

Abstract
Background
A genome-wide association study for upper aerodigestive tract cancers identified 19 candidate single-nucleotide polymorphisms (SNPs). We used these SNPs to investigate the potential gene-gene and gene-environment interactions in head and neck squamous cell carcinoma (HNSCC) risk.
Methods
The 19 variants were genotyped using Taqman (Applied Biosystems) assays among 575 cases and 676 controls in our population-based case-control study.
Results
A restricted cubic spline model suggested both ADH1B and HEL308 modified the association between smoking pack-years and HNSCC. Classification and regression tree analysis demonstrated a higher order interaction between smoking status, ADH1B, FLJ13089 and FLJ35784 in HNSCC risk. Compared with ever smokers carrying ADH1B T/C+T/T genotypes, smokers carrying ADH1B C/C genotype and FLJ13089 A/G+A/A genotypes had a highest risk of HNSCC (OR=1.84).
Conclusions
Our results suggest that the risk associated with these variants may be specifically important amongst specific exposure groups.
Keywords: post-genome wide association study, head and neck cancer, gene and environment interaction
Introduction
Head and neck cancer is a common cancer in the U.S, accounting for 3% to 5% of all cancers (1). Tobacco and alcohol use play a prominent role in the etiology of the majority of head and neck squamous cell carcinoma (HNSCC), and human papilloma virus (HPV) is considered causal in about 25% of the disease. At the same time, not all smokers and alcohol users develop HNSCC, suggesting that individual variation in genetic susceptibility plays a critical role. Candidate gene studies with moderate success have focused on variation in genes involved in carcinogen metabolism, DNA repair, and cell cycle control and their interactions with tobacco and alcohol exposure (2), with the hypothesis that variations in these genes may alter the ability to metabolize or eliminate tobacco or alcohol-associated carcinogens, cooperating with a dysfunctional DNA repair and dysfunctional cellular responses to DNA damage (3–6).
To identify novel genes and genetic variations associated with HNSCC risk, a genome wide association study (GWAS) involving a consortium of studies of HNSCC has been undertaken, with the data from this study included in confirmation of the initial discovery phase of the GWAS. GWAS provide a powerful approach to identify lower penetrance alleles that cannot be detected by genetic linkage studies, through typing hundreds of thousands of single-nucleotide polymorphisms (SNPs) simultaneously, and scanning for associations without prior knowledge of function or position (7). This approach does require large sample sizes, requiring the collaboration of multiple studies across often varied populations to provide the appropriate statistical power to identify these variants. Thus, GWAS have challenges in investigating gene-gene and gene-environment interactions due to the heterogeneity of population structure, different strategies of data collection and exposure information ascertainment and particularly in the case of HNSCC, different prevalences of the different locations of the cancers of interest. Our study, focused on a single study population can better address these questions by examining the genes identified in the GWAS in combination with the consistently collected information and more homogeneous population structure. Thus, in this study, we selected the significant variants identified from an initial GWAS and investigated the possible gene-gene and gene-environment interaction in HNSCC risk within our population-based case-control study of HNSCC in the greater Boston metropolitan area.
Materials and methods
Study subjects
Cases in this study were head and neck squamous cell carcinoma patients identified from head and neck clinics and departments of otolaryngology or radiation oncology at nine medical facilities in Greater Boston, MA between December 1999 and December 2003 (for further details see (8–10)). HNSCC cases included diagnosis codes 141–146, 148, 149, and 161 according to International Classification of Disease, Ninth Revision (ICD-9). Eligible cases were residents in the study area aged 18 years or older and with a pathologically confirmed diagnosis of HNSCC no more than 6 months before the time of patient contact. The cancer registry was queried to insure that all eligible cases in the area were identified. Cases presenting with recurrent disease were excluded. Controls were randomly selected from Massachusetts town books and frequency-matched to cases on age (± 3 years), gender, and town of residence (for further details see (8–10)). Study protocols and materials for recruitment of cases and controls were approved by the Institutional Review Boards at the nine medical facilities (Boston University Medical Center Institutional Review Board, Dana-Farber/Harvard Cancer Center Office of the Protection of Research Subjects (covers four study sites: Beth Israel Deaconess Medical Center, Brigham and Women’s Hospital, Dana-Farber Cancer Institute, and Massachusetts General Hospital), Harvard Pilgrim Health Care Human Studies Committee, Massachusetts Eye and Ear Infirmary Human Studies Committee, Tufts-New England Medical Center Institutional Review Board, Veteran’s Administration Boston Healthcare System Institutional Review Board), and Brown University Research Protections Office. Written informed consent was obtained from all study subjects.
SNPs selection and genotyping
There were 19 variants selected from initial GWAS for upper aero-digestive tract (UADT) cancers. These included: 10 variants that achieved a p-value of <10−5, 6 non synonymous SNPs that achieved a p-value of <10−4 and 2 variants that achieved a p-value of <5×10−7 in restricting to UADT cancer subsite, or heavy drinkers/smokers. Finally, a non-synonmous ADH1B SNP rs1229984 was also included, which previously was indicated to be associated with UADT cancers but not genotyped or tagged through Linkage disequilibrium (LD) by a proxy SNP on the HumanHap300 beadchip (11).
For the genotyping of 19 SNPs, Taqman (Applied Biosystems) genotyping assays were designed and reaction conditions were optimized at International Association for Research on Cancer (IARC). The robustness of the assays was confirmed at IARC by re-genotyping the CEPH HapMap (CEU) trios and confirming concordance with HapMap genotypes (http://www.hapmap.org). Any discordance between hapmap and Taqman generated genotypes were evaluated based on the sequences from prior GWAS (11). All Taqman assays were found to be performing robustly. Once genotyping was completed, we conducted a routine series of systematic quality control steps. Assays that had a success rate of <90% and that deviated from Hardy-Weinberg Equilibrium (HWE) among controls were excluded.
Measurement of other covariates
A self-administered questionnaire was used to collect information about demographic characteristics and the standard risk factors for HNSCC, including medical history, family history of cancer, detailed smoking and drinking habits, detailed marijuana use history, occupational history and residency history. Questionnaires were distributed to cases during an initial clinic visit and to controls by mail. All the subject responses were reviewed by study personnel and research coordinators during in-person visits with cases or controls. To elicit the history of tobacco and alcohol use, subjects were first asked to report whether or not they ever used tobacco or alcohol drinks. The subjects who reported having ever used tobacco or alcohol drinks were asked to specify their ages at starting and stopping use, amount, frequency and duration of use for 8 time periods in their life (ages 10–19, 20–29, 30–39, 40–49, 50–59, 60–69, 70–79, and 80+). The calculation of pack-years and alcohol consumption per week were described previously (8).
The HPV16 serologic status of case and control subjects was ascertained as described previously (8, 9). Venous blood samples were obtained from cases and controls at enrollment. Serum was separated from plasma within 24 hours of collection and stored at −80°C. The HPV Competitive Luminex Immunoassay was used to determine the presence of antibodies to the L1 protein of HPV16. Positive and negative controls were used for quality control, and all samples were tested in duplicate.
Statistical methods
To investigate the difference in characteristics distribution between case and controls, chi-square tests for categorical variables and t-tests for continuous variables were applied in SAS 9.13. Hardy-Weinberg equilibrium was tested for each SNP among controls in STATA 10. To examine the association between each SNP and HNSCC risk, we use unconditional logistic regression to calculate the crude odds ratio (OR) as well as 95% confidence intervals (CIs) in SAS 9.13. Bonferroni correction was used to adjust for multiple comparisons.
We examined the multiplicative association between continuous smoking or alcohol drinking and genotypes using a restricted cubic spline model to account for possible nonlinearity in dose-response. To test the linearity assumption of the relationship between smoking pack-year or alcohol use and the log odds of HNSCC, We fitted the restricted cubic spline models in R using rcspline.plot. A Wald chi-square test showed there was a linear relationship between smoking pack-years and the log odds of HNSCC (Wald χ2=3.51, p=0.1726) while the Wald test showed nonlinearity between alcohol use and the log odds of HNSCC. (Wald χ2 =32.74, p<0.0001). Therefore, we performed non-parametric logistic regression analyses with b-spline expansions of alcohol use separately by genotypes to determine whether the dose-response for alcohol and HNSCC risk differed by genotypes. We also used this method to investigate the interaction between smoking and SNPs as smoking was measured as a continuous variable. To fit the linear association between smoking pack years and HNSCC, the non-linear terms were set as zero in the spline model. To fit the non-linear relationship between alcohol drinking and SNPs, we used four knots as the curves were not sensitive to the changes with the number of knots. The degree of freedom (df) was equal to 3 selected according to the smallest Akaike’s Information Criterion (AIC). All these models adjusted for age (continuous), gender, race (African American, Caucasian and other), HPV16 serologic status (negative and positive), smoking pack-years (continuous) or average drinks per week (continuous) as appropriate. It was noted that about 295 subjects were excluded in this analysis due to missing data in HPV16 (221), alcohol drinks (68) and race (6). These models were generated using SAS macro %regspline, ORs were calculated using %regspline_or and smooth plots were drawn in R. SAS macros were provided by Gregory (12).
To explore the high-order gene-environment interactions, classification and regression tree (CART) analysis was performed using rpart package in R 2.7.2. CART is a binary recursive partitioning method that produces a tree structure (13). The product of this analysis is a dendrogram defining risk subgroup based on SNP genotype, smoking or alcohol drinking.
Results
Characteristics of subjects
There were 1,280 eligible subjects successfully recruited in this study. Details regarding participation rates and reasons for refusal were described previously (8). Of these 1,280 subjects, 29 were excluded due to unavailable genotypes of SNPs. Among the 1251 subjects, 575 were cases with HNSCC (223 oral cavity cancers, 243 pharyngeal cancers, and 109 laryngeal cancers) and 676 were control subjects. The distribution of descriptive characteristics for cases and controls were presented in Table 1. As cases and controls were frequency matched on age, gender, and race, there were no significant difference in the distribution of age, gender and race between cases and controls. The mean ages of the cases and controls were 60.0 and 61.2, respectively. Most subjects were males (73%) and Caucasian (90%). Significant differences in the distribution of smoking pack-years, alcohol drinks per week and HPV16 serology were observed. Cases were more likely to smoke or drink heavily than controls (p<0.001). Also, we observed a greater prevalence of HPV16 seropositivity in cases than in controls (p<0.001).
Table 1.
Distribution of descriptive characteristics for cases and controls
| Characteristic | No. of Cases (%) (n=575) | No. of Controls (%) (n=676) | P value |
|---|---|---|---|
| Age (years) | 0.8 | ||
| Mean ± SD | 60.04 ± 11.50 | 61.22 ± 11.37 | |
| Gender | 0.7 | ||
| Female | 155(26.96) | 188(27.81) | |
| Male | 420(73.04) | 488(72.19) | |
| Race | 0.8 | ||
| Caucasian | 515(90.51) | 617(91.27) | |
| African-American | 22(3.87) | 22(3.25) | |
| Other | 32(5.62) | 37(5.47) | |
| Missing | 6 | 0 | |
| Pack-years of tobacco use | <0.0001 | ||
| Never | 105 (18.26) | 234 (34.62) | |
| 0 to<20 | 112 (19.48) | 183 (27.07) | |
| >=20 | 358 (62.26) | 259 (38.31) | |
| Alcohol consumption, average drinks per week | <0.0001 | ||
| <8 | 191 (37.3) | 395 (58.87) | |
| >=8 | 321 (62.7) | 276 (41.13) | |
| Missing | 63 | 5 | |
| HPV 16 | <0.0001 | ||
| Negative | 338(70.12) | 490(89.42) | |
| Positive | 144(29.88) | 58(10.58) | |
| Missing | 93 | 128 | |
| Tumor sites | |||
| Oral | 223 (38.78) | ||
| Pharynx | 243 (42.26) | ||
| Larynx | 109 (18.96) | ||
Association between each SNP and HNSCC
Among the 19 SNPs, one (CWF19L1, rs7924284), was excluded because the allele frequencies deviated from HWE in controls. The remaining 18 SNPs were used in the statistical analyses, including eight higher priority markers and ten lower priority makers according to initial GWAS results. Table 2 shows the associations between SNPs and HNSCC. For ADH1B (rs1229984), an inverse association was observed for the heterozygous genotype (T/C) (OR=0.56; 95% CI: 0.37–0.84) when compared with the homozygous wild-type reference group. For HEL308 (rs1494961), we observed marginally inverse associations for the C/T (OR=0.78, 95%CI: 0.60–1.02) and T/T genotypes (OR=0.78, 95%CI: 0.57–1.06) when compared with C/C genotype, respectively. For ASPH (rs1431918), a marginally increased association was indicated for homozygous A/A genotype when compared with homozygous G/G genotype (OR=1.41; 95%CI: 1.01–1.97). For FHOD3 (rs4799863), the model showed an inverse association for homozygous variant type (G/G) (OR=0.67; 95%CI: 0.49–0.93) and a marginal inverse association for A/G genotype (OR=0.80; 95%CI: 0.61–1.03) when compared with G/G genotype, respectively. For FLJ13089 (rs4767364), compared with homozygous G/G genotype, the heterozygous A/G genotype had a 1.32–fold increased risk (95%CI: 1.04–1.68) and the association reached 1.95-fold (95%CI: 1.32–2.87) for the homozygous variant A/A genotype. No associations were observed for the remaining SNPs, but most of them demonstrated marginal association with HNSCC. After Bonferroni adjustment, only FLJ13089 (rs4767364) indicated a significant association with HNSCC.
Table 2.
Associations between SNPs and HNSCC
| Gene | No. of Cases | No. of Controls | Crude OR | 95% CI | |
|---|---|---|---|---|---|
| ADH1B (rs1229984) | |||||
| C/C | 530 | 593 | Ref. | ||
| T/C | 38 | 76 | 0.56 | 0.37 | 0.84 |
| T/T | 5 | 5 | 1.12 | 0.32 | 3.89 |
| ADH7 (rs1573496) | |||||
| G/G | 471 | 536 | Ref. | ||
| C/G | 97 | 131 | 0.84 | 0.63 | 1.13 |
| C/C | 4 | 7 | 0.65 | 0.19 | 2.24 |
| Missing | 3 | 2 | |||
| CHRN (rs16969968) | |||||
| G/G | 274 | 302 | Ref. | ||
| A/G | 247 | 298 | 0.91 | 0.72 | 1.16 |
| A/A | 51 | 74 | 0.76 | 0.51 | 1.13 |
| Missing | 3 | 2 | |||
| COL5A3 (rs2287802) | |||||
| A/A | 211 | 255 | Ref. | ||
| A/G | 292 | 331 | 1.07 | 0.84 | 1.36 |
| G/G | 69 | 88 | 0.95 | 0.66 | 1.36 |
| Missing | 3 | 2 | |||
| HEL308 (rs1494961) | |||||
| C/C | 165 | 161 | Ref. | ||
| C/T | 264 | 330 | 0.78 | 0.60 | 1.02 |
| T/T | 143 | 179 | 0.78 | 0.57 | 1.06 |
| Missing | 3 | 6 | |||
| FCRL5 (rs2012199) | |||||
| T/T | 407 | 492 | Ref. | ||
| C/T | 143 | 161 | 1.07 | 0.83 | 1.39 |
| C/C | 14 | 11 | 1.54 | 0.69 | 3.43 |
| Missing | 11 | 12 | |||
| ASPH (rs1431918) | |||||
| G/G | 196 | 240 | Ref. | ||
| A/G | 269 | 338 | 0.98 | 0.76 | 1.25 |
| A/A | 108 | 94 | 1.41 | 1.01 | 1.97 |
| Missing | 2 | 4 | |||
| IL1RL1 (rs1041973) | |||||
| C/C | 294 | 337 | Ref. | ||
| A/C | 230 | 282 | 0.94 | 0.74 | 1.18 |
| A/A | 48 | 52 | 1.06 | 0.69 | 1.61 |
| Missing | 3 | 5 | |||
| FHOD3 (rs4799863) | |||||
| A/A | 179 | 173 | Ref. | ||
| A/G | 284 | 345 | 0.80 | 0.61 | 1.03 |
| G/G | 106 | 152 | 0.67 | 0.49 | 0.93 |
| Missing | 6 | 6 | |||
| RBMS3 (rs7431530) | |||||
| C/C | 300 | 329 | Ref. | ||
| C/T | 228 | 282 | 0.89 | 0.70 | 1.12 |
| T/T | 41 | 56 | 0.80 | 0.52 | 1.24 |
| Missing | 6 | 9 | |||
| TBX3 (rs11067362) | |||||
| T/T | 460 | 552 | Ref. | ||
| C/T | 102 | 109 | 1.12 | 0.83 | 1.51 |
| C/C | 7 | 7 | 1.20 | 0.42 | 3.45 |
| Missing | 6 | 8 | |||
| OPRD1 (rs16837730) | |||||
| C/C | 509 | 610 | Ref. | ||
| C/T | 58 | 57 | 1.22 | 0.83 | 1.79 |
| Missing | 8 | 9 | |||
| PRIC285 (rs3810481) | |||||
| G/G | 397 | 485 | Ref. | ||
| A/G | 148 | 168 | 1.08 | 0.83 | 1.39 |
| A/A | 21 | 16 | 1.60 | 0.83 | 3.11 |
| Missing | 9 | 7 | |||
| FLJ13089 (rs4767364) | |||||
| G/G | 247 | 351 | Ref. | ||
| A/G | 240 | 258 | 1.32 | 1.04 | 1.68 |
| A/A | 74 | 54 | 1.95 | 1.32 | 2.87 |
| Missing | 14 | 13 | |||
| ZNF326 (rs10801805) | |||||
| G/G | 229 | 281 | Ref. | ||
| A/G | 257 | 293 | 1.08 | 0.85 | 1.37 |
| A/A | 78 | 94 | 1.02 | 0.72 | 1.44 |
| Missing | 11 | 8 | |||
| MSH5 (rs2299851) | |||||
| G/G | 470 | 571 | Ref. | ||
| A/G | 92 | 91 | 1.23 | 0.90 | 1.68 |
| A/A | 4 | 4 | 1.22 | 0.30 | 4.88 |
| Missing | 9 | 10 | |||
| FLJ35784 (rs484870) | |||||
| A/A | 221 | 263 | Ref. | ||
| A/G | 263 | 316 | 0.99 | 0.78 | 1.26 |
| G/G | 82 | 89 | 1.10 | 0.77 | 1.56 |
| Missing | 9 | 8 | |||
| C6orf15 (rs2517452) | |||||
| C/C | 162 | 220 | Ref. | ||
| C/T | 291 | 319 | 1.24 | 0.96 | 1.60 |
| T/T | 112 | 127 | 1.20 | 0.87 | 1.66 |
| Missing | 10 | 10 | |||
Abbreviations: SNPS= single-nucleotide polymorphisms; HNSCC=head and neck squamous cell carcinoma
Two-way interaction
To investigate the interactions between SNPs and smoking pack years (continuous) or alcohol drinks per week (continuous) in HNSCC risk, non-parametric logistic regression models with b-spline expansions were fitted separated by different genotypes. Figure 1 shows the dose-response relationship between alcohol use and HNSCC stratified by HEL308 genotypes (C/C+C/T and T/T). The plot was truncated at 80 drinks per week because few participants drank greater than that amount. Although both genotypes presented increased risk of HNSCC with increase of alcohol consumption over 30 drinks/week, subjects with T/T genotypes tended to have a lower risk relative to those carrying C/C+C/T genotypes with the same amount of alcohol consumption. However, there was a cross between the intervals for the two genotypes. Likelihood ratio test did not suggest a significant departure from multiplicative interaction. Figure 2 displays the trend of HNSCC risk with the increase of smoking pack years separated by HEL308 genotypes (C/C+C/T and T/T). The plot was truncated at 100 pack years due to the small number of subjects smoking beyond that amount. We observed no difference in HNSCC risk between the two genotypes for those who smoked less than 70 pack years. However, a marked deviation was observed beyond 70 pack years. A likelihood ratio test for multiplicative interaction between smoking pack-years and HEL308 genotypes suggested significance (P interaction=0.026). Figure 3 presents the dose-response relationship between smoking and HNSCC risk stratified by ADH1B genotypes (C/C and T/C). The risk of HNSCC increased with increasing smoking pack years among those with ADH1B homozygous wild-type genotype (C/C), whereas a flat trend of HNSCC risk was observed for those with ADH1B heterozygous genotype (T/C). Here, the likelihood ratio test for multiplicative interaction between smoking pack years and ADH1B genotypes also suggested a significant departure from multiplicative interaction (P interaction=0.0016). In addition, we investigated the interactions between smoking and ADH1B or HEL308 by tumor sites. The multiplicative interactions between ADH1B and smoking were significant consistently across tumor sites (oral, pharynx and larynx). However, the multiplicative interactions between HEL308 and smoking were only detected in oral cavity cancers and pharyngeal cancers (data not shown).
Figure 1. The association between alcohol consumption and head and neck squamous cell carcinoma (HNSCC) stratified by HEL308 genotypes.
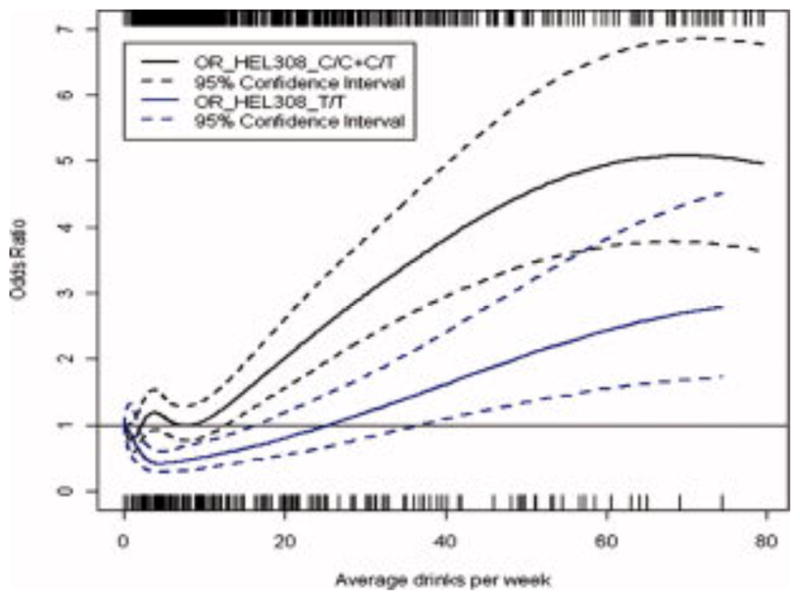
This figure shows the dose-response relationship between alcohol use and HNSCC stratified by HEL308 genotypes (C/C+C/T and T/T). The plot was truncated at 80 drinks per week because few consumers drink greater than that amount. Subjects with T/T genotypes tended to have a lower risk relative to those carrying C/C+C/T genotypes with the same amount of alcohol consumption. A cross between the intervals for the two genotypes was oberved. Likelihood ratio test did not suggest a significant departure from multiplicative interaction.
Figure 2. The association between smoking and HNSCC stratified by HEL308 genotypes.
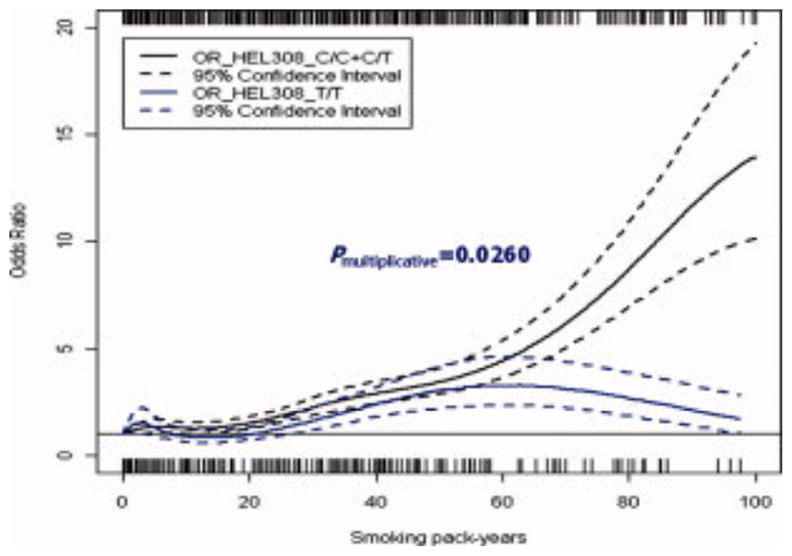
This figure displays the trend of HNSCC risk with the increase of smoking pack years separated by HEL308 genotypes (C/C+C/T and T/T). The plot was truncated at 100 pack years due to the small number of subjects smoking beyond that amount. A marked deviation between the two genotypes was observed beyond 70 smoking pack years. A likelihood ratio test for multiplicative interaction between smoking pack-years and HEL308 genotypes suggested significance (P interaction=0.026).
Figure 3. The association between smoking and HNSCC stratified by ADH1B genotypes.
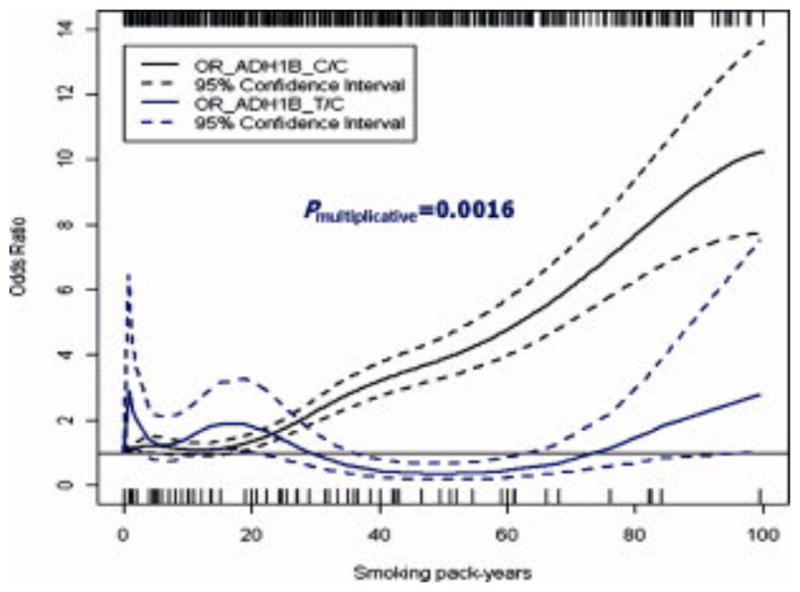
This figure presents the dose-response relationship between smoking and HNSCC risk stratified by ADH1B genotypes (C/C and T/C). The risk of HNSCC increased with increasing smoking pack years among those with ADH1B homozygous wild-type genotype (C/C), whereas a flat trend of HNSCC risk was observed for those with ADH1B heterozygous genotype (T/C). Here, the likelihood ratio test for multiplicative interaction between smoking pack years and ADH1B genotypes suggested a significant departure from multiplicative interaction (P interaction=0.0016).
High-order interaction
To explore the high-order interaction between genes and environmental factors, we performed a CART analysis incorporating both genetic and smoking status variables. Figure 4 displays the tree structure generated using CART analysis. Smoking status was the initial split, suggesting that smoking was a major risk factor for HNSCC. ADH1B subsequently separated the ever smokers into two subgroups (C/C and T/C+T/C). Subsequent splits were FLJ13089 and FLJ35784. The final tree structure contained 5 terminal nodes, representing a range of low- versus high-risk subgroups as defined by the different combination of smoking status and genotypes. Among ever smokers, gene-gene interactions for the three SNPs (ADH1B, FLJ3089 and FLJ35784) were identified. To calculate the ORs as defined by the terminal nodes among ever smokers, we selected ever smokers with ADH1B T/C+T/T genotypes as the reference group. Compared with the reference group, smokers carrying ADH1B wild-type (C/C) and FLJ13089 A/G+A/A genotypes had a highest risk of HNSCC (OR=1.84). The combination of ADH1B wild-type, FLJ13089 G/G and FLJ35784 any genotypes had an attenuated positive association with HNSCC. The permutation test from random forest suggested the high order interaction is reasonable with significance for the overall tree structure (P=0.017). To examine whether this association was driven by smoking status, we fitted the gene-gene interaction and permutation test suggested that this association (with tobacco smoking) was non-significant.
Figure 4. Classification and regression tree (CART) analysis of genetic polymorphisms and smoking status.

This figure displays the tree structure generated using CART analysis. Smoking status was the initial split, and ADH1B subsequently separated the ever smokers into two subgroups (C/C and T/C+T/C). Subsequent splits were FLJ13089 and FLJ35784. The final tree structure contained 5 terminal nodes, representing a range of low- versus high-risk subgroups as defined by the different combination of smoking status and genotypes. Smokers carrying ADH1B wild-type (C/C) and FLJ13089 A/G+A/A genotypes had a highest risk of HNSCC (OR=1.84). The combination of ADH1B wild-type, FLJ13089 G/G and FLJ35784 any genotypes had an attenuated positive association with HNSCC.
Discussion
In this study, we explored the potential gene and environment interactions in HNSCC risk using 19 SNPs identified from a prior GWAS of upper aerodigestive tract cancers. Generally through consortia, GWAS can provide novel genetic variants associated independently with disease. At the same time, the ability to examine gene-gene and gene-environment interaction in these consortia, which include studies using various designs, across heterogeneous populations, and with various measurements of exposures and confounders are limited. Thus, it is important to follow up these GWAS in more homogeneously defined study populations to examine the impact of the environment on the association between genetic variant and disease.
Our results suggested that only FLJ13089 exhibited a significant, main effect, association with HNSCC after Bonferroni adjustment. We also observed that ADH1B and HEL308 interacted with smoking in HNSCC risk. Subanalyses found that the departure from multiplicative interactions between ADH1B and smoking pack-years were consistent across all tumor sites, whereas the departure from multiplicative interaction between HEL308 and smoking was only detected in oral cavity cancers and pharyngeal cancers but not in laryngeal cancers. CART analysis demonstrated the higher order interactions between smoking status, ADH1B, FLJ13089 and FLJ35784 in HNSCC risk.
The ADH1B*2 allele encodes an enzyme approximately 40 times more active in ethanol metabolism to acetaldehyde than the enzyme encoded by the ADH1B*1 allele (14). However, the possible mechanisms behind ADH1B polymorphisms and the increased risk of HNSCC have remained indistinct. Actually, the effect of the fast-metabolizing ADH1B allele has not been definitively established (15, 16). Hashibe et al. (16) showed that subjects carrying ADH1B fast alleles compared with those carrying common allele homozygous genotype have 0.56 times the risk of aerodigestive cancers (95% CI: 0.47–0.66). Consistent with this work, we found a lower risk of HNSCC for subjects possessing ADH1B (T/C+T/T) genotypes compared with those carrying C/C genotypes (OR=0.59; 95% CI: 0.40–0.88). This lower risk was also observed in the initial GWAS (OR=0.69 for UADT, preplication=3×10−10) (submitted). The possible mechanism to explain this association is that subjects with the highly active ADH1B*2 allele rapidly convert ethanol to acetaldehyde, which leads to acetaldehyde accumulation and results in toxic side effects, such as a flushing syndrome with sweating, accelerated heart rate, nausea, and vomiting. Thus, these adverse symptoms may exert a protective effect by altering alcohol use behavior, reducing acute and chronic alcohol consumption leading to protection from alcohol associated cancer development(14). This syndrome is enhanced in Asians as fast alleles are predominant in Asian population (17).
The initial GWAS also showed that the association between ADH1B and HNSCC was observed in never smokers, ever drinkers, but absent in never drinkers, and ever smokers, suggesting these effects are modulated through alcohol exposure rather than smoking (submitted). These results are similar to those obtained by prior studies (16), in which the ADH1B variants have little or no effect on aerodigestive cancer risk among nondrinkers, whereas the protective effect is more apparent among alcohol drinkers with higher alcohol intake, indicating interaction between ADH1B and alcohol consumption. Here, we did not observe an interaction between alcohol use and ADH1B. Instead, departure from multiplicative interaction was identified between smoking pack years and ADH1B. One possible reason is that we lack enough power to detect the interaction between ADH1B and alcohol use due to the small sample size among subjects with T/C and T/T genotypes (fast alleles). About 90% of subjects in our study were Caucasians and the ADH1B*2/2 genotype was rarely observed among Caucasians but more common among Asians(18). In contrast, the ADH1B*1 “slow” allele was very common among Caucasians, with approximately 95 percent having the homozygous ADH1B*1/1 genotype and 5 percent having the heterozygous ADH1B*1/2 genotype (19). On the other hand, when analyzing interaction, we used a continuous measure (packyears) of smoking instead of smoking status (categorical), which provided statistical efficiency. This could be the possible explanation that prior replication studies did not detect an interaction between ADH1B and smoking. Finally, we could not exclude the possibility that the interaction between ADH1B and smoking may actually represent the interaction between ADH1B and alcohol drinking, as alcohol drinking is highly correlated with smoking.
We also found that HEL308 (rs1494961) was associated with HNSCC. Heterozygous C/T genotype was inversely associated with HNSCC compared with homozygous C/C genotypes. To our knowledge, there were no studies focusing on HEL308 and the risk of cancer, especially head and neck cancer. The prior GWAS and its replication studies both suggested HEL308 was significantly associated with HNSCC. The subanalysis in the prior replication studies indicated that the effect of HEL308 on HNSCC risk tended to be more pronounced in younger ages and smokers, which were consistent with our results that HEL308 interacted with smoking in HNSCC risk. HEL308 is a single-stranded DNA-dependent ATPase and DNA helicase (20). Evidence from biochemistry and genetics implicates HEL308 in the early stages of recombination following replication fork arrest and demonstrates a specificity for removal of the lagging strand in model replication forks (21). Therefore, HEL308 is likely involved in DNA repair, recombination, and genome stability. The subsites analysis suggested the interaction between HEL308 and smoking was only present in oral cavity and pharyngeal cancers. This could suggest that HEL308 variation may be related to the incorporation of HPV into the genome, as these are the sites where risk can be mediated by HPV infection.
To explore the high-order interaction in HNSCC risk, we applied CART analysis. A multifactor dimensionality reduction (MDR) approach was also employed to replicate the results produced by CART. We obtained the same results by using these two methods. The CART analysis exhibited the existence of gene-smoking and gene-gene interactions in HNSCC risk. The findings showed that smokers carrying ADH1B (C/C) wild-type genotype, FLJ13089 (A/G+A/A) variant genotypes have highest risk of HNSCC compared with smokers carrying ADH1B (T/C+T/T) variant genotypes. The mechanisms underlying these high-order interactions among genetic polymorphisms and smoking in modulating HNSCC remain to be elucidated. Few studies reported these gene-gene and gene-environment interactions for HNSCC. The prior replication studies failed to detect any gene-gene interactions. The possible reasons could be that the combined samples from different studies and the use of traditional logistic regression models made the detection of these higher-order interactions difficult.
The CART results were consistent with our two-way interaction, which also suggested that ADH1B interacted with smoking in HNSCC risk. Although CART analysis did not identify the interaction between smoking and HEL308 on HNSCC, it is possible that the magnitude of HEL308 and smoking interaction is not as strong as the interaction between ADH1B and smoking. It could also be that HEL308 is not related to other genes (such as FLJ13089 and FLJ35784). Another possible reason is the small sample size in the terminal node restricting the ability to detect further modest interaction. Given the small sample size in each node, the results should be interpreted with caution. Further validations from independent populations are necessary to confirm these results.
Interestingly, our study confirmed another locus rs4767364 (FLJ13089) at 12q24.13a reported in the prior GWAS. The chromosome regions: 12q24 was found to be associated with type I diabetes in the Wellcome Trust Case Control Consortium (WTCCC) primary genome-wide association scan and its follow-up analysis (7, 22). It appears that few studies reported the role of this gene on disease. Further studies are needed to replicate this result. Our study also identified other two potential genes (ASPH, and FHOD3) that could be associated with HNSCC. ASPH (aspartyl beta-hydroxylase) is a highly conserved enzyme that hydroxylates epidermal growth factor-like domains in transformation-associated proteins (23). It is unregulated in many human malignancies and is thought to play an important role in cell motility or invasiveness (24). The polymorphisms in ASPH have been studied for their associations with other malignancies. One study reported overexpression of ASPH plays a role in the development and progression of hepatocellular carcinoma (25). FHOD3 (formin homology 2 domain containing 3) also known as FHOS2 or FLJ22717 was reported to has a function in regulation of the actin cytoskeleton (26), which is vital for many cellular processes, including movement, adhesion, polarity establishment, intracellular trafficking, and modulation of mechanical strength. This gene was only reported to be associated with methotrexate polyglutamate accumulation in leukemia (27, 28).
Our study could be susceptible to a number of limitations. First, small sample size limited the ability to detect the associations between genes and HNSCC and the interactions especially high-order interactions. Also, this was a retrospective case-control study, and so issues of selection bias and recall bias as well as misclassification were unavoidable. However, the hypotheses tested in this study were genotype-driven rather than environment-driven, and it is unlikely to have improperly selected individuals related to genotype. Genotyping was performed with strict quality control procedures in place. In addition, this is a population-based study, and our comparisons with the Massachusetts Cancer Registry suggest that we are performing equal to or better than the registry at identifying incident cases of the disease. Recall bias would be a concern for the assessment of smoking and alcohol consumption, but our data regarding smoking and alcohol drinks were collected decade-specifically, which to some extent reduces misclassification. As a result, these biases in our study would not have a great influence on our findings.
In summary, our study suggested that ADH1B and HEL308 are associated with HNSCC risk, and both significantly modified the association between smoking and HNSCC. The findings in our study inform understanding of the genetic basis of HNSCC and provide insights relevant to the pursuit of a post-GWAS in general. Due to the moderate sample size of the current study, these findings (especially the findings obtained for the high-order interaction) merits further replications in different population with large sample size.
References
- 1.Jemal A, Siegel R, Ward E, Hao Y, Xu J, Thun MJ. Cancer statistics, 2009. CA Cancer J Clin. 2009;59:225–49. doi: 10.3322/caac.20006. [DOI] [PubMed] [Google Scholar]
- 2.Sturgis EM, Wei Q. Genetic susceptibility--molecular epidemiology of head and neck cancer. Curr Opin Oncol. 2002;14:310–7. doi: 10.1097/00001622-200205000-00010. [DOI] [PubMed] [Google Scholar]
- 3.Cheng L, Eicher SA, Guo Z, Hong WK, Spitz MR, Wei Q. Reduced DNA repair capacity in head and neck cancer patients. Cancer Epidemiol Biomarkers Prev. 1998;7:465–8. [PubMed] [Google Scholar]
- 4.Ho T, Wei Q, Sturgis EM. Epidemiology of carcinogen metabolism genes and risk of squamous cell carcinoma of the head and neck. Head Neck. 2007;29:682–99. doi: 10.1002/hed.20570. [DOI] [PubMed] [Google Scholar]
- 5.Varela-Lema L, Taioli E, Ruano-Ravina A, et al. Meta-analysis and pooled analysis of GSTM1 and CYP1A1 polymorphisms and oral and pharyngeal cancers: a HuGE-GSEC review. Genet Med. 2008;10:369–84. doi: 10.1097/GIM.0b013e3181770196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Asakage T, Yokoyama A, Haneda T, et al. Genetic polymorphisms of alcohol and aldehyde dehydrogenases, and drinking, smoking and diet in Japanese men with oral and pharyngeal squamous cell carcinoma. Carcinogenesis. 2007;28:865–74. doi: 10.1093/carcin/bgl206. [DOI] [PubMed] [Google Scholar]
- 7.Genome-wide association study of 14,000 cases of seven common diseases and 3, 000 shared controls. Nature. 2007;447:661–78. doi: 10.1038/nature05911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Applebaum KM, Furniss CS, Zeka A, et al. Lack of association of alcohol and tobacco with HPV16-associated head and neck cancer. J Natl Cancer Inst. 2007;99:1801–10. doi: 10.1093/jnci/djm233. [DOI] [PubMed] [Google Scholar]
- 9.Peters ES, McClean MD, Liu M, Eisen EA, Mueller N, Kelsey KT. The ADH1C polymorphism modifies the risk of squamous cell carcinoma of the head and neck associated with alcohol and tobacco use. Cancer Epidemiol Biomarkers Prev. 2005;14:476–82. doi: 10.1158/1055-9965.EPI-04-0431. [DOI] [PubMed] [Google Scholar]
- 10.Peters ES, McClean MD, Marsit CJ, Luckett B, Kelsey KT. Glutathione S-transferase polymorphisms and the synergy of alcohol and tobacco in oral, pharyngeal, and laryngeal carcinoma. Cancer Epidemiol Biomarkers Prev. 2006;15:2196–202. doi: 10.1158/1055-9965.EPI-06-0503. [DOI] [PubMed] [Google Scholar]
- 11.McKay J, Truong T, Gaborieau V, et al. A genome-wide association study of upper aerodigestive tract cancers conducted within the INHANCE consortium. PLoS genetics. 2011 doi: 10.1371/journal.pgen.1001333. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gregory M, Ulmer H, Pfeiffer KP, Lang S, Strasak AM. A set of SAS macros for calculating and displaying adjusted odds ratios (with confidence intervals) for continuous covariates in logistic B-spline regression models. Comput Methods Programs Biomed. 2008;92:109–14. doi: 10.1016/j.cmpb.2008.05.004. [DOI] [PubMed] [Google Scholar]
- 13.Breiman L, Friedman JH, Olshen RA, Stone CJ. Classification and Regression Trees. Belmont, CA: Wadsworth; 1984. [Google Scholar]
- 14.Seitz HK, Becker P. Alcohol metabolism and cancer risk. Alcohol Res Health. 2007;30:38–41. 4–7. [PMC free article] [PubMed] [Google Scholar]
- 15.Hashibe M, Boffetta P, Zaridze D, et al. Evidence for an important role of alcohol- and aldehyde-metabolizing genes in cancers of the upper aerodigestive tract. Cancer Epidemiol Biomarkers Prev. 2006;15:696–703. doi: 10.1158/1055-9965.EPI-05-0710. [DOI] [PubMed] [Google Scholar]
- 16.Hashibe M, McKay JD, Curado MP, et al. Multiple ADH genes are associated with upper aerodigestive cancers. Nat Genet. 2008;40:707–9. doi: 10.1038/ng.151. [DOI] [PubMed] [Google Scholar]
- 17.Lee CH, Lee JM, Wu DC, et al. Carcinogenetic impact of ADH1B and ALDH2 genes on squamous cell carcinoma risk of the esophagus with regard to the consumption of alcohol, tobacco and betel quid. Int J Cancer. 2008;122:1347–56. doi: 10.1002/ijc.23264. [DOI] [PubMed] [Google Scholar]
- 18.Brennan P, Lewis S, Hashibe M, et al. Pooled analysis of alcohol dehydrogenase genotypes and head and neck cancer: a HuGE review. Am J Epidemiol. 2004;159:1–16. doi: 10.1093/aje/kwh003. [DOI] [PubMed] [Google Scholar]
- 19.Borras E, Coutelle C, Rosell A, et al. Genetic polymorphism of alcohol dehydrogenase in europeans: the ADH2*2 allele decreases the risk for alcoholism and is associated with ADH3*1. Hepatology. 2000;31:984–9. doi: 10.1053/he.2000.5978. [DOI] [PubMed] [Google Scholar]
- 20.Marini F, Wood RD. A human DNA helicase homologous to the DNA cross-link sensitivity protein Mus308. J Biol Chem. 2002;277:8716–23. doi: 10.1074/jbc.M110271200. [DOI] [PubMed] [Google Scholar]
- 21.Woodman IL, Bolt EL. Molecular biology of Hel308 helicase in archaea. Biochem Soc Trans. 2009;37:74–8. doi: 10.1042/BST0370074. [DOI] [PubMed] [Google Scholar]
- 22.Joo J, Kwak M, Ahn K, Zheng G. A Robust Genome-Wide Scan Statistic of the Wellcome Trust Case-Control Consortium. Biometrics. 2009 doi: 10.1111/j.1541-0420.2009.01185.x. [DOI] [PubMed] [Google Scholar]
- 23.Xian ZH, Zhang SH, Cong WM, Yan HX, Wang K, Wu MC. Expression of aspartyl beta-hydroxylase and its clinicopathological significance in hepatocellular carcinoma. Mod Pathol. 2006;19:280–6. doi: 10.1038/modpathol.3800530. [DOI] [PubMed] [Google Scholar]
- 24.Lavaissiere L, Jia S, Nishiyama M, et al. Overexpression of human aspartyl(asparaginyl)beta-hydroxylase in hepatocellular carcinoma and cholangiocarcinoma. J Clin Invest. 1996;98:1313–23. doi: 10.1172/JCI118918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.de la Monte SM, Tamaki S, Cantarini MC, et al. Aspartyl-(asparaginyl)-beta-hydroxylase regulates hepatocellular carcinoma invasiveness. J Hepatol. 2006;44:971–83. doi: 10.1016/j.jhep.2006.01.038. [DOI] [PubMed] [Google Scholar]
- 26.Kanaya H, Takeya R, Takeuchi K, Watanabe N, Jing N, Sumimoto H. Fhos2, a novel formin-related actin-organizing protein, probably associates with the nestin intermediate filament. Genes Cells. 2005;10:665–78. doi: 10.1111/j.1365-2443.2005.00867.x. [DOI] [PubMed] [Google Scholar]
- 27.French D, Yang W, Cheng C, et al. Acquired variation outweighs inherited variation in whole genome analysis of methotrexate polyglutamate accumulation in leukemia. Blood. 2009;113:4512–20. doi: 10.1182/blood-2008-07-172106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Amos CI, Wu X, Broderick P, et al. Genome-wide association scan of tag SNPs identifies a susceptibility locus for lung cancer at 15q25. 1. Nat Genet. 2008;40:616–22. doi: 10.1038/ng.109. [DOI] [PMC free article] [PubMed] [Google Scholar]