

Abstract
OBJECTIVE
Diabetes is associated with oxidative stress and increased mortality, but a possible correlation between leukocyte-endothelium interactions, oxidative stress, and silent myocardial ischemia (SMI) is yet to be confirmed.
RESEARCH DESIGN AND METHODS
Mitochondrial dysfunction and interactions between leukocytes and human umbilical vein endothelial cells were evaluated in 200 type 2 diabetic patients (25 with SMI) and 60 body composition– and age-matched control subjects. A possible correlation between these parameters and the onset of SMI was explored, and anthropometric and metabolic parameters were also analyzed.
RESULTS
Waist, levels of triglycerides, proinflammatory cytokines (interleukin-6 and tumor necrosis factor-α), HbA1c, high-sensitivity C-reactive protein (hs-CRP), glucose, and insulin, and homeostasis model assessment of insulin resistance were higher in diabetic patients than in control subjects. However, no statistical differences in hs-CRP and insulin levels were detected when the data were adjusted for waist. None of these parameters varied between SMI and non-SMI patients. Mitochondrial function was impaired and leukocyte-endothelium interactions were more frequent among diabetic patients, which was evident in the lower mitochondrial O2 consumption, membrane potential, polymorphonuclear cell rolling velocity, and GSH/GSSG ratio, and in the higher mitochondrial reactive oxygen species production and rolling flux, adhesion, and vascular cell adhesion molecule-1 (VCAM-1) and E-selectin molecules observed in these subjects. Moreover, these differences correlated with SMI. Statistical differences were maintained after adjusting the data for BMI and waist, with the exception of VCAM-1 levels when adjusted for waist.
CONCLUSIONS
Oxidative stress, mitochondrial dysfunction, and endothelium-inducing leukocyte-endothelium interactions are features of type 2 diabetes and correlate with SMI.
Adiagnosis of diabetes worsens the prognosis of cardiovascular diseases (CVDs) to the extent that postinfarction mortality among these patients is double that of nondiabetic patients. In this sense, coronary artery disease is the major cause of morbidity and mortality in patients with diabetes, in which circumstances the prognosis is unfavorable (1), and silent myocardial ischemia (SMI) is thought to be a predictor of subclinical CVD (2).
Evidence points to the involvement of impaired mitochondrial energetics in cardiac dysfunction in obesity and diabetes (3,4). Multiple abnormalities associated with diabetes, such as hyperglycemia, hyperlipidemia, and insulin resistance, are thought to contribute to adverse outcomes in diabetes after myocardial ischemia (5).
The pathogenesis of mitochondrial dysfunction in obesity or diabetes-related heart disease is multifactorial and includes oxidative damage (4,6). In experimental models of obesity and diabetes, insulin resistance is commonly detected in tissues such as those in the heart (7).
Oxidative stress is implicated in the etiology of the insulin resistance associated with type 2 diabetes (8). Numerous studies of nonspecific antioxidant treatments provide indirect evidence of a link between oxidative stress and insulin resistance (9). Mitochondria are the main source of reactive oxygen species (ROS), which are important markers of mitochondrial function (10). Several studies have shown that the rate of mitochondrial H2O2 emission is significantly greater when basal respiration is supported by fatty acids (11), raising the possibility that mitochondrial H2O2 emission is a primary factor in the etiology of insulin resistance.
Peripheral polymorphonuclear leukocytes (PMNs) are one of the main inflammatory cell types. Once activated, PMNs release ROS, which contributes to oxidative stress and the inflammation and endothelial damage that follow. Oxidative stress occurs in the PMNs of insulin-resistant patients and is related to an impairment of mitochondrial function (8,12).
The onset of many CVDs is heralded by the movement and accumulation of leukocytes in the vessel wall. These processes are mediated by an interaction between the adhesion molecules expressed on white blood and/or endothelial cells. Different cellular adhesion molecules have been implicated in atherogenesis, including vascular cell adhesion molecule-1 (VCAM-1), intercellular adhesion molecule-1 (ICAM-1), and selectins (13).
The current study was performed to throw light on the relationship between mitochondrial dysfunction, leukocyte activation, and the increase of leukocyte-endothelium interactions observed in type 2 diabetes and to explore a possible correlation between these factors and different clinical features, including SMI. We detected an increase in ROS production and PMN rolling flux, adhesion, and adhesion molecules, and a decrease in GSH levels, GSH/GSSG ratio, mitochondrial mass, membrane potential, and PMN rolling velocity, all of which are signs of an impairment of mitochondrial function and enhanced leukocyte-endothelium interaction. Our results endorse the hypothesis of an association between type 2 diabetes, mitochondrial dysfunction, and leukocyte-endothelium interaction. Additionally, they point to a correlation between these parameters and SMI, in the presence of which they were more affected.
RESEARCH DESIGN AND METHODS
Subjects
Our study population consisted of 200 patients (25 with SMI, 56.1 ± 2.0 years of age; those without SMI, 55.3 ± 1.7 years of age) consecutively attending the Department of Endocrinology’s outpatient clinic of the University Hospital Dr. Peset and who were diagnosed with type 2 diabetes according to the criteria of the American Diabetes Association. Diagnosis was confirmed when a patient fulfilled one or more of the following criteria: 1) levels of fasting serum glucose ≥7.0 mmol/L (126 mg/dL) or random serum glucose ≥11.1 mmol/L (200 mg/dL) on at least two occasions; 2) HbA1c ≥6.5%; or 3) antidiabetic medication. Patients taking allopurinol or any oral antioxidant supplements were excluded from the study.
Sixty volunteer control subjects (54.2 ± 1.8 years of age) were enrolled in the study and in any case they did not have the following criteria: 1) levels of fasting serum glucose ≥7.0 mmol/L (126 mg/dL) or random serum glucose ≥11.1 mmol/L (200 mg/dL) on at least two occasions; 2) HbA1c ≥6.5%; or 3) antidiabetic medication. An additional inclusion criterion was the absence of any documented history of vascular disease (ischemic cardiopathy, peripheral arteriopathy, or cerebrovascular accident) and diabetes. Exclusion criteria were physical disability or alterations in the electrocardiogram (ECG) that made it impossible to perform an exercise stress test, or clinical signs of coronary artery, peripheral, or cerebrovascular disease. In accordance with the Declaration of Helsinki, all participants were informed of the purpose, risks, procedures, and possible benefits of the study and gave their expressed consent to take part. Approval was obtained from the hospital’s ethics committee.
Study procedure
During their first visit, subjects provided their case history and underwent a physical examination that included measurement of BMI and waist circumference. Subjects were given an appointment at the Department of Endocrinology’s functional testing unit for collection of blood for biochemical determinations. The appointment took place between 8:00 and 10:00 a.m. and patients were instructed to have fasted for at least 10 h. During the appointment, subjects were fitted with a Holter in order to monitor blood pressure over 24 h.
Evaluation of SMI
During the basal visit, a baseline ECG was performed and patients were monitored over the following 30 days. If a Q wave of previous myocardial necrosis was detected (i.e., <0.04 s with a depth greater than one-third the height of the QRS waves of the ECG in at least two anatomically consecutive leads), a Doppler echocardiography was performed. If this test also showed segmentary contractibility disorders, the subject was classified as positive for SMI. Otherwise, the subsequent step was 24-h Holter monitoring of the ECG, with any decrease in the ST segment ≥1 mV or any increase ≥1.5 mV and continuing for at least 1 min considered a positive result, even in the absence of symptoms of angina. If the result was negative, the patient was submitted to an exercise stress test consisting of the Bruce protocol and according to the recommendations of the American College of Cardiology (ACC) and American Heart Association (AHA) (14). When the result of this test was negative, SMI was ruled out. If inconclusive, a nuclear stress test was performed using the criteria for positivity established by the ACC, AHA, and American Society of Nuclear Cardiology (15). If the result of the Bruce protocol or nuclear stress was positive, the patient was offered a coronary angiograph in order to confirm the diagnosis.
Biochemical determinations
Blood samples were collected and centrifuged at 23,000g for 15 min at 4°C. Aliquots of serum were obtained to determine total cholesterol, glucose, and triglyceride (TG) levels using an enzymatic method and HDL levels using a Beckman LX20 analyzer (Beckman Corp., Brea, CA). The intraseries coefficient of variation (CV) was <3.5% for all these determinations. LDL content was calculated with the Friedewald formula. Insulin and lipoprotein(a) levels were determined by enzyme-linked immunosorbent assay (intratest CV <4%). Insulin resistance in patients not taking insulin was calculated using homeostasis model assessment of insulin resistance (HOMA-IR). High-sensitivity C-reactive protein (hs-CRP) levels were quantified by a latex-enhanced immunonephelometric assay (intra-assay CV of 8.7%).
Cells
PMNs were isolated from citrated blood samples and incubated with dextran (3%) for 45 min. The supernatant was obtained from the Ficoll-Hypaque gradient and centrifuged at 250g for 25 min, and the pellet was resuspended in lysis buffer after centrifugation at room temperature (100g, 5 min). PMNs were counted, washed in Hanks’ balanced salt solution, and resuspended in complete RPMI medium.
Human umbilical vein endothelial cell culture
Human umbilical vein endothelial cells (HUVECs) were harvested from umbilical cords by means of collagenase treatment (16). Passage 1 from primary cultures was used in subsequent experiments. For adhesion studies, HUVECs were cultured on fibronectin (5 mg/mL)-coated, 25-mm plastic coverslips until confluent (∼48 h).
Measurements of O2 consumption, membrane potential, and mitochondrial mass
PMNs were resuspended (5 × 106 cells/mL) in Hanks’ balanced salt solution and placed in a gas-tight chamber. O2 consumption was then measured with a Clark-type O2 electrode (Rank Brothers, Bottisham, U.K.) (12). Sodium cyanide (10−3 mol/L) was used to confirm whether or not O2 consumption was mainly mitochondrial (95–99%). Tetramethylrhodamine methyl ester (TMRM; 5 × 10−6 mol/L) fluorescent dye was used to assess ΔΨm. Mitochondrial mass was measured in cells treated with the fluorescent dye 10-N-nonyl acridine orange (NAO; 5 × 10−6 mol/L), which specifically binds to cardiolipin independently of ΔΨm (17).
Measurement of ROS production
Three different methods were used to measure ROS. Total ROS production was evaluated by fluorimetry using a Fluoroskan plate reader (Thermo Labsystems, Thermo Scientific, Rockford, IL) after a 30-min incubation with the fluorescent probe 2’,7’-dichlorodihydrofluorescein diacetate (DCFH-DA; 5 × 10−6 mol/L), as described elsewhere (16). H2O2 was assessed with the Amplex RedR H2O2/Peroxidase Assay kit. H2O2 (0.5 × 10−4 mol/L) was used as a positive control. The fluorescent probe MitoSOX (5 × 10−6 mol/L, 30 min) was used to detect mitochondrial superoxide.
Glutathione content, adhesion assay, and levels of cytokines and adhesion molecules
The determination of glutathione content and the parallel plate flow chamber in vitro model have been described in detail previously (8,18,19). These techniques and the drugs and solutions used are described in the Supplementary Data.
Data analysis
Statistical analysis was carried out with R version 2.15.1 (The R Foundation for Statistical Computing, Vienna, Austria). Quantitative variables are expressed as mean and SD if normally distributed; if not, they are expressed as median and quartiles. The parametric data were compared with a one-way ANOVA, and nonparametric data were compared with a Kruskal-Wallis, followed in each case by a post hoc test (Student-Newman-Keuls or Dunn multiple comparison, respectively). Adjustment of variables was carried out using a univariate general linear model; the variable to be adjusted was the dependent variable and the group (control, SMI, or absence of SMI) the independent variables. Possible confounding variables (waist circumference or BMI) were defined as covariates. The χ2 test was used to compare proportions among groups of subjects. Pearson correlation or Spearman correlation coefficients were used to measure the strength of the association between two variables for parametric and nonparametric data, respectively. All the tests used a confidence interval of 95% and differences were considered significant when P < 0.05.
RESULTS
Clinical and metabolic characteristics
The anthropometric characteristics of type 2 diabetic patients (with or without SMI) and control subjects are presented in Table 1, which shows a higher (P < 0.05) waist circumference among the former subjects. Fasting levels of serum TGs, HbA1c, hs-CRP, glucose, insulin, and HOMA-IR were higher in type 2 diabetic patients (P < 0.05) (Table 1), whereas levels of HDL and LDL were lower (P < 0.05). No changes were found with respect to type of treatment between type 2 diabetic patients with or without SMI. Statistically significant differences remained after adjustment for BMI and waist, with the exception of those between hs-CRP and insulin levels after adjustment for waist alone.
Table 1.
Anthropometric, clinical, and metabolic characteristics of type 2 diabetic patients (with and without SMI) and control subjects
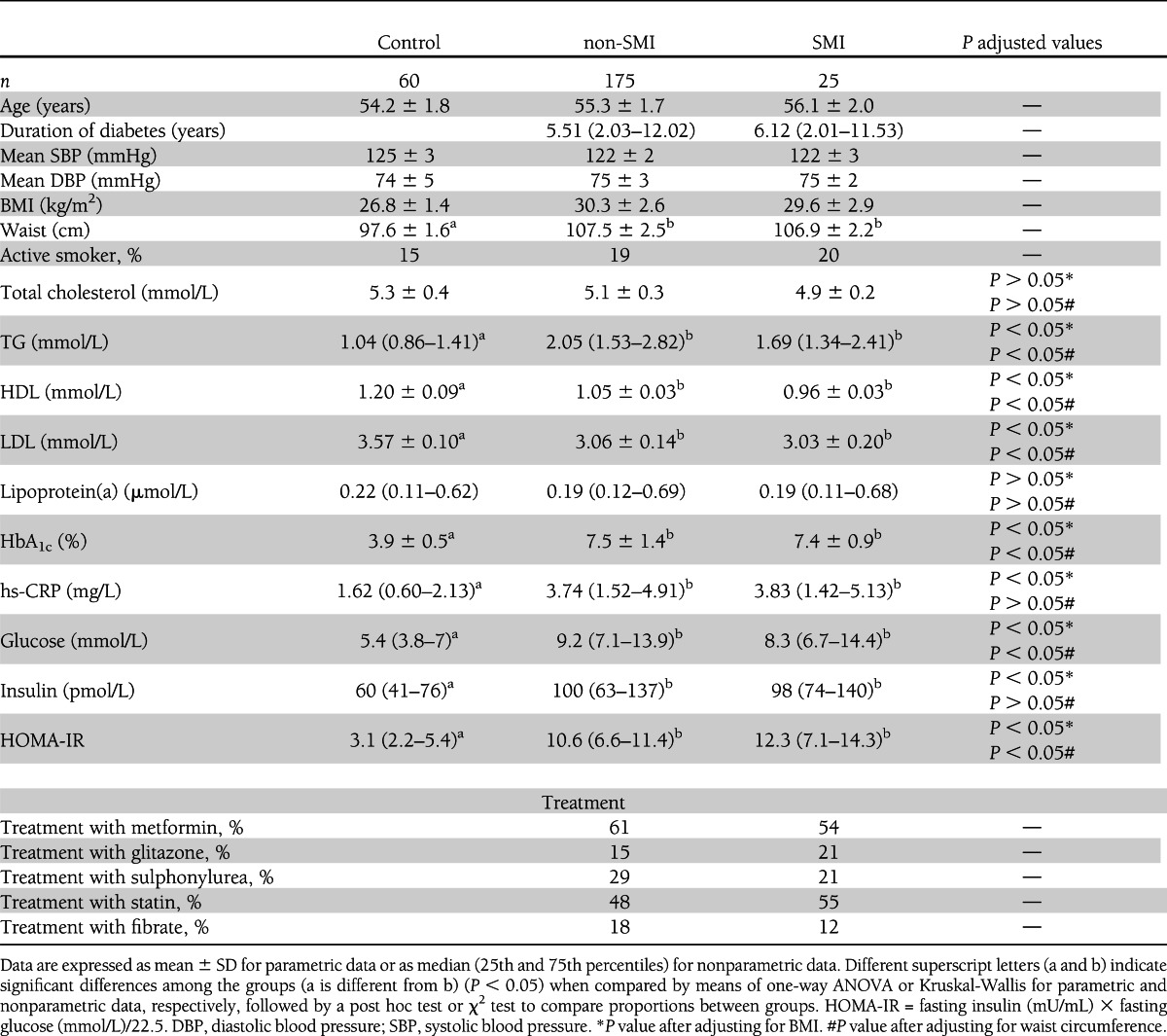
Twenty-five patients presented SMI, of which 23 were diagnosed by exercise stress testing, 1 by Holter-ECG, and 1 due to a Q wave of previous myocardial necrosis. Nineteen patients agreed to undergo the coronary angiography test.
Mitochondrial O2 consumption, membrane potential, and mitochondrial mass
An O2-tight chamber was used to monitor the rate of O2 consumption in PMNs from the blood of control subjects and type 2 diabetic patients with and without SMI. The O2 requirement of the cells was mainly mitochondrial, since addition of sodium cyanide resulted in almost complete (95–99%) inhibition of O2 consumption (not shown). The rate of O2 consumption was lower in type 2 diabetic patients and more significant among those with SMI. Figure 1A represents the reduction in O2 consumption in type 2 diabetic patients with SMI (P < 0.001) versus those without (P < 0.01). Both TMRM (P < 0.01 without SMI and P < 0.001 with SMI) and NAO (P < 0.05) fluorescence were diminished in diabetic patients (P < 0.05 without SMI and P < 0.05 with SMI), the former indicating a reduction in Δψm (Fig. 1B) and the latter a reduction in mitochondrial mass (Fig. 1C).
Figure 1.

Effects of type 2 diabetes on oxygen consumption and mitochondrial membrane potential and mass. A: O2 consumption in control subjects (n = 30) and type 2 diabetic patients with (n = 25) or without SMI (n = 47) in a closed respiration chamber measured as nmol O2/min/million cells. B: Mitochondrial membrane potential (TMRM fluorescence, % of control) in control subjects (n = 29) and type 2 diabetic patients with (n = 25) or without SMI (n = 68). C: Mitochondrial mass (NAO fluorescence, % of control) in control subjects (n = 60) and type 2 diabetic patients with (n = 25) or without SMI (n = 80). Effects of type 2 diabetes on ROS. D: Changes in the fluorescence of DCFH in control subjects (n = 37) and type 2 diabetic patients with (n = 25) or without SMI (n = 50). E: H2O2 production in control subjects (n = 28) and type 2 diabetic patients with (n = 25) or without SMI (n = 25). F: Mitochondrial ROS production (MitoSOX fluorescence, % of control) in control subjects (n = 25) and type 2 diabetic patients with (n = 25) or without SMI (n = 38). Data are expressed as mean + SEM. Different letters (a, b, and c) indicate significant differences among groups (a is different from b, a is different from c, and b is different from c) (P < 0.05) when compared by means of one-way ANOVA followed by a post hoc test.
ROS production
DCFH-DA fluorescence was significantly higher among type 2 diabetes patients but was more evident in the group with SMI (P < 0.01 without SMI, and P < 0.001 in the group with SMI) (Fig. 1D). Diabetic patients presented higher levels of H2O2 (P < 0.01 without SMI and P < 0.001 with SMI) (Fig. 1E) and mitochondrial ROS production (P < 0.05 without SMI and P < 0.001 with SMI) (Fig. 1F). In general, the patients with SMI exhibited a higher level of ROS than those without SMI.
GSH levels
As shown in Fig. 2A, levels of GSH were significantly lower in diabetic patients (P < 0.01 without SMI and P < 0.001 with SMI). Figure 2B shows how the GSH/GSSG ratio was also lower among these patients (P < 0.01 without SMI and P < 0.001 with SMI). Type 2 diabetic patients with SMI exhibited lower levels of GSH and GSH/GSSG than those without SMI (P < 0.001), which is of great relevance with respect to oxidative stress.
Figure 2.

A: The levels of GSH in control subjects (n = 36) and type 2 diabetic patients with (n = 25) or without SMI (n = 36). B: The GSH/GSSG ratio in control subjects (n = 37) and type 2 diabetic patients with (n = 25) or without SMI (n = 38). Effects of type 2 diabetes on C: PMN rolling velocity (μm sec−1) (n = 10 per group), D: rolling flux (PMN/min) (n = 10 per group), and E: PMN adhesion (PMN/mm2) (n = 10 per group). Data are expressed as mean + SEM. Different letters (a, b, and c) indicate significant differences among the groups (a is different from b, a is different from c, and b is different from c) (P < 0.05) when compared by means of one-way ANOVA followed by a post hoc test.
Adhesion assay under flow conditions
Type 2 diabetes was related to lower rolling velocity of PMN (P < 0.01 without SMI and P < 0.001 with SMI) (Fig. 2C) and a significantly higher rolling flux (cells per minute; P < 0.01 without SMI and P < 0.001 with SMI) (Fig. 2D) and PMN adhesion (cells per square millimeter; P < 0.01 without SMI and P < 0.001 with SMI) (Fig. 2E). These results provide evidence that type 2 diabetes is characterized by a higher frequency of leukocyte-endothelium interactions that is even more pronounced among patients with SMI than those without SMI (P < 0.05) (Fig. 2C–E). After adding tumor necrosis factor-α (TNF-α; 10 ng/mL, 4 h) and PAF (1 μmol/L, 1 h) (positive controls) to HUVECs and leukocytes, respectively, an increase in rolling flux (P < 0.001) and adhesion (P < 0.001) and a decrease of rolling velocity (P < 0.05) were observed (Supplementary Fig. 1).
Levels of cytokines and adhesion molecules
Type 2 diabetes was related to a significant increase in the levels of proinflammatory cytokines interleukin-6 (IL-6; P < 0.05 in both groups) and TNF-α (P < 0.05 without SMI and P < 0.01 with SMI) (Table 2) and higher levels of the adhesion molecules VCAM-1 and E-selectin (P < 0.05 without SMI and P < 0.01 with SMI). Type 2 diabetes was characterized by higher levels of proinflammatory cytokines and adhesion molecules, which were even higher in diabetic patients with SMI than those without SMI, with the exception of IL-6 (P < 0.05).
Table 2.
Cytokines and adhesion molecules in the serum of type 2 diabetic patients (with and without SMI) and control subjects
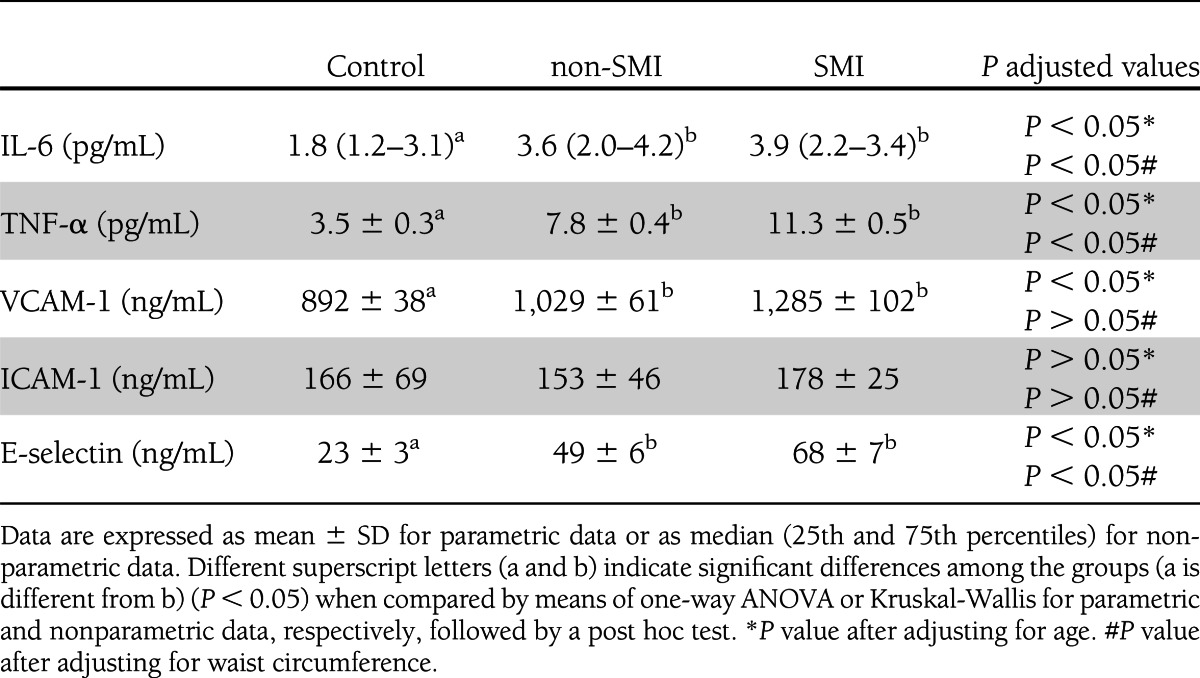
After adjusting for BMI and waist circumference, significant differences remained between the groups in relation to IL-6, TNF-α, and E-selectin but disappeared in the case of VCAM-1. No significant differences in VCAM-1 levels were observed between the groups.
Correlation studies
We studied possible correlations between leukocyte mitochondrial function, ROS (both total and mitochondrial), and leukocyte-endothelial interactions and type 2 diabetic patients with and without SMI (Supplementary Table 1).
Mitochondrial O2 consumption was positively correlated with rolling velocity in SMI patients (r = 0.954, P < 0.001) and negatively correlated with PMN rolling in non-SMI patients (r = −0.770, P < 0.05) and with PMN adhesion in both groups (r = −0.979 and P < 0.001, r = −0.873 and P < 0.05, in non-SMI and SMI patients, respectively). Membrane potential was negatively correlated with PMN rolling in SMI patients (r = −0.765, P < 0.05) and with PMN adhesion in both groups (r = −0.865 and P < 0.01, r = −0.929 and P < 0.01, in non-SMI and SMI patients, respectively). ROS was positively correlated with PMN rolling (r = 0.820 and P < 0.05, r = 0.777 and P < 0.05 in non-SMI and SMI patients, respectively) and with PMN adhesion (r = 0.965 and P < 0.001, r = 0.838 and P < 0.05, in non-SMI and SMI patients, respectively). Interestingly, mitochondrial ROS production was negatively correlated with rolling velocity (r = −0.814, P < 0.05) and positively correlated with PMN rolling (r = 0.848, P < 0.05) and with PMN adhesion (r = 0.989, P < 0.001) in SMI patients but not in non-SMI patients.
Regarding mitochondrial parameters, O2 consumption was negatively correlated with ROS production in both groups (r = −0.407 and P < 0.05, r = −0.647 and P < 0.01, in non-SMI and SMI patients, respectively). Mitochondrial membrane potential was negatively correlated with mitochondrial ROS in SMI patients (r = −0.468, P < 0.05) but not in non-SMI patients.
CONCLUSIONS
We can report an increase in fasting levels of TG, HbA1c, hs-CRP, glucose, insulin, and HOMA-IR in type 2 diabetic patients. Given that waist circumference and BMI were also higher in this group (but significantly only in the former case), we adjusted the data for these parameters. However, statistical differences remained, except between hs-CRP and insulin levels after adjustment for waist.
We observed a prevalence of SMI of 12.5%, which falls within the range of 10 to 20% attributed to the diabetic population (20). Furthermore, mitochondrial dysfunction (defined as a decrease in O2 consumption and membrane potential and an increase in mitochondrial ROS production) in PMNs was identified as a characteristic of type 2 diabetes and was found to correlate with the presence of SMI. This effect was evident in the decrease in mitochondrial O2 consumption, membrane potential, mitochondrial mass, GSH levels, and GSH/GSSG ratio and the increase in mitochondrial ROS production observed in these patients. Furthermore, we have observed how type 2 diabetes induces leukocyte-endothelium interactions by undermining PMN rolling velocity and increasing rolling flux and adhesion, which is accompanied by a simultaneous increase in proinflammatory cytokines IL-6 and TNF-α and adhesion molecules. These effects on the parameters under study were more evident in the SMI group, which may partially explain the increase of SMI and cardiovascular events during type 2 diabetes.
Our results provide evidence that the rate of mitochondrial dysfunction correlates with SMI in type 2 diabetes. It has been reported that leukocytes from patients with oxidative stress are in a proinflammatory state expressed by a heightened sensitivity to physiologic hyperglycemia and elevated plasma CRP (21).
It has been described that insulin signaling per se also regulates myocardial mitochondrial O2 consumption and rates of ATP synthesis (22,23). The studies in question demonstrated a coordinated reduction in fatty acid enzymes and tricarboxilic acid that appeared to impair the delivery of reducing equivalents to the electron transport chain. Additionally, mitochondria from insulin receptor–deficient myocytes were found to be more susceptible to fatty acid–induced oxidative stress and mitochondrial uncoupling. These data highlight the important role of the insulin signaling pathway in modulating mitochondrial bioenergetics and integrity. Our results show that mitochondrial dysfunction may be related to the onset of SMI. Given that SMI seems to be a predictor of CVD driven by increased cardiovascular risk markers, it may be of relevance to monitor mitochondrial function as a marker of the appearance and prevalence of SMI.
In addition to mitochondrial dysfunction, insulin resistance in patients with type 2 diabetes is characterized by reduced maximal O2 consumption (24). This parameter is positively correlated with insulin sensitivity and is considered to be a strong determinant of the insulin sensitivity index (25–27). In skeletal muscle of obese subjects and patients with type 2 diabetes, mitochondrial dysfunction appears to be caused by a lower number and decreased functional capacity of mitochondria (25,28).
Oxidative stress and ROS-mediated mitochondrial dysfunction are both major mechanisms of mitochondrial dysfunction. In the present work, we have demonstrated a higher production of mitochondrial ROS by leukocytes of type 2 diabetic patients in basal conditions, which is more pronounced among those with SMI.
ROS can be released from the endothelium and inflammatory cells, inducing oxidative damage (29). Indeed, excessive amounts of ROS are generally harmful to cells, as they can provoke lipid peroxidation and apoptosis. The antioxidant system of organisms neutralizes the damaging effects of ROS and, together with GSH, plays a vital role in protecting cells from oxidative stress (30). In this way, an increase in the production of ROS, a decrease in GSH levels and the GSH/GSSG ratio, and a reduction in the mitochondrial membrane potential and mitochondrial mass, all of which are characteristics of diabetes, are likely to lead to dysfunction within the respiratory chain, which subsequently compromises the functioning of mitochondria as a source of energy. Mitochondria are known to be an important site for the generation of ROS, which are highly toxic to various sites of the mitochondrial respiratory chain. The inhibition of complex I is the most likely consequence of this toxicity.
Pathophysiological states such as atherosclerosis and hypertension are characterized by leukocyte recruitment to the arterial wall. To study this process, we have used an in vitro model in which human leukocytes flow over a monolayer of human endothelial cells with a shear stress similar to that observed in vivo (31). This reproduces the process that precedes inflammation in vivo (rolling and adhesion) and that is critical to homeostasis and vascular cell integrity. If these interactions are exacerbated, the vascular dysfunction and injury associated with many CVDs can occur (32). Our experimental system has been widely applied to visualize and analyze the multistep recruitment of leukocytes in these diseases, and allows the mechanisms of action implicated in this recruitment to be assessed (33,34). In the current study, we have observed how type 2 diabetes induces a significant increase in rolling flux and PMN adhesion and a decrease in the rolling velocity of PMN. These effects seem to directly correlate with SMI, as they were more pronounced in SMI patients in all cases. Thus, we can demonstrate that mitochondrial ROS production fully correlates with adhesion, rolling, and rolling velocity in type 2 diabetic patients with SMI. In relation to this, it is known that the causal link between elevated glucose and hyperglycemic damage is the increased production of ROS by mitochondria (35). Furthermore, increases in leukocyte-endothelium interactions have been related to oxidative stress in a model of insulin resistance (19). The present findings demonstrate a higher increase of ROS in diabetic patients with SMI. This is relevant, as ROS promotes a proinflammatory/prothrombogenic phenotype within the vasculature by different mechanisms (36), including the inactivation of nitric oxide, the activation of redox-sensitive transcription factors (e.g., nuclear factor-κB) that govern the expression of endothelial cell adhesion molecules, and the activation of enzymes (e.g., phospholipase A[2]) that produce leukocyte-stimulating inflammatory mediators.
Our data are in accordance with the evidence associating capillary occlusion by leukocytes with extravascular macrophage accumulation in animal models of diabetes (34). Additionally, an intravital microscopy study in which rats were made diabetic by streptozotocin injection has provided evidence that the diabetic state induces lower venular shear rates and an increased number of rolling leukocytes in mesenteric venules with respect to nondiabetic animals (37).
Endothelial activation can be assessed by measurement of the soluble adhesion molecules E-selectin, VCAM-1, and ICAM-1. In general, circulating levels of adhesion molecules are elevated in patients with type 2 diabetes (38) and are upregulated in healthy individuals in response to systemic inflammation (39). In the current study, we have observed an increase in adhesion molecules in accordance with a rise in the number of leukocyte-endothelium interactions and selectins that mediate the initial tethering and subsequent rolling of leukocytes. The elevated levels of adhesion molecules observed in the serum of type 2 diabetic patients are of particular interest given that they provide strong evidence of an ongoing inflammatory process in the endothelium that could, theoretically, lead to CVD. Furthermore, we have observed an increase in proinflammatory levels of the cytokines TNF-α and IL-6 in type 2 diabetic patients, and an enhanced release of TNF-α from leukocytes after activation by ROS-induced oxidative stress may inhibit insulin signaling and impair glucose uptake (40).
In conclusion, the current study provides evidence of oxidative stress, mitochondrial dysfunction, and endothelium-inducing leukocyte-endothelium interactions in type 2 diabetes. These alterations are more pronounced in patients with SMI, which suggests a correlation between the rate of mitochondrial dysfunction and leukocyte-endothelium interactions on the one hand and the prevalence/appearance of SMI in diabetes on the other. Future exploration of this oxidative stress may help to clarify the nature of the molecular mechanisms involved and the physiological significance of insulin resistance in type 2 diabetic patients with SMI. Such knowledge would no doubt help to develop strategies to reduce the risk of the onset of type 2 diabetes.
Acknowledgments
This study was financed by grants PI10/1195, PI09/01025, SAF2010-16030, CIBERehd CB06/04/0071, PROMETEO 2010/060, ACOMP/2012/042, and ACOMP/2012/045 and by the European Union from the European Regional Development Fund. C.d.P. is funded by an FPI grant (BES-2008-04338), and A.A. is the beneficiary of the Ramón y Cajal (RYC2005-02295) and I3 programs from the Ministry of Science and Innovation. V.M.V. and M.R. are recipients of contracts from the Regional Ministry of Health of the Valencian Regional Government and Carlos III Health Institute (CES10/030 and CP10/0360, respectively).
No potential conflicts of interest relevant to this article were reported.
A.H.-M. contributed to the discussion and wrote, reviewed, and edited the manuscript. M.R. researched data, contributed to the discussion, and reviewed and edited the manuscript. S.R.-L. researched data and contributed to the discussion. C.B. and L.B. researched data. C.d.P., I.R.-T., and E.S.-I. reviewed and edited the manuscript. A.A. contributed to the discussion and reviewed and edited the manuscript. V.M.V. wrote, reviewed, and edited the manuscript. A.H.-M. is the guarantor of this work and, as such, had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
The authors thank B. Normanly for his editorial assistance (University of Valencia) and Isabel Soria for her work in the extraction of the biological samples (University Hospital Dr. Peset).
Footnotes
This article contains Supplementary Data online at http://care.diabetesjournals.org/lookup/suppl/doi:10.2337/dc12-1224/-/DC1.
References
- 1.Kannel WB, McGee DL. Diabetes and cardiovascular disease. The Framingham study. JAMA 1979;241:2035–2038 [DOI] [PubMed] [Google Scholar]
- 2.Fava S, Azzopardi J, Muscat HA, Fenech FF. Factors that influence outcome in diabetic subjects with myocardial infarction. Diabetes Care 1993;16:1615–1618 [DOI] [PubMed] [Google Scholar]
- 3.Scheuermann-Freestone M, Madsen PL, Manners D, et al. Abnormal cardiac and skeletal muscle energy metabolism in patients with type 2 diabetes. Circulation 2003;107:3040–3046 [DOI] [PubMed] [Google Scholar]
- 4.Boudina S, Sena S, O’Neill BT, Tathireddy P, Young ME, Abel ED. Reduced mitochondrial oxidative capacity and increased mitochondrial uncoupling impair myocardial energetics in obesity. Circulation 2005;112:2686–2695 [DOI] [PubMed] [Google Scholar]
- 5.Capes SE, Hunt D, Malmberg K, Gerstein HC. Stress hyperglycaemia and increased risk of death after myocardial infarction in patients with and without diabetes: a systematic overview. Lancet 2000;355:773–778 [DOI] [PubMed] [Google Scholar]
- 6.Shen X, Zheng S, Metreveli NS, Epstein PN. Protection of cardiac mitochondria by overexpression of MnSOD reduces diabetic cardiomyopathy. Diabetes 2006;55:798–805 [DOI] [PubMed] [Google Scholar]
- 7.Park SY, Cho YR, Kim HJ, et al. Unraveling the temporal pattern of diet-induced insulin resistance in individual organs and cardiac dysfunction in C57BL/6 mice. Diabetes 2005;54:3530–3540 [DOI] [PubMed] [Google Scholar]
- 8.Hernandez-Mijares A, Rocha M, Apostolova N, et al. Mitochondrial complex I impairment in leukocytes from type 2 diabetic patients. Free Radic Biol Med 2011;50:1215–1221 [DOI] [PubMed] [Google Scholar]
- 9.Bonnard C, Durand A, Peyrol S, et al. Mitochondrial dysfunction results from oxidative stress in the skeletal muscle of diet-induced insulin-resistant mice. J Clin Invest 2008;118:789–800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schafer FQ, Buettner GR. Redox environment of the cell as viewed through the redox state of the glutathione disulfide/glutathione couple. Free Radic Biol Med 2001;30:1191–1212 [DOI] [PubMed] [Google Scholar]
- 11.Anderson EJ, Yamazaki H, Neufer PD. Induction of endogenous uncoupling protein 3 suppresses mitochondrial oxidant emission during fatty acid-supported respiration. J Biol Chem 2007;282:31257–31266 [DOI] [PubMed] [Google Scholar]
- 12.Victor VM, Rocha M, Bañuls C, et al. Mitochondrial complex I impairment in leukocytes from polycystic ovary syndrome patients with insulin resistance. J Clin Endocrinol Metab 2009;94:3505–3512 [DOI] [PubMed] [Google Scholar]
- 13.Jude EB, Douglas JT, Anderson SG, Young MJ, Boulton AJ. Circulating cellular adhesion molecules ICAM-1, VCAM-1, P- and E-selectin in the prediction of cardiovascular disease in diabetes mellitus. Eur J Intern Med 2002;13:185–189 [DOI] [PubMed] [Google Scholar]
- 14.Gibbons RJ, Balady GJ, Bricker JT, et al. American College of Cardiology/American Heart Association Task Force on Practice Guidelines. Committee to Update the 1997 Exercise Testing Guidelines ACC/AHA 2002 guideline update for exercise testing: summary article. A report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Committee to Update the 1997 Exercise Testing Guidelines). J Am Coll Cardiol 2002;40:1531–1540 [DOI] [PubMed] [Google Scholar]
- 15.Klocke FJ, Baird MG, Lorell BH, et al. American College of Cardiology. American Heart Association Task Force on Practice Guidelines. American Society for Nuclear Cardiology ACC/AHA/ASNC guidelines for the clinical use of cardiac radionuclide imaging—executive summary: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (ACC/AHA/ASNC Committee to Revise the 1995 Guidelines for the Clinical Use of Cardiac Radionuclide Imaging). Circulation 2003;108:1404–1418 [DOI] [PubMed] [Google Scholar]
- 16.Esplugues JV, Rocha M, Nuñez C, et al. Complex I dysfunction and tolerance to nitroglycerin: an approach based on mitochondrial-targeted antioxidants. Circ Res 2006;99:1067–1075 [DOI] [PubMed] [Google Scholar]
- 17.Apostolova N, Gomez-Sucerquia LJ, Moran A, Alvarez A, Blas-Garcia A, Esplugues JV. Enhanced oxidative stress and increased mitochondrial mass during efavirenz-induced apoptosis in human hepatic cells. Br J Pharmacol 2010;160:2069–2084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.De Pablo C, Orden S, Apostolova N, Blanquer A, Esplugues JV, Alvarez A. Abacavir and didanosine induce the interaction between human leukocytes and endothelial cells through Mac-1 upregulation. AIDS 2010;24:1259–1266 [DOI] [PubMed] [Google Scholar]
- 19.Victor VM, Rocha M, Bañuls C, et al. Induction of oxidative stress and human leukocyte/endothelial cell interactions in polycystic ovary syndrome patients with insulin resistance. J Clin Endocrinol Metab 2011;96:3115–3122 [DOI] [PubMed] [Google Scholar]
- 20.Scheidt-Nave C, Barrett-Connor E, Wingard DL. Resting electrocardiographic abnormalities suggestive of asymptomatic ischemic heart disease associated with non-insulin-dependent diabetes mellitus in a defined population. Circulation 1990;81:899–906 [DOI] [PubMed] [Google Scholar]
- 21.Kelly CC, Lyall H, Petrie JR, Gould GW, Connell JMC, Sattar N. Low grade chronic inflammation in women with polycystic ovarian syndrome. J Clin Endocrinol Metab 2001;86:2453–2455 [DOI] [PubMed] [Google Scholar]
- 22.Boudina S, Bugger H, Sena S, et al. Contribution of impaired myocardial insulin signaling to mitochondrial dysfunction and oxidative stress in the heart. Circulation 2009;119:1272–1283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sena S, Hu P, Zhang D, et al. Impaired insulin signaling accelerates cardiac mitochondrial dysfunction after myocardial infarction. J Mol Cell Cardiol 2009;46:910–918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Højlund K, Beck-Nielsen H. Impaired glycogen synthase activity and mitochondrial dysfunction in skeletal muscle: markers or mediators of insulin resistance in type 2 diabetes? Curr Diabetes Rev 2006;2:375–395 [DOI] [PubMed] [Google Scholar]
- 25.Fleg JL, Piña IL, Balady GJ, et al. Assessment of functional capacity in clinical and research applications: An advisory from the Committee on Exercise, Rehabilitation, and Prevention, Council on Clinical Cardiology, American Heart Association. Circulation 2000;102:1591–1597 [DOI] [PubMed] [Google Scholar]
- 26.Rodnick KJ, Haskell WL, Swislocki AL, Foley JE, Reaven GM. Improved insulin action in muscle, liver, and adipose tissue in physically trained human subjects. Am J Physiol 1987;253:E489–E495 [DOI] [PubMed] [Google Scholar]
- 27.Rosenthal M, Haskell WL, Solomon R, Widstrom A, Reaven GM. Demonstration of a relationship between level of physical training and insulin-stimulated glucose utilization in normal humans. Diabetes 1983;32:408–411 [DOI] [PubMed] [Google Scholar]
- 28.Kelley DE, He J, Menshikova EV, Ritov VB. Dysfunction of mitochondria in human skeletal muscle in type 2 diabetes. Diabetes 2002;51:2944–2950 [DOI] [PubMed] [Google Scholar]
- 29.Diao FY, Xu M, Hu Y, et al. The molecular characteristics of polycystic ovary syndrome (PCOS) ovary defined by human ovary cDNA microarray. J Mol Endocrinol 2004;33:59–72 [DOI] [PubMed] [Google Scholar]
- 30.Victor VM, Rocha M, De la Fuente M. Immune cells: free radicals and antioxidants in sepsis. Int Immunopharmacol 2004;4:327–347 [DOI] [PubMed] [Google Scholar]
- 31.Ibiza S, Alvarez A, Romero W, Barrachina MD, Esplugues JV, Calatayud S. Gastrin induces the interaction between human mononuclear leukocytes and endothelial cells through the endothelial expression of P-selectin and VCAM-1. Am J Physiol Cell Physiol 2009;297:C1588–C1595 [DOI] [PubMed] [Google Scholar]
- 32.Krieglstein CF, Granger DN. Adhesion molecules and their role in vascular disease. Am J Hypertens 2001;14:44S–54S [DOI] [PubMed] [Google Scholar]
- 33.Rao RM, Yang L, Garcia-Cardena G, Luscinskas FW. Endothelial-dependent mechanisms of leukocyte recruitment to the vascular wall. Circ Res 2007;101:234–247 [DOI] [PubMed] [Google Scholar]
- 34.Yang XD, Michie SA, Mebius RE, Tisch R, Weissman I, McDevitt HO. The role of cell adhesion molecules in the development of IDDM: implications for pathogenesis and therapy. Diabetes 1996;45:705–710 [DOI] [PubMed] [Google Scholar]
- 35.Nishikawa T, Edelstein D, Du XL, et al. Normalizing mitochondrial superoxide production blocks three pathways of hyperglycaemic damage. Nature 2000;404:787–790 [DOI] [PubMed] [Google Scholar]
- 36.Cooper D, Stokes KY, Tailor A, Granger DN. Oxidative stress promotes blood cell-endothelial cell interactions in the microcirculation. Cardiovasc Toxicol 2002;2:165–180 [DOI] [PubMed] [Google Scholar]
- 37.Panés J, Kurose I, Rodriguez-Vaca D, et al. Diabetes exacerbates inflammatory responses to ischemia-reperfusion. Circulation 1996;93:161–167 [DOI] [PubMed] [Google Scholar]
- 38.Meigs JB, Hu FB, Rifai N, Manson JE. Biomarkers of endothelial dysfunction and risk of type 2 diabetes mellitus. JAMA 2004;291:1978–1986 [DOI] [PubMed] [Google Scholar]
- 39.Lemaire LC, de Kruif MD, Giebelen IA, van Zoelen MA, van’t Veer C, van der Poll T. Differential dose-dependent effects of prednisolone on shedding of endothelial adhesion molecules during human endotoxemia. Immunol Lett 2008;121:93–96 [DOI] [PubMed] [Google Scholar]
- 40.González F, Rote NS, Minium J, Kirwan JP. Reactive oxygen species-induced oxidative stress in the development of insulin resistance and hyperandrogenism in polycystic ovary syndrome. J Clin Endocrinol Metab 2006;91:336–340 [DOI] [PubMed] [Google Scholar]