

Abstract
Inflammatory bowel disease (IBD) is a group of inflammatory disorders in the small and large intestines. Several studies have proved that persistent and disregulated host/microbial interactions are required for the development of IBD. It is well known that chronic IBD is strongly associated with an increased risk of developing colorectal cancer by 0.5–1% annually, 8–10 years after the initial diagnosis. To detect the tiny dysplasia or early stage of cancer in chronic IBD patients, a tremendous amount of effort is currently directed for improving colonoscopic technology and noninvasive serological marker development. However, there is only a limited amount of data available to understand the exact mechanism of how long term chronic colitis is connected to the development of colorectal tumors. Recently, our group has identified that significantly increased expression of chitinase 3-like 1 (CHI3L1) molecule in non-dysplastic mucosa from patients with IBD and remote dysplasia/cancer, compared to patients with IBD without dysplasia or healthy controls. CHI3L1 seems to contribute to the proliferation, migration, and neoplastic progression of colonic epithelial cells (CECs) under inflammatory conditions. Furthermore, the CHI3L1-mediated intracellular signaling cascade is likely to interact with TLR4 signaling in CECs. In this review article, we have concisely summarized the cellular and molecular mechanisms underlining the development of IBD and colitis-associated cancer, with particular focus on the CHI3L1-and TLR4-signaling pathways in CECs.
Keywords: mammalian chitinase, inflammation, microbiota, colitis-associated cancer, autoimmunity
Introduction
Inflammatory bowel disease (IBD), including Crohn's disease (CD) and ulcerative colitis (UC), is a complex disorder characterized by chronic inflammation of the gastrointestinal tract1. Involvement of bacteria, in particular the presence of commensal bacteria is required for the development of IBD2, 3. Data from animal models of colitis suggests that the fundamental mechanism of IBD is much more complicated than previously predicted, and several factors including cell types, tissue specificity, and genetic/environmental factors are tightly involved in the pathogenesis of IBD4, 5. Immunological abnormalities are also key factors in this pathogenesis6. IBD is most common in developed countries, affecting the quality of life of 1.4 million individuals in the United States7, and the affected patients will suffer from the chronic inflammation throughout their lives. IBD is characterized by excess immune responses to the intestinal microbiota but differs in the site and nature of the inflammatory pathology depending on epithelial defense, IL-23/Th17 axis, and immune regulation8–10.
Toll like receptors (TLRs) are type I transmembrane glycoprotein receptors. So far, thirteen TLRs (TLR1-TLR13) have been identified in humans and mice11. TLR-mediated adapter proteins and kinases play crucial roles in the following signaling pathways, which is divided into two major pathways, the MyD88 (Myeloid differentiation primary response gene 88)-dependent and TRIF (TIR-domain-containing adapter-inducing interferon-β)-dependent pathways. MyD88 and MyD88-like adapter mediate an early response, while TRIF and TRIF-related adapter molecule leads to the rather delayed cascade12. On cell surface, TLR2 forms heterodimer with TLR1 to recognize triacylated lipoproteins or TLR6 to recognize diacylated lipoproteins from Gram-positive bacteria, mycobacteria, or mycoplasma13. In contrast, TLR4 homodimer is the main receptor for Gram-negative bacterial LPS14, of which ligation is conjugated with 3 accessory molecules, including CD14, LPS-binding protein (LBP) and MD-215, 16. TLR4/MD2 expression on intestinal epithelial cells is negatively regulated and is kept at low levels under normal conditions, but is significantly upregulated during the development of IBD17. Recently, Ferwerda et al identified two polymorphisms of TLR4 Asp299Gly (D299G) and Thr399Ile (T399I) in populations from Europe, Asia and Africa, which have been positively associated with susceptibility to Gram-negative bacterial infections and septic shock 18. Furthermore, a recent report revealed that breast cancer patients harboring the TLR4- D299G mutation is related to an increased frequency of cancer metastasis after conventional chemotherapy19. The same group also found that the TLR4 mutation in mice reduces the efficacy of both radiation as well as chemotherapy, suggesting a crucial role of TLR4-mediated signaling not only in inflammatory responses but also in cancer development.
Chitinase 3-like 1 (CHI3L1, also known as YKL-40) is classified in the glycosyl hydrolase 18 family based on the structural similarity with other chitinases such as plant and bacterial chitinases20. CHI3L1 is produced by restricted types of cells, including osteosarcoma cells, chondrocytes, smooth muscle cells, macrophages, neutrophils and Colonic epithelial cells (CECs)21. This protein is frequently found in inflammatory environments; however the factors determining its production in these pathological conditions are particularly unknown22. Our group has reported previously that CHI3L1 is highly induced in CECs and macrophages with intestinal inflammation and enhances potentially pathogenic, but not non-pathogenic, bacterial adhesion and invasion on/into CECs23. A Recent report from our group also suggests that CHI3L1 plays a major role in inflammation-associated neoplastic changes in CECs, and CHI3L1 may effectively promotes tumor development by enhancing cell proliferation, migration, angiogenesis, and cell survival in CECs and macrophages24, 25. In this review article, we will discuss the potential linkage between TLR4- and CHI3L1-signaling pathways on CECs in inflammatory bowel disease and the subsequent colitis-associated neoplastic processes in epithelial cells.
1. TLR4 expression on colonic epithelial cells (CECs)
TLRs, are a class of transmembrane, non-catalytic pattern recognition receptors whose role is to discern self from non-self by broadly conserved molecular patterns26. They are directly involved in the induction of pro/anti-inflammatory genes and the activation of immune responses27. TLRs comprise a total of 13 mammalian transmembrane proteins, 10 in humans and 12 in mice, which each contain multiple leucine rich repeat motifs in a large extracellular domain, and a highly conserved region in the intracellular tail named the toll-interleukin receptor (TIR) domain. This TIR domain contains homo and hetero-dimmer sub-units, which interact with receptor ligands, and recruit adapter proteins for downstream cell signaling28, 29.
TLR4 is the major intracellular signaling complex for lipopolysaccharides (LPS), which are found in gram negative bacteria cell membranes and act as a direct ligand to the receptor30. TLR4 is also capable of binding to other exogenous stimuli such as fusion proteins from respiratory syncytial virus31, envelope proteins from mouse mammary tumor virus32 and bacterial HSP6033. TLR4 is found in a receptor complex requiring several accessory molecules: CD14, LPS-binding-protein (LBP), and MD-234. LPS is transferred to cell surface CD14 by LBP, which then presents the ligand to TLR4 which is bound to MD2. Without interacting with MD2, TLR4 is nonfunctional and unable to induce the subsequent signaling cascade35. Recently, it was shown that CD14 is required for the microbe-induced endocytosis of TLR4 to the various cell organelles36.
TLR4 induces a signaling cascade primarily dependent on MyD88, which is a universal adapter protein used by all TLRs except TLR3. Downstream, TLR4-mediated signaling leads to activation of nuclear factor kappa-light-chain-enhancer of activated B cells (NF- κB), activator protein-1 (AP-1), mitogen-activated protein kinase (MAPK) and other transcription factors. However, it is also capable of signaling through MyD88 independent TRIF pathway27. Through both MyD88 dependent NF- κB and MAPK activation, and TRIF pathway activation, transcription of inflammatory cytokines are initiated37.
Under normal conditions within the colonic epithelium, TLR4 expression in human colonic mucosa samples is barely detectable through immunohistochemical staining38. TLR4 is down regulated at the cell surface, and the mechanism of hyporesponsiveness in CECs to gut lumen LPS was found to be due to a down-regulation of MD2 and TLR4 expressions. However intracellular TLR4 remained functionally intact, perhaps as a potential defense against pathogenic stimulus, capable of rapidly upregulating TLR4 and MD2 without causing chronic over-activation39. Another group found that CECs do not express mRNA or protein for CD14 in the human CEC lines HT-29, Caco-2, and T-84, and mouse CEC line CMT93, and this has been proposed as another mechanism behind CEC hyporesponsiveness40. TLR inhibition is required to prevent inappropriate activation despite large amounts of LPS in the gut lumen. It has also been shown that the CECs transfer TLR4 from the cell surface to the cytoplasm to reduce lumenal sensing, as well as up-regulate the TLR downstream inhibitory molecule toll interacting protein (Tollip) to prevent over stimulation41. Other inhibitory controls include single immunoglobulin IL-1R -related molecule (SIGIRR) which inhibits TLR4's interaction with MyD88 through its TIR domain42. Triad domain-containing protein 3 variant A (TRIAD3A) is a E3 ubiquitin protein ligase which interacts with TIR-domain containing proteins such as toll-interleukin 1 receptor domain containing adaptor protein (TIRAP), toll-like receptor adaptor molecule 1 (TRIF), and receptor interacting protein-1 (RIP-1)43. Knockdown of TRIAD3A upregulates NF-κB, while over-expression down regulates NF-κB44. Interleukin-1 receptor-associated kinase 1-M (IRAK-M) inhibits MyD88 associated signaling by preventing the dissociation of IRAKs to MyD88 and the formation of IRAK-TRAF6 (Tumor necrosis factor receptor associated factor 6) complexes45. A20 is a negative regulator of TLR signaling and a de-ubiquitinating enzyme, which removes K63-linked ubiquitin molecules from TRAF6 to abrogate downstream signaling to NF-κB and other pro-inflammatory cytokines46. A recent study has proved that A20 is an early response negative regulator of TLR5 signaling in intestinal epithelial cells during inflammation that regulates the innate immune system in the intestine47. Additional negative regulatory molecules include microRNA miR-21, which suppresses programmed cell death protein 4 (PDCD4), an activator of NF-κB, thus regulating the inflammatory response to LPS48. Vaccinia virus protein (VACV) A52R interferes with the association of IRAK2 and TRAF6 to prevent complex formation, with this protein being a viral mechanism to suppress host defense49. TRAF family member-associated NF-κB activator (TANK) contrary to its name, negatively regulates TLR4 signaling through binding to TRAF6 and preventing its ubiquitination, unlike A20, which removes ubiquintin molecules after they have been attached50. ST2825, a synthetic peptido-mimetic compound and a possible strategy for clinical treatment, prevents recruitment of IRAKs by MyD8851. In the MyD88 independent pathway, Deubiquitinating enzyme A (DUBA) cleaves the polyubiquitin chains on TRAF3, suppressing production of type I interferons52. We have summarized the regulatory mechanisms of TLR4 signaling in Figure 1.
Figure 1. Schematic of TLR4 negative regulators.

Arrows indicate sites of inhibition. Sigirr inhibits the initiation of this pathway by preventing TLR4's interaction with MyD88. IRAK-M and TOLLIP both prevent the dissociation of IRAKs from MyD88. TRIAD3A has several mechanisms of inhibition by interacting within the Toll-interleukin-1 receptor domains of TIRAP, TRIF, and RIP1. Overexpression of TRIAD3 also degrades TLR4. A20 is a de-ubiquitinating enzyme which removes ubiquitin moieties from TRAF6 to prevent downstream signaling. miR-21 is a microRNA targeting the protein PDCD4 and preventing activation of NF-κB. VACV A52R is a viral protein that prevents the formation of IRAK TRAF6 complexes. TANK prevents the ubiquitination of TRAF6 suppressing further downstream signaling. ST2825 a synthetic compound, prevents MyD88's association with IRAKs. DUBA cleaves polyubiquitin chains from TRAF3 to suppress type I interferons.
In the mouse colon, TLR4 is located in the apical side of epithelial crypts and in lamina propria mononuclear cells to simplify the task of sensing gut contents53. In contrast, in the T84 human CEC line, TLR4 and/or MD-2 were found localized to the basolateral surface membrane. Potentially-pathogenic and pathogenic bacteria that transverse the colonic epithelium may efficiently encounter TLR4 and/or MD2 at this location. This is proposed as a physical mechanism for commensal hyporesponsiveness, while still maintaining the ability to activate pro-inflammatory genes in response to invading pathogens54.
TLR4 expression is strongly upregulated and its presence detectable through immunohistochemistry in CECs of colonic biopsies obtained from patients with inflammatory bowel disease (IBD) including ulcerative colitis (UC) and Crohn's disease (CD)17. In both active and inactive regions of the terminal ileum of CD and UC patients, TLR4 expression was highly upregulated, which reflects a state of hyper-activation and maximization of responsiveness to the gut environment. Presumably, this abnormal expression of TLR4 on CECs may be associated with the exacerbation of chronic inflammation in IBD. Interestingly, differential TLR4-staining pattern was observed in the basolateral and apical surfaces of the colon in UC and CD patients, respectively. Furthermore, TLR4-positive intestinal epithelial cells were observed in inactive UC and CD14. It has been suggested that host tolerance to luminal bacterial components (e.g. LPS) tends to be disregulated in IBD patients55, 56. This disregulation is likely to enhance the LPS recognition as a result of upregulated TLR expression on CECs under inflammatory conditions in the gut.
2. TLR4 polymorphism in inflammation and cancer
To date, at least 8 TLR4 receptor single nucleotide polymorphisms (SNP) have been identified57. Of these, the A896G (D299G) and C1196T (T399I) missense mutations are the most widely studied. They cause a conformation change in the extracelluar domain of TLR and a blunted response to LPS58, 59. These two SNPs have been implicated in several inflammatory disorders as well as cancer.
Both the D299G and T399I mutations have been associated with increased risk of gram negative bacterial infection with septic shock 18, 60, Helicobacter pylori-mediated gastric carcionoma61, and head and neck squamous cell carcinomas in conjunction with increased chemotherapy resistance62. Interestingly, these mutations have been implicated in Malaria disease manifestation; with the D299G and T399I conferring, a 1.5- to-2.6-fold increased chance of severe malaria infection respectively. This has been hypothesized to be the result of reduced responsiveness to the Plasmodium falciparum glycosylphosphatidyl-inositol63.
Specifically, The D299G mutation has been shown to inhibit LPS-mediated signaling in airway epithelial cells, and increase the risk of Crohn's disease but not ulcerative colitis64 [Figure 2]. Recently it has been reported to induce neoplastic progression in intestinal epithelial cells, increase expression of pro-inflammatory genes and proteins, including alpha-2-macroglobulin (A2M), complement component 5 (CC5), CHI3L1, and tissue factor pathway inhibitor (TFPI); and was associated with more advanced and aggressive colon cancers in humans65. In breast cancer patients, the D299G mutation was associated with decreased binding of TLR4 to High-mobility group protein B1 (HMGB1); an “alarm” signal released by dying cancer cells that mediates the immune response against cancer, as well as quicker relapse after radiotherapy and chemotherapy66. Alzheimer's disease, in which inflammation plays a key role, the D299G mutation was associated with an increased risk of the disease, possibly due to increased production of inflammatory cytokines in the brain and decreased beta-amyloid clearance67, 68. An eight-fold increased risk of endometriosis was found in women carrying the D299G allele versus wild-type TLR4 due to increased peritoneal inflammation69. In the context of metabolic syndrome, D299G was associated with increased serum insulin levels, homeostatic model assessment of insulin resistance, and family history of type 2 diabetes70.
Figure 2. Amino acid locations of TLR4 SNPs and Histidine residues involved in nickel hypersensitivity within the TLR4 peptide sequence.
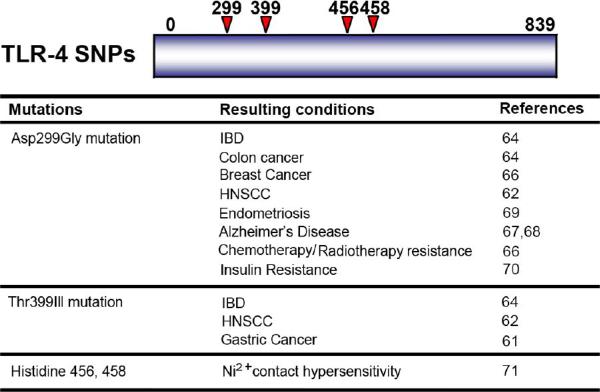
Below is a list of conditions associated with the TLR4 SNPs and histidine residues. Abbreviations: IBD, inflammatory bowel disease; HNSCC, head and neck squamous cell carcinomas.
Contact hypersensitivity to nickel containing jewelry, piercing, and coins has been shown to be mediated through TLR4 receptor binding, specifically to the Histidine 456 and 458 residues71. Mouse macrophages transfected with mutant Histidine 456 and 458 residues showed Ni2+ binding only occurs in human TLR4. This effect was independent of LPS signaling and as such, site-specific inhibition of TLR4 could be a potential treatment in patients with this allergy without negatively affecting the immune response.
Surprisingly, these mutations have been associated with a protective role in several conditions including myocardial infarction72, increased survival after melanoma metastasis57, and decreased levels of pro-inflammatory cytokines following cardiac surgery73 and may even confer longevity by decreasing systemic inflammation74. Taken together, TLR4 mutations may have a variable role depending on the disease or condition.
3. Mammalian chitinases and chitinase-like proteins
Mammalian chitinases and chitinase-like proteins (CLPs) are members of the glycohydrolase family 18 enzymes20,21. Family 18 chitinases are characterized by an eight-fold alpha/beta barrel structure and are represented by bacteria, plants, fungi, insects, viruses, protozoan parasites, and mammals75,76. This family includes chitotriosidase and acidic mammalian chithnase (AMCase) which both possess chitinase enzymatic activity77. Chitinase activity appears to play an important role in various diseases such as malaria, parasitic and fungal infections78. CLPs include CHI3L1, which is also one of the members of glycohydrolase 18 family79. CHI3L1 selectively and strongly binds to chitin (a polymer of N-acetylglucosamine) but does not possess chitinase activity80. Mammalian chitinases and CLPs are produced by a small number of cell types including human synovial cells, osteosarcoma cells, chondrocytes, smooth muscle cells, macrophages, neutrophils, and CECs81. Chitinases are potent stimulators of the innate immune response. It has been shown that stimulation with LPS upregulates chitinase activity in human-monocytes and macrophages. Chitotriosidase is used as a marker for macrophage differentiation because it is expressed by the late stage of activated macrophages82, 83. AMCase is highly expressed in the glandular cells of the stomach and intestinal tissues under normal physiological conditions purportedly to aid in host defense and food processing84. CHI3L1 plays an important role in the processes of inflammation and it must be a crucial factor during the development of colitis under the controls of pro-inflammatory cytokines and chemokines on CECs23, 85. The chitin-binding motif (CBM) within CHI3L1 allows it to bind with chitin or chito-oligosaccharide. This CBM has been shown to be critical in activating the Akt pathway, which is closely associated with exacerbation and chronicity of IBD, as well as stimulating IL8 production, a pro-inflammatory cytokine, in SW480 cells80. CHI3L1 in serum has been used as a sensitive biomarker for early detection of several inflammatory disorders including IBD86. CHI3L1 mRNA level was found to be significantly upregluated after stimulating with pro-inflammatory cytokines such as tumor necrosis factor-alpha (TNFα), IL-1β and IL-6 in CEC lines23, 87, 88. CHI3L1 is known to be a growth factor for connective tissue cells and adhesion molecule for endothelial cells89. Biological activities of CHI3L1 include regulation of cell proliferation, migration, and activation24. CHI3L1 has been upregulated in not only inflammatory conditions but also various solid tumors and is associated with the disease severity. Elevated level of CHI3L1 in serum was found in breast cancer, colorectal cancer, glioblastoma and malignant melanoma, extracellular myxoid-chondrosarcoma, and Hodgkin's lymphoma90 and is positively associated with the poor prognosis of the cancers. CHI3L1-mediated signaling actively enhances the pro-inflammatory cytokine productions and cellular proliferation in CECs91. AMCase also possesses the ability to exacerbate local inflammation (e.g. allergic conjunctivitis) by facilitating the production of chemical mediators such as monocyte chemoattractant protein-1 and eotaxin-121, 92. From the results, AMCase and CHI3L1 are likely to be used as major and/or supplementary biomarkers for active inflammations as well as variety of cancers, including breast, ovarian, gastric, prostate, and colon cancer93.
4. Potential role of CHI3L1 in colitis and colitis-associated cancer
CHI3L1 has been extensively studied in the context of rheumatoid arthritis94 and asthma95, two conditions characterized by excess inflammation conditions in joints and lung, respectively. However, its role in colitis and colitis-associated cancer is beginning to be elucidated. There are several types of colitis clinically, including autoimmune mediated- (Crohn's disease and ulcerative Colitis), idiopathic-, iatrogenic-, and infectious-types of colitis. Of note, patients with chronic IBD have a 0.5% to 1% increased risk of developing colitis-associated cancer after 10 years after the initial diagnosis of the colitis96. Colonic CHI3L1 mRNA expression is approximately 20-fold increased in patients with UC who harbored remote neoplastic lesions as compared to healthy individuals24. This result strongly suggests that CHI3L1 plays a major/direct role in inflammation-associated neoplastic changes in CECs.
CHI3L1 has been found to be increased in murine models of colitis including dextran sulfate sodium (DSS)-induced colitis, IL-10- or TCRα- deficiency-mediated chronic colitis, as well as in human patients with IBD. CHI3L1 was also found to be required for the adhesion and invasion of the pathogenic bacteria strains (e.g. Salmonella typhimurium and potentially pathogenic Escherichia coli) on/into CECs23. CHI3L1 was found to exacerbate the infectious colitis induced by S. typhimurium as well as DSS-induced acute colitis in C57Bl/6 wildtype mice. Blocking of CHI3L1 activity utilizing anti- CHI3L1 neutralizing antibodies resulted in improved recovery from the acute phase of DSS-induced-colitis as shown by improved clinical scores and percent body weight loss compared with the normal rabbit IgG-treated control group. Histologically, the antibody-treated group showed significantly less epithelial damage/proliferation and inflammatory cell infiltration in colonic lamina propria. Taken together, these data suggest that the neutralization of CHI3L1 suppresses the colonic inflammation in DSS-induced colitis by reducing the adhesion and internalization of luminal bacteria into the colonic mucosa and eventually suppresses their translocations into the mesenteric lymph nodes, spleen, and liver23. Interestingly, CHI3L1 has been associated with poor prognosis and decreased overall mortality in all types of cancer, in particular gastrointestinal neoplasia92, 97. CHI3L1 is increased in both IBD and colorectal cancer. Patients diagnosed with Crohn's disease have a 5.6 fold increased risk of colonic adenocarcinoma development98, suggesting that CHI3L1 may play a pivotal role in the initiation and/or progression of IBD into colon cancer. CHI3L1 has been found to be over expressed in several types of solid cancer (breast-, colon-, lung-, kidney-, pancreas-, ovarian-, prostate-, and uterine carcinoma, osteosarcoma, oligodendroglioma, glioblastoma and germ cell tumors)89. Recently, CHI3L1 was found to be increased in the visceral fat biopsies of patients with diagnosed colon cancer. This result suggests that CHI3L1 is not only produced at the site of inflammation and released into serum, but is also secreted as an adipokine by visceral fat99.
It has been reported that CHI3L1 plays a critical role in tumor angiogenesis by coordinating membrane-bound receptor syndecan-1 and integrin αv β3 as well as stimulating focal adhesion kinase (FAK) and MAPK, both which are involved in angiogenesis100. In human breast cancer, CHI3L1 was correlated with blood vessel density101. The molecule was also found to have an effect on extracellular tissue remodeling by binding specifically to collagen types I, II, and III102, suggesting that CHI3L1 is involved in the processes of fibrillogenesis and tissue remodeling, both which are associated with tumor progression and fibrosis.
It has been previously demonstrated that intestinal epithelial cells express TLR4 and respond to LPS in a time-, dose-, and serum-dependent manner103. To examine whether CHI3L1 protein potentially enhances the TLR4 expression on CECs, we stimulated SW480 human colon cancer cells with LPS from E. coli O55:B5 (Enzo Life Sciences, Farmingdale, NY) or purified CHI3L1 (Quidel Corporation, San Diego, CA), which was purified from the culture supernatant of MG-63 cells in serum-free medium as previously described104. We selected SW480 CEC line for this experiment since it constitutively expresses TLR4 on the cell surface105. As shown in Figure 3, CHI3L1 protein does not stimulate the upregulated expression of TLR4 in SW480 cells after 30 minutes [Figure 3A] or 3 hours [Figure 3B] stimulation. In contrast, LPS significantly activates the expressions of TLR4 at the both time points [Figure 3]. Although CHI3L1 stimulation at the dose of 100 ng/ml does not alter the TLR4 expression, it has been found to activate NF-κB24 and MAPK p42/p4480 and phosphoinositide 3-kinase (PI3K)106 mediated pathways in human synovial cells, fibroblasts, articular chondrocytes or CECs at the same dose. Both the MAPK and PI3K pathways are highly involved in cell growth, proliferation, survival, mitogenesis and apoptosis, as well as in the progress of cancer cellular transformation. It has been suggested that G-protein (guanine nucleotide-binding proteins), which are involved in transmitting chemical signals outside the cells and regulates MAPK-signaling networks, are involved in the action of most non-nuclear oncogenes and subsequent carcinogenesis107. These networks may enhance carcinogenic changes of epithelial cells with increased CHI3L1 expression under inflammatory conditions.
Figure 3. No enhanced activation of TLR4 in colonic epithelial cells by the stimulation with purified Chitinase 3-like 1 (CHI3L1) protein.

SW480 human colon cancer cells at 95 % confluency were cultured without (none) or with low (10 ng/ml) or high (100 ng/ml) concentrations of purified CHI3L1 protein for 30 minutes (A) or 3 hours (B). As an internal control, cells were stimulated with LPS O55:B5 at high dose (≥10 μg/ml). Quantitative RT-PCR analysis for human TLR4 (Forward: 5'-AGACCTGTCCCTGAACCCTAT; Reverse: 5'- CGATGGACTTCTAAACCAGCCA) mRNA was performed in each group (n=6). CT values were normalized to the housekeeping gene (GAPDH) and shown as average ± standard error. *P<0.05.
The canonical Wnt/β-catenin pathway is known to play a crucial role in UC-associated carcinogenic progression108. Our group has identified significantly increased expression and nuclear translocation of β-catenin in SW480 colonic cancer cell line after stimulating with a medium dose (50 ng/ml) of purified CHI3L1 protein90. This result suggests that CHI3L1 may play a direct role in inflammation based carcinogenesis by continuously activating the β-catenin pathway.
Recently, it was found that CHI3L1 mRNA was increased by 20-fold in non-dysplastic regions of human colonic biopsies of patients with IBD who had remote dysplasia and/or adenocarcinoma compared with healthy controls as detected by DNA-microarray analysis and RT-PCR21. The CHI3L1 message was also significantly increased, but by a lower extent, when compared to quiescent IBD patients without dysplasia. This suggests that CHI3L1 may be a useful and sensitive biomarker in detecting an early stage of colonic dysplasia in IBD patients. Specific expression of CHI3L1 was found in the specific types of CECs including Lgr5+ stem-like cells, Paneth cells and neuroendocrine-type cells in UC patients with dysplasia. In contrast, CHI3L1 expressing cells were not observed in UC patients without dysplasia. This finding strongly suggests that the increased CHI3L1 expression in specific cell types within the colonic crypts must be associated with the neoplastic transformation of CECs in UC patients. One case in particular, a surgical resection from a patient with UC with mucinous adenocarcinoma and high-grade neuroendocrine carcinoma, had both tumors positively stained with anti-CHI3L1 antibody. In particular, as compared to the mucinous adenocarcinoma, the neuroendocrine tumor showed much more intense CHI3L1 staining in colon, which showed a large CHI3L1-positive metastatic niche formation to the liver. This finding strongly suggests that CHI3L1 may be associated with invasiveness and metastatic ability of malignant tumor cells.
The pro-inflammatory cytokine IL-6 has been shown to be a critical tumor promoter in the early stages of colitis-associated cancer, by enhancing proliferation and suppressing apoptosis109. These effects were found to be largely mediated by the activation of STAT3 transcription factor, which is also associated with cancer progression by increasing proliferation and inhibiting apoptosis. CHI3L1 production is enhanced by IL-6 stimulation23, and as such, blocking IL-6-mediated CHI3L1 may be useful in preventing inflammation and subsequent inflammation-mediated carcinogenesis in epithelial cells90.
Activation of NF-κB is associated with cell survival, inflammation, and inflammation-associated carcinogenesis110. Stimulated SW480 cells with 80 ng/ml of CHI3L1 cells showed activation in select genes including IRAK1, IκB, NF-κB p65, MyD88, and increased NF- κB pathway activation in a dose dependent manner. Phosphorylation of IkBα was also found to be dose-dependent with highest expression at 80 ng/ml by Western blot analysis. Enhancement of the pro-inflammatory cytokine TNFα and chemokine IL-8 were also found to be dose-dependent at the levels of the previous stimulations. CHI3L1 has also shown potent epithelial growth stimulating effects in vitro. After treatment with CHI3L1 Colo205 and SW480 cell lines showed increased proliferation evaluated by BrdU (bromodeoxyuridine)-incorporation index21. This growth stimulating effect was found to be similar to IGF-1, a well-characterized growth factor for CECs. CHI3L1 was also found to increase cellular migration of SW480 cells in a Boyden chamber assay by six-fold. This effect was significantly diminished after incubating the cells in a rabbit anti-CHI3L1 polyclonal antibody, suggesting a CHI3L1-mediated cell migratory effect.
Taken together, it appears CHI3L1 plays a pivotal role in enhancing the adhesion and invasion of potentially pathogenic bacteria on/into CECs and in initiating/perpetuating colitis. CHI3L1 has also been discovered to be a valuable biomarker for cancer progression and prognosis. The recent works by our group have bridged this information in the context of colitis-associated cancer. CHI3L1 induces the production of inflammatory mediators, which are crucial in colitis-associated neoplasia formation as a result of the activation of MAPK- and NF- κB-signaling pathways24, 25. CHI3L1 highly contributes to cellular proliferation, survival, migration, and angiogenesis, which all play a critical role in maintaining the tumor-microenvironment.
5. Differential linkage between TLR4 and CHI3L1
The link between CHI3L1 and TLR4 has not been fully understood. Treatment of SW480 cells with TLR4 ligand LPS for 2 and 6 hours both exhibited a down-regulation of CHI3L1 expression [E. Mizoguchi, unpublished observation]. The 299th and 399th amino acids mutations in TLR4 protein has been described as a “loss-of-function” mutation which leads to a hypo-responsive effect with blunted activation of NF- κB and decreased secretions of pro-inflammatory cytokine IL-1α after stimulation of airway epithelial cells with LPS59. It has been shown that Caco2 cells transfected with the D299G or T399I mutation both had significant increases in CHI3L1 mRNA65. This demonstrates a direct link between these TLR4 mutations and enhanced production of CHI3L1. In fact, we have identified the D299G/T399I mutations in SW480 CEC cells, and this could explain the downregulated responsiveness against LPS and upregulated endogenous expression of CHI3L1 in this cell line23.
The D299G mutation in TLR4 was found to constitutively increase the expressions of Wnt target genes Connexin 43 and Dickkopf-related protein 162. Similarly, the nucleic translocation of β-catenin after stimulating with CHI3L1 in SW480 cells was increased90. This suggests there is an indirect or perhaps direct, co-stimulatory effect of Wnt and/or PI3K/Akt signaling pathway activation111. In addition, Stat3 phosphorylation was found to be induced in Caco-2 cells65, which have the TLR4 D299G mutation. As our group previously reported, CHI3L1 is induced by the proinflammatory cytokine IL-6, one of the potent signal transducers of STAT323. It appears the D299G mutation in TLR4 and the upregulated CHI3L1 expression synergistically activate the β-catenin signaling pathway, which will be associated with the neoplastic change of CECs, subsequently. The two may work indirectly but additively to activate many of the proinflammatory pathways or other pathways that result in the disregulated function of epithelial cells. It can be speculated that colon cancer patients with elevated CHI3L1 that are also carriers of the D299G/T399I mutations in TLR4 will have a worse cancer prognosis and increased metastasis versus patients with elevated CHI3L1 and wild-type TLR4.
Another important and critical proinflammatory cytokine, TNFα, was found to increase the CHI3L1 expression in SW480 and T84 CEC lines23. Recent reports have shown that the T399I mutation in TLR4 enhances the expression of TNFα message, which is associated with the risk of nasopharyngeal carcinoma development112. This represents a potentially more direct link between the T399I mutation in TLR4 and excessive CHI3L1 expression in nasopharyngeal epithelial cells via TNFα.
In order to further elucidate the linkage between CHI3L1 expression and TLR4 mutation, our group has recently examined the presence of the TLR4 D299G and T399I mutations in various CEC lines by sequencing the PCR products, which were amplified with D299G and T399I specific primers as previously published113. As summarized in Table 1, we examined four separate human CEC lines, since these SNPs are associated with neoplastic changes, and thus cancerous cells would more likely exhibit these mutations. This information is also pertinent for IBD patients who may be placed at an increased risk for colorectal cancer. Both D299G and T399I mutations were identified in Caco2 and SW480 cells, which express the message of CHI3L1 endogenously20. In contrast, HT-29 cells do not have D299G mutation and lack the endogenous expression of CHI3L1. Therefore, we hypothesized that the presence of TLR4 mutations may directly or indirectly modulate the endogenous expression of CHI3L1. Presumably TLR4 D299G and T399I mutations keep the endogenous CHI3L1 levels aberrantly high, which may prevent the negative regulation of CHI3L1 expression after LPS/TLR4 ligation on CECs.
Table 1.
CHI3L1 expression and TLR4 mutations in CEC lines
| Cell Line | CHI3L1 Expression | D299G Mutation | T399I Mutation |
|---|---|---|---|
| SW-480 | (+)* | (+) | (+) |
| Caco-2 | (+) | (+) | (+) |
| Colo-205 | (-)** | (+) | (+) |
| HT-29 | (-) | (-) | (+) |
Presence of CHI3L1 expression or TLR4 D299G/T399I mutations.
absence of CHI3L1 expression or TLR4 D299G/T399I mutations.
6. Future prospective
The D299G and T399I mutations in TLR4 seem to constitutively increase the expressions of CHI3L1 in CECs. It has been demonstrated that these mutations are associated with the carcinogenic changes of CECs62. However, their role in conjunction with CHI3L1 has yet to be completely unrevealed. It is likely that the coexistence of one or more TLR4 mutations and elevated CHI3L1 work synergistically to further promote carcinogenic changes of CECs under inflammatory conditions. Since SW480 cells show a blunted down regulation of CHI3L1 after stimulation with LPS, and patients with the D299G mutation were found with more advanced tumor grade and metastasis, perhaps these factors play together in a positive feedback loop increasing CHI3L1 expression in CECs, which potentially promotes the carcinogenic change of CECs in IBD. Further in vitro studies will be required to solidify this connection. Subsequent in vivo studies could be employed to determine the systemic effects of this mutation with CHI3L1-associated carcinogenic change of CECs. The presence of both TLR4 mutation and elevated CHI3L1 expression may represent co-morbidities, and could have important prognostic consequences in the diagnosis/treatment for patients with inflammatory disorders including IBD and colitis-associated cancer.
Conclusion
TLR4 represents a critical component of innate immune responses, of which its main function is sensing the existence of gram-negative bacteria in the gut. It is known that TLR4 expression is barely detectable under normal physiological conditions, but is strongly upregulated under inflammatory conditions including IBD. Two common missense mutations, D299G and T399I, alter the function of TLR4. In particular, D299G mutation leads to a hypo-responsive effect to LPS and both are associated with increased inflammation and risk of infection in several disorders. However, these mutations may also be protective in preventing systemic inflammation in several other disorders including myocardial infarction. D299G and T399I mutations have been associated with radio-resistance of head and neck squamous carcinoma cells as well as promoting the epithelial-mesenchymal transition, increased tumor grade, and metastasis in colon cancer. We here hypothesize that one of the potential mechanisms by which these mutations may increase the rate and severity of carcinogenesis is by increasing the expression of CHI3L1, which is involved in tissue remodeling, angiogenesis, and tumor progression. CHI3L1 is upregulated in many inflammatory disorders and cancers including IBD and colorectal cancer. The D299G mutation and cellular CHI3L1 stimulation were both found to independently activate the β-catenin trans-nucleic localization, which is associated with tumor initiation and progression.
Further research is required to determine and clarify the complete relationship between the TLR4 mutations and the CHI3L1 overexpression and their role in inflammatory disorders and cancer formation. We strongly hope that this mini-review would provide us some clues in developing the diagnostic as well as therapeutic strategies for patients with IBD and colitis-associated cancer in the near future.
Acknowledgements
We would like to thank Mr. Terry Danford Lott for his excellent assistance in preparing this manuscript. This work has been supported by National Institute of Health (DK 80070, DK74454, DK64289 and DK43351), and grants from the Eli and Edythe L. Broad Medical Foundation and American Gastroenterological Association Foundation to E. Mizoguchi. I.A. Lee has been supported by the National Research Foundation of Korea Fellowship Award.
Abbreviations used
- AMCase
acidic mammalian chitinase
- CECs
colonic epithelial cells
- CLPs
chitinase-like proteins
- CD
Crohn's disease
- IBD
inflammatory bowel disease
- IECs
intestinal epithelial cells
- TLRs
toll-like receptors
- UC
ulcerative colitis
- WT
wild-type
- LPS
lipopolysaccharide
- CHI3L1
Chitinase 3-like-1
- TIR
toll-interleukin receptor
- LPD
Lipopolysaccharide binding protein
- MYD88
myeloid differentiation primary response gene 88
- NF- κB
nuclear factor kappa-light-chain-enhancer of activated B cells
- AP-1
activator protein-1
- MAPK
mitogen-activated protein kinase
- TRIF
TIR-domain-containing adaptor-inducing interferon-γ
- Tollip
toll-interacting protein
- SIGIRR
single immunoglobulin IL-1R -related molecule
- TRIAD3
triad domain-containing protein 3 variant A
- TIRAP
toll-interleukin 1 receptor domain containing adaptor protein
- RIP-1
receptor interacting protein-1
- IRAK
Interleukin-1 receptor-associated kinase
- TRAF6
Tumor necrosis factor receptor associated factor 6
- SNP
single nucleotide polymorphism
- A2M
alpha-2-macroglobulin
- CC5
complement component 5
- TFPI
tissue factor pathway inhibitor
- HMGB1
High-mobility group protein B1
- TNF-α
tumor necrosis factor alpha
- DSS
dextran sulfate sodium
- TCRα
T-cell receptor alpha
- Brd-U
Bromodeoxyuridine
- FAK
focal adhesion kinase
- PI3K
phosphoinositide 3-kinase
- STAT3
Signal transducer and activator of transcription 3
- IκBα
nuclear factor of kappa light polypeptide gene enhancer in B-cells inhibitor, alpha
- PCR
polymerase chain reaction
Footnotes
Conflict of interest: The authors have no financial conflicts of interest.
References
- 1.Podolsky DK. Inflammatory Bowel Disease I. N. Engl. J. Med. 1991;325:928–937. doi: 10.1056/NEJM199109263251306. [DOI] [PubMed] [Google Scholar]
- 2.Xavier RJ, Podolsky DK. Unravelling the pathogenesis of inflammatory bowel disease. Nature. 2007;448:427–34. doi: 10.1038/nature06005. [DOI] [PubMed] [Google Scholar]
- 3.Brest P, Corcelle EA, Cesaro A, et al. Autophagy and Crohn's disease: at the crossroads of infection, inflammation, immunity, and cancer. Curr. Mol. Med. 2010;10:486–502. doi: 10.2174/156652410791608252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mizoguchi A, Mizoguchi E, Bhan AK. Immune networks in animal models of inflammatory bowel disease. Inflamm. Bowel Dis. 2003;9:246–259. doi: 10.1097/00054725-200307000-00005. [DOI] [PubMed] [Google Scholar]
- 5.Mizoguchi A. Animal models of inflammatory bowel disease. Prog. Mol. Biol. Transl. Sci. 2012;105:263–320. doi: 10.1016/B978-0-12-394596-9.00009-3. [DOI] [PubMed] [Google Scholar]
- 6.Khor B, Gardet A, Xavier RJ. Genetics and pathogenesis of inflammatory bowel disease. Nature. 2011;15474:307–317. doi: 10.1038/nature10209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Saleh M, Trinchieri G. Innate immune mechanisms of colitis and colitis-associated colorectal cancer. Nat. Rev. Immunol. 2011;11:9–20. doi: 10.1038/nri2891. [DOI] [PubMed] [Google Scholar]
- 8.Mizoguchi A, Mizoguchi E. Animal model of IBD: linkage to human disease. Curr. Opin. Pharmacol. 2010;10:578–587. doi: 10.1016/j.coph.2010.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Monteleone I, Sarra M, Pallone F, et al. Th17-related cytokines in inflammatory bowel disease: friends or foes? Curr. Mol. Med. 2012;12P:592–597. doi: 10.2174/156652412800620066. [DOI] [PubMed] [Google Scholar]
- 10.Caruso R, Stolfi C, De Nitto D, et al. The dual role of interleukin-25 in the control of immune-mediated pathologies. Curr. Mol. Med. 2011;11:26–30. doi: 10.2174/156652411794474365. [DOI] [PubMed] [Google Scholar]
- 11.Morales C, Wu S, Yang Y, et al. Drosophila Glycoprotein 93 Is an Ortholog of Mammalian Heat Shock Protein gp96 (grp94, HSP90b1, HSPC4) and Retains Disulfide Bond-Independent Chaperone Function for TLRs and Integrins. J. Immunol. 2009;183:5121–5128. doi: 10.4049/jimmunol.0900811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Palsson-McDermott EM, O'Neill LA. Signal transduction by the lipopolysaccharide receptor, Toll-like receptor-4. Immunology. 2004;113:153–162. doi: 10.1111/j.1365-2567.2004.01976.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Takeuchi O, Sato S, Horiuchi T, et al. Cutting edge: role of Toll-like receptor 1 in mediating immune response to microbial lipoproteins. J. Immunol. 2002;169:10–14. doi: 10.4049/jimmunol.169.1.10. [DOI] [PubMed] [Google Scholar]
- 14.Xiong Y, Medvedev AE. Induction of endotoxin tolerance in vivo inhibits activation of IRAK4 and increases negative regulators IRAK-M, SHIP-1, and A20. J. Leukoc. Biol. 2011;90:1141–1148. doi: 10.1189/jlb.0611273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Takeda K, Kaisho T, Akira S. Toll-like receptors. Annu. Rev. Immunol. 2003;21:335–376. doi: 10.1146/annurev.immunol.21.120601.141126. [DOI] [PubMed] [Google Scholar]
- 16.Beutler B, Jiang Z, Georgel P, et al. Genetic analysis of host resistance: Toll-like receptor signaling and immunity at large. Annu. Rev. Immunol. 2006;24:353–389. doi: 10.1146/annurev.immunol.24.021605.090552. [DOI] [PubMed] [Google Scholar]
- 17.Cario E, Podolsky DK. Toll-like receptor signaling and its relevance to intestinal inflammation. Ann. N.Y. Acad. Sci. 2006;1072:332–338. doi: 10.1196/annals.1326.006. [DOI] [PubMed] [Google Scholar]
- 18.Ferwerda B, McCall MB, Alonso S, et al. Proc. Natl. Acad. Sci. USA. 2007;104:16645–16650. doi: 10.1073/pnas.0704828104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Apetoh L, Tesniere A, Ghiringhelli F, et al. Molecular interactions between dying tumor cells and the innate immune system determine the efficacy of conventional anticancer therapies. Cancer Res. 2008;68:4026–4030. doi: 10.1158/0008-5472.CAN-08-0427. [DOI] [PubMed] [Google Scholar]
- 20.Hakala BE, White C, Recklies AD. Human cartilage gp-39, a major secretory product of articular chondrocytes and synovial cells, is a mammalian member of a chitinase protein family. J. Biol. Chem. 1993;268:25803–25810. [PubMed] [Google Scholar]
- 21.Kawada M, Hachiya Y, Arihiro A, et al. Role of mammalian chitinases in inflammatory conditions. Keio J. Med. 2007;56:21–27. doi: 10.2302/kjm.56.21. [DOI] [PubMed] [Google Scholar]
- 22.Recklies AD, Ling H, White C, et al. Inflammatory cytokines induced production of CHI3L1 by particular chondrocytes. J. Biol. Chem. 2005;280:41213–41221. doi: 10.1074/jbc.M510146200. [DOI] [PubMed] [Google Scholar]
- 23.Mizoguchi E. Chitinase 3-like-1 exacerbates intestinal inflammation by enhancing bacterial adhesion and invasion in colonic epithelial cells. Gastroenterol. 2006;130:398–411. doi: 10.1053/j.gastro.2005.12.007. [DOI] [PubMed] [Google Scholar]
- 24.Chen CC, Pekow J, Llado V, et al. Chitinase 3-like-1 expression in colonic epithelial cells as a potentially novel marker for colitis-associated neoplasia. Am. J. Pathol. 2011;179:1494–1503. doi: 10.1016/j.ajpath.2011.05.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kawada M, Seno H, Kanda K, et al. Chitinase 3-like 1 promotes macrophage recruitment and angiogenesis in colorectal cancer. Oncogene. 2012;31:3111–3123. doi: 10.1038/onc.2011.498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Janeway CA, Jr, Goodnow CC, Medzhitov R. Danger - pathogen on the premises! Immunological tolerance. Curr. Biol. 1996;6:519–522. doi: 10.1016/s0960-9822(02)00531-6. [DOI] [PubMed] [Google Scholar]
- 27.Medzhitov R, Preston-Hurlburt P, Janeway CA. A human homologue of the Drosophila Toll protein signals activation of adaptive immunity. Nature. 1997;388:394–397. doi: 10.1038/41131. [DOI] [PubMed] [Google Scholar]
- 28.Gay NJ, Keith FJ. Drosophila Toll and IL-1 receptor. Nature. 1991;351:355–356. doi: 10.1038/351355b0. [DOI] [PubMed] [Google Scholar]
- 29.Belvin MP, Anderson KV. A conserved signaling pathway: The Drosophila Toll-Dorsal Pathway. Ann. Rev. Cell Dev. Biol. 1996;12:393–416. doi: 10.1146/annurev.cellbio.12.1.393. [DOI] [PubMed] [Google Scholar]
- 30.Hoshino K, Takeuchi O, Kawai T, et al. Cutting Edge: Toll-Like Receptor 4 (TLR4)-Deficient Mice Are Hyporesponsive to Lipopolysaccharide: Evidence for TLR4 as the Lps Gene Product. J. Immunol. 1999;162:3749–3752. [PubMed] [Google Scholar]
- 31.Kurt-Jones EA, Popova L, Kwinn L, et al. Pattern recognition receptors TLR4 and CD14 mediate response to respiratory syncytial virus. Nat. Immunol. 2000;1:398–401. doi: 10.1038/80833. [DOI] [PubMed] [Google Scholar]
- 32.Rassa JC, Meyers JL, Zhang Y, et al. Murine retroviruses activate B cells via interaction with toll-like receptor 4. Proc. Natl. Acad. Sci. U.S.A. 2002;99:2281–2286. doi: 10.1073/pnas.042355399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ohashi K, Burkart V, Flohé S, et al. Cutting Edge: Heat Shock Protein 60 Is a Putative Endogenous Ligand of the Toll-Like Receptor-4 Complex. J. Immunol. 2000;164:558–561. doi: 10.4049/jimmunol.164.2.558. [DOI] [PubMed] [Google Scholar]
- 34.Fitzgerald KA, Rowe DC, Golenbock DT. Endotoxin recognition and signal transduction by the TLR4/MD2-complex. Microbes. Infect. 2004;6(15):1361–1367. doi: 10.1016/j.micinf.2004.08.015. [DOI] [PubMed] [Google Scholar]
- 35.Shimazu R, Akashi S, Ogata H, et al. MD-2, a molecule that confers lipopolysaccharide responsiveness on Toll-like receptor 4. J. Exp. Med. 1999;189:1777–1782. doi: 10.1084/jem.189.11.1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zanoni I, Ostuni R, Marek LR, et al. CD14 controls the LPS-induced endocytosis of Toll-like receptor 4. Cell. 2011;147(4):868–880. doi: 10.1016/j.cell.2011.09.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kumar H, Kawai T, Akira S. Toll-like receptors and innate immunity. Biochem. Biophys. Res. Commun. 2009;388:621–625. doi: 10.1016/j.bbrc.2009.08.062. [DOI] [PubMed] [Google Scholar]
- 38.Cario E, Podolsky DK. Differential alteration in intestinal epithelial cell expression of toll-like receptor 3 (TLR3) and TLR4 in inflammatory bowel disease. Infect. Immun. 2000;68:7010–7017. doi: 10.1128/iai.68.12.7010-7017.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Abreu MT, Vora P, Faure E, et al. Decreased expression of Toll-like receptor-4 and MD-2 correlates with intestinal epithelial cell protection against dysregulated proinflammatory gene expression in response to bacterial lipopolysaccharide. J. Immunol. 2001;167:1609–1616. doi: 10.4049/jimmunol.167.3.1609. [DOI] [PubMed] [Google Scholar]
- 40.Cario E, Rosenberg IM, Brandwein SL, et al. Lipopolysaccharide Activates Distinct Signaling Pathways in Intestinal Epithelial Cell Lines Expressing Toll-Like Receptors. J Immunol. 2000;164:966–972. doi: 10.4049/jimmunol.164.2.966. [DOI] [PubMed] [Google Scholar]
- 41.Otte J-M, Cario E, Podolsky DK. Mechanisms of cross hyporesponsiveness to toll-like receptor bacterial ligands in intestinal epithelial cells. Gastroenterology. 2004;126:1054–1070. doi: 10.1053/j.gastro.2004.01.007. [DOI] [PubMed] [Google Scholar]
- 42.Qin J, Qian Y, Yao J, et al. SIGIRR Inhibits Interleukin-1 Receptor- and Toll-like Receptor 4-mediated Signaling through Different Mechanisms. J. Biol. Chem. 2005;280:25233–25241. doi: 10.1074/jbc.M501363200. [DOI] [PubMed] [Google Scholar]
- 43.Fearns C, Pan Q, Mathison JC, et al. Triad3A regulates ubiquitination and proteasomal degradation of RIP1 following disruption of Hsp90 binding. J. Biol. Chem. 2006;281:34592–34600. doi: 10.1074/jbc.M604019200. [DOI] [PubMed] [Google Scholar]
- 44.Chuang T-H, Ulevitch RJ. Triad3A, an E3 ubiquitin-protein ligase regulating Toll-like receptors. Nat. Immunol. 2004;5:495–502. doi: 10.1038/ni1066. [DOI] [PubMed] [Google Scholar]
- 45.Kobayashi K, Hernandez LD, Galán JE, et al. IRAK-M is a negative regulator of Toll-like receptor signaling. Cell. 2002;110:191–202. doi: 10.1016/s0092-8674(02)00827-9. [DOI] [PubMed] [Google Scholar]
- 46.Boone DL, Turer EE, Lee EG, et al. The ubiquitin-modifying enzyme A20 is required for termination of Toll-like receptor responses. Nat. Immunol. 2004;5:1052–1060. doi: 10.1038/ni1110. [DOI] [PubMed] [Google Scholar]
- 47.Oshima N, Ishihara S, Rumi MAK, et al. A20 is an early responding negative regulator of Toll-like receptor 5 signalling in intestinal epithelial cells during inflammation. Clin. Exp. Immunol. 2010;159:185–198. doi: 10.1111/j.1365-2249.2009.04048.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sheedy FJ, Palsson-McDermott E, Hennessy EJ, et al. Negative regulation of TLR4 via targeting of the proinflammatory tumor suppressor PDCD4 by the microRNA miR-21. Nat. Immunol. 2010;11:141–7. doi: 10.1038/ni.1828. [DOI] [PubMed] [Google Scholar]
- 49.Harte MT, Haga IR, Maloney G, et al. The poxvirus protein A52R targets Toll-like receptor signaling complexes to suppress host defense. J. Exp. Med. 2003;197:343–51. doi: 10.1084/jem.20021652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kawagoe T, Takeuchi O, Takabatake Y, et al. TANK is a negative regulator of Toll-like receptor signaling and is critical for the prevention of autoimmune nephritis. Nat. Immunol. 2009;10:965–72. doi: 10.1038/ni.1771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Loiarro M, Capolunghi F, Fantò N, et al. Pivotal Advance: Inhibition of MyD88 dimerization and recruitment of IRAK1 and IRAK4 by a novel peptidomimetic compound. J. Leukoc. Biol. 2007;82:801–10. doi: 10.1189/jlb.1206746. [DOI] [PubMed] [Google Scholar]
- 52.Kayagaki N, Phung Q, Chan S, et al. DUBA: a deubiquitinase that regulates type I interferon production. Science. 2007;318:1628–32. doi: 10.1126/science.1145918. [DOI] [PubMed] [Google Scholar]
- 53.Ortega-Cava CF, Ishihara S, Rumi MAK, et al. Strategic compartmentalization of Toll-like receptor 4 in the mouse gut. J. Immunol. 2003;170:3977–3985. doi: 10.4049/jimmunol.170.8.3977. [DOI] [PubMed] [Google Scholar]
- 54.Vamadevan AS, Fukata M, Arnold ET, et al. Regulation of Toll-like receptor 4-associated MD-2 in intestinal epithelial cells: a comprehensive analysis. Innate. Immun. 2010;16:93–103. doi: 10.1177/1753425909339231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Duchmann R, Kaiser I, Hermann E, et al. Tolerance exists towards resident intestinal flora but is broken in active inflammatory bowel disease (IBD) Clin. Exp. Immunol. 1995;102:448–455. doi: 10.1111/j.1365-2249.1995.tb03836.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hotta T, Yoshida N, Yoshikawa T, et al. Lipopolysaccharide-induced colitis in rabbits. Res. Exp. Med. (Berl) 1986;186:61–69. doi: 10.1007/BF01851834. [DOI] [PubMed] [Google Scholar]
- 57.Gast A, Bermejo JL, Claus R, et al. Association of inherited variation in Toll-like receptor genes with malignant melanoma susceptibility and survival. PLoS. ONE. 2011;6:e24370. doi: 10.1371/journal.pone.0024370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Rallabhandi P, Bell J, Boukhvalova MS, et al. Analysis of TLR4 Polymorphic Variants: New Insights into TLR4/MD-2/CD14 Stoichiometry, Structure, and Signaling. J. Immunol. 2006;177:322–332. doi: 10.4049/jimmunol.177.1.322. [DOI] [PubMed] [Google Scholar]
- 59.Arbour NC, Lorenz E, Schutte BC, et al. TLR4 mutations are associated with endotoxin hyporesponsiveness in humans. Nat. Genet. 2000;25:187–191. doi: 10.1038/76048. [DOI] [PubMed] [Google Scholar]
- 60.Lorenz E, Mira JP, Frees KL, et al. Relevance of mutations in the TLR4 receptor in patients with gram-negative septic shock. Arch. Intern. Med. 2002;162:1028–1032. doi: 10.1001/archinte.162.9.1028. [DOI] [PubMed] [Google Scholar]
- 61.Hold GL, Rabkin CS, Chow W-H, et al. A functional polymorphism of toll-like receptor 4 gene increases risk of gastric carcinoma and its precursors. Gastroenterology. 2007;132:905–912. doi: 10.1053/j.gastro.2006.12.026. [DOI] [PubMed] [Google Scholar]
- 62.Bergmann C, Bachmann HS, Bankfalvi A, et al. Toll-like receptor 4 single-nucleotide polymorphisms Asp299Gly and Thr399Ile in head and neck squamous cell carcinomas. J. Transl. Med. 2011;9:139. doi: 10.1186/1479-5876-9-139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Mockenhaupt FP, Cramer JP, Hamann L, et al. Toll-like receptor (TLR) polymorphisms in African children: Common TLR-4 variants predispose to severe malaria. Proc. Natl. Acad. Sci. USA. 2006;103:177–182. doi: 10.1073/pnas.0506803102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.De Jager PL, Franchimont D, Waliszewska A, et al. The role of the Toll receptor pathway in susceptibility to inflammatory bowel diseases. Genes. Immun. 2007;8:387–397. doi: 10.1038/sj.gene.6364398. [DOI] [PubMed] [Google Scholar]
- 65.Eyking A, Ey B, Rünzi M, et al. Toll-like Receptor 4 Variant D299G Induces Features of Neoplastic Progression in Caco-2 Intestinal Cells and Is Associated With Advanced Human Colon Cancer. Gastroenterology. 2011;141:2154–2165. doi: 10.1053/j.gastro.2011.08.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Apetoh L, Ghiringhelli F, Tesniere A, et al. Toll-like receptor 4-dependent contribution of the immune system to anticancer chemotherapy and radiotherapy. Nat. Med. 2007;13:1050–1059. doi: 10.1038/nm1622. [DOI] [PubMed] [Google Scholar]
- 67.Balistreri CR, Grimaldi MP, Chiappelli M, et al. Association between the polymorphisms of TLR4 and CD14 genes and Alzheimer's disease. Curr. Pharm. Des. 2008;14:2672–2677. doi: 10.2174/138161208786264089. [DOI] [PubMed] [Google Scholar]
- 68.Wang L-Z, Yu J-T, Miao D, et al. Genetic association of TLR4/11367 polymorphism with late-onset Alzheimer's disease in a Han Chinese population. Brain Res. 2011;1381:202–207. doi: 10.1016/j.brainres.2011.01.007. [DOI] [PubMed] [Google Scholar]
- 69.Latha M, Vaidya S, Movva S, et al. Molecular pathogenesis of endometriosis; Toll-like receptor-4 A896G (D299G) polymorphism: a novel explanation. Genet. Test. Mol. Biomarkers. 2011;15:181–184. doi: 10.1089/gtmb.2010.0178. [DOI] [PubMed] [Google Scholar]
- 70.Cuda C, Badawi A, Karmali M, et al. Polymorphisms in Toll-like receptor 4 are associated with factors of the metabolic syndrome and modify the association between dietary saturated fat and fasting high-density lipoprotein cholesterol. Metab. Clin. Exp. 2011;60:1131–1135. doi: 10.1016/j.metabol.2010.12.006. [DOI] [PubMed] [Google Scholar]
- 71.Schmidt M, Raghavan B, Muller V, et al. Crucial role for human Toll-like receptor 4 in the development of contact allergy to nickel. Nat. Immunol. 2010;11:814–819. doi: 10.1038/ni.1919. [DOI] [PubMed] [Google Scholar]
- 72.Balistreri CR, Candore G, Colonna-Romano G, et al. Role of Toll-like Receptor 4 in Acute Myocardial Infarction and Longevity. J. Am. Med. Association. 2004;292:2339–2340. doi: 10.1001/jama.292.19.2339. [DOI] [PubMed] [Google Scholar]
- 73.Koch A, Hamann L, Schott M, et al. Genetic variation of TLR4 influences immunoendocrine stress response: an observational study in cardiac surgical patients. Crit. Care. 2011;15:R109. doi: 10.1186/cc10130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Balistreri CR, Caruso C, Listì F, et al. LPS-mediated production of pro/anti-inflammatory cytokines and eicosanoids in whole blood samples: Biological effects of +896A/G TLR4 polymorphism in a Sicilian population of healthy subjects. Mechan. Age. Develop. 2011;132:86–92. doi: 10.1016/j.mad.2010.12.005. [DOI] [PubMed] [Google Scholar]
- 75.Henrissat B, Davies G. Structural and sequence-based classification of glycoside hydrolases. Curr. Opin. Struct. Biol. 1997;7:637–644. doi: 10.1016/s0959-440x(97)80072-3. [DOI] [PubMed] [Google Scholar]
- 76.Kzhyshkowska J, Gratchev A, Goerdt S. Human Chitinases and Chitinase-like protein as indicators for inflammation and cancer. Biomark. Insights. 2007;2:128–146. [PMC free article] [PubMed] [Google Scholar]
- 77.Boot RG, Blommaart FC, Swart E. Identification of a novel acidic mammalian chitinase distinct from chitoriosidase. J. Biol. Chem. 2001;276:6770–6778. doi: 10.1074/jbc.M009886200. [DOI] [PubMed] [Google Scholar]
- 78.Kalkum M, Vega K. Chitin, chitinase responses, and invasive fungal infections. Int. J. Microbiol. 2012;2012:1–10. doi: 10.1155/2012/920459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Bussink AP, Speijer D, Aerts JMFG, et al. Evolution of mammalian chitinase(-like) members of family 18 glycosyl hydrolases. Genetics. 2007;177:959–970. doi: 10.1534/genetics.107.075846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Chen CC, Llado V, Eurich K, et al. Carbohydrate-binding motif in chitinase 3-like 1 (CHI3L1/YKL-40) specifically activates Akt signaling pathway in colonic epithelial cells. Clin. Immunol. 2011;140:268–275. doi: 10.1016/j.clim.2011.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kawada M, Chen CC, Arihiro A, Nagataini K, et al. Chitinase 3-like-1 enhances bacterial adhesion to colonic epithelial cells through the interaction with bacterial chitin-binding protein. Lab. Invest. 2008;2008:1–13. doi: 10.1038/labinvest.2008.47. [DOI] [PubMed] [Google Scholar]
- 82.Malaguarnera L, Musumeci M, Di Rosa M, et al. Interferon-gamma, tumor necrosis factor-alpha, and lipopolysaccharide promote chitoriosidase gene expression in human macrophages. J. Clin. Lab. Anal. 2005;19:128–132. doi: 10.1002/jcla.20063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Di Rosa M, Musumeci M, Scuto A, et al. Effect of interferon-gamma, interleukin-10, lipopolysaccharide and tumor necrosis factor-alpha on chitotriosidase synthesis in human macrophages. Clin. Chem. Lab. Med. 2005;43:499–502. doi: 10.1515/CCLM.2005.088. [DOI] [PubMed] [Google Scholar]
- 84.Boot RG, Bussink AP, Verhoek M, et al. Marked differences in tissue-specific expression of chitinases in mouse and man. J. Histochem. Cytochem. 2005;53:1283–1292. doi: 10.1369/jhc.4A6547.2005. [DOI] [PubMed] [Google Scholar]
- 85.Homer RJ, Zhu Z, Cohn L, et al. Differential expression of chitinases identify subsets of murine airway epithelial cells in allergic inflammation. Am. J. Physiol. Lung Cell. Mol. Physiol. 2006;291:L502–511. doi: 10.1152/ajplung.00364.2005. [DOI] [PubMed] [Google Scholar]
- 86.Vind I, Johansen JS, Price PA, et al. Serum YKL-40, a potential new marker of disease activity in patients with inflammatory bowel disease. Scand. J. Gastroenterol. 2003;38:599–605. doi: 10.1080/00365520310000537. [DOI] [PubMed] [Google Scholar]
- 87.Lee CG, Da Silva CA, Lee JY. Chitin regulation of immune response: an old molecule with new roles. Curr. Opin. Immunol. 2008;20:684–689. doi: 10.1016/j.coi.2008.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Kawada M, Hachiya Y, Mizoguchi E. Chitinase 3-like-1 (CHI3L1) elicits the production of proinflammatory cytokines and chemokines in colonic epithelial cells and exacerbates TNBS-induced colitis. Gastroenterology. 2006;130:A699. [Google Scholar]
- 89.Johansen JS. Studies on serum YKL-40 as a biomarker in diseases with inflammation, tissue remodeling, fibroses and cancer. Dan. Med. Bull. 2006;53:172–209. [PubMed] [Google Scholar]
- 90.Eurich K, Segawa M, Toei-shimizu S, et al. Potential role of chitinase 3-like-1 in inflammation-associated carcinogenic changes of epithelial cells. World J. Gastroenterol. 2009;15:5249–5259. doi: 10.3748/wjg.15.5249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Brasso K, Christensen IJ, Johansen JS, et al. Prognostic value of PINP, bone alkaline phosphatase, CTX-1, and YKL-40 in patients with metastatic prostate carcinoma. Prostate. 2006;66(5):503–513. doi: 10.1002/pros.20311. [DOI] [PubMed] [Google Scholar]
- 92.Bucolo C, Musumeci M, Maltese A, et al. Effect of chitinase inhibitors on endotoxin-induced uveitis (EIU) in rabbits. Pharmacol. Res. 2008;57:247–252. doi: 10.1016/j.phrs.2008.02.002. [DOI] [PubMed] [Google Scholar]
- 93.Qureshi AM, Hannigan A, Campbell D, et al. Chitinase-like proteins are autoantigens in a model of inflammation-promoted incipient neoplasia. Genes. Cancer. 2011;2:74–87. doi: 10.1177/1947601911402681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Johansen JS, Stoltenberg M, Hansen M, et al. Serum YKL-40 concentrations in patients with rheumatoid arthritis: relation to disease activity. Rheumatology. 1999;38:618–626. doi: 10.1093/rheumatology/38.7.618. [DOI] [PubMed] [Google Scholar]
- 95.Chupp GL, Lee CG, Jarjour N, et al. A chitinase-like protein in the lung and circulation of patients with severe asthma. N. Engl. J. Med. 2007;357:2016–2027. doi: 10.1056/NEJMoa073600. [DOI] [PubMed] [Google Scholar]
- 96.Eaden JA, Abrams KR, Mayberry JF. The risk of colorectal cancer in ulcerative colitis: a meta-analysis. Gut. 2001;48:526–535. doi: 10.1136/gut.48.4.526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Johansen JS, Bojesen SE, Mylin AK, et al. Elevated plasma YKL-40 predicts increased risk of gastrointestinal cancer and decreased survival after any cancer diagnosis in the general population. J. Clin. Oncol. 2009;27:572–578. doi: 10.1200/JCO.2008.18.8367. [DOI] [PubMed] [Google Scholar]
- 98.Ekbom A, Adami H-O, Helmick C, et al. Increased risk of large-bowel cancer in Crohn's disease with colonic involvement. Lancet. 1990;336:357–359. doi: 10.1016/0140-6736(90)91889-i. [DOI] [PubMed] [Google Scholar]
- 99.Catalán V, Gómez-Ambrosi J, Rodríguez A, et al. Up-regulation of the novel proinflammatory adipokines lipocalin-2, chitinase-3 like-1 and osteopontin as well as angiogenic-related factors in visceral adipose tissue of patients with colon cancer. J. Nutr. Biochem. 2011;22:634–641. doi: 10.1016/j.jnutbio.2010.04.015. [DOI] [PubMed] [Google Scholar]
- 100.Hood JD, Frausto R, Kiosses WB, et al. Differential alpha v integrin-mediated Ras-ERK signaling during two pathways of angiogenesis. J. Cell Biol. 2003;162:933–943. doi: 10.1083/jcb.200304105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Shao R, Hamel K, Petersen L, et al. YKL-40, a secreted glycoprotein, promotes tumor angiogenesis. Oncogene. 2009;28:4456–4468. doi: 10.1038/onc.2009.292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Bigg HF, Wait R, Rowan AD, et al. The mammalian chitinase-like lectin, YKL-40, binds specifically to type I collagen and modulates the rate of type I collagen fibril formation. J. Biol. Chem. 2006;281:21082–21095. doi: 10.1074/jbc.M601153200. [DOI] [PubMed] [Google Scholar]
- 103.Cario E, Podolsky DK. Differential alteration in intestinal epithelial cell expression of Toll-like receptor 3 (TLR3) and TLR4 in inflammatory bowel disease. Infect. Immun. 2000;68:7010–7017. doi: 10.1128/iai.68.12.7010-7017.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Harvey S, Weisman M, O'Dell J, et al. Chondrex: new marker of joint disease. Clin. Chem. 1998;44:509–516. [PubMed] [Google Scholar]
- 105.Suzuki M, Hisamatsu T, Podolsky DK. Gamma interferon augment the intracellular pathway for lipopolysaccharide (LPS) recognition in human intestinal epithelial cells through coordinated up-regulation of LPS uptake and expression of the intracellular toll-like receptor 4-MD-2 complex. Infect. Immun. 2002;71:3503–3511. doi: 10.1128/IAI.71.6.3503-3511.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Recklies AD, White C, Ling H. The chitinase 3-like protein human cartilage glycoprotein 39 (HC-gp39) stimulates proliferation of human connective-tissue cells and activates both extracellular signal-regulated kinase- and protein kinase B-mediated signalling pathways. Biochem. J. 2002;365:119–126. doi: 10.1042/BJ20020075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Pearson G, Robinson F, Beers Gibson T, et al. Mitogen-activated protein (MAP) kinase pathways: regulation and physiological functions. Endocr. Rev. 2001;22:153–183. doi: 10.1210/edrv.22.2.0428. [DOI] [PubMed] [Google Scholar]
- 108.van Dekken H, Wink JC, Vissers KJ, et al. Wnt pathway-related gene expression during malignant progression in ulcerative colitis. Acta Histochem. 2007;109:266–272. doi: 10.1016/j.acthis.2007.02.007. [DOI] [PubMed] [Google Scholar]
- 109.Grivennikov S, Karin E, Terzic J, et al. IL-6 and Stat3 are required for survival of intestinal epithelial cells and development of colitis-associated cancer. Cancer Cell. 2009;15:103–113. doi: 10.1016/j.ccr.2009.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Sethi G, Sung B, Aggarwal BB. Nuclear factor-kappaB activation: from bench to bedside. Exp. Biol. Med. (Maywood) 2008;233(1):21–31. doi: 10.3181/0707-MR-196. [DOI] [PubMed] [Google Scholar]
- 111.Kanneganti M, Mino-Kenudson M, Mizoguchi E. Animal models of colitis-associated carcinogenesis. J. Biomed. Biochem. 2011;2011:342637. doi: 10.1155/2011/342637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Yang Z-H, Dai Q, Gu Y-J, et al. Cytokine and chemokine modification by Toll-like receptor polymorphisms is associated with nasopharyngeal carcinoma. Cancer Science. 2012;103:653–658. doi: 10.1111/j.1349-7006.2012.02210.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Davoodi H, Seow HF. Variant Toll-like receptor 4 (Asp299Gly and Thr300Ile alleles) and Toll-like receptor2 (Arg753Gln and Arg677Trp alleles) in colorectal cancer. Iran. J. Allergy Asthma Immunol. 2011;10:91–99. [PubMed] [Google Scholar]