

Abstract
The base excision repair system is vital to the repair of endogenous and exogenous DNA damage. This pathway is initiated by one of several DNA glycosylases that recognizes and excises specific DNA lesions in a coordinated fashion. Methyl-CpG Domain Protein 4 (MBD4) and Thymine DNA Glycosylase (TDG) are the two major G:T glycosylases that remove thymine generated by the deamination of 5-methylcytosine. Both of these glycosylase also remove a variety of other base lesions, including G:U and preferentially act at CpG sites throughout the genome. Many have questioned the purpose of seemingly redundant glycosylases, but new information has emerged to suggest MBD4 and TDG have diverse biological functions. MBD4 has been closely linked to apoptosis, while TDG has been clearly implicated in transcriptional regulation. This article reviews all these developments, and discusses the consequences of germline and somatic mutations that lead to non-synonymous amino acid substitutions on MBD4 and TDG protein function. In addition, we report the finding of alternately spliced variants of MBD4 and TDG and the results of functional studies of a tumor-associated variant of MBD4.
Keywords: Base excision repair, DNA glycosylase, DNA methylation, polymorphisms
1. Introduction
Endogenous DNA damage occurs at a rate upwards of 20,000 lesions per cell per day [1]. This damage often occurs in the form of deamination, alkyation and oxidation of bases in the DNA and can result from hydrolysis or exposure to reactive oxygen species or metabolites [2]. The hydrolytic deamination of 5-methylcytosine (5-meC) and cytosine (C) to thymine (T) and uracil (U), respectively, poses a constant challenge to the integrity of the genome. These spontaneous events occur at a rate of 2–300 lesions/cell/day and if not corrected, lead to C:G to T:A transitions in the following round of DNA replication [3]. These transitions comprise the most frequent mutations in human cancer and other diseases [4] with nearly 50% of somatic mutations of the tumor suppressor gene TP53 in colorectal cancer (CRC) being C:G to T:A within CpG sites [5]. Such findings suggest these mutations have an impact on tumorigenesis making efficient correction of DNA damage essential.
The cell has developed several mechanisms to combat DNA damage. The base excision repair (BER) pathway is mainly responsible for the lesion-specific removal of nonbulky-DNA adducts and has been proposed to act as a tumor suppressor mechanism (for detailed review see [6]). Short-patch BER is initiated by a monofunctional DNA glycosylase that recognizes and excises a damaged base, generating an abasic site. AP endonuclease (APE1) converts the abasic site into a single nucleotide gap by incising the backbone, generating a 3′-OH and a 5′-deoxyribose phosphate (dRP) group. DNA polymerase β then catalyzes removal of the dRP group and fills in the gap. Bifunctional glycosylases possess AP lyase activity and therefore catalyze the backbone incision themselves. APE1 is then needed to remodel the ends and generate a 3′OH group and Pol β fills in the gap. In both cases, the XRCC1-Ligase IIIα complex seals the nick, completing repair. Additionally, long-patch BER involves the excision of 2–12 nucleotides which are replaced by the action of DNA polymerase ɗ or ε. This pathway is less understood and requires additional proteins including PCNA, RFC, FEN1 and DNA ligase I.
The focus of this review is on two of the main DNA glycosylases responsible for initiating the repair of deaminated C and 5-meC within CpG sites: Methyl-Binding Domain Protein 4 (MBD4) and Thymine DNA Glycosylase (TDG). Evidence is emerging for a role for these glycosylases in active demethylation, a process that is important for gene regulation. Interestingly, single nucleotide polymorphisms (SNPs) that result in non-synonymous codon alterations have been identified in both MBD4 and TDG, and these genes have also been found to be mutated in human tumors, as shown in Table 1. These SNPs and tumor-associated mutations result in single amino acid changes that have the potential to affect protein structure and function. Because these glycosylases are vital to repair of G:T/U lesions, subtle alterations in protein function of MBD4 or TDG could disrupt repair of these mutagenic lesions resulting in increased mutations, most likely at CpG sites, altered transcription profiles and/or genomic instability and lead to cancer.
Table 1.
MBD4 and TDG variants in the normal population and in tumors
2. MBD4 Genomic and Protein Structure
The human MBD4 gene is located on chromosome 3q21.3 and spans a region of 9 kbs. Its coding DNA sequence (CDS) is 1,743 bps and the full-length protein is coded for by 8 exons (NCB Gene ID: 8930; Figure 1).
Figure 1. Alternatively spliced forms of MBD4 that are reported in the NCBI database.<.

br>Using SpliceMiner, there are several forms of alternatively spliced variants of MBD4 reported in NCBI database [7].
2.1. MBD4 alternatively spliced variants
According to SpliceMiner [7], a web interface for querying Evidence Viewer Database based on data obtained from NCBI Entrez Gene and NCBI Evidence Viewer, there are several splice variants of the MBD4 reported (Figure 1). This includes four mRNA variant submissions which are translated into two forms of full-length MBD4 proteins with sizes equaling 580 aa (Accession number AF072250, AF114784, NM_003925) or 574 aa (Accession number BC011752) long proteins (Figure 1). There are also three short versions of alternatively spliced MBD4 reported. These are translated into protein with a size of 540 aa (Accession number AF532602), 572 aa (Accession number AK303013), and 262 aa (Accession number AM180876). Results from further studies of the short 262 aa version of MBD4, which was identified in HeLa cells, indicated that the protein possessed uracil DNA glycosylase but not thymine DNA glycosylase activity [8].
2.2. Full length MBD4 proteins with 580 or 574 amino acids are found in humans
All of the biochemical studies that have used full-length MBD4 are based on the 580 aa long protein [9–11]. We recently isolated and cloned a full-length MBD4 encoding gene. During the cloning process, we found it was missing 18 nts. This encodes a 574 aa protein similar to the one reported previously (Accession number BC011752). The result from further investigation revealed both the full-length MBD4 encoding mRNA with and without the 18 nts was expressed in several tissues tested (Figure 2). The 18 nts is before exon 4 and is located between the two major domains of MBD4, the amino-terminal methyl-CpG-binding domain and a carboxy-terminal glycosylase domain, which may have been formed from fusion [9]. The altered spacing between the two domains might provide extra level of flexibility to the protein to access its target.
Figure 2. Human cells express two forms of full length MBD4 protein with size of 574 aa and 580 aa.

Panel A: Total RNA from several human tissues and A549 cell line were isolated and transcribed into cDNA. Using primers specific to the beginning of the first and last exon of MBD4 several potential splicing variants were detected of which the full length MBD4 encoding cDNA (top band) appeared to be the abundant copy. The full-length copy of the MBD4 encoding DNA (top band) was isolated and used as a template for further sequencing analysis. Panel B: As sequencing results show, all three tissues tested (lung, pancreas, and breast) as well as the A549 cells expressed both the 1743 nts and 1725 nucleotide variants. The 18 nucleotide piece of DNA with the sequence of “GTGAGAAAATATTTCAAG” found in the longer copy is located before exon 4.
2.3. MBD4 is a multidomain protein
MBD4 is unique from other thymine and uracil-processing glycosylases in that it contains two DNA binding domains, an N-terminal methyl-binding domain (MBD) and a C-terminal glycosylase domain (Figure 3). MBD4 is 580 amino acids in length with the MBD spanning residues 82–147 and the glycosylase domain spanning residues 426 to 580. The long spacer domain that separates these two functional domains contains two serine residues targeted for phosphorylation by protein-kinase C (PKC). This phosphorylation may alter the structure of MBD4 to allow a shift in substrate specificity ultimately resulting in MBD4-mediated DNA demethylation [12]. So far, MBD4 has evaded efforts to crystallize the full-length protein, but partial crystal structures, along with modeling of MBD4, have given some insight into MBD4 substrate specificity and function.
Figure 3. Schematic of MBD4 and TDG structures and location of SNPs.<.

br>Not drawn to scale. MBD4 includes an amino-terminal methyl-binding domain, carboxy-terminal glycosylase domain and a long spacer domain with putative nuclear localization signals. TDG includes a core catalytic domain flanked by less conserved amino and carboxy-terminal ends. The relative location of single amino acid alterations is shown.
MBD4 was first identified in a bioinformatics study by its amino-terminal methyl-CpG binding domain (MBD), which shares sequence homology with other MBD proteins, such as methyl CpG binding protein 2 (MeCP2) and methyl-CpG binding domain protein 2 (MBD2) [13]. This domain includes four anti-parallel beta-sheets that generate a wedge-shaped structure [14]. The two longer beta-sheets are thought to interact with the major groove of DNA where methylation specifically takes place. This domain directs binding to hemi-methylated or fully methylated DNA in vitro and is thought to be responsible for localizing MBD4 to methylation-rich genomic areas where base deamination is likely to occur. It has been proposed that the severe bending of the DNA by the glycosylase domain prevents simultaneous binding of both domains to a single mCpG:T lesion, and therefore it is likely that the MBD binds neighboring mCpG sites. A solution structure of the MBD of human MBD4 bound to DNA will be vital to understanding how the methylation signal is read by MBD4 and how this glycosylase accesses CpG sites on nucleosome cores.
The glycosylase domain of MBD4 belongs to the helix-hairpin-helix (HhH) DNA glycosylase superfamily, which includes AlkA, MutY and OGG1 [15–17]. This domain contains ten alpha helices that come together to form a cleft in the middle. Modeling of the glycosylase domain of mouse MBD4 suggested it binds DNA via the minor groove and bends it ~70° at the damaged base [15]. This orientation may allow for a base flipping mechanism to occur where the damaged base is swung out of the DNA helix by torsional rotation of the sugar-phosphate backbone and docked into the active-site cleft [15]. Additional modeling of the glycosylase domain of human MBD4 revealed residues Arg468-Thr469-Ser470 are important for DNA binding and base flipping, while residues Gln449-Tyr540-Asp560 are essential for thymine recognition and catalysis [16,17].
Manvilla and colleagues were the first to succeed in crystallizing the glycosylase domain of MBD4 bound to DNA containing an abasic site analog paired with G [17]. This structure supports previous models of MBD4 but also revealed new information. The crystal structure confirmed MBD4 binds the minor groove of DNA at the target site, but that the DNA is bent only about 57°. The structure revealed that the opposing G remains intrahelical but is nestled into a recognition pocket where it makes several electrostatic interactions with residues of the active site [17]. These contacts are not compatible with A, thus providing an explanation for the strict preference for excising T from G:T versus A:T. In addition, the crystal structure showed MBD4 do not form an interaction with the G:C pair at the 3′ side of the lesion, thus explaining why the glycosylase domain has little specificity for acting at CpG sites by itself [17]. The MBD therefore must be necessary for conferring the substrate specificity seen with purified full-length MBD4 [9].
3. Substrates of MBD4
3.1. MBD4 removes T and U paired with G in DNA
There are several DNA glycosylases expressed in human cells that function to remove specific types of base lesions. MBD4 specifically catalyzes the removal of T and U paired with guanine (G) within CpG sites [9,18,19]. These bases often arise in DNA due to the hydrolytic deamination of 5-meC and C to T and U, respectively, but U can also arise due to misincorporation of dUMP from the nucleotide pool. Efficient removal of T and U is critical as failure to excise these lesions leads to mutagenesis.
3.2. MBD4 removes halogenated pyrimidines
MBD4 is active on several halogenated pyrimidines, including 5-chlorouracil and 5-bromouracil paired with G that result from peroxidase-mediated inflammatory processes [20,21], as well as chemotherapy-induced 5-fluorouracil (5-FU) and 5-iododeoxyuracil lesions [22,23]. Furthermore, MBD4 exhibits weak glycosylase activity on 3,N4-ethenocytosine (εC), a product of lipid peroxidation and metabolite of vinyl chloride and ethyl carbamate [18]. MBD4 preferentially binds lesions within methylated or hemimethylated CpG sites, but also processes lesions at unmethylated CpG sites in DNA [9].
3.3. MBD4 and active demethylation
More recent work has revealed MBD4 is capable of excising 5-hydroxymethyluracil (5-hmU) from a double-stranded CpG dinucleotide in vitro [24,25]. 5-hmU is thought to arise as an intermediate in a multistep active demethylation pathway whereby 5-meC is first hydroxylated by the ten-eleven translocation (TET) family of dioxygenases, deaminated by activation-induced deaminase (AID) and finally removed by MBD4 or TDG in a BER process [26,27]. Additional work also revealed a novel role for MBD4 as a demethylase itself. Zhu and colleagues showed MBD4 is capable of removing 5-meC from hemimethylated DNA in an in vitro system [11], while another group reported that PKC phosphorylation of MBD4 potentiated its 5-meC glycosylase activity [12]. These were the first works supporting the existence of a human 5-meC glycosylase.
4. MBD4 Biological Features
4.1. MBD4 is important for suppression of mutagenesis
MBD4 has been shown to play a central role in the suppression of CpG mutability and tumorigenesis in vivo. Two groups have successfully generated MBD4-deficient mice using gene targeting techniques to introduce a null mutation into the murine Mbd4 gene. Both heterozygous and homozygous mice are fertile and develop normally. Whereas loss of MBD4 did not lead to tumor formation in mice by itself, mice deficient for MBD4 were shown to have a two to three-fold increase in the number of C to T transition mutations at CpG sites in the small intestine compared to wildtype mice [28,29]. In addition, MBD4-deficient mice show accelerated intestinal tumor formation and increased multiplicity on the adenomatous polyposis coli (Apc) min/+background. Examination of these tumors revealed an increase in somatic Apc mutations compared to WT, 82% of which were C to T transitions at CpG sites [28]. Although inactivation of MBD4 was not enough to initiate tumorigenesis by itself, these results provide evidence that MBD4 functions in vivo to suppress mutations at CpG sites in mammalian genomes, and that loss of MBD4 can have an accelerating effect on tumor formation in cancer-predisposing backgrounds.
4.2. MBD4 interacts with MLH1
In addition to its role as a DNA glycosylase in BER, MBD4 was identified in a yeast two-hybrid screen to interact with the mismatch repair (MMR) protein MLH1 [10]. Due to the fact that MBD4 is mutated in a high percentage of MMR-deficient colorectal tumors with MSI, as well as the fact that loss of MLH1 in mice enhances tumor formation [30,31], Sansom and colleagues addressed the question of whether or not loss of MBD4 contributes to tumorigenesis by generating mice doubly deficient for MBD4 and MLH1 [31]. Interestingly, the additional loss of MBD4 did not accelerate tumorigenesis or increase mutation frequency in these mice, suggesting MBD4 may not play a direct role in MMR-deficient tumorigenesis.
4.3. MBD4 is linked to apoptosis
Many MMR proteins, including MLH1, have been functionally linked to apoptosis, and mammalian cell lines defective in MMR are resistant to treatment with methylating agents [32,33]. These cell lines accumulate damage, mainly in the form of methylation of the O6 position of guanine generating O6-methylguanine (O6-meG). If not repaired, these base lesions direct misincorporation of T during replication generating O6-meG:T mispairs. These mispairs are usually recognized by MMR leading to cell cycle arrest and apoptosis, but MBD4 was also shown remove the T opposite O6-meG [32]. Due to the fact that MBD4 interacts with MLH1, several groups have investigated the biological consequence of loss of MBD4 on the DNA damage response and apoptosis in mice [31,32,34]. Interestingly, mouse embryonic fibroblasts (MEFs) deficient in MBD4 had reduced levels of several MMR proteins, as well as diminished apoptotic response to several cytotoxic agents, such as MNNG, 5-FU, cisplatin and temozolomide [32,34]. Intriguingly though, MBD4−/− mice did not display microsatellite instability (MSI), a key feature of compromised MMR [28], and mice exposed to 5-FU and temozolomide that were doubly mutant for MBD4 and MLH1 did not exhibit an additive effect. These results suggest MBD4 and MLH1 act in the same apoptotic pathway.
Further evidence for a role of MBD4 in apoptotic signaling or control came when MBD4 was shown to directly interact with proapoptotic Fas-associated death domain protein (FADD) in a complex with MLH1 [35]. More specifically, MBD4 was found to regulate DNA damage-, Fas ligand- and cell detachment-induced apoptosis in cells. Interestingly, MBD4 was also shown to interact with DNA methyltransferase (DNMT1) in a complex with MLH1 in Xenopus embryos [36]. Ruzov and colleagues found depletion of xMBD4 or xMLH1 increased the survival rate of xDMNT-depleted embryos while over-expression of these proteins induced programmed cell death at the onset of gastrulation. They went on to show MBD4 recruits both DNMT1 and MLH1 to sites of heterochromatin in mouse embryonic cells, and that these proteins accumulate at site of IR-induced DNA damage in mammalian cells. Together, these data strongly support a link between genome surveillance/DNA repair and apoptosis.
4.4. MBD4 and DNA methylation
In the mammalian genome, about 70% of all CpG sites are methylated [37]. These dinucleotides are often found clustered in CpG-islands within promoter regions and are essential for the regulation of gene expression, genomic stability, genomic imprinting and X-inactivation. Because this epigenetic mark is important for so many cellular processes, careful regulation of DNA methylation and demethylation is crucial. DNA methyltransferases (DNMTs) are responsible for establishing and maintaining DNA methylation in mammalian cells. DNMT3a/b mediates de novo DNA methylation, while DNMT1 acts on newly synthesized DNA to maintain methylation marks [38]. DNA demethylation can occur passively through replication in the absence of remethylation, or actively in a coordinated multistep process.
There has long been evidence of active demethylation in mammals, mainly during embryonic development, as well as during somatic hypermutation/class-switch recombination in immune cells [39]. More recently, new evidence has emerged supporting an active demethylation process occurring at hormonally-regulated gene promoters and enhancers that involves deamination of methylated cytosine followed by a base excision repair process. Interestingly, both MBD4 and TDG have been implicated in this process suggesting DNA repair mechanisms, in particular BER, are critical in transcriptional regulation [12,26,40].
4.4.1. Role of MBD4 in active demethylation
Rai and colleagues first linked MBD4 to active DNA demethylation using a zebrafish model system. They presented evidence that demethylation was a coordinated system involving a deaminase (AID/Apobec), an MBD4-related G:T glycosylase, and a Gadd45 family member [27]. They showed that overexpression of AID and MBD4 caused widespread demethylation, and that knockdown of these proteins led to remethylation of specific genes. In addition, co-injection of a catalytically inactive MBD4 mutant with a methylated DNA fragment led to the accumulation of G:T intermediates and a small number of 5-meC to T transition mutations, suggesting that the catalytic activity of MBD4 is critical for proper demethylation. Finally, they demonstrated that that non-enzymatic factor Gadd45 promoted demethylation in zebrafish embryos and enhanced the functional interactions between MBD4 and AID/Apobec in human cells. It was proposed that Gadd45 may serve as a scaffold to physically and/or functionally couple DNA demethylation components, and that MBD4 may promote 5-meC replacement through a base excision repair process.
4.4.2. Phosphorylation of MBD4 promotes 5-meC glycosylase activity
Further evidence emerged to support the involvement of MBD4 in active demethylation. Protein-kinase C phosphorylation of MBD4 at two specific serine residues (165 and 262) following parathyroid hormone stimulation was shown to promote demethylation within the CYP27B1 gene promoter [12]. This was achieved by excision of 5-meC through the glycosylase activity of MBD4 followed by a BER process. In vitro kinetic assays showed that while unmodified MBD4 was able to remove 5-meC to a small degree, phosphorylation enhanced its 5-meC glycosylase activity significantly. Overall, this work raised the possibility that MBD4 may function to derepress other hormone-regulated genes and that loss of MBD4 expression or function may lead to altered gene expression, potentially impacting the integrity of the genome.
4.5. MBD4 is mutated in tumors
MBD4 was found to be mutated in 26 to 43% of human gastric, colorectal, endometrial and pancreatic cancers that exhibit MSI (Table 1, [41–44]). It is important to note these were small scale studies with sample size ranging from 19 to 54 tumors, and therefore it is possible that mutations could have been over or underrepresented. There is evidence for biallelic inactivation of MBD4 as well as heterozygous mutations in CRC tumors. A majority of these mutations occur within the polyA10 tract of exon 3 resulting in the production of a truncated protein that is able to bind to meCpG sites but lacks its glycosylase activity. This variant, called MBD4tru, exerts a dominant negative effect by competitively inhibiting wt MBD4 activity on T:G and U:G mismatches in vitro [45]. In addition, expression of MBD4tru in human MSI colon cancer cells more than doubled the mutation frequency and affected the whole mutation spectrum. These findings suggest mutation of MBD4 has a widespread effect on genomic stability and may be of significance in tumor progression.
4.6. SNPs of MBD4
Several polymorphisms of MBD4 have been identified in the normal population in addition to a tumor-associated variant (Table 1). Most of these variants have not been characterized for their catalytic or DNA binding activity and few predictions can be made given their locations in MBD4 and the lack of a complete crystal structure. Two variants of MBD4, D568H and C61R were each identified in 1 out of 42 gastric tumors with MSI [46]. In the same study, C61R was also found in 4 out of 95 normal control samples, indicating it was a novel germline variant or SNP of MBD4. Cytosine residue 61 is located adjacent to the MBD. Mutation of a nucleophillic Cys to a large and basic Arg could affect the structure of the MBD ultimately affecting the affinity of MBD4 for DNA. Aspartic acid residue 568 is located within helix-K of the glycosylase domain. Helix-K contains several residues important for DNA binding and base flipping, and helps form the catalytic pocket of MBD4. Mutation of acidic Asp to basic His may affect the catalytic ability of D560 or the docking ability of K562 if the switch in charge causes steric hindrance that alters the structure of helix-K. Both SIFT and PolyPhen, two programs that predict the possible impact of an amino acid substitution on the structure and function of a human protein, predicted that mutation of D568 to H would be damaging and affect MBD4 function. Conversely, these programs predicted C61R would be a tolerated or benign substitution.
Biochemical studies in our lab have recently shown that variant D568H, but not C61R, has both reduced catalytic activity and binding affinity for its two predominant substrates, T and U paired with G within a CpG context (Figure 4 and data not shown). More specifically, D568H forms 6-fold less product as WT in an in vitro glycosylase assay and exhibits a 2 to 3-fold reduced binding affinity for its DNA substrates compared to WT. The results seen are not due to major global changes in protein structure, as circular dichroism revealed WT and D568H have overall similar tertiary structures (unpublished data). These data suggest D568H may likely have a functional phenotype in vivo that could alter DNA repair capacity and contribute to cancer progression.
Figure 4. D568H has reduced catalytic activity and binding affinity for its two predominant substrates in vitro. :<.

br>Panel AIncreasing amounts of WT or D568H protein were incubated with 5nM 32P-labeled 38-mer oligonucleotides containing a G:U or G:T lesion. Reactions were placed at 37°C and samples were collected over time up to 20 minutes. Samples were treated with NaOH at 90°C for 30mins. Samples were resolved on a polyacrylamide denaturing gel, detected, quantitated, fit to a first-order rate equation and the catalytic rates were derived. Panel B: Increasing concentrations of WT or D568H (0–1000nM) were incubated with 32P-labeled oligonucleotides carrying a G:T or G:U lesion for 15 mins at RT. Binding reactions were resolved on a 6% non-denaturing polyacrylamide gel, visualized, quantified and fit to a nonlinear regression to derive a dissociation constant (Kd) for each substrate. Panel C: Summary of dissociation constants (Kd) and catalytic rates (kcat).
Thymine DNA Glycosylase
5. TDG Genomic Structure
The human TDG gene is located on chromosome 12q24.1 and spans a region of 23 kbs. Its CDS is 3,251 bps long with the full-length protein coded for by 10 exons (NCB Gene ID: 6996; Figure 5).
Figure 5.

Alternatively spliced forms of the TDG reported in the NCBI database that are detected by SpliceMiner [7].
5.1. TDG alternatively spliced variants
Whereas the full-length TDG protein is 410 amino acids (aa) in length (Accession numbers BC037557, and NM_003211), there are a few shorter spliced variants of human TDG reported with lengths equaling 206 aa (Accession number AK295387), 161 aa (Accession number AK299832), and 267 aa (Accession number AK303631) (Figure 5). Furthermore, another alternative splicing variant of TDG has been reported of which the entire exon 2 is missing. The excision of exon 2 from this TDG transcript causes a frameshift in the TDG open reading frame [47–49]. It was reported that the presence of this TDG variant is either a result of aberrant splicing and does not code for any functional protein, or its translation starts from a non-AUG codon [48].
5.2. Identification of a novel alternatively spliced version of TDG
The full-length human thymine DNA glycosylase gene, TDG, encodes for a protein with a size of 410 aa [50,51]. During cloning and analysis of this gene, we identified a novel alternatively spliced variant of TDG which is missing exons 2–4 (Figure 6). The results from quantification of this variant estimated that it could be found in several tissues tested at levels 35–65% lower than that of the 1233 nts variant (Figure 6). The role of this variant, if any, is not known.
Figure 6. Identification of a new splicing variant of TDG mRNA in human cell.<.
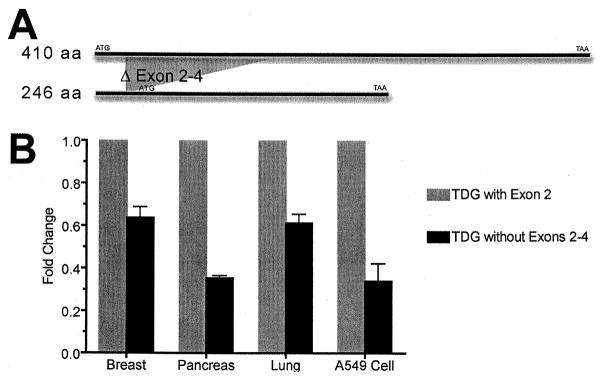
br>Panel A: The full-length copy of the TDG encoding gene is 1233 nucleotides long (top band). Total RNA from several human tissues and the A549 cell line was isolated and transcribed into cDNA. We used primers specific to the beginning and end of the gene to amplify and clone the full-length TDG gene using the cDNA as a template. During the cloning and PCR process we identified a new splicing variant of TDG that is 788 nucleotides long. As shown above, the new TDG variant is missing exon 2–4. If the first ATG is used for translation there will be a truncated protein of 16 aa long whereas if the second ATG acts as translation starting point, it will lead to a protein with a size of 246 aa. Panel B: We used a forward primer specific to the first exon and reverse primers specific to exon 2 and exon 5 to evaluate the amounts of these two variants in several human tissues. Results from quantification analysis indicate some level of the new short variant present in all tissues tested. We calculated the fold changes using the ddCT method by which the fold change represented as 2−ddCT, and found that the variant missing exons 2–4 was present at levels approximately 35–65% of the variant with exon 2.
6. TDG Structure and Biochemistry
6.1. TDG is a member of the MUG superfamily
TDG is a member of the Mismatch Uracil Glycosylase (MUG) branch of the monofunctional uracil DNA glycosylase superfamily, named for the E. coli Mug protein [51]. This group of glycosylases shares a common α/β structural fold and all have a conserved catalytic core flanked by less-conserved C- and N-terminal ends of variable length. TDG itself is 410 amino acids in length with its catalytic core spanning residues 123–300, referred to as the glycosylase domain (Figure 1). Its divergent terminal domains have been shown to modulate the enzymatic activity of TDG as well as mediate interactions with other proteins, such as nuclear receptors and other transcriptional regulators [52–55]. In addition, the C-terminal domain contains a SUMO conjugation site Lys330 while the N-terminal domain contains a regulatory domain that is subject to acetylation at several lysine residues [56,57]. All of these interactions are thought to modify the function of TDG and some help determine substrate specificity, substrate interaction and the kinetics of base release.
6.2. TDG/MUG proteins use a wedge for catalysis
Initial insight into the structure-function of TDG and other MUG family proteins came with the resolution of the 3D structure of the E. coli Mug (eMug) protein [58]. This structure confirmed an intercalation/nucleotide flipping mechanism for base recognition and removal, as well as revealed a large hydrophobic catalytic pocket that would accommodate a broad range of substrates including more bulky adducts, such as ethenoadducts of C and A, in addition to its preferred G:T substrate. This structure also explained the strict double-strand substrate dependency of MUG proteins. The conserved structure of the catalytic core forms a ‘wedge’ that intercalates into the DNA from the minor groove and occupies the space from where the substrate base has been flipped out. This wedge then establishes hydrogen bonds with the widowed base that are specific for G, thus explaining the substrate specificity of Mug proteins. Mutational analyses are in alignment with this Mug-based model [59,60] and more recent crystal structures of TDG have revealed new information about the structure and function of this protein.
6.3. Structural basis of sumoylation
Crystal structures of Small Ubiquitin-like Modifiers (SUMO)-1 and -2/3-conjugated TDG have allowed for a better understanding of the C-terminal region in TDG function. TDG is sumoylated at Lys330 via an isopeptide bond that forms between the ε-amino group of Lys and the C-terminus of the SUMO protein [61,62]. The structures show there are two major domains, a catalytic core domain of TDG and a SUMO-containing domain, that are connected by a short crossover loop comprising residues 301–307 of TDG. This crossover allows for a small molecular interface at the C-terminal region of TDG that makes noncovalent contacts with the SUMO domain in addition to its covalent link. Conjugation of either SUMO-1 or SUMO-2/3 to TDG does not cause any major structural changes but modeling of TDG-SUMO-1 bound to AP-site DNA showed the C-terminus of the catalytic core would now encounter steric clash with the sugar-phosphate backbone at positions +1 and +2 from the unpaired G [61]. It was proposed that this steric clash helps stimulate dissociation of sumolyated TDG from its abasic product.
6.4. Two subunits of TDG can bind DNA lesions
Additional insight into TDG binding and release came with the resolution of a crystal structure of TDG in complex with abasic DNA [63]. This structure revealed that TDG can bind in a 2:1 or 1:1 complex with DNA and provides an explanation for the strict specificity for G versus A as the pairing partner opposite the lesion. In both structures one subunit of TDG is bound to the abasic site (referred to as the product complex) and makes many specific interactions with its substrate including contacts with the abasic strand, the abasic sugar and both guanines of the CpG site. The structures show the abasic nucleotide is completely flipped out of the helix and into the active site and the DNA is bent at a 43° angle. Interestingly, while this structure supported previous models with which the overall structure of TDG mimics that of eMug, it also revealed unexpected differences in the protein-DNA interactions.
Of particular note was the finding that a second subunit of TDG can be bound at an undamaged or nonspecific site next to the product complex. This nonspecific (NS) subunit makes less extensive interactions with DNA that are mostly restricted to the complementary strand. While it is unclear if the NS subunit flips an undamaged nucleotide out of the helix, it does make contacts with a “target” base, which may or may not be specific for cytosine [63,64]. This interaction would provide a nice explanation for the CpG sequence specificity that is seen with TDG binding. Interestingly, it was recently shown that the NS subunit is dispensable for locating and processing of G:T and G:U lesions by TDG in vitro [64], thus suggesting the NS complex may rather be important for the ability of TDG to modulate the activity of transcription factors and transcriptional co-activators. Additional studies are needed to explore these ideas further.
6.5. TDG processes many DNA lesions
A common feature of the MUG proteins is their broad range of substrates [51], TDG being no exception. TDG actively processes lesions that result from oxidation, alkylation and deamination of C, 5-meC, T and A [65]. TDG removes 5′ halogenated derivatives of U and C, such as 5-fluorouracil and 5-bromouracil [66], as well as εC, a product of lipid peroxidation [67]. In addition, TDG is able to actively remove 5-formylcytosine(5-fC) and 5-carboxylcytosine(5-caC), two oxidation products of TET protein modification [68,69]. TDG exhibits highest processing efficiency on lesions opposite G within CpG sites, but can also remove several bases opposite A [65,70]. This broad range of substrates allows TDG to eliminate a variety of mutagenic bases and thus help stabilize the genome.
Whereas TDG is best known for its ability to remove T from G:T lesions, it actually processes U mispaired with G most efficiently [65]. The lower catalytic activity on G:T relative to G:U substrates may be explained by steric hindrance involving the bulky C-5 methyl group. This steric hindrance also likely accounts for the 30-fold weaker binding affinity of TDG for G:T relative to G:U substrates [70]. Recent resolution of a crystal structure of TDG bound to 2′-deoxy-2′-fluoroarabinouridine (UF) revealed mutation of two active site residues (Ala145 and His151) increased G:T glycosylase activity but also enhanced the aberrant removal of T from A:T base pairs [71]. It was shown that Ala145 destabilizes nucleotide flipping for dT, but not dU, via steric hindrance, and that His151 forms a repulsive electrostatic interaction with the base that slows the chemical step of the reaction. This feature of TDG may reflect a compromise between the need for efficient processing of deaminated 5-meC in order to prevent C→T transitions and the need to avoid aberrant removal of T from A:T pairs.
It has been revealed that the N-terminus of TDG also plays a role in conferring G:T/G:U specificity [56]. Snet-Nocca et al. identified a regulatory domain (RD) spanning residues 51–111 that is able to adopt a distinct conformation that enhances the DNA binding affinity of TDG for G:T DNA while preventing G:U processivity and enzymatic turnover [56]. This domain exhibits a small degree of structural organization and is able to directly interact with the catalytic domain. It was proposed that the RD may provide additional substrate recognition surfaces for the methyl group of T that allow TDG to bind substrates containing a G:T mispair. Further work needs to be done in order to understand the careful regulation of the RD conformation and its inter- and intramolecular interaction that help mediate G:T versus G:U processing activity.
6.6. TDG exhibits a slow rate of turnover
TDG is distinct in that it exhibits extremely tight binding to its abasic product after base removal. Studies have shown that TDG binds to AP-site:G DNA with a higher affinity than it binds to any of its preferred substrates [72]. Because of this, the rate of dissociation of TDG from its abasic product is the rate-limiting step of the glycosylase reaction. Direct interaction of TDG with APE1 significantly increases the rate of dissociation of TDG from AP sites and allows downstream processing of the lesion [72,73]. Both of these features of TDG provide safeguards for lesion processing because they prevent the release of unstable and cytotoxic BER intermediates until the proper downstream repair machinery has assembled.
The enzymatic turnover of TDG is also regulated by sumoylation [57]. Small Ubiquitin-like Modifiers (SUMO) are small polypeptides that can interact with or be covalently linked to other proteins. Covalent SUMO modification requires its own set of E1, E2 and E3 conjugating enzymes and is an ATP-dependent process [74]. In general, these modifications affect protein localization, stability or enzymatic activity. TDG can be sumoylated by SUMO-1 or SUMO-2/3 at lysine residue 330 [57]. This residue is located within a sumoylation consensus motif (VKEE) located in the C-terminal domain of TDG, as previously discussed. Upon sumoylation, TDG is no longer able to stably bind to AP-sites or to interact with free SUMO. In addition, sumoylation of TDG enhances the stimulating effects of APE1 in order to significantly increase enzymatic turnover of bound TDG [57,73]; as well as increases G:U repair activity while impairing G:T processivity [57]. Intriguingly, sumoylation was also found to inhibit the interaction of TDG with histone acetyltransferase CBP [75], a finding that will be expanded on later in this review. All together, structural and biochemical evidence supports the idea that sumoylation of TDG, through both covalent and noncovalent interactions, induces changes in protein conformation that are required for its functionality. TDG can be de-modified through the action of a SUMO-specific isopeptidase to allow continued action of TDG on G:U and G:T mispairs that may be generated.
7. TDG Biological Features
7.1. TDG is not expressed during S phase
Mammalian cells possess several enzymes with uracil and thymine glycosylase activity, namely nuclear uracil-DNA glycosylase (UNG2), single-strand selective monofunctional uracil DNA glycosylase (SMUG1), TDG and MBD4. The fact that these enzymes have co-evolved suggests they each fulfill distinct biological functions. In accordance with this idea, expression of TDG was found to be strictly regulated according to the cell cycle [76]. While TDG is highly expressed during G2-M and G1 phases; expression rapidly declines at the G1-S transition [76], just when UNG2 expression begins to rise [77]. This change in TDG protein level is facilitated through ubiquitylation and proteosome degradation [76]. Interestingly, it was shown that incomplete degradation of exogenously expressed TDG impeded S-phase progression and cell proliferation, suggesting expression of TDG during S-phase is disadvantageous to the cell [76]. This tight regulation may prevent TDG from interfering with coordinated DNA repair events already set in place during replication, such as mismatch repair or action by UNG2. Moreover, because TDG binds tightly to AP sites and requires sumoylation to induce turnover, removal of misincorporated U or other lesions by TDG during replication may lead to stalling and eventual collapse of the replication fork. This, in turn, could induce the formation of SS and DSBs, posing an even greater repair challenge to the cell.
7.2. TDG is essential for embryogenesis
TDG was found to be uniformly and ubiquitously expressed early during embryonic development, with expression seen as early as mouse embryonic day (E) 7.5 [78]. High levels of mRNA were established by E14.5, particularly in the developing nervous system, thymus, lung, liver, kidney and intestine. The role of TDG in embryogenesis was found to be essential, as knockout of TDG in mice led to embryonic lethality by E12.5 [26,79]. Surprisingly, evaluation of TDG null embryos revealed TDG status did not affect cell survival following ionizing radiation or H2O2, indicating the role of TDG in repair of base damage is minor during development [79]. Instead, the authors discovered that this lethal phenotype was associated with epigenetic alterations, namely aberrant promoter methylation and imbalanced histone modification, impairing the expression of developmental genes in TDG knockout mice. TDG was shown to directly associate with the promoters of these aberrantly regulated genes, along with two downstream components of BER, XRCC1 and APE1. Moreover, TDG was required for targeting H3K4-specific methyltransferase MLL1 and transcription-activating histone acetyltransferase CBP/p300 to these promoters. These data support the formation of a transcription regulatory complex that maintains proper epigenetic states at developmentally regulated gene promoters and it is likely that TDG protects these promoters from aberrant CpG methylation through a BER process.
7.3. TDG plays a role in toxicity of 5-FU
Work from the Schar laboratory using TDG−/− MEFs generated from knockout embryos, along with siRNA studies, revealed a major role of TDG in mediating the cytotoxic effects of the chemotherapeutic drug 5-FU [80]. 5-FU is used in the first line treatment of many human cancers, in particular colorectal, gastrointestinal, breast and ovarian cancer. Once it penetrates the cell, 5-FU is converted into several active metabolites that have been shown to disrupt RNA synthesis through incorporation into RNA [81]. In addition, 5FU can inhibit thymidylate synthase (TS) leading to a nucleotide pool imbalance, and consequentially, the misincorporation of U and fluorodeoxyuridine monophosphate (FdUMP) into DNA. It was originally proposed that the excision of misincorporated U by UDGs resulted in cell death through the induction of destructive DNA fragmentation but work by Kunz and colleagues questions this idea. They showed massive incorporation of 5-FU into DNA followed by excision by TDG actually mediates 5-FU cytotoxicity [80]. More specifically, they showed that TDG is the major and rate-limiting 5-FU:A glycosylase and that inactivation of this glycosylase in both mouse and human cancer cells significantly increased resistance to 5-FU. They revealed TDG mediates 5-FU cytotoxicity by binding tightly to abasic sites after lesion removal and preventing downstream processing of the repair intermediate. This ultimately leads to the accumulation of DSBs, delays S-phase progression and activates a DNA damage response in these cells. This work brings to light the possibility of TDG being an important prognostic marker in patients diagnosed with cancer treated with 5-FU-based chemotherapy, and that slight alterations in TDG function could either facilitate or impair the effectiveness of this treatment.
7.4. Transcriptional Regulation and TDG
TDG was originally identified as a G/T-specific DNA glycosylase involved in BER [50,82] but since has been shown to process numerous other lesions and participate in a variety of cellular processes. TDG has been shown to interact with several transcription factors (TFs)[54,83], transcriptional coactivators [53,84] and a DNA methyltransferase [55], supporting a role for TDG in transcriptional regulation. In addition, recent work has linked TDG to an active DNA demethylation process at gene promoters [26,40], further supporting a role for TDG in the regulation of gene expression and epigenetics.
7.4.1. TDG interacts with NR superfamily
The initial indication that TDG played a role in transcription came in 1992 when TDG was identified as an interacting factor of the sequence-specific DNA-binding protein c-Jun [85]. TDG was then shown to interact with two members of the nuclear receptor (NR) superfamily, RAR (retinoic acid receptor) and RXR (retinoid X receptor) in a yeast two-hybrid system [83]. RXRs bind to genomic response elements (RAREs) as either homodimers or heterodimers with RARs in order to modulate transcription in a promoter-specific and ligand-dependent manner [86]. TDG was found to interact with RXR/RAR through its central domain (residues 122–346) and was shown to enhance RXR and RXR/RAR binding to DNA substrates containing RAREs in vitro [83]. In addition, transfection of TDG into mammalian cells increased RXR- and RXR/RAR-mediated transactivation from RARE-containing reporter genes. These data provided the first concrete evidence that TDG functioned as a coactivator of transcription.
Work studying thyroid transcription factor 1 (TTF-1) revealed TDG can also play a role in the suppression of transcription [52]. TTF-1 is a member of the NK2 family of homeobox transcription factors and is vital for the normal development of forebrain, lung and thyroid during embryogenesis as well as for gene expression in adult lung and thyroid tissues. TDG was found to interact with TTF-1 in both yeast and mammalian cells specifically through the C-terminal activation domain of TTF1. This interaction strongly inhibited TTF-1 transcriptional activity in several cell lines, but the mechanism responsible for this repression remains unclear.
TDG was subsequently found to interact with the estrogen receptor α (ERα), another member of the NR superfamily [54]. ERα and ERβ are responsible for the major estrogenic responses in mammals, with ERα being most important in the development and regulation of reproductive tissues, as well as in bone development and homeostasis [87]. Similar to other NR, ERs are composed of two major domains, a ligand-binding domain (LBD) that binds estradiol (E2) and a core DNA-binding domain (DBD) that binds cognate DNA sequences, known as estrogen responsive elements (EREs). Upon ligand binding, ERs form homo- or heterodimers at EREs and stimulate gene expression through the recruitment of transcriptional cofactors and direct interaction with the transcription machinery [87]. A majority of these cofactors bind to the LBD of ERα and can play a role in ligand binding, signal transduction and formation of active transcription complexes. These cofactors can help turn on transcription (coactivators) or repress it (corepressors).
Chen et al. identified TDG as a novel coactivator of ERα. TDG was shown to directly interact with ERα in a ligand-dependent manner in vivo and stimulated ERα activity in a reporter gene assay [54]. Biochemical studies revealed this interaction is established through helix 12 of the LBD and a putative α-helical motif between amino acids 115–146 of the GD of TDG. This motif is related to the LXXLL signature found in other cofactors and is known to mediate interaction with NR. Interestingly, while the glycosylase domain was required for this interaction, the catalytic activity of TDG was dispensable as mutation of catalytic residue Asn140 to Ala had little effect on the stimulation of ERα activity. These findings suggested TDG may act as a protein scaffold for TFs and other coregulators at hormonally-regulated genes.
7.4.2. TDG associates with transcriptional coregulators
Further support for this idea came when TDG was found to be associated with SRC1 and NCoR3, both coactivators of the p160 family [84,88]. p160 proteins, or nuclear receptor coactivators (NCoAs), interact with the LBD of ligand-bound ERα in a similar fashion to TDG [87]. They contain LXXLL motifs that orient within a hydrophobic pocket of helix 12, and once bound, recruit other proteins required for transcription activation, including methyltransferases and histone acetyltransferases.
TDG and SRC1 were found to localize to the same promoters of estrogen-responsive genes upon E2 stimulation of cells, suggesting these proteins associated in vivo [84]. Both in vitro GTS-binding assays and mammalian two-hybrid confirmed TDG interacts directly with SRC1 via residues 989–1240 of SRC1. Further deletion mapping in yeast revealed a tyrosine repeat motif within residues 334–346 of TDG responsible for binding to a similar tyrosine repeat in SRC1. TDG was also found to interact with NCoR3 using a mammalian two-hybrid system [88]. This interaction was mediated through a DXXD motif spanning residues 294–297 of TDG and the LLXXL domain of NCoR3. The array of novel protein interaction motifs indentified in TDG raised the possibility that TDG could interact with additional transcriptional cofactors and supported the role of TDG as a scaffold for transcription protein complexes.
This idea was supported by the discovery that TDG associates with CREB-binding protein (CBP) and its paralog p300 [53].
CBP/p300 is a transcriptional coactivator that interacts with sequence-specific TFs such as tumor suppressor p53, CREB, AP-1 (Fos and Jun) and NRs [89]. When recruited to promoters, CBP/p300 mediates transcriptional activation by inducing changes in chromatin structure through the acetylation of histone tails. This change in structure is thought to make promoter regions more accessible for TF binding and CBP/p300 helps facilitate interaction with the basal transcription machinery. TDG was found to directly associate with CBP/p300 through both its C- and N-terminal regions and serve as a substrate for acetylation [53]. This complex was shown to retain the enzymatic activities of both proteins and enhanced CBP-dependent transcription of a reporter gene in cotransfection experiments. CBP/p300 bound TDG via its histone acetyltransferase (HAT) and CH3 domains and targeted four distinct lysine residues within the N-terminal RD of TDG for acetylation. This acetylation consequently led to inhibition of the CBP-TDG interaction as well as prevented downstream recruitment of APE1 [53]. These findings provide a link between transcription and DNA repair and suggest TDG acetylation may act as a molecular switch to coordinate BER and transcriptional function.
7.4.3. TDG is directly linked to methylation
An additional mechanistic link between DNA repair and transcriptional regulation came with the discovery that TDG interacts with DNA methyltransferase DNMT3a [55]. This interaction was seen both in vitro and in vivo, and was facilitated by the PWWP and catalytic domains of DNMT3a and the N-terminus of TDG. The interaction served to inhibit the methylation activity of DNMT3a while simultaneously enhanced the catalytic activity of TDG in vitro. The purpose of this interaction may be to increase the repair of deaminated 5-meC while preventing aberrant methylation of nearby sequences. Once repair is complete and TDG is displaced by APE1, DNMT3a may remain at the repair site via interactions with other BER components, such as LIG3. At this point, DNMT3a activity would no longer be repressed thus allowing cytosine remethylation after repair and restoration of the correct epigenetic signal at promoter regions.
7.4.4. TDG localizes to gene promoters
There is clear evidence to support a role of TDG in transcriptional regulation but questions still remain about why a DNA glycosylase would localize to promoters and whether or not the catalytic activity of TDG is important for its role in transcription. It is well known that CpG dinucleotide methylation is a central feature of gene regulation. It can serve to impede the binding of transcription factors to a gene while simultaneously acting to recruit MBD proteins that help regulate and stabilize chromatin structure through the recruitment of additional proteins such as histone deacetylases and chromatin remodeling proteins. While methylated CpGs restrict transcription, unmethylated CpGs in the vicinity of a gene allow that gene to be expressed. Unfortunately, this regulation can be corrupted by deamination of 5-meC, a phenomenon known to contribute to as much as 30% of all germline mutations [4]. Because promoter regions commonly contain CpG islands, they are targets for spontaneous deamination and therefore considered mutational hotspots. It is possible that TDG may be responsible for the removal of G:T lesions that arise from the deamination of 5-meC in these regions, and perhaps also for the repair of G:U lesions arising from cytosine deamination. In this context, TFs or other transcriptional regulators may serve to “sense” DNA damage and recruit TDG to promoters of active genes as a means of maintaining the integrity of such sequences.
7.4.5. Role of TDG in active demethylation
Alternatively, TDG has recently been implicated in epigenetic regulation through a process of active demethylation at gene promoters, similar to the process seen for MBD4. The first concrete evidence that TDG was directly involved in this process came from the Bellacosa laboratory when they generated knockin mice with a mutation that inactivated the glycosylase function of TDG [26]. They found this mutation to lead to embryonic lethality in mice, thus demonstrating that the catalytic activity of TDG is essential for proper development. In addition, they showed that TDG forms a complex with AID and the DNA damage response protein GADD45a, supporting a two-step catalytic mechanism for DNA demethylation during mammalian development. Furthermore, they showed TDG was required for preventing hypermethylation of CpG islands, recruitment of p300 to RA-regulated promoters and active demethylation of tissue-specific developmentally and hormonally regulated promoters [26].
The authors proposed a model for the role of TDG in DNA demethylation whereby 5-meC is first hydroxylated by the TET family to form 5-hmC followed by deamination to 5hmU by AID/APOBEC [26]. Alternatively, 5-meC may be directly deaminated to T. The resulting lesions can then be excised by TDG followed by BER to ultimately replace the original 5-meC with C (for extensive review see [39]). This model has further been supported by the finding that TDG is able to actively remove 5fC and 5caC, two additional oxidation products of TET protein modification [68,69]. These TDG substrates, in addition to T and 5hmU, are all potential lesions generated during DNA demethylation pathways [39,65].
Additional work revealed that TDG localizes to the hormonally-regulated pS2 gene, which undergoes transcriptional cycling through a methylation-demethylation mechanism [40,90]. pS2 is an estrogen-inducible gene that was originally found to be expressed in breast cancer cells [91]. The promoter of pS2 was found to have 13 CpGs that undergo cyclical variation in methylation status in conjunction with E2 treatment of ER positive breast cancer cells [40]. Not surprisingly, this dynamic demethylation/methylation process was found to involve coordinated recruitment of DNMT3a/b, p68 (a coactivator of ERα), TDG and BER proteins. Interestingly, the authors described a novel function of DNMT3a/b as a deaminase of C and 5-meC in vitro. Bearing in mind the ability of TDG to inhibit the methyltransferase activity of DNMT3a [55], it is worth considering the possibility that such an interaction might influence the deaminase function of DNMT3a/b while also increasing the glycosylase activity of TDG. It is also important to note that strict timing and coordination of protein function are critical to transcriptional regulation. Inappropriate demethylation or failure to methylate a promoter region could lead to altered transcriptional profiles in cells. In addition, altered function of TDG or BER proteins could generate mispairs or SSBs that, if repaired incorrectly, could lead to mutations or DSBs. Identification of other genes that undergo cyclical DNA methylation might reveal additional information about how these processes are regulated and provide information about the role of TDG in transcription commitment.
7.5. SNPs of TDG
Polymorphisms G199S and V367M are the most common germline variants in human TDG (Table 1). They have been identified in a variety of populations, including Asian, African and European. G199S has a minor allele frequency (MAF) of 0.10 and has been identified in Asian, Sub-Saharan African and European populations [92]. V367M has a MAF of 0.13 and was identified in Asian, African American and European populations [92]. One study identified V367M in 10 CRC patients but no control cases were evaluated [93]. Another study conducted on a Polish population found no statistically significant association of these two variants with respect to lung cancer [48]. Unfortunately, due to the limited number of association studies and the lack of any functional studies, it is difficult to conclude whether these variants contribute to tumorigenesis.
Glycine residue 199 is located within the active site pocket of TDG and displays a 4.5 Å distance from the THF residue [personal communication Doublié S., University of Vermont]. G199 helps stabilize the flipped abasic nucleotide into the active site pocket and poses a barrier to its retrograde flipping back into the helix [63]. It is possible that mutation of Gly199 to a larger and nucleophillic Ser could either disrupt or strengthen this interaction with the abasic site. Valine residue 367 is located outside the glycosylase domain of TDG and has not been included in any of the crystal structures published thus far. It is possible that the mutation of Val to Met, an amino acid that now contains a sulfur residue, may lead to the inappropriate formation of intra- or intermolecular disulfide bonds. This could ultimately affect protein structure and/or function. Not surprisingly, both SIFT and PolyPhen predict this non-synonymous substitution to be damaging.
There have been a few studies conducted to identify cancer-associated variants of TDG, but the functional significance of these polymorphisms has not been studied. Variant R66G was identified in 1 out of 94 heterozygous CRC patients and was not found in the case controls [93]. A more recent study looked at the mutational profile of a rectal cancer patient with biallelic germline inactivating mutations in PMS2, a mismatch repair gene [94]. They found a high number of somatic C:G to T:A transition mutations within several tumor suppressor genes which prompted them to sequence several DNA glycosylases active on G:T/U lesions. Interestingly, analyses of these genes revealed a heterozygous mutation in TDG that results in a non-synonymous substitution of amino acid 284 from Asp to Tyr. This mutation was associated with TDG protein loss in the tumor. These findings suggest that loss of TDG function may contribute to the supermutator phenotype seen and provide evidence that TDG plays an important role in protecting the genome against the mutagenic effects of 5-meC deamination.
8. Summary/Concluding Remarks
MBD4 and TDG are multifaceted enzymes that were originally recognized for their ability to remove T and U lesions arising due to the spontaneous deamination of 5-meC and C, respectively. They have since been shown to act on a variety of other DNA lesions, including products of lipid peroxidation, hydroxylation and halogenated bases, and to play a role in gene regulation through active demethylation.
MBD4 preferentially binds to meCpG sites through its MBD and serves to protect the integrity of CpG sites throughout the genome. It also appears to be important for the suppression of tumorigenesis, as loss of MBD4 in mice led to increased tumor formation on an Apc min/+ background. In addition, MBD4 has been functionally linked to apoptosis and plays a role in the effective response to a variety of chemotherapeutic and other cytotoxic drugs [32,34].
TDG may also help maintain genomic stability by repair of mutagenic DNA base damage, but it seems to have a greater impact on epigenetic stability. TDG is vital to embryonic development, a time of dynamic change in CpG methylation, and was shown to protect against hypermethylation of CpG islands in association with AID. TDG has also been shown to interact with DNMT3a and CBP/p300, both important players in epigenetic regulation. TDG is able to recognize and excise deamination and hydroxylation products of 5-meC, and has been shown to participate in transcriptional cycling in conjunction with ERα through a methylation-demethylation process at an E2-regulated gene [40]. The fact that TDG has been shown to interact with a long list of additional TFs and cofactors suggests it may function similarly at other hormonally-regulated genes. Together, this experimental evidence suggests TDG plays an active role in DNA demethylation at promoter regions as a way to regulate gene expression.
Because MBD4 and TDG play such important and versatile roles in the cell, mutation of either of these proteins could have detrimental affects. The fact that a tumor-associated variant of MBD4 was found to have significantly altered biochemical function suggests it may have contributed to tumorigenesis or affected the response to cancer therapy. This idea is supported by studies that have shown tumor-associated and germline variants of other DNA glycosylases and BER proteins can alter DNA repair capacity, induce mutations in vivo and lead to a transformed phenotype [95–97]. It is feasible that polymorphisms affecting MBD4 or TDG increase the probability of developing cancer by inducing mutagenesis or affecting other cellular mechanisms, such as MMR or active demethylation. It would be of interest to assess the role of MBD4 and TDG variants, in particular D568H, in disease susceptibility and drug response in an in vivo model.
Moreover, additional mutational analysis of MBD4 and TDG in tumors could provide insight into tumor development in a particular organ. It is possible other protein variants have functional phenotypes that alter DNA repair capacity and contribute to cancer progression. Assessing the ability of MBD4 TDG variants to induce cellular transformation and characterizing their biochemical properties will provide insight into their mechanism of action as well as cancer biology.
Acknowledgments
We apologize in advance if relevant articles were missed or not included in this extensive review. This research was supported by the National Institutes of Health (P01 CA129186).
Abbreviations
- 5-FU
5-fluorouracil
- 5-hmU
5-hydroxymethyluracil
- 5-meC
5-methylcytosine
- A
adenine
- AID
activation-induced deaminase
- Apc
adenomatous polyposis coli
- APE1
AP endonuclease 1
- BER
base excision repair
- CDS
coding DNA sequence
- CRC
colorectal carcinoma
- DBD
DNA-binding domain
- DNMT
DNA methyltransferases
- dRP
5′-deoxyribose phosphate
- DSB
double-strand break
- E2
estadiol
- ER
estrogen receptor
- EREs
estrogen responsive elements
- FADD
Fas-associated death domain protein
- FdUMP
fluorodeoxyuridine monophosphatep
- HhH
helix-hairpin-helix
- LBD
ligand-binding domain
- MAF
minor allele frequency
- MBD
methyl-CpG binding domain
- MBD2
methyl-CpG binding domain protein 2
- MBD4
methyl-CpG binding domain protein 4
- MeCP2
methyl-CpG binding protein 2
- MEF
mouse embryonic fibroblast
- MMR
mismatch repair
- MSI
microsatellite instability
- MUG
mismatch uracil glycosylase
- NCoAs
nuclear receptor coactivators
- NR
nuclear receptor superfamily
- RXR/RAR
retinoid X receptor/retinoic acid receptor
- SMUG1
single-strand selective monofunctional uracil DNA glycosylase
- SNPs
single nucleotide polymorphisms
- SUMO
Small Ubiquitin-like Modifiers
- TDG
thymine DNA glycosylase
- TET
ten-eleven translocation protein
- TFF-1
thyroid transcription factor 1
- TF
transcription factor
- TS
thymidylate synthase
- UNG2
nuclear uracil-DNA glycosylase
- εC
3,N4-ethenocytosine
References
- 1.Barnes DE, Lindahl T. Repair and genetic consequences of endogenous DNA base damage in mammalian cells. Annu Rev Genet. 2004;38:445–476. doi: 10.1146/annurev.genet.38.072902.092448. [DOI] [PubMed] [Google Scholar]
- 2.De Bont R, van Larebeke N. Endogenous DNA damage in humans: a review of quantitative data. Mutagenesis. 2004;19:169–185. doi: 10.1093/mutage/geh025. [DOI] [PubMed] [Google Scholar]
- 3.Duncan BK, Miller JH. Mutagenic deamination of cytosine residues in DNA. Nature. 1980;287:560–561. doi: 10.1038/287560a0. [DOI] [PubMed] [Google Scholar]
- 4.Cooper DN, Youssoufian H. The CpG dinucleotide and human genetic disease. Hum Genet. 1988;78:151–155. doi: 10.1007/BF00278187. [DOI] [PubMed] [Google Scholar]
- 5.Kleihues P, Schäuble B, Zur Hausen A, Estève J, Ohgaki H. Tumors associated with p53 germline mutations: A synopsis of 91 families. American Journal of Pathology. 1997;150:1–13. [PMC free article] [PubMed] [Google Scholar]
- 6.Starcevic D, Dalal S, Sweasy JB. Is there a link between DNA polymerase beta and cancer? Cell Cycle. 2004;3:998–1001. [PubMed] [Google Scholar]
- 7.Kahn AB, Ryan MC, Liu H, Zeeberg BR, Jamison DC, Weinstein JN. SpliceMiner: a high-throughput database implementation of the NCBI Evidence Viewer for microarray splice variant analysis. BMC Bioinformatics. 2007;8:75. doi: 10.1186/1471-2105-8-75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Owen RM, Baker RD, Bader S, Dunlop MG, Nicholl ID. The identification of a novel alternatively spliced form of the MBD4 DNA glycosylase. Oncol Rep. 2007;17:111–116. [PubMed] [Google Scholar]
- 9.Hendrich B, Hardeland U, Ng HH, Jiricny J, Bird A. The thymine glycosylase MBD4 can bind to the product of deamination at methylated CpG sites. Nature. 1999;401:301–304. doi: 10.1038/45843. [DOI] [PubMed] [Google Scholar]
- 10.Bellacosa A, Cicchillitti L, Schepis F, Riccio A, Yeung AT, Matsumoto Y, Golemis EA, Genuardi M, Neri G. MED1, a novel human methyl-CpG-binding endonuclease, interacts with DNA mismatch repair protein MLH1. Proc Natl Acad Sci U S A. 1999;96:3969–3974. doi: 10.1073/pnas.96.7.3969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhu B, Zheng Y, Angliker H, Schwarz S, Thiry S, Siegmann M, Jost JP. 5-Methylcytosine DNA glycosylase activity is also present in the human MBD4 (G/T mismatch glycosylase) and in a related avian sequence. Nucleic Acids Res. 2000;28:4157–4165. doi: 10.1093/nar/28.21.4157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kim MS, Kondo T, Takada I, Youn MY, Yamamoto Y, Takahashi S, Matsumoto T, Fujiyama S, Shirode Y, Yamaoka I, Kitagawa H, Takeyama KI, Shibuya H, Ohtake F, Kato S. DNA demethylation in hormone-induced transcriptional derepression. Nature. 2009;461:1007–1012. doi: 10.1038/nature08456. [DOI] [PubMed] [Google Scholar]
- 13.Hendrich B, Bird A. Identification and characterization of a family of mammalian methyl-CpG binding proteins. Mol Cell Biol. 1998;18:6538–6547. doi: 10.1128/mcb.18.11.6538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wakefield RID, Smith BO, Nan X, Free A, Soteriou A, Uhrin D, Bird AP, Barlow PN. The solution structure of the domain from MeCP2 that binds to methylated DNA. Journal of Molecular Biology. 1999;291:1055–1065. doi: 10.1006/jmbi.1999.3023. [DOI] [PubMed] [Google Scholar]
- 15.Wu P, Qiu C, Sohail A, Zhang X, Bhagwat AS, Cheng X. Mismatch repair in methylated DNA. Structure and activity of the mismatch-specific thymine glycosylase domain of methyl-CpG-binding protein MBD4. J Biol Chem. 2003;278:5285–5291. doi: 10.1074/jbc.M210884200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang W, Liu Z, Grombet L, Amaya MF, Liu Y, Zhang X, Kuang W, Ma P, Niu L, Qi C. Crystal structure of the mismatch-specific thymine glycosylase domain of human methyl-CpG-binding protein MBD4. Biochemical and Biophysical Research Communications. 2011;412:425–428. doi: 10.1016/j.bbrc.2011.07.091. [DOI] [PubMed] [Google Scholar]
- 17.Manvilla BA, Maiti A, Begley MC, Toth EA, Drohat AC. Crystal Structure of Human Methyl-Binding Domain IV Glycosylase Bound to Abasic DNA. J Mol Biol. 2012 doi: 10.1016/j.jmb.2012.04.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Petronzelli F, Riccio A, Markham GD, Seeholzer SH, Genuardi M, Karbowski M, Yeung AT, Matsumoto Y, Bellacosa A. Investigation of the substrate spectrum of the human mismatch-specific DNA N-glycosylase MED1 (MBD4): fundamental role of the catalytic domain. J Cell Physiol. 2000;185:473–480. doi: 10.1002/1097-4652(200012)185:3<473::AID-JCP19>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 19.Petronzelli F, Riccio A, Markham GD, Seeholzer SH, Stoerker J, Genuardi M, Yeung AT, Matsumoto Y, Bellacosa A. Biphasic kinetics of the human DNA repair protein MED1 (MBD4), a mismatch-specific DNA N-glycosylase. J Biol Chem. 2000;275:32422–32429. doi: 10.1074/jbc.M004535200. [DOI] [PubMed] [Google Scholar]
- 20.Henderson JP, Byun J, Takeshita J, Heinecke JW. Phagocytes produce 5-chlorouracil and 5-bromouracil, two mutagenic products of myeloperoxidase, in human inflammatory tissue. J Biol Chem. 2003;278:23522–23528. doi: 10.1074/jbc.M303928200. [DOI] [PubMed] [Google Scholar]
- 21.Valinluck V, Liu P, Kang JI, Jr, Burdzy A, Sowers LC. 5-halogenated pyrimidine lesions within a CpG sequence context mimic 5-methylcytosine by enhancing the binding of the methyl-CpG-binding domain of methyl-CpG-binding protein 2 (MeCP2) Nucleic Acids Res. 2005;33:3057–3064. doi: 10.1093/nar/gki612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Turner DP, Cortellino S, Schupp JE, Caretti E, Loh T, Kinsella TJ, Bellacosa A. The DNA N-glycosylase MED1 exhibits preference for halogenated pyrimidines and is involved in the cytotoxicity of 5-iododeoxyuridine. Cancer Res. 2006;66:7686–7693. doi: 10.1158/0008-5472.CAN-05-4488. [DOI] [PubMed] [Google Scholar]
- 23.Aziz MA, Schupp JE, Kinsella TJ. Modulation of the activity of methyl binding domain protein 4 (MBD4/MED1) while processing iododeoxyuridine generated DNA mispairs. Cancer Biol Ther. 2009;8:1156–1163. doi: 10.4161/cbt.8.12.8536. [DOI] [PubMed] [Google Scholar]
- 24.Hashimoto H, Liu Y, Upadhyay AK, Chang Y, Howerton SB, Vertino PM, Zhang X, Cheng X. Recognition and potential mechanisms for replication and erasure of cytosine hydroxymethylation. Nucleic Acids Research. 2012 doi: 10.1093/nar/gks155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hashimoto H, Zhang X, Cheng X. Excision of thymine and 5- hydroxymethyluracil by the MBD4 DNA glycosylase domain: structural basis and implications for active DNA demethylation. Nucleic Acids Res. 2012 doi: 10.1093/nar/gks628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cortellino S, Xu J, Sannai M, Moore R, Caretti E, Cigliano A, Le Coz M, Devarajan K, Wessels A, Soprano D, Abramowitz LK, Bartolomei MS, Rambow F, Bassi MR, Bruno T, Fanciulli M, Renner C, Klein-Szanto AJ, Matsumoto Y, Kobi D, Davidson I, Alberti C, Larue L, Bellacosa A. Thymine DNA glycosylase is essential for active DNA demethylation by linked deamination-base excision repair. Cell. 2011;146:67–79. doi: 10.1016/j.cell.2011.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rai K, Huggins IJ, James SR, Karpf AR, Jones DA, Cairns BR. DNA demethylation in zebrafish involves the coupling of a deaminase, a glycosylase, and gadd45. Cell. 2008;135:1201–1212. doi: 10.1016/j.cell.2008.11.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wong E, Yang K, Kuraguchi M, Werling U, Avdievich E, Fan K, Fazzari M, Jin B, Brown AM, Lipkin M, Edelmann W. Mbd4 inactivation increases Cright-arrowT transition mutations and promotes gastrointestinal tumor formation. Proc Natl Acad Sci U S A. 2002;99:14937–14942. doi: 10.1073/pnas.232579299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Millar CB, Guy J, Sansom OJ, Selfridge J, MacDougall E, Hendrich B, Keightley PD, Bishop SM, Clarke AR, Bird A. Enhanced CpG mutability and tumorigenesis in MBD4-deficient mice. Science. 2002;297:403–405. doi: 10.1126/science.1073354. [DOI] [PubMed] [Google Scholar]
- 30.Prolla TA, Baker SM, Harris AC, Tsao JL, Yao X, Bronner CE, Zheng B, Gordon M, Reneker J, Arnheim N, Shibata D, Bradley A, Liskay RM. Tumour susceptibility and spontaneous mutation in mice deficient in M1h1, Pms1 and Pms2 DNA mismatch repair. Nat Genet. 1998;18:276–279. doi: 10.1038/ng0398-276. [DOI] [PubMed] [Google Scholar]
- 31.Sansom OJ, Bishop SM, Bird A, Clarke AR. MBD4 deficiency does not increase mutation or accelerate tumorigenesis in mice lacking MMR. Oncogene. 2004;23:5693–5696. doi: 10.1038/sj.onc.1207767. [DOI] [PubMed] [Google Scholar]
- 32.Cortellino S, Turner D, Masciullo V, Schepis F, Albino D, Daniel R, Skalka AM, Meropol NJ, Alberti C, Larue L, Bellacosa A. The base excision repair enzyme MED1 mediates DNA damage response to antitumor drugs and is associated with mismatch repair system integrity. Proc Natl Acad Sci U S A. 2003;100:15071–15076. doi: 10.1073/pnas.2334585100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Branch P, Hampson R, Karran P. DNA mismatch binding defects, DNA damage tolerance, and mutator phenotypes in human colorectal carcinoma cell lines. Cancer Res. 1995;55:2304–2309. [PubMed] [Google Scholar]
- 34.Sansom OJ, Zabkiewicz J, Bishop SM, Guy J, Bird A, Clarke AR. MBD4 deficiency reduces the apoptotic response to DNA-damaging agents in the murine small intestine. Oncogene. 2003;22:7130–7136. doi: 10.1038/sj.onc.1206850. [DOI] [PubMed] [Google Scholar]
- 35.Screaton RA, Kiessling S, Sansom OJ, Millar CB, Maddison K, Bird A, Clarke AR, Frisch SM. Fas-associated death domain protein interacts with methyl-CpG binding domain protein 4: A potential link between genome surveillance and apoptosis. PNAS. 2003;100:5211–5216. doi: 10.1073/pnas.0431215100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ruzov A, Shorning B, Mortusewicz O, Dunican DS, Leonhardt H, Meehan RR. MBD4 and MLH1 are required for apoptotic induction in xDNMT1- depleted embryos. Development. 2009;136:2277–2286. doi: 10.1242/dev.032227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Doerfler W. DNA methylation and gene activity. Annu Rev Biochem. 1983;52:93–124. doi: 10.1146/annurev.bi.52.070183.000521. [DOI] [PubMed] [Google Scholar]
- 38.Law JA, Jacobsen SE. Establishing, maintaining and modifying DNA methylation patterns in plants and animals. Nat Rev Genet. 2010;11:204–220. doi: 10.1038/nrg2719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bhutani N, Burns DM, Blau HM. DNA demethylation dynamics. Cell. 2011;146:866–872. doi: 10.1016/j.cell.2011.08.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Metivier R, Gallais R, Tiffoche C, Le Peron C, Jurkowska RZ, Carmouche RP, Ibberson D, Barath P, Demay F, Reid G, Benes V, Jeltsch A, Gannon F, Salbert G. Cyclical DNA methylation of a transcriptionally active promoter. Nature. 2008;452:45–50. doi: 10.1038/nature06544. [DOI] [PubMed] [Google Scholar]
- 41.Riccio A, Aaltonen LA, Godwin AK, Loukola A, Percesepe A, Salovaara R, Masciullo V, Genuardi M, Paravatou-Petsotas M, Bassi DE, Ruggeri BA, Klein-Szanto AJ, Testa JR, Neri G, Bellacosa A. The DNA repair gene MBD4 (MED1) is mutated in human carcinomas with microsatellite instability. Nat Genet. 1999;23:266–268. doi: 10.1038/15443. [DOI] [PubMed] [Google Scholar]
- 42.Miquel C, Jacob S, Grandjouan S, Aime A, Viguier J, Sabourin JC, Sarasin A, Duval A, Praz F. Frequent alteration of DNA damage signalling and repair pathways in human colorectal cancers with microsatellite instability. Oncogene. 2007;26:5919–5926. doi: 10.1038/sj.onc.1210419. [DOI] [PubMed] [Google Scholar]
- 43.Bader S, Walker M, Hendrich B, Bird A, Bird C, Hooper M, Wyllie A. Somatic frameshift mutations in the MBD4 gene of sporadic colon cancers with mismatch repair deficiency. Oncogene. 1999;18:8044–8047. doi: 10.1038/sj.onc.1203229. [DOI] [PubMed] [Google Scholar]
- 44.Yamada T, Koyama T, Ohwada S, Tago K, Sakamoto I, Yoshimura S, Hamada K, Takeyoshi I, Morishita Y. Frameshift mutations in the MBD4/MED1 gene in primary gastric cancer with high-frequency microsatellite instability. Cancer Lett. 2002;181:115–120. doi: 10.1016/s0304-3835(02)00043-5. [DOI] [PubMed] [Google Scholar]
- 45.Bader SA, Walker M, Harrison DJ. A human cancer-associated truncation of MBD4 causes dominant negative impairment of DNA repair in colon cancer cells. Br J Cancer. 2007;96:660–666. doi: 10.1038/sj.bjc.6603592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pinto M, Wu Y, Suriano G, Mensink RG, Duval A, Oliveira C, Carvalho B, Hamelin R, Seruca R, Hofstra RM. MBD4 mutations are rare in gastric carcinomas with microsatellite instability. Cancer Genet Cytogenet. 2003;145:103–107. doi: 10.1016/s0165-4608(03)00062-1. [DOI] [PubMed] [Google Scholar]
- 47.Schmutte C, Baffa R, Veronese LM, Murakumo Y, Fishel R. Human thymine-DNA glycosylase maps at chromosome 12q22-q24.1: a region of high loss of heterozygosity in gastric cancer. Cancer Res. 1997;57:3010–3015. [PubMed] [Google Scholar]
- 48.Krzesniak M, Butkiewicz D, Samojedny A, Chorazy M, Rusin M. Polymorphisms in TDG and MGMT genes - epidemiological and functional study in lung cancer patients from Poland. Ann Hum Genet. 2004;68:300–312. doi: 10.1046/j.1529-8817.2004.00079.x. [DOI] [PubMed] [Google Scholar]
- 49.Yatsuoka T, Furukawa T, Abe T, Yokoyama T, Sunamura M, Kobari M, Matsuno S, Horii A. Genomic analysis of the thymine-DNA glycosylase (TDG) gene on 12q22-q24.1 in human pancreatic ductal adenocarcinoma. Int J Pancreatol. 1999;25:97–102. doi: 10.1385/IJGC:25:2:97. [DOI] [PubMed] [Google Scholar]
- 50.Neddermann P, Gallinari P, Lettieri T, Schmid D, Truong O, Hsuan JJ, Wiebauer K, Jiricny J. Cloning and expression of human G/T mismatch-specific thymine-DNA glycosylase. J Biol Chem. 1996;271:12767–12774. doi: 10.1074/jbc.271.22.12767. [DOI] [PubMed] [Google Scholar]
- 51.Cortazar D, Kunz C, Saito Y, Steinacher R, Schar P. The enigmatic thymine DNA glycosylase. DNA Repair (Amst) 2007;6:489–504. doi: 10.1016/j.dnarep.2006.10.013. [DOI] [PubMed] [Google Scholar]
- 52.Missero C, Pirro MT, Simeone S, Pischetola M, Di Lauro R. The DNA glycosylase T:G mismatch-specific thymine DNA glycosylase represses thyroid transcription factor-1-activated transcription. J Biol Chem. 2001;276:33569–33575. doi: 10.1074/jbc.M104963200. [DOI] [PubMed] [Google Scholar]
- 53.Tini M, Benecke A, Um SJ, Torchia J, Evans RM, Chambon P. Association of CBP/p300 acetylase and thymine DNA glycosylase links DNA repair and transcription. Mol Cell. 2002;9:265–277. doi: 10.1016/s1097-2765(02)00453-7. [DOI] [PubMed] [Google Scholar]
- 54.Chen D, Lucey MJ, Phoenix F, Lopez-Garcia J, Hart SM, Losson R, Buluwela L, Coombes RC, Chambon P, Schar P, Ali S. T:G mismatch-specific thymine-DNA glycosylase potentiates transcription of estrogen-regulated genes through direct interaction with estrogen receptor alpha. J Biol Chem. 2003;278:38586–38592. doi: 10.1074/jbc.M304286200. [DOI] [PubMed] [Google Scholar]
- 55.Li YQ, Zhou PZ, Zheng XD, Walsh CP, Xu GL. Association of Dnmt3a and thymine DNA glycosylase links DNA methylation with base-excision repair. Nucleic Acids Res. 2007;35:390–400. doi: 10.1093/nar/gkl1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Smet-Nocca C, Wieruszeski JM, Chaar V, Leroy A, Benecke A. The Thymine-DNA Glycosylase Regulatory Domain: Residual Structure and DNA. Binding Biochemistry. 2008;47:6519–6530. doi: 10.1021/bi7022283. [DOI] [PubMed] [Google Scholar]
- 57.Hardeland U, Steinacher R, Jiricny J, Schar P. Modification of the human thymine-DNA glycosylase by ubiquitin-like proteins facilitates enzymatic turnover. Embo J. 2002;21:1456–1464. doi: 10.1093/emboj/21.6.1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Barrett TE, Scharer OD, Savva R, Brown T, Jiricny J, Verdine GL, Pearl LH. Crystal structure of a thwarted mismatch glycosylase DNA repair complex. Embo J. 1999;18:6599–6609. doi: 10.1093/emboj/18.23.6599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hardeland U, Bentele M, Jiricny J, Schar P. The versatile thymine DNA- glycosylase: a comparative characterization of the human, Drosophila and fission yeast orthologs. Nucleic Acids Res. 2003;31:2261–2271. doi: 10.1093/nar/gkg344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Maiti A, Morgan MT, Drohat AC. Role of Two Strictly Conserved Residues in Nucleotide Flipping and N-Glycosylic Bond Cleavage by Human Thymine DNA Glycosylase. Journal of Biological Chemistry. 2009;284:36680–36688. doi: 10.1074/jbc.M109.062356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Baba D, Maita N, Jee JG, Uchimura Y, Saitoh H, Sugasawa K, Hanaoka F, Tochio H, Hiroaki H, Shirakawa M. Crystal structure of thymine DNA glycosylase conjugated to SUMO-1. Nature. 2005;435:979–982. doi: 10.1038/nature03634. [DOI] [PubMed] [Google Scholar]
- 62.Baba D, Maita N, Jee JG, Uchimura Y, Saitoh H, Sugasawa K, Hanaoka F, Tochio H, Hiroaki H, Shirakawa M. Crystal Structure of SUMO-3-modified Thymine-DNA Glycosylase. Journal of Molecular Biology. 2006;359:137–147. doi: 10.1016/j.jmb.2006.03.036. [DOI] [PubMed] [Google Scholar]
- 63.Maiti A, Morgan MT, Pozharski E, Drohat AC. Crystal structure of human thymine DNA glycosylase bound to DNA elucidates sequence-specific mismatch recognition. Proc Natl Acad Sci U S A. 2008;105:8890–8895. doi: 10.1073/pnas.0711061105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Maiti A, Drohat AC. Dependence of substrate binding and catalysis on pH, ionic strength, and temperature for thymine DNA glycosylase: Insights into recognition and processing of G.T mispairs. DNA Repair (Amst) 2011;10:545–553. doi: 10.1016/j.dnarep.2011.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hardeland U, Bentele M, Jiricny J, Schär P. The versatile thymine DNA--glycosylase: a comparative characterization of the human, Drosophila and fission yeast orthologs. Nucleic Acids Research. 2003;31:2261–2271. doi: 10.1093/nar/gkg344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Morgan MT, Bennett MT, Drohat AC. Excision of 5-halogenated uracils by human thymine DNA glycosylase. Robust activity for DNA contexts other than CpG. J Biol Chem. 2007;282:27578–27586. doi: 10.1074/jbc.M704253200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Saparbaev M, Laval J. 3,N4-ethenocytosine, a highly mutagenic adduct, is a primary substrate for Escherichia coli double-stranded uracil-DNA glycosylase and human mismatch-specific thymine-DNA glycosylase. Proc Natl Acad Sci U S A. 1998;95:8508–8513. doi: 10.1073/pnas.95.15.8508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.He YF, Li BZ, Li Z, Liu P, Wang Y, Tang Q, Ding J, Jia Y, Chen Z, Li L, Sun Y, Li X, Dai Q, Song CX, Zhang K, He C, Xu GL. Tet-mediated formation of 5-carboxylcytosine and its excision by TDG in mammalian DNA. Science. 2011;333:1303–1307. doi: 10.1126/science.1210944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Maiti A, Drohat AC. Thymine DNA glycosylase can rapidly excise 5-formylcytosine and 5-carboxylcytosine: potential implications for active demethylation of CpG sites. J Biol Chem. 2011;286:35334–35338. doi: 10.1074/jbc.C111.284620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Morgan MT, Maiti A, Fitzgerald ME, Drohat AC. Stoichiometry and affinity for thymine DNA glycosylase binding to specific and nonspecific DNA. Nucleic Acids Res. 2011;39:2319–2329. doi: 10.1093/nar/gkq1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Maiti A, Noon MS, Mackerell AD, Jr, Pozharski E, Drohat AC. Lesion processing by a repair enzyme is severely curtailed by residues needed to prevent aberrant activity on undamaged DNA. Proc Natl Acad Sci U S A. 2012;109:8091–8096. doi: 10.1073/pnas.1201010109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Waters TR, Gallinari P, Jiricny J, Swann PF. Human thymine DNA glycosylase binds to apurinic sites in DNA but is displaced by human apurinic endonuclease 1. J Biol Chem. 1999;274:67–74. doi: 10.1074/jbc.274.1.67. [DOI] [PubMed] [Google Scholar]
- 73.Fitzgerald ME, Drohat AC. Coordinating the Initial Steps of Base Excision Repair. Journal of Biological Chemistry. 2008;283:32680–32690. doi: 10.1074/jbc.M805504200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hay RT. SUMO: a history of modification. Mol Cell. 2005;18:1–12. doi: 10.1016/j.molcel.2005.03.012. [DOI] [PubMed] [Google Scholar]
- 75.Mohan RD, Rao A, Gagliardi J, Tini M. SUMO-1-dependent allosteric regulation of thymine DNA glycosylase alters subnuclear localization and CBP/p300 recruitment. Mol Cell Biol. 2007;27:229–243. doi: 10.1128/MCB.00323-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Hardeland U, Kunz C, Focke F, Szadkowski M, Schar P. Cell cycle regulation as a mechanism for functional separation of the apparently redundant uracil DNA glycosylases TDG and UNG2. Nucleic Acids Res. 2007;35:3859–3867. doi: 10.1093/nar/gkm337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Fischer JA, Muller-Weeks S, Caradonna S. Proteolytic degradation of the nuclear isoform of uracil-DNA glycosylase occurs during the S phase of the cell cycle. DNA Repair (Amst) 2004;3:505–513. doi: 10.1016/j.dnarep.2004.01.012. [DOI] [PubMed] [Google Scholar]
- 78.Niederreither K, Harbers M, Chambon P, Dolle P. Expression of T:G mismatch-specific thymidine-DNA glycosylase and DNA methyl transferase genes during development and tumorigenesis. Oncogene. 1998;17:1577–1585. doi: 10.1038/sj.onc.1202072. [DOI] [PubMed] [Google Scholar]
- 79.Cortazar D, Kunz C, Selfridge J, Lettieri T, Saito Y, MacDougall E, Wirz A, Schuermann D, Jacobs AL, Siegrist F, Steinacher R, Jiricny J, Bird A, Schar P. Embryonic lethal phenotype reveals a function of TDG in maintaining epigenetic stability. Nature. 2011;470:419–423. doi: 10.1038/nature09672. [DOI] [PubMed] [Google Scholar]
- 80.Kunz C, Focke F, Saito Y, Schuermann D, Lettieri T, Selfridge J, Schar P. Base excision by thymine DNA glycosylase mediates DNA-directed cytotoxicity of 5-fluorouracil. PLoS Biol. 2009;7:e91. doi: 10.1371/journal.pbio.1000091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Longley DB, Harkin DP, Johnston PG. 5-fluorouracil: mechanisms of action and clinical strategies. Nat Rev Cancer. 2003;3:330–338. doi: 10.1038/nrc1074. [DOI] [PubMed] [Google Scholar]
- 82.Neddermann P, Jiricny J. The purification of a mismatch-specific thymine- DNA glycosylase from HeLa cells. J Biol Chem. 1993;268:21218–21224. [PubMed] [Google Scholar]
- 83.Um S, Harbers M, Benecke A, Pierrat B, Losson R, Chambon P. Retinoic acid receptors interact physically and functionally with the T:G mismatch-specific thymine-DNA glycosylase. J Biol Chem. 1998;273:20728–20736. doi: 10.1074/jbc.273.33.20728. [DOI] [PubMed] [Google Scholar]
- 84.Lucey MJ, Chen D, Lopez-Garcia J, Hart SM, Phoenix F, Al-Jehani R, Alao JP, White R, Kindle KB, Losson Rg, Chambon P, Parker MG, SchAnr P, Heery DM, Buluwela L, Ali S. T:G mismatch-specific thymine-DNA glycosylase (TDG) as a coregulator of transcription interacts with SRC1 family members through a novel tyrosine repeat motif. Nucl Acids Res. 2005;33:6393–6404. doi: 10.1093/nar/gki940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Chevray PM, Nathans D. Protein interaction cloning in yeast: identification of mammalian proteins that react with the leucine zipper of Jun. Proc Natl Acad Sci U S A. 1992;89:5789–5793. doi: 10.1073/pnas.89.13.5789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Bastien J, Rochette-Egly C. Nuclear retinoid receptors and the transcription of retinoid-target genes. Gene. 2004;328:1–16. doi: 10.1016/j.gene.2003.12.005. [DOI] [PubMed] [Google Scholar]
- 87.Nilsson S, Mäkelä S, Treuter E, Tujague M, Thomsen J, Andersson Gr, Enmark E, Pettersson K, Warner M, Gustafsson Jk. Mechanisms of Estrogen Action. Physiological Reviews. 2001;81:1535–1565. doi: 10.1152/physrev.2001.81.4.1535. [DOI] [PubMed] [Google Scholar]
- 88.Chiang S, Burch T, Van Domselaar G, Dick K, Radziwon A, Brusnyk C, Edwards M, Piper J, Cutts T, Cao J, Li X, He R. The interaction between thymine DNA glycosylase and nuclear receptor coactivator 3 is required for the transcriptional activation of nuclear hormone receptors. Molecular and Cellular Biochemistry. 2010;333:221–232. doi: 10.1007/s11010-009-0223-1. [DOI] [PubMed] [Google Scholar]
- 89.Goodman RH, Smolik S. CBP/p300 in cell growth, transformation, and development. Genes Dev. 2000;14:1553–1577. [PubMed] [Google Scholar]
- 90.Metivier R, Penot G, Hubner MR, Reid G, Brand H, Kos M, Gannon F. Estrogen receptor-alpha directs ordered, cyclical, and combinatorial recruitment of cofactors on a natural target promoter. Cell. 2003;115:751–763. doi: 10.1016/s0092-8674(03)00934-6. [DOI] [PubMed] [Google Scholar]
- 91.Jakowlew SB, Breathnach R, Jeltsch JM, Masiakowski P, Chambon P. Sequence of the pS2 mRNA induced by estrogen in the human breast cancer cell line MCF-7. Nucleic Acids Res. 1984;12:2861–2878. doi: 10.1093/nar/12.6.2861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Sherry ST, Ward MH, Kholodov M, Baker J, Phan L, Smigielski EM, Sirotkin K. dbSNP: the NCBI database of genetic variation. Nucleic Acids Res. 2001;29:308–311. doi: 10.1093/nar/29.1.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Broderick P, Bagratuni T, Vijayakrishnan J, Lubbe S, Chandler I, Houlston RS. Evaluation of NTHL1, NEIL1, NEIL2, MPG, TDG, UNG and SMUG1 genes in familial colorectal cancer predisposition. BMC Cancer. 2006;6:243. doi: 10.1186/1471-2407-6-243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Vasovcak P, Krepelova A, Menigatti M, Puchmajerova A, Skapa P, Augustinakova A, Amann G, Wernstedt A, Jiricny J, Marra G, Wimmer K. Unique mutational profile associated with a loss of TDG expression in the rectal cancer of a patient with a constitutional PMS2 deficiency. DNA Repair (Amst) 2012 doi: 10.1016/j.dnarep.2012.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Lang T, Dalai S, Chikova A, DiMaio D, Sweasy JB. The E295K DNA polymerase beta gastric cancer-associated variant interferes with base excision repair and induces cellular transformation. Mol Cell Biol. 2007;27:5587–5596. doi: 10.1128/MCB.01883-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Sweasy JB, Lang T, Starcevic D, Sun KW, Lai CC, Dimaio D, Dalai S. Expression of DNA polymerase {beta} cancer-associated variants in mouse cells results in cellular transformation. Proc Natl Acad Sci U S A. 2005;102:14350–14355. doi: 10.1073/pnas.0505166102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Bravard A, Vacher M, Moritz E, Vaslin L, Hall J, Epe B, Radicella JP. Oxidation status of human OGG1-S326C polymorphic variant determines cellular DNA repair capacity. Cancer Res. 2009;69:3642–3649. doi: 10.1158/0008-5472.CAN-08-3943. [DOI] [PubMed] [Google Scholar]
- 98.Consortium TGP. A map of human genome variation from population-scale sequencing. Nature. 2011;467:1061–1073. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]