097/NEN.0b013e31825e77de. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McKhann G, Drachman D, Folstein M, et al. Clinical diagnosis of Alzheimer's disease: Report of the NINCDS-ADRDA Work Group under the auspices of the Department of Health and Human Services Task Force on Alzheimer's Disease. Neurology. 1984;34:939–44. doi: 10.1212/wnl.34.7.939. [DOI] [PubMed] [Google Scholar]
- 24.Consensus recommendations for the postmortem diagnosis of Alzheimer's disease. The National Institute of Aging, and Reagan Institute Working Group on Diagnostic Criteria for the Neuropathological Assessment of Alzheimer's Disease. Neurobiol Aging. 1997;18:S1–S2. [PubMed] [Google Scholar]
- 25.Arnold SE, Hyman BT, Flory J, et al. The topographical and neuroanatomical distribution of neurofibrillary tangles and neuritic plaques in the cerebral cortex of patients with Alzheimer's disease. Cereb Cortex. 1991;1:103–16. doi: 10.1093/cercor/1.1.103. [DOI] [PubMed] [Google Scholar]
- 26.Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991;82:239–59. doi: 10.1007/BF00308809. [DOI] [PubMed] [Google Scholar]
- 27.Beach TG, Walker R, McGeer EG. Patterns of gliosis in Alzheimer's disease and aging cerebrum. Glia. 1989;2:420–36. doi: 10.1002/glia.440020605. [DOI] [PubMed] [Google Scholar]
- 28.Ito D, Imai Y, Ohsawa K, et al. Microglia-specific localization of a novel calcium binding protein, Iba1. Mol Brain Res. 1998;57:1–9. doi: 10.1016/s0169-328x(98)00040-0. [DOI] [PubMed] [Google Scholar]
- 29.Verbeek MM, Otte-Höller I, Wesseling P, et al. A lysosomal marker for activated microglial cells involved in Alzheimer classic senile plaques. Acta Neuropathol. 1995;90:493–503. doi: 10.1007/BF00294811. [DOI] [PubMed] [Google Scholar]
- 30.Hyman BT, Gómez-Isla T, Irizarry MC. Stereology: a practical primer for neuropathology. J Neuropathol Exp Neurol. 1998;57:305–10. doi: 10.1097/00005072-199804000-00001. [DOI] [PubMed] [Google Scholar]
- 31.Ingelsson M, Shin Y, Irizarry MC, et al. Genotyping of apolipoprotein E: comparative evaluation of different protocols. Curr Protoc Hum Genet. 2003;9.14:1–13. doi: 10.1002/0471142905.hg0914s38. [DOI] [PubMed] [Google Scholar]
- 32.Hyman BT, Marzloff K, Arriagada PV. The lack of accumulation of senile plaques or amyloid burden in Alzheimer's disease suggests a dynamic balance between amyloid deposition and resolution. J Neuropathol Exp Neurol. 1993;52:594–600. doi: 10.1097/00005072-199311000-00006. [DOI] [PubMed] [Google Scholar]
- 33.Hyman BT, West HL, Rebeck GW, et al. Quantitative analysis of senile plaques in Alzheimer disease: observation of log-normal size distribution and molecular epidemiology of differences associated with apolipoprotein E genotype and trisomy 21 (Down syndrome). Proc Natl Acad Sci USA. 1995;92:3586–90. doi: 10.1073/pnas.92.8.3586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Datta S, Satten GA. Rank-sum tests for clustered data. J Am Stat Assoc. 2005;100:908–15. [Google Scholar]
- 35.Schmechel DE, Saunders AM, Strittmatter WJ, et al. Increased amyloid β-peptide deposition in cerebral cortex as a consequence of apolipoprotein E genotype in late-onset Alzheimer disease. Proc Natl Acad Sci USA. 1993;90:9649–53. doi: 10.1073/pnas.90.20.9649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Polvikoski T, Sulkava R, Haltia M, et al. Apolipoprotein E, dementia, and cortical deposition of beta-amyloid protein. N Engl J Med. 1995;333:1242–7. doi: 10.1056/NEJM199511093331902. [DOI] [PubMed] [Google Scholar]
- 37.Gómez-Isla T, West HL, Rebeck GW, et al. Clinical and pathological correlates of apolipoprotein E ε4 in Alzheimer's disease. Ann Neurol. 1996;39:62–70. doi: 10.1002/ana.410390110. [DOI] [PubMed] [Google Scholar]
- 38.McNamara MJ, Gómez-Isla T, Hyman BT. Apolipoprotein E genotype and deposits of Aβ40 and Aβ42 in Alzheimer disease. Arch Neurol. 1998;55:1001–4. doi: 10.1001/archneur.55.7.1001. [DOI] [PubMed] [Google Scholar]
- 39.Koistinaho M, Lin S, Wu X, et al. Apolipoprotein E promotes astrocyte colocalization and degradation of deposited amyloid-beta peptides. Nat Med. 2004;10:719–26. doi: 10.1038/nm1058. [DOI] [PubMed] [Google Scholar]
- 40.Terwell D, Steffensen KR, Verghese PB, et al. Critical role of astroglial apolipoprotein E and liver X receptor-α expression for microglial Aβ phagocytosis. J Neurosci. 2011;31:7049–59. doi: 10.1523/JNEUROSCI.6546-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jiang Q, Lee CY, Mandrekar S, et al. ApoE promotes the proteolytic degradation of Aβ. Neuron. 2008;58:681–93. doi: 10.1016/j.neuron.2008.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhao L, Lin S, Bales KR, et al. Macrophage-mediated degradation of β-amyloid via an apolipoprotein E-dependent mechanism. J Neurosci. 2009;29:3603–12. doi: 10.1523/JNEUROSCI.5302-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhu Y, Nwabuisi-Heath E, Dumanis SB, et al. APOE genotype alters glial activation and loss of synaptic markers in mice. Glia. 2012;60:559–69. doi: 10.1002/glia.22289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cudaback E, Li X, Montine KS, et al. Apolipoprotein E isoform-dependent microglia migration. FASEB J. 2011;25:2082–91. doi: 10.1096/fj.10-176891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ishikuza K, Kimura T, Igata-yi R, et al. Identification of monocyte chemoattractant protein-1 in senile plaques and reactive microglia of Alzheimer's disease. Psychiatry Clin Neurosci. 1997;51:135–8. doi: 10.1111/j.1440-1819.1997.tb02375.x. [DOI] [PubMed] [Google Scholar]
- 46.Stoltzner SE, Grenfell TJ, Mori C, et al. Temporal accrual of complement proteins in amyloid plaques in Down's syndrome with Alzheimer's disease. Am J Pathol. 2000;156:489–99. doi: 10.1016/S0002-9440(10)64753-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wyss-Coray T, Loike JD, Brionne TC, et al. Adult mouse astrocytes degrade amyloid-β in vitro and in situ. Nat Med. 2003;9:453–7. doi: 10.1038/nm838. [DOI] [PubMed] [Google Scholar]
- 48.Ryu JK, Cho T, Choi HB, et al. Microglial VEGF receptor response is an integral chemotactic component in Alzheimer's disease pathology. J Neurosci. 2009;29:3–13. doi: 10.1523/JNEUROSCI.2888-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Davis JB, McMurray HF, Schubert D. The amyloid beta-protein of Alzheimer's disease is chemotactic for mononuclear phagocytes. Biochem Biophys Res Comm. 1992;189:1096–1100. doi: 10.1016/0006-291x(92)92317-q. [DOI] [PubMed] [Google Scholar]
- 50.Yan SD, Chen X, Fu J, et al. RAGE and amyloid-β peptide neurotoxicity in Alzheimer's disease. Nature. 1996;382:685–91. doi: 10.1038/382685a0. [DOI] [PubMed] [Google Scholar]
- 51.El Khoury J, Hickman SE, Thomas CA, et al. Scavenger receptor-mediated adhesion of microglia to β-amyloid fibrils. Nature. 1996;382:716–9. doi: 10.1038/382716a0. [DOI] [PubMed] [Google Scholar]
- 52.Le Y, Gong W, Tiffany HL, et al. Amyloid β42 activates a G-protein-coupled chemoattractant receptor, FPR-like-1. J Neurosci. 2001;21:RC123(1-5). doi: 10.1523/JNEUROSCI.21-02-j0003.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Shankar GM, Li S, Mehta TH, et al. Amyloid-beta protein dimers isolated directly from Alzheimer's brains impair synaptic plasticity and memory. Nat Med. 2008;14:837–42. doi: 10.1038/nm1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hong S, Quintero-Monzon O, Ostaszewski BL, et al. Dynamic analysis of amyloid β-protein in behaving mice reveals opposing changes in ISF versus parenchymal Aβ during age-related plaque formation. J Neurosci. 2011;31:15861–9. doi: 10.1523/JNEUROSCI.3272-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hickman SE, Allison EK, El Khoury J. Microglial dysfunction and defective β-amyloid clearance pathways in aging Alzheimer's disease mice. J Neurosci. 2008;28:8354–60. doi: 10.1523/JNEUROSCI.0616-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bolmont T, Haiss F, Eicke D, et al. Dynamics of the microglial/amyloid interaction indicate a role in plaque manteinance. J Neurosci. 2008;28:4283–92. doi: 10.1523/JNEUROSCI.4814-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Grathwohl SA, Kälin RE, Bolmont T, et al. Formation and manteinance of Alzheimer's disease β-amyloid plaques in the absence of microglia. Nat Neurosci. 2009;12:1358–60. doi: 10.1038/nn.2432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Heneka MT, Nadrigny F, Regen T, et al. Locus ceruleus controls Alzheimer's disease pathology by modulating microglial functions through norepinephrine. Proc Natl Acad Sci USA. 2010;107:6058–63. doi: 10.1073/pnas.0909586107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Liu Z, Condello C, Schain A, et al. CX3CR1 in microglia regulates brain amyloid deposition through selective protofibrillar amyloid-β phagocytosis. J Neurosci. 2010;30:17091–101. doi: 10.1523/JNEUROSCI.4403-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Simard AR, Soulet D, Gowing G, et al. Bone marrow-derived microglia play a critical role in restricting senile plaque formation in Alzheimer's disease. Neuron. 2006;49:489–502. doi: 10.1016/j.neuron.2006.01.022. [DOI] [PubMed] [Google Scholar]
- 61.El Khoury J, Toft M, Hickman SE, et al. Ccr2 deficiency impairs microglial accumulation and accelerates progression of Alzheimer-like disease. Nat Med. 2007;13:432–8. doi: 10.1038/nm1555. [DOI] [PubMed] [Google Scholar]
- 62.Wu H, Hudry E, Hashimoto T, et al. Amyloid beta induces the morphological neurodegenerative triad of spine loss, dendritic simplification, and neuritic dystrophies through calcineurin activation. J Neurosci. 2010;30:2636–49. doi: 10.1523/JNEUROSCI.4456-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ingelsson M, Fukumoto H, Newell KL, et al. Early Aβ accumulation and progressive synaptic loss, gliosis, and tangle formation in AD brain. Neurology. 2004;62:925–31. doi: 10.1212/01.wnl.0000115115.98960.37. [DOI] [PubMed] [Google Scholar]
- 64.Egensperger R, Kösel S, von Eitzen U, et al. Microglial activation in Alzheimer disease: association with APOE genotype. Brain Pathol. 1998;8:439–47. doi: 10.1111/j.1750-3639.1998.tb00166.x. [DOI] [PMC free article] [PubMed] [Google Scholar]