

Abstract
Proliferation of pancreatic β-cells is an important mechanism underlying β-cell mass adaptation to metabolic demands. Increasing β-cell mass by regeneration may ameliorate or correct both type 1 and type 2 diabetes, which both result from inadequate production of insulin by β-cells of the pancreatic islet. Transforming growth factor β (TGF-β) signaling is essential for fetal development and growth of pancreatic islets. In this study, we exposed HIT-T15, a clonal pancreatic β-cell line, to TGF-β signaling. We found that inhibition of TGF-β signaling promotes proliferation of the cells significantly, while TGF-β signaling stimulation inhibits proliferation of the cells remarkably. We confirmed that this proliferative regulation by TGF-β signaling is due to the changed expression of the cell cycle regulator p27. Furthermore, we demonstrated that there is no observed effect on transcriptional activity of p27 by TGF-β signaling. Our data show that TGF-β signaling mediates the cell-cycle progression of pancreatic β-cells by regulating the nuclear localization of CDK inhibitor, p27. Inhibition of TGF-β signaling reduces the nuclear accumulation of p27, and as a result this inhibition promotes proliferation of β-cells.
Keywords: TGF-β signaling, HIT-T15 cells, inhibition, pancreatic β-cell proliferation, p27
I. Introduction
Pancreatic β-cells in the islets of Langerhans have a central role in glucose homeostasis, and display the property of sensing the prevailing blood glucose and secreting insulin in a manner dependent on the glucose concentration [8, 21]. The β-cell mass is dynamic and responds to variations in metabolic demand on insulin production. Inability of the endocrine pancreas to adapt to changing insulin demand is found in both type 1 and type 2 diabetes mellitus. In type 1 and type 2 diabetes, there are a near-complete and partial loss, respectively, of β-cells [6]. Transplantation of β-cells was anticipated to be the most promising approach to restore endogenous insulin secretion in diabetes [3, 6]. However, its application on a large scale is limited by the availability of pancreas donors. Therefore, strategies that lead to increased β-cell mass, as well as retained or enhanced function of islets, would be a promising approach for treating diabetes.
In adults, β-cell mass is stably maintained, and low levels of proliferation suggest that most β-cells are quiescent. In adult humans, there are approximately 0.05 Ki-67-positive β-cells per islet [6, 8], whereas in adult mice the corresponding frequency is tenfold greater in lean adult mice (~0.50 β-cells per islet), and much greater in obese animals [7, 8, 43].
Multiple intracellular signaling pathways participate in β-cell proliferation. These pathways include protein kinase A (PKA), mitogen-activated protein kinase (MAPK), janus kinase (JAK)-signal transducer and activator of transcription (STAT), phosphatidyl inositol-3 (PI3) kinase/protein kinase B (AKT) and insulin-receptor-substrate pathways [10], and epidermal growth factor (EGF)-receptor signaling [19, 31]. These signaling cascades leads to G1/S transition, mainly by stimulating the activity of the cyclin-dependent kinase (CDK) 4 (CDK4)-cyclin D complex [5, 11, 18]. Eukaryotic cell cycle progression, particularly the G1/S transition, is controlled by a series of protein complexes composed of cyclins and CDKs, the activity of which is regulated by a group of CDK inhibitors (CKIs). Two groups, the INK4 family (p15, p16, p18, p19) and the Cip/Kip family (p21, p27, p57), have been described. A member of the INK4 family, p16, inhibits formation of the CDK4-cyclin D2 complex, and can inhibit cell cycle progression and regeneration of β-cells [24, 28]. A member of the Cip/Kip family, p27, has a pivotal role in the control of cell proliferation, and reducing p27 levels in quiescent β-cells promotes β-cell proliferation and leads to increased β-cell mass [17, 23].
Transforming growth factor β (TGF-β) regulates cell proliferation and differentiation of diverse cell types, including epithelial, endothelial and hematopoietic cells, by autocrine and paracrine mechanisms [29, 33, 38]. Disruption of TGF-β signaling impairs embryonic pancreatic β-cell differentiation [13, 38, 40], suggesting essential roles for this pathway in establishing and maintaining defining features of mature pancreatic β-cells.
In this study, we investigated the effects of TGF-β signaling on β-cell proliferation by using the hamster pancreatic β-cell line HIT-T15. Inhibition of TGF-β signaling by TGF-β receptor I (TβRI/ALK5) inhibitor, SB-431542, led to instability of p27, and as a result, enhanced β-cell proliferation.
II. Materials and Methods
Materials
SB-431542 was purchased from Tocris Bioscience (Ballwin, MO). TGF-β1, anti-p16 INK4a/CDKN2 antibody and anti-p19 INK4 antibody were purchased from Sigma Aldrich (St. Louis, MO). Anti-p18 INK4C antibody, anti-p21 Waf1/Cip1 antibody and anti-p57 Kip2 antibody were obtained from Cell Signaling Technology (Beverly, MA). Anti-p15 INK4B antibody, anti-GAPDH antibody and anti-p27 Kip1 antibodies were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). Alexa Fluor 488 goat anti-mouse, Alexa Fluor 488 goat anti-rabbit, Alexa Fluor 594 goat anti-rabbit antibodies, OPTI-MEM and Lipofectamine 2000 were obtained from Invitrogen (Carlsbad, CA). Dual luciferase reporter assay system was purchased from Promega Co. (Madison, WI).
Cell culture and cell proliferation
A hamster pancreatic β-cell line, HIT-T15, purchased from Dainippon Pharmaceutical Corporation (Osaka, Japan), was cultured in RPMI 1640 medium containing 10% FBS, 2 mM glutamine, 100 U/ml penicillin and 100 µg/ml streptomycin at 37°C in a humidified atmosphere of 5% CO2. In order to investigate the effect of TGF-β signaling on the proliferation of HIT-T15 cells, for TGF-β treatment, cells were treated with or without 5 ng/ml TGF-β1 dissolved in 1 mg/ml bovine serum albumin containing 4 mM HCl for different lengths of time, as indicated in the legend for Fig. 1A, and for TβRI/ALK5 inhibitor treatment, cells were treated with or without 2 µM SB-431542 dissolved in dimethyl sulfoxide (DMSO) (Sigma) for different lengths of time, as indicated in the legend for Fig. 1B. For cell count, cells were trypsinized and enumerated with a hemocytometer.
Fig. 1.
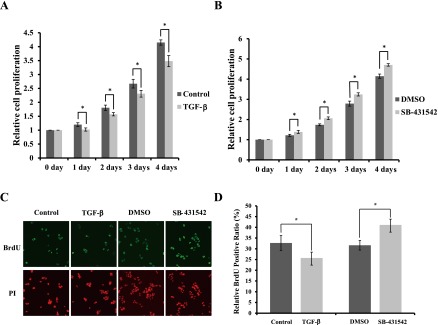
Effects of TGF-β signaling on cell proliferation of HIT-T15 cells. (A) HIT-T15 cells were cultured in 10% FBS RPMI 1640 medium with or without 5 ng/ml TGF-β1 for 1 day, 2 days, 3 days and 4 days (6 independent repeats of experiments). (B) HIT-T15 cells were cultured in 10% FBS RPMI 1640 medium containing DMSO or 2 µM SB-431542 for 1 day, 2 days, 3 days and 4 days. Six repeats of the experiment were done independently. The data are mean±SD (error bars) of a representative experiment. *p<0.01 (n=8). (C, D) Cell cycle distribution of HIT-T15 cells was analyzed by BrdU incorporation assay after treatment with or without 5 ng/ml TGF-β1, and/or with DMSO or 2 µM SB-431542, for 48 hr. (C) Upper row: BrdU staining (green). Lower row: Merged with nuclear staining (PI; red). (D) Results are displayed as the percentage of cells in S phase of the cell cycle. Six repeats of the experiment were done independently. The data are mean±SD (error bars) of a representative experiment. *p<0.01 (n=5).
5-Bromo-2'-Deoxyuridine incorporation
5-Bromo-2'-Deoxyuridine (BrdU) incorporation was assessed as a measure of cells in the S phase using the BrdU Labeling and Detection Kit (Roche Applied Science, Penzberg, Germany). HIT-T15 cells were incubated in the RPMI 1640 medium with or without 5 ng/ml TGF-β1 and/or with or without 2 µM SB-431542 for 46 hr, followed by cultured in medium containing 10 µM BrdU labeling reagent for 2 hr before fixation. BrdU incorporation assay was performed according to instruction manual. The stained cells were mounted with VECTASHIELD mounting medium with propidium iodide (PI) (Vector Laboratories, Burlingame, CA) and observed with a confocal microscope (FV1000; Olympus, Tokyo, Japan). Both the BrdU-positive S phase cells and the PI-stained total cells were counted.
Immunofluorescence staining and image analysis
Cells were incubated for 2 days and fixed with 2% paraformaldehyde in PBS for more than 1 hr. After fixation, cells were washed 3 times with PBS for 5 min and permeabilized by incubation with 0.1% Triton X-100 in PBS for 10 min. Then, cells were washed 3 times with PBS for 5 min and blocked with 3% skim milk in PBS for 2 hr and incubated with primary antibody (1:250) in 3% skim milk solution overnight. After 5 washes with PBS for 5 min each time, cells were incubated with secondary antibody (1:1000) in 3% skim milk solution for 2 hr. DNA was stained with VECTASHIELD mounting medium with PI or TO-PRO-3 iodide (Invitrogen-Molecular Probes) for 30 min. Stained cells were observed with a FV1000 confocal microscope.
Fluorescence intensities were quantified by using FV10-ASW software (Olympus).
Whole cell extract and western blotting
Cells treated as indicated in the legends for Fig. 2H and Fig. 4A were washed in PBS. Cells were lysed in the lysis buffer (50 mM Hepes, pH 7.5, 250 mM NaCl, 0.2 mM EDTA, 10 mM NaF, 0.5% Nonidet P-40), and whole cell lysates were prepared. Lysates were separated on 10% SDS-polyacrylamide gels, and analyzed by Western blotting using anti-p15, anti-p16, anti-p18, anti-p19, anti-p21, anti-p27 and anti-p57 antibodies.
Fig. 2.
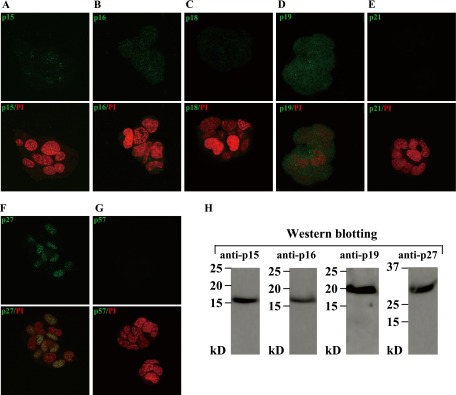
Expression of CKIs in HIT-T115 cells. Cells were incubated for 2 days and fixed, and then stained with different CKI antibodies. Upper row: Expression of different indicated CKIs (green). Lower row: Merged with nuclear staining (PI; red). (A) p15; (B) p16; (C) p18; (D) p19; (E) p21; (F) p27; (G) p57. (H) Western blotting of the whole cell extracts for p15, p16, p19 and p27.
Luciferase reporter assays
HIT-T15 cells (1×105/ml) were seeded in 35-mm dishes. The next day, using Lipofectamine 2000 (Invitrogen, CA), cells were transfected with 20 ng of the internal control plasmid pRL-CMV (Promega), and either 100 ng of p27 full-length promoter luciferase reporter plasmid, p27PF [32], or 100 ng of empty vector, pGVB2. For TGF-β signaling stimulation, cells were transfected together with either 100 ng of ALK5* (a constitutively active form of TβRI) plasmid or 100 ng of empty vector pcDNA3.1 (Invitrogen). For TβRI/ALK5 inhibitor treatment, cells were treated with either 2 µM SB-431542 or DMSO for 40 hr. For luciferase assays, all samples were harvested together at 40 hr after transfection. Luciferase activity in cell lysates was measured using the dual luciferase reporter assay system (Promega) in a Berthold Lumat LB 9705 luminometer (Pforzheim, Germany).
Statistical analysis
Data are expressed as the mean±SD. Differences between groups were evaluated by using Student’s t-test. Differences of p<0.05 were considered statistically significant. For all experiments, at least 3 replicates were included, and all experiments were repeated at least 3 times.
III. Results
TGF-β signaling affects proliferation of HIT-T15 cells
TGF-β signaling regulates cell proliferation and differentiation of diverse cell types by autocrine and paracrine mechanisms [2, 29, 45]. To estimate the effect of TGF-β signaling on proliferation of HIT-T15 cells [37], we exposed HIT-T15 cells to culture with or without TGF-β1 (5 ng/ml), and counted the number of the cells in a time-dependent manner. Compared with non-TGF-β1-stimulated groups, the TGF-β1 stimulation significantly inhibited cell proliferation, by 15% at 1 day, by 13% at 2 days, by 14% at 3 days, and by 16% at 4 days, respectively (Fig. 1A).
Next, we used a highly specific and potent TβRI/ALK5 inhibitor, SB-431542, which instantaneously inhibits ALK5 kinase activity [22], and treated the cells with 2 µM SB-431542 or DMSO, which served as a control, for different lengths of time. A marked increase in cell proliferation, by 14% at 1 day, by 19% at 2 days, by 16% at 3 days, and by 15% at 4 days, was observed in SB-431542-treated groups compared with DMSO-treated groups, respectively (Fig. 1B). These experimental data demonstrated that TGF-β signaling regulates cell proliferation of HIT-T15 β-cells.
TGF-β signaling affects proportion of cell phases
To confirm that TGF-β signaling affects cell proliferation by shifting the proportion of HIT-T15 cell phases, we investigated the proportion of cells in S phase by using BrdU incorporation assay (Fig. 1C). Compared with non-TGF-β1-stimulated groups, the TGF-β1 stimulation significantly decreased the proportion of cells at S phase, from 32.7% to 25.7% (Fig. 1D). However, a marked increase in S phase, from 31.6% to 41.1%, was observed in SB-431542-treated groups compared with DMSO-treated groups at 48 hr (Fig. 1D). Taken together, these results show that inhibition of TGF-β signaling enhances the cell cycle progression, and as a result, promotes the proliferation of HIT-T15 β-cells.
Expression of CKIs in HIT-T15 cells
Cell cycle progression, particularly the G1/S transition, is negatively controlled by CKIs. We examined which CKIs are expressed in HIT-T15 cells by immunofluorescence staining and western blotting (Fig. 2). The CKI proteins, p15, p16, p19 and p27, were expressed in HIT-T15 cells (Fig. 2A, 2B, 2D, 2F and 2H), whereas p18, p21 and p57 were under detectable levels both by immunofluorescence staining (Fig. 2C, 2E, and 2G) and western blotting (data not shown). In addition, p15 was localized in nuclei at a low level (Fig. 2A), whereas p16 (Fig. 2B) and p19 (Fig. 2D) were uniformly present both in cytoplasm and nuclei at a low level and a high level, respectively. p27 was localized and present in nuclei at various levels (Fig. 2F). These data suggest that p27 may serve as a key regulator in controlling the progression of cell cycle in HIT-T15 β-cells.
TGF-β signaling affects subcellular localization of p27
We investigated whether the increased cell proliferation of HIT-T15 cells produced by the inhibition of TGF-β signaling was due to the shifted p27 subcellular localization. To stimulate TGF-β signaling, cells were transfected with a constitutively active form of TRβ-I, ALK5*[27], or empty vector, pcDNA3.1, which served as a control. The nuclear protein level of p27 was increased in the ALK5*-transfected cells (Fig. 3B and 3C). In contrast, no marked decrease or increase of p27 protein level was observed in the control (Fig. 3A and 3C). On the other hand, HIT-T15 β-cells treated with 2 µM SB-431542 showed significant decrease of the nuclear protein level of p27 (Fig. 3E and 3F) compared with the control, DMSO-treated cells (Fig. 3D and 3F). These data demonstrated that TGF-β signaling affects cell proliferation of HIT-T15 β-cells via regulating p27 subcellular localization.
Fig. 3.

Effects of TGF-β signaling on nuclear localization of p27 (green). To stimulate TGF-β signaling, HIT-T15 cells were transfected with pcDNA.3.1.mRFP1 (red), which served as a transfectant indicator, and either empty vector, pcDNA3.1 (A) or a constitutively active form of TRβ-I, ALK5* (B). (C) Nuclear p27 integrated density (A. U., arbitrary unit) in (A) and (B). To inhibit TGF-β signaling, HIT-T15 cells were treated with DMSO (D) or 2 µM SB-431542 (E). (F) Nuclear p27 integrated density (A. U.) in (D) and (E). Nuclei were detected with TO-PRO-3 iodide (blue) (A, B) or PI (red) (D, E). The data are mean±SD (error bars) of a representative experiment. *p<0.01.
TGF-β signaling affects expression of p27
To examine whether TGF-β signaling influences expression of p27 post-transcriptionally, the protein level of p27 in HIT-T15 was analyzed by using western blotting. Compared with non-TGF-β1-stimulated groups, the TGF-β1 stimulation significantly increased the protein level of p27, by 28%, while in SB-431542-treated groups compared with DMSO-treated groups, inhibition of TGF-β signaling decreased the protein level of p27, by 40%, at 48 hr (Fig. 4A and 4B). We further examined whether TGF-β signaling also influences the transcriptional activity of p27 or not. p27 promoter luciferase reporter assay [9] revealed no significant difference in p27 transcriptional activity in the comparison of treatment with or without TGF-β1 stimulation (Fig. 4C) and/or of treatment with or without SB-431542 (Fig. 4D). Collectively, these findings suggest that TGF-β signaling can affect the proliferation of HIT-T15 β-cells through regulating the expression of p27.
Fig. 4.

Effects of TGF-β signaling on expression of p27. (A) HIT-T15 cells were treated with or without 5 ng/ml TGF-β1, and/or with DMSO or 2 µM SB-431542, for 2 days. Western blotting of the whole cell extracts for p27, and GAPDH, which was used as a control. (B) Results are displayed as the relative protein level of p27. Six repeats of the Western blotting were done independently. The data are mean±SD (error bars) of a representative experiment. *p<0.001. (C, D) Luciferase reporter assays in HIT-T15 cells. Cells were transfected with either p27 luciferase reporter p27PF or empty luciferase reporter pGVB2. In addition, transfection was performed, to stimulate TGF-β signaling, together with or without ALK5* in (C), or to inhibit TGF-β signaling, together with treatment with either DMSO or 2 µM SB-431542 in (D). All data shown are mean±SD (error bars) of a representative experiment performed in at least triplicate. n.s.: not significant.
IV. Discussion
TGF-β signaling regulates the developmental programs of many, if not all, diverse cell types [1, 29, 30]. Disrupted TGF-β signaling impairs embryonic pancreatic β-cell differentiation and development [4, 25, 40, 44]. However, the role of this pathway in pancreatic β-cell proliferation has remained largely obscure. Here, we demonstrated that TGF-β signaling regulates pancreatic β-cell proliferation via the cell cycle regulator p27. We showed that inhibition of TGF-β signaling promotes proliferation of the β-cells, and that this enhanced β-cell proliferation induced by the inhibition of TGF-β signaling is due to the down-regulated nuclear expression of p27.
During embryogenesis, β-cells arise from differentiation of pancreatic progenitor cells; these differentiated β-cells are mitotically quiescent and do not proliferate, and p27 accumulates in the differentiated β-cells [10, 17]. Disruption of p27 in mice results in such effects as increased body size and multiorgan hyperplasia [15, 26]. In the absence of p27, the differentiated β-cells are no longer quiescent and reenter the cell cycle to proliferate, and thus down-regulation of p27 levels in β-cells may play a key role in promoting β-cell proliferation in vitro and in vivo [17]. We have shown using HIT-T15 cells that TGF-β signaling regulates β-cell proliferation via controlling protein level of p27 (Figs. 1, 3 and 4A), and not via regulating transcriptional activity of p27, which thereby regulates β-cell growth (Fig. 4). Several studies using animal models of diabetes showed the importance of down-regulation or loss of p27 in β-cells for promoting proliferation of β-cells and thereby expansion of β-cell mass for correcting diabetes [17, 41, 46].
In mammalian cells, two CDK enzymes, CDK4 or CDK6, combined with three D-type cyclins (D1, D2, D3), and CDK2 associated with cyclin E play distinct key roles in regulating G1 progression. CDK4/6-cyclin D couples extracellular growth signals to the cell cycle, while CDK2-cyclin E controls the initiation of DNA replication [39]. p27 inhibits functions of cell cycle regulators like CDK2 and CDK4 [35, 36]. For pancreatic islets, this view is consistent with reported work showing that regulation of CDK4 is required for islet growth control [10, 11, 16]. Replacement of the endogenous CDK4 gene with INK4-resistant activated CDK4 allele resulted in mice with hyperplastic islets, comprised mainly of insulin-producing β-cells [14]. The interplay between TGF-β-inducible inhibitors and their targets involves CDK inhibitors, p21 [12], p15 [20] and p27 [36]. In normal cells, the amount of p27 is large during the quiescent G0 phase of the cell cycle, while it is rapidly decreased on reentry of cells from G0 into G1 phase [42]. In proliferating cells, p27 is degraded in the nucleus during S and G2 phases by Skp2, the F-box protein component of the SCF ubiquitin ligase (E3) complex [34]. In fact, p27 was the first identified mediator of the CDK inhibitory effect of TGF-β [36]. Upon addition of TGF-β to Mv1Lu mink lung epithelial cells, induced p15 expression causes a replacement of active p27-cyclin D-CDK4 complexes with inactive cyclin D-CDK4-p15 [35]. Concomitantly, cyclin E-CDK2 complexes bind p27 and become inhibited, and lead to G1 arrest [23, 36]. Thus, further studies are required to confirm how TGF-β signaling controls expression of p15, cyclin E, CDK2, and CDK4 in pancreatic β-cells.
Taken together with previous reports and our data, it is strongly argued that Skp2-dependent p27 degradation regulated by TGF-β signaling is one of the mechanisms by which pancreatic β-cells accumulated p27 in the nuclei, and, as a result, controlling proliferative activity of pancreatic β-cells.
Our data show that TGF-β signaling regulates pancreatic β-cell proliferation through control of cell cycle regulator p27 expression. Inhibition of TGF-β signaling reduces the expression of p27, and as a result this inhibition promotes proliferation of β-cells.
V. Acknowledgments
We thank T. Sakai (Kyoto Prefectural University of Medicine) for plasmid of p27 PF, K. Miyazono (The University of Tokyo) for plasmid of ALK5*, and R. Y. Tsien (University of California, San Diego) for mRFP1 cDNA.
VI. References
- 1.Arai H., Furusu A., Nishino T., Obata Y., Nakazawa Y., Nakazawa M., Hirose M., Abe K., Koji T., Kohno S. Thalidomide prevents the progression of peritoneal fibrosis in mice. Acta Histochem. Cytochem. 2011;44:51–60. doi: 10.1267/ahc.10030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Attisano L., Wrana J. L. Signal transduction by the TGF-beta superfamily. Science. 2002;296:1646–1647. doi: 10.1126/science.1071809. [DOI] [PubMed] [Google Scholar]
- 3.Bar Y., Russ H. A., Knoller S., Ouziel-Yahalom L., Efrat S. HES-1 is involved in adaptation of adult human beta-cells to proliferation in vitro. Diabetes. 2008;57:2413–2420. doi: 10.2337/db07-1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bottinger E. P., Jakubczak J. L., Roberts I. S., Mumy M., Hemmati P., Bagnall K., Merlino G., Wakefield L. M. Expression of a dominant-negative mutant TGF-beta type II receptor in transgenic mice reveals essential roles for TGF-beta in regulation of growth and differentiation in the exocrine pancreas. EMBO J. 1997;16:2621–2633. doi: 10.1093/emboj/16.10.2621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brennand K., Melton D. Slow and steady is the key to beta-cell replication. J. Cell. Mol. Med. 2009;13:472–487. doi: 10.1111/j.1582-4934.2008.00635.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Butler A. E., Janson J., Bonner-Weir S., Ritzel R., Rizza R. A., Butler P. C. Beta-cell deficit and increased beta-cell apoptosis in humans with type 2 diabetes. Diabetes. 2003;52:102–110. doi: 10.2337/diabetes.52.1.102. [DOI] [PubMed] [Google Scholar]
- 7.Butler A. E., Janson J., Soeller W. C., Butler P. C. Increased beta-cell apoptosis prevents adaptive increase in beta-cell mass in mouse model of type 2 diabetes: evidence for role of islet amyloid formation rather than direct action of amyloid. Diabetes. 2003;52:2304–2314. doi: 10.2337/diabetes.52.9.2304. [DOI] [PubMed] [Google Scholar]
- 8.Butler P. C., Meier J. J., Butler A. E., Bhushan A. The replication of beta cells in normal physiology, in disease and for therapy. Nat. Clin. Pract. Endocrinol. Metab. 2007;3:758–768. doi: 10.1038/ncpendmet0647. [DOI] [PubMed] [Google Scholar]
- 9.Cen B., Deguchi A., Weinstein I. B. Activation of protein kinase G Increases the expression of p21CIP1, p27KIP1, and histidine triad protein 1 through Sp1. Cancer Res. 2008;68:5355–5362. doi: 10.1158/0008-5472.CAN-07-6869. [DOI] [PubMed] [Google Scholar]
- 10.Cozar-Castellano I., Fiaschi-Taesch N., Bigatel T. A., Takane K. K., Garcia-Ocana A., Vasavada R., Stewart A. F. Molecular control of cell cycle progression in the pancreatic beta-cell. Endocr. Rev. 2006;27:356–370. doi: 10.1210/er.2006-0004. [DOI] [PubMed] [Google Scholar]
- 11.Cozar-Castellano I., Harb G., Selk K., Takane K., Vasavada R., Sicari B., Law B., Zhang P., Scott D. K., Fiaschi-Taesch N., Stewart A. F. Lessons from the first comprehensive molecular characterization of cell cycle control in rodent insulinoma cell lines. Diabetes. 2008;57:3056–3068. doi: 10.2337/db08-0393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Datto M. B., Li Y., Panus J. F., Howe D. J., Xiong Y., Wang X. F. Transforming growth factor beta induces the cyclin-dependent kinase inhibitor p21 through a p53-independent mechanism. Proc. Natl. Acad. Sci. U S A. 1995;92:5545–5549. doi: 10.1073/pnas.92.12.5545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dichmann D. S., Miller C. P., Jensen J., Scott Heller R., Serup P. Expression and misexpression of members of the FGF and TGFbeta families of growth factors in the developing mouse pancreas. Dev. Dyn. 2003;226:663–674. doi: 10.1002/dvdy.10270. [DOI] [PubMed] [Google Scholar]
- 14.Dor Y., Brown J., Martinez O. I., Melton D. A. Adult pancreatic beta-cells are formed by self-duplication rather than stem-cell differentiation. Nature. 2004;429:41–46. doi: 10.1038/nature02520. [DOI] [PubMed] [Google Scholar]
- 15.Fero M. L., Rivkin M., Tasch M., Porter P., Carow C. E., Firpo E., Polyak K., Tsai L. H., Broudy V., Perlmutter R. M., Kaushansky K., Roberts J. M. A syndrome of multiorgan hyperplasia with features of gigantism, tumorigenesis, and female sterility in p27(Kip1)-deficient mice. Cell. 1996;85:733–744. doi: 10.1016/s0092-8674(00)81239-8. [DOI] [PubMed] [Google Scholar]
- 16.Georgia S., Bhushan A. Beta cell replication is the primary mechanism for maintaining postnatal beta cell mass. J. Clin. Invest. 2004;114:963–968. doi: 10.1172/JCI22098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Georgia S., Bhushan A. p27 Regulates the transition of beta-cells from quiescence to proliferation. Diabetes. 2006;55:2950–2956. doi: 10.2337/db06-0249. [DOI] [PubMed] [Google Scholar]
- 18.Georgia S., Hinault C., Kawamori D., Hu J., Meyer J., Kanji M., Bhushan A., Kulkarni R. N. Cyclin D2 is essential for the compensatory beta-cell hyperplastic response to insulin resistance in rodents. Diabetes. 2010;59:987–996. doi: 10.2337/db09-0838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hakonen E., Ustinov J., Mathijs I., Palgi J., Bouwens L., Miettinen P. J., Otonkoski T. Epidermal growth factor (EGF)-receptor signalling is needed for murine beta cell mass expansion in response to high-fat diet and pregnancy but not after pancreatic duct ligation. Diabetologia. 2011;54:1735–1743. doi: 10.1007/s00125-011-2153-1. [DOI] [PubMed] [Google Scholar]
- 20.Hannon G. J., Beach D. p15INK4B is a potential effector of TGF-beta-induced cell cycle arrest. Nature. 1994;371:257–261. doi: 10.1038/371257a0. [DOI] [PubMed] [Google Scholar]
- 21.Heit J. J., Karnik S. K., Kim S. K. Intrinsic regulators of pancreatic beta-cell proliferation. Annu. Rev. Cell Dev. Biol. 2006;22:311–338. doi: 10.1146/annurev.cellbio.22.010305.104425. [DOI] [PubMed] [Google Scholar]
- 22.Inman G. J., Nicolas F. J., Callahan J. F., Harling J. D., Gaster L. M., Reith A. D., Laping N. J., Hill C. S. SB-431542 is a potent and specific inhibitor of transforming growth factor-beta superfamily type I activin receptor-like kinase (ALK) receptors ALK4, ALK5, and ALK7. Mol. Pharmacol. 2002;62:65–74. doi: 10.1124/mol.62.1.65. [DOI] [PubMed] [Google Scholar]
- 23.Karnik S. K., Hughes C. M., Gu X., Rozenblatt-Rosen O., McLean G. W., Xiong Y., Meyerson M., Kim S. K. Menin regulates pancreatic islet growth by promoting histone methylation and expression of genes encoding p27Kip1 and p18INK4c. Proc. Natl. Acad. Sci. U S A. 2005;102:14659–14664. doi: 10.1073/pnas.0503484102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kassem S. A., Ariel I., Thornton P. S., Scheimberg I., Glaser B. Beta-cell proliferation and apoptosis in the developing normal human pancreas and in hyperinsulinism of infancy. Diabetes. 2000;49:1325–1333. doi: 10.2337/diabetes.49.8.1325. [DOI] [PubMed] [Google Scholar]
- 25.Kim S. K., Hebrok M., Li E., Oh S. P., Schrewe H., Harmon E. B., Lee J. S., Melton D. A. Activin receptor patterning of foregut organogenesis. Genes Dev. 2000;14:1866–1871. [PMC free article] [PubMed] [Google Scholar]
- 26.Kiyokawa H., Kineman R. D., Manova-Todorova K. O., Soares V. C., Hoffman E. S., Ono M., Khanam D., Hayday A. C., Frohman L. A., Koff A. Enhanced growth of mice lacking the cyclin-dependent kinase inhibitor function of p27(Kip1) Cell. 1996;85:721–732. doi: 10.1016/s0092-8674(00)81238-6. [DOI] [PubMed] [Google Scholar]
- 27.Kokura K., Kim H., Shinagawa T., Khan M. M., Nomura T., Ishii S. The Ski-binding protein C184M negatively regulates tumor growth factor-beta signaling by sequestering the Smad proteins in the cytoplasm. J. Biol. Chem. 2003;278:20133–20139. doi: 10.1074/jbc.M210855200. [DOI] [PubMed] [Google Scholar]
- 28.Krishnamurthy J., Ramsey M. R., Ligon K. L., Torrice C., Koh A., Bonner-Weir S., Sharpless N. E. p16INK4a induces an age-dependent decline in islet regenerative potential. Nature. 2006;443:453–457. doi: 10.1038/nature05092. [DOI] [PubMed] [Google Scholar]
- 29.Massague J., Blain S. W., Lo R. S. TGFbeta signaling in growth control, cancer, and heritable disorders. Cell. 2000;103:295–309. doi: 10.1016/s0092-8674(00)00121-5. [DOI] [PubMed] [Google Scholar]
- 30.Massague J., Gomis R. R. The logic of TGFbeta signaling. FEBS Lett. 2006;580:2811–2820. doi: 10.1016/j.febslet.2006.04.033. [DOI] [PubMed] [Google Scholar]
- 31.Miettinen P. J., Ustinov J., Ormio P., Gao R., Palgi J., Hakonen E., Juntti-Berggren L., Berggren P. O., Otonkoski T. Downregulation of EGF receptor signaling in pancreatic islets causes diabetes due to impaired postnatal beta-cell growth. Diabetes. 2006;55:3299–3308. doi: 10.2337/db06-0413. [DOI] [PubMed] [Google Scholar]
- 32.Minami S., Ohtani-Fujita N., Igata E., Tamaki T., Sakai T. Molecular cloning and characterization of the human p27Kip1 gene promoter. FEBS Lett. 1997;411:1–6. doi: 10.1016/s0014-5793(97)00660-1. [DOI] [PubMed] [Google Scholar]
- 33.Miyazono K., Suzuki H., Imamura T. Regulation of TGF-beta signaling and its roles in progression of tumors. Cancer Sci. 2003;94:230–234. doi: 10.1111/j.1349-7006.2003.tb01425.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nakayama K. I., Nakayama K. Ubiquitin ligases: cell-cycle control and cancer. Nat. Rev. Cancer. 2006;6:369–381. doi: 10.1038/nrc1881. [DOI] [PubMed] [Google Scholar]
- 35.Reynisdottir I., Massague J. The subcellular locations of p15(Ink4b) and p27(Kip1) coordinate their inhibitory interactions with cdk4 and cdk2. Genes Dev. 1997;11:492–503. doi: 10.1101/gad.11.4.492. [DOI] [PubMed] [Google Scholar]
- 36.Reynisdottir I., Polyak K., Iavarone A., Massague J. Kip/Cip and Ink4 Cdk inhibitors cooperate to induce cell cycle arrest in response to TGF-beta. Genes Dev. 1995;9:1831–1845. doi: 10.1101/gad.9.15.1831. [DOI] [PubMed] [Google Scholar]
- 37.Santerre R. F., Cook R. A., Crisel R. M., Sharp J. D., Schmidt R. J., Williams D. C., Wilson C. P. Insulin synthesis in a clonal cell line of simian virus 40-transformed hamster pancreatic beta cells. Proc. Natl. Acad. Sci. U S A. 1981;78:4339–4343. doi: 10.1073/pnas.78.7.4339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sanvito F., Herrera P. L., Huarte J., Nichols A., Montesano R., Orci L., Vassalli J. D. TGF-beta 1 influences the relative development of the exocrine and endocrine pancreas in vitro. Development. 1994;120:3451–3462. doi: 10.1242/dev.120.12.3451. [DOI] [PubMed] [Google Scholar]
- 39.Sherr C. J., Roberts J. M. CDK inhibitors: positive and negative regulators of G1-phase progression. Genes Dev. 1999;13:1501–1512. doi: 10.1101/gad.13.12.1501. [DOI] [PubMed] [Google Scholar]
- 40.Smart N. G., Apelqvist A. A., Gu X., Harmon E. B., Topper J. N., MacDonald R. J., Kim S. K. Conditional expression of Smad7 in pancreatic beta cells disrupts TGF-beta signaling and induces reversible diabetes mellitus. PLoS Biol. 2006;4:e39. doi: 10.1371/journal.pbio.0040039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Stein J., Milewski W. M., Hara M., Steiner D. F., Dey A. GSK-3 inactivation or depletion promotes beta-cell replication via down regulation of the CDK inhibitor, p27 (Kip1) Islets. 2011;3:21–34. doi: 10.4161/isl.3.1.14435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Susaki E., Nakayama K. I. Multiple mechanisms for p27(Kip1) translocation and degradation. Cell Cycle. 2007;6:3015–3020. doi: 10.4161/cc.6.24.5087. [DOI] [PubMed] [Google Scholar]
- 43.Teta M., Long S. Y., Wartschow L. M., Rankin M. M., Kushner J. A. Very slow turnover of beta-cells in aged adult mice. Diabetes. 2005;54:2557–2567. doi: 10.2337/diabetes.54.9.2557. [DOI] [PubMed] [Google Scholar]
- 44.Tulachan S. S., Tei E., Hembree M., Crisera C., Prasadan K., Koizumi M., Shah S., Guo P., Bottinger E., Gittes G. K. TGF-beta isoform signaling regulates secondary transition and mesenchymal-induced endocrine development in the embryonic mouse pancreas. Dev. Biol. 2007;305:508–521. doi: 10.1016/j.ydbio.2007.02.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yue J., Mulder K. M. Transforming growth factor-beta signal transduction in epithelial cells. Pharmacol. Ther. 2001;91:1–34. doi: 10.1016/s0163-7258(01)00143-7. [DOI] [PubMed] [Google Scholar]
- 46.Zhong L., Georgia S., Tschen S. I., Nakayama K., Bhushan A. Essential role of Skp2-mediated p27 degradation in growth and adaptive expansion of pancreatic beta cells. J. Clin. Invest. 2007;117:2869–2876. doi: 10.1172/JCI32198. [DOI] [PMC free article] [PubMed] [Google Scholar]