

Abstract
Myeloid-derived suppressor cells (MDSC) are one of the major factors limiting the efficacy of immune therapy. In a clinical trial of patients with extensive stage small cell lung cancer (SCLC), we tested the possibility that targeting MDSC can improve the induction of immune responses by a cancer vaccine. Forty-one patients with extensive stage SCLC were randomized into three arms: arm A—control, arm B—vaccination with dendritic cells transduced with wild-type p53, and arm C—vaccination in combination with MDSC targeted therapy with all-trans-retinoic acid (ATRA). Interim results of the ongoing clinical trial are presented. Pre-treatment levels of MDSC populations in patients from all three arms were similar. Vaccine alone did not affect the proportion of MDSC, whereas in patients treated with ATRA, the MDSC decreased more than twofold (p = 0.02). Before the start of treatment, no patients had detectable p53-specific responses in IFN-γ ELISPOT. Sequential measurements did not show positive p53 responses in any of the 14 patients from arm A. After immunization, only 3 out of 15 patients (20 %) from arm B developed a p53-specific response (p = 0.22). In contrast, in arm C, 5 out of 12 patients (41.7 %) had detectable p53 responses (p = 0.012). The proportion of granzyme B-positive CD8+ T cells was increased only in patients from arm C but not in arm B. Depletion of MDSC substantially improved the immune response to vaccination, suggesting that this approach can be used to enhance the effect of immune interventions in cancer.
Keywords: Cancer vaccine, Myeloid-derived suppressor cells, Small cell lung cancer, All-trans-retinoic acid, Dendritic cells
Introduction
The efforts in cancer immune therapy are focused on increasing the frequency of CTL-mediated tumor destruction by applying cancer vaccines, adoptively transferred antigen-specific T cells, inhibitors of checkpoint blockades (CTLA4, PD1), or other immune therapeutics [1]. However, even in tumor-bearing mice, once tumors are established, these interventions have relatively limited efficacy, and the rate of clinical responses in cancer patients, although encouraging, remains relatively low [2, 3].
It is now fully appreciated that tumor-specific immune responses in cancer are inhibited. Large numbers of different factors have been implicated in this process, including regulatory T cells (Treg), myeloid cells, various soluble factors and cytokines, inhibitory molecules expressed by immune and tumor cells, etc. [4–6]. Among these factors, myeloid-derived suppressor cells (MDSC) play a prominent role. MDSC are a heterogeneous group of pathologically activated immature myeloid cells and myeloid precursors with potent immune suppressive activity [7–10]. Two major groups of MDSC have been identified: polymorphonuclear (PMN) and mononuclear (M) MDSC. These cells share common features (immature myeloid cells with immune suppressive activity) but differ in morphology, phenotype, and mechanisms of suppressive activity [8]. In cancer patients, these MDSC have a partially overlapping phenotype that can be identified using several markers, which largely depends on the type of tumor. In most tumors, immune suppressive MDSC are defined as Lin− HLA-DR− CD33+ or CD33+CD14− CD11b+ cells that can be further sub-divided into CD15+ PMN-MDSC and CD15− M-MDSC. In some tumors, most notably melanoma, M-MDSC are defined as CD14+HLA-DR−/lo [11, 12]. MDSC are characterized by potent immune suppressive activities in both antigen-specific and non-specific experimental systems.
The association between tumor progression and the presence of MDSC in cancer patients was demonstrated in a number of studies. In patients with solid tumors, a significant correlation between circulating MDSC and clinical cancer stage was observed. Among stage IV patients, those with extensive metastatic tumor burden had the highest percent and absolute number of MDSC [13]. Patients with lower levels of circulating MDSC at baseline and on the last cycle of chemotherapy had a significantly higher probability of a pathologic complete response [14]. Increased circulating MDSC correlated with clinical stage and pathological grade in patients with bladder cancer [15, 16] and pancreatic cancer [17]. Recently, there was the first report linking clinical response to vaccination to accumulation of MDSC in patients with renal cell cancer [18]. Experiments in mice demonstrated that elimination of MDSC with antibodies or different compounds could substantially improve an antitumor immune response, which resulted in antitumor effect [8]. However, no data are available testing the hypothesis that depletion of MDSC can improve the effect of cancer vaccines or other immune therapeutic modalities in cancer patients.
In this study, we addressed this question in the settings of a randomized phase II trial in patients with extensive stage small cell lung cancer (SCLC). As a vaccine, we used dendritic cells (DC) transduced with an adenovirus expressing p53 (Ad.p53) [19]. This vaccine was used in our previous phase I/II trial of patients with SCLC and demonstrated immunological activity with induction of p53-specific response in almost 50 % of the treated patients [20]. Moreover, the immune response to vaccination was associated with substantial objective clinical response to second-line chemotherapy in those patients. To target MDSC, we chose to use all-trans-retinoic acid (ATRA). In previous studies, ATRA showed a direct effect on MDSC by causing apoptosis of PMN-MDSC and differentiating M-MDSC to mature myeloid cells (macrophages and DC) [21–23]. Treatment of tumor-bearing mice with ATRA substantially decreased the presence of MDSC in spleens [21, 24–26]. Treatment of patients with renal cell carcinoma showed a substantial reduction in the level of MDSC in peripheral blood [27].
The current study was designed to address the question of whether the combination of DC-p53 with ATRA improved the clinical outcome of the disease. An optimal two-stage design with early termination rule was used to compare the effect of ATRA on clinical outcome. Although this clinical trial is still ongoing, the first stage is completed. This allowed us to perform interim immunological analysis of the data and evaluate the effect of MDSC depletion on induction of antigen-specific immune responses in cancer patients.
Materials and methods
Trial design, patient selection, and treatment
Patients with extensive stage SCLC were enrolled on this study. The trial was registered at The ClinicalTrials.gov as NCT00617409. All patients provided a written informed consent, and the treatment protocol was approved by University of South Florida Institutional Review Board. The treatment schema is shown in Fig. 1. Patients received 4–6 cycles of a standard platinum/etoposide regimen as first-line chemotherapy. Patients with stable disease (SD) or better with Eastern Cooperative Oncology Group (ECOG) performance status of 0–2, and adequate organ function were screened for initial registration approximately 4–6 weeks after the completion of first-line chemotherapy. Prophylactic cranial irradiation (PCI) was permitted and was administered at the discretion of treating oncologist. Eligible patients were randomized at 1:1:1 ratio to one of three arms: arm A (standard of care control patients or observation), arm B: (patients treated with p53 vaccine only), or arm C (patients treated with vaccine in combination with ATRA) at the time when clinical response to initial chemotherapy was documented. The primary objective was to evaluate the efficacy of the two experimental regimens. An optimal two-stage design was used that has been successfully applied in randomized phase II trials to rank experimental agents [28, 29]. The control arm was used to examine consistency with historical control data to assure that patients entering the phase II trial were comparable to historical controls.
Fig. 1.

Treatment schema. Schema for patients’ enrollment, treatment, and analysis
Each vaccine consisted of 2–5 × 106 autologous DC expressing p53 after infection with adenovirus encoding full-length wild-type p53 gene (Ad-p53 DC). Cells were injected intradermally, at two-week intervals three times (three vaccine doses). Patients were restaged approximately 2 weeks after the 3rd vaccine dose. Patients without disease progression (PD) underwent a second leukapheresis and were then vaccinated 3 more times at 4-week intervals. Patients on arm C also received 150 mg/m2 ATRA for 3 days beginning 1-day prior to each vaccine. This scheduling was based on our previous data [27] that demonstrated persistence of the ATRA effect on MDSC for a minimum of 2 weeks. All patients were followed until evidence of PD at which time, they received salvage or second-line therapy with single agent paclitaxel.
Preparation and administration of DC vaccine
DC were prepared from mononuclear cells (MNC) collected by leukapheresis in the Cell Therapy Core at H. Lee Moffitt Cancer Center. Preparation of the DC and their characterization was described previously [20]. Briefly, after thawing, MNC were placed in X-VIVO-15 medium (Biowhittaker, Walkersville, MD, USA) in tissue culture flasks at a concentration of 1.3–1.7 × 106 cells per cm2. After 2-h culture, non-adherent cells were removed and the flasks were recharged with X-VIVO-15 medium supplemented with 5 ng/ml GM-CSF (Immunex), and 5 ng/ml IL-4 (R&D Systems, Minneapolis, MN, USA). The flasks were incubated for 48 h, at which time additional cytokine supplemented medium was added to the flasks. The flasks were then incubated for an additional 72 h. The non-adherent and loosely adherent cells were collected and infected with Ad.p53 [20] and incubated for 2 h after which X-VIVO-15 medium was added for a 106 cells/ml concentration, and cells were incubated for an additional 46 h. The optimal viral particle to cell ratio used was 15,000:1 as determined for our previous study [20]. The vaccine release criteria included (a) negative Gram’s staining; (b) negative Mycoplasma test by PCR analysis; (c) maximum endotoxin concentration of 5 EU/mL; and (d) a mature DC-p53 expressing phenotype. DC phenotype was defined as lineage (CD3, CD14, CD19, CD20, CD56) negative, HLA-DR positive, CD86 positive, and p53 positive cells. Intracellular staining for p53 was performed using a kit from Caltag, Burlingame, CA. DC vaccines in 1 ml were injected intradermally into 4 separate sites (0.25 ml at each site) in bilateral proximal upper and lower extremities (in the regions of the axillary and inguinal nodal basins) three times after the baseline blood samples were drawn and at 2-week intervals.
Evaluation of immune responses
Blood samples were collected in heparin tubes and processed immediately usually within 1 h. For MNC isolation, a density gradient protocol was used (Ficoll-Paque™ PLUS, GE Healthcare Biosciences, PA). MNC were collected from patients at different time points during the treatment (Fig. 1) and kept frozen in liquid nitrogen. All samples from one patient were analyzed simultaneously to reduce inter-experimental variability. MNC were thawed, incubated overnight in complete medium (RPMI-1640 supplemented with antibiotics and 10 % FBS) and then used in experiments. Cell viability was greater than 80 %. T cell responses were assessed using IFN-γ ELISPOT after the addition of a recombinant canarypox virus (ALVAC) containing wild-type p53 or empty vector (obtained from Aventis Pasteur, Toronto, Canada) to accomplish infection of APC in the culture and incubated for 48 h. Empty ALVAC virus served as a control. The initial infection step was performed in serum-free media (supplemented with cytokines) for 90 min after which complete media was added. In the IFN-γ ELISPOT assay, 2 × 105 MNC were seeded in triplicates or quadruplicates in 96-well plates pre-coated with an anti-IFN-γ antibody. To ascertain that T cells were functionally competent, for each sample, additional controls were prepared (unstimulated or PHA (5 μg/ml) stimulated cells), and the plate was further incubated for 36 h. The number of IFN-γ producing cells was evaluated as described previously [20] using an automated ELISPOT reader (Cellular Technology, Ltd, OH).
After start of the trial, we developed a more effective method of T cell stimulation, and it was used in addition to the one described above for most of the patients. This method included the generation of DC from MNC in vitro from one of the patients’ samples with GM-CSF and IL-4 using cytokines and serum-free media from CellGenix Technologie Transfer GmbH, Freiburg, Germany. DC were infected with ALVAC-p53 or ALVAC-control (20,000 viral particles per DC) and used for T cell stimulation at DC:T cell ratio 1:10. IFN-γ ELISPOT assays were performed as described above.
Criteria used to determine the presence of immune responses
An individual patient was considered a responder if at any time point the response in the IFN-γ ELISPOT assay was higher than 30 spots per 2 × 105 and the response to ALVAC p53 was more than 2 SD higher than the response to corresponding ALVAC-control at the same time point and 2 SD higher than the corresponding response at the base line (before start of the treatment).
Analysis of cell phenotype
Cell phenotype was evaluated by multicolor flow cytometry using a LSR II flow cytometer and monoclonal antibodies obtained from Becton–Dickinson. The following antibodies were used: CD4-Alexa 700; CD25-PE; CD127-Alexa 647; CD45RA-PerCP-Cy5.5; Foxp3-Pacific Blue (BioLegend, San Diego, CA); CD3-FITC (for the Treg panel); CCR7-PE-Cy7 (for the Treg, and CTL panels); CD3-PE; CD14-PE; CD19-PE; CD56-PE (as lineage-PE); HLA-DR-APC; CD33-PE-Cy7; CD86-FITC; CD11b-APC. Additional antibodies were used for the CTL panel: CD107a-PE; IFN-γ-APC; granzyme B-FITC; CD3-PerCP; CD8-APC-H7; CD4-Pacific Blue.
The following combinations of antibodies were used to identify cell populations:
Total population of DC: Lineage− (Lin) (CD3, 14, 19, 56) HL-DR+; Mature DC: Lin− HLA-DR+CD86+; MDSC: Lin− HLA-DR− CD33+ and CD11b+ CD14− CD33+; Treg: CD4+CD25+Foxp3+; Cytokine secreting CTLs: CD3+ CD8+ (IFN-γ+, or granzyme B+).
Dead cells were eliminated from the analysis by using DAPI staining, and in samples with intracellular staining by using Live/Dead Yellow solution (Invitrogen, Carlsbad, CA).
Intracellular cytokine staining
For the antigen-specific CD8+ and CD4+ T cell, characterization cells were cultured for 24 h in the presence of ALVAC-p53 or ALVAC-control. Golgi-stop (0.7 μl/ml) and Golgi-plug (1μl/ml) (monensin and brefeldin from BD Biosciences) were added to each well. After 6 h of incubation at 37 °C in a 5 % CO2 incubator, cells were harvested and the surface and intracellular staining was performed. For all intracellular staining steps, the fixation and permeabilization reagents used were from eBiosciences according to the manufacturer’s instructions. To determine p53-specific responses, the proportion of cytokine-producing cells cultured in the presence of ALVAC-control was subtracted from the proportion of cytokine-positive cells cultured with ALVAC-p53.
Statistical analysis
An optimal two-stage design was used in this study [28]. From historical and preliminary data, we considered <25 % response rate to second-line (salvage) chemotherapy as not warranting further study [30]. We were interested in at least 25 % (25 vs. 50 %) improvement in treatment efficacy for arms B and C. Therefore, 50 % response rate was used as a promising result to pursue further study. Using this design with a 5 % type I error rate and 20 % type II error rate, 9 patients were to be enrolled in the first stage of the trial with 5 % rejection error. If 2 or fewer of the 9 patients responded, the treatment was to be stopped. If 3 or more patients showed a response, 15 additional patients (a total of 24 patients per group) would be enrolled.
We utilized GraphPad Prism software for statistical analyses. Antigen-specific immune responses to immunization were analyzed by descriptive statistics. The Wilcoxon Mann–Whitney test was used to determine a relationship between two groups with continuous variables. Univariate associations between frequency of responders and groups were analyzed by Fisher’s exact test considering small sample sizes. Two-sided tests were used for all calculations. The statistical significance for all analyses was determined with p < 0.05.
Results
Fifty-six patients were enrolled to stage one of the trial: 18 to arm A (control), 19 to arm B (vaccine alone), and 19 to arm C (vaccine plus ATRA). Patient dropouts and rapid disease progression preventing patients from completing at least one round of salvage (paclitaxel) chemotherapy were the reason for the larger number of patients enrolled in stage one. Objective clinical response to paclitaxel was observed only in 2 patients in arm B. Therefore, arm B was closed due to the lack of efficacy rule. In contrast, patients in arm C met the criteria for further investigation and arm C continued to stage II.
Accrual to the clinical trial continues to address the clinical endpoints; however, sufficient patients were treated to assess the immunological endpoints of the trial. Therefore, we report the analysis of immunological parameters for 41 patients (14 to arm A, 15 to arm B, and 12 to arm C) for whom paired blood samples before and after completion of three vaccinations were available. Patients’ clinical characteristics were mostly comparable in all arms as described in Table 1. Peripheral blood was collected before the start of the first vaccination and 1–2 weeks after the completion of three rounds of vaccination (arms B, C) and at similar time points after enrollment in arm A. In all cases, samples were collected before the start of second-line chemotherapy.
Table 1.
Clinical characteristics of patients evaluated immunologically in the study
| Arm A | Arm B | Arm C | Total | |
|---|---|---|---|---|
| Subtotal | 14 | 15 | 12 | 41 |
| Gender | ||||
| M | 8 | 6 | 9 | 23 |
| F | 6 | 9 | 3 | 18 |
| Age | ||||
| Median | 63 | 63 | 63 | 63 |
| Range | 48–73 | 51–74 | 48–73 | 48–74 |
| Race | ||||
| White | 12 | 15 | 12 | 39 |
| AA/other | 1/1 | 0/0 | 0/0 | 1/1 |
| Performance status | ||||
| 0 | 2 | 4 | 3 | 9 |
| 1 | 12 | 11 | 9 | 32 |
| First-line chemotherapy | ||||
| ≤4 cycles | 3 | 3 | 2 | 8 |
| >4 cycles | 11 | 12 | 10 | 33 |
| Leukapheresis | ||||
| Total | 22 | 16 | 38 | |
| 0/1 | 1/14 | 0/12 | 1/26 | |
| 2 | 4 | 2 | 6 | |
| Vaccinations | ||||
| Median | 3 | 3 | 3 | |
| Range | 0–6 | 1–6 | 0–6 | |
| <3/3/>3 | 3/8/4 | 2/8/2 | 5/16/6 | |
| Total | 44 | 39 | 83 | |
First, we asked whether treatment with ATRA resulted in a decrease of MDSC in the patients. In many tumor types, MDSC are defined as Lin− HLA-DR− CD33+ or CD11b+CD14− CD33+ cells [8, 31]. We previously demonstrated that these cells have potent immune suppressive activity [32] and observed accumulation of these cell populations in patients with SCLC [20]. Gating strategy used in this study to define these cells is shown in Fig. 2a. Pre-treatment levels of MDSC populations in patients from all three arms were similar (p > 0.07). Treatment of patients from arm B with vaccine alone did not affect the proportion of MDSC (Fig. 2b, c), whereas in patients treated with ATRA the presence of Lin− HLA-DR− CD33+ MDSC decreased more than twofold (p = 0.02) (Fig. 2b). The proportion of CD11b+CD14− CD33+ MDSC decreased less dramatically but also significantly (p = 0.03) (Fig. 2c). No significant differences were observed in the proportion of conventional DC and Treg (Fig. 2d, e).
Fig. 2.

ATRA reduced presence of MDSC in patients with SCLC. Peripheral blood of patients was evaluated before and 1–2 weeks after completion of vaccination. For arm A, blood was collected during enrollment. a Gating strategy used to define cell populations shown in figures (b–e). In all analysis, doublets and dead cells were excluded from the calculation. For gating, matched isotypes controls were used. b–e Cell populations indicated on the graphs were analyzed among live PBMC. Values for individual patients are shown. p values for differences between pre- and post-treatment samples are shown
Next, we evaluated p53-specific immune responses using IFN-γ ELISPOT assays and the criteria, described in Methods. Prior to the start of treatment, no patients had detectable p53-specific responses. Consecutive measurements did not show positive p53 responses in any of the 14 patients from arm A. After three immunizations, 3 out of 15 tested patients (20 %) from arm B met the criteria for a p53-specific immune response, which was not significantly different from arm A (p = 0.22). In contrast, 5 out of 12 patients (41.7 %) in arm C had detectable p53 responses, which was significantly higher than in control group (p = 0.012) (Fig. 3a).
Fig. 3.

Association between treatment with ATRA and immune response to vaccination. a The proportion of patients that developed p53-specific immune response after completion of vaccination; b–d The proportion of cytokine-producing CD4+ or CD8+ T cells in patients treated with vaccine with and without ATRA. The graphs show intracellular cytokines values calculated within live CD4+ or CD8+ T cells. For each parameter, the values of samples stimulated with control virus (ALVAC-c) were subtracted from the values of samples incubated with ALVAC-p53 virus. p values for differences between pre- and post-treatment samples are shown. b IFN-γ+CD8+ T cells, c IFN-γ+CD4+ T cells; d GrzB+CD8+ T cells
The proportion of p53-specific IFN-γ positive cells CD4+ and CD8+ T cells was evaluated using intracellular cytokine staining. Prior to the treatment, no statistically significant differences between the arms were seen (p > 0.07) (Fig. 3b, c). The immunization increased the proportion IFN-γ positive CD8+ (Fig. 3b) and CD4+ (Fig. 3c) T cells. The proportion of granzyme B (GrzB) positive CD8+ T cells was increased only in patients from arm C but not from arm B (Fig. 3d).
Further, we assessed the kinetics of immune responses in 8 p53-responsive patients. In three patients, only two time points (before and after vaccination) were available, and in one patient, the immune response disappeared after the start of chemotherapy. In two patients, the immune response generated by the vaccine persisted for more than a year, and in other two patients, it lasted 6–8 months. In all these cases, the p53-specific response declined without chemotherapy (patients had no progressive disease and were not treated with second-line chemotherapy) (Fig. 4).
Fig. 4.
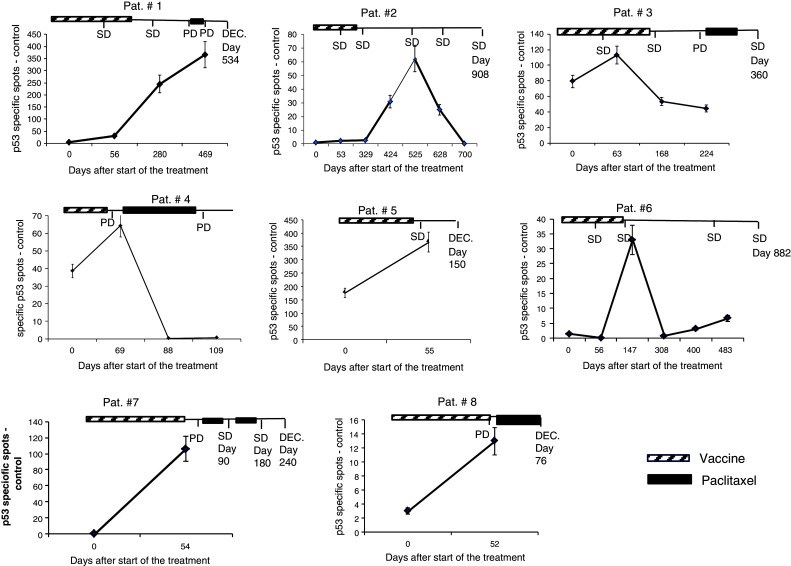
The kinetic of immune response to vaccination. P53-specific IFN-γ ELISPOT in patients who developed immune response to vaccination. SD stable disease, PD progressive disease. The days were counted from start of the treatment. The last day when information was available is shown on the graph. DEC designates patient deceased
Discussion
Among many factors involved in negative regulation of immune responses in cancer, MDSC play a very prominent role due to their ability to substantially expand and inhibit immune responses in tumor-bearing hosts. This suggested that targeting MDSC could be an attractive therapeutic strategy.
There are currently a number of drugs that have a toxic effect on MDSC [8]. ATRA was historically the first therapeutic compound that was used to target MDSC [22]. Its use was based on the initial observation that in patients with solid tumors, MDSC comprised of immature myeloid cells with some features of promyelocytes [33]. The promyelocytic nature of human MDSC was recently confirmed in patients with hematologic malignancies [34]. ATRA exerts its effect via binding in the nucleus to specific retinoic acid receptors (RAR) α, β, or γ. These RARs can bind to one of the retinoid X receptors (RXRs) α, β, or γ, and then in the presence of retinoic acid (RA), this complex binds to DNA and activates transcription of numerous RA response genes (reviewed in [35]). ATRA was known to have clinical activity in patients with acute promyelocytic leukemia (APL). APL is associated with a chromosomal translocation involving chromosomes 15 (PML gene) and 17 (RARα gene), which results in the expression of PML-RARα fusion protein. Treatment of APL with ATRA displaces the chimeric PML-RARα protein and restores PML and RARα overcoming the differentiation block and committing APL cells to differentiation.
In previous studies, ATRA showed a direct effect on MDSC, causing apoptosis of PMN-MDSC and differentiating M-MDSC to mature myeloid cells [21–23, 33]. The effect of ATRA on MDSC was linked to up-regulation of glutathione synthase and glutathione (GSH) in these cells. GSH dampened the level of reactive oxygen species (ROS) in MDSC and thus promoted their differentiation [36]. ATRA has also been shown to improve the differentiation of DC in cancer patients [37] and to drive the differentiation of monocytes into DC-like cells [38]. Treatment of tumor-bearing mice with ATRA substantially decreased the number of MDSC in spleens and enhanced the effect of cancer vaccines [21, 24–26]. Treatment of patients with renal cell carcinoma showed a substantial reduction in the level of MDSC in peripheral blood [27].
Previously, in a non-randomized phase II trial, we demonstrated that p53 vaccine alone had rather limited antitumor effect. However, patients who were treated with second-line chemotherapy at progression demonstrated remarkably strong clinical response when compared to historic controls [20]. These results contributed to the development of a new paradigm, suggesting that immune therapy could augment the effect of chemotherapy [39–41]. The current trial was designed to test this concept prospectively in newly diagnosed patients in a randomized trial. Patients that progressed after vaccine were treated with second-line chemotherapy. This clinical trial included early stoppage rule, which required assessment of clinical response to chemotherapy when at least 9 patients in each treatment group completed at least one round of chemotherapy. Not all patients that were treated with vaccine received the second-line chemotherapy. Some patients had stable disease or better after completion of the vaccination that persists, some patients progressed rapidly and could not be treated, while some patients refused further treatment. Therefore, a total of 56 patients were enrolled in first stage of the trial. Patients in arm C (vaccine + ATRA) met the response criteria that warranted continuation to the second stage. The response of patients on arm B (vaccine alone), which was 20 %, did not meet the response criteria, and this arm was discontinued. Stage two of the trial is currently under way. However, the completed portion of the trial allowed us to address the question whether ATRA has an impact on the development of p53-specific immune responses.
This study was not designed to power a formal comparison between the two treatment arms (B and C) because it will require a much larger sample size. Because of this limitation, we used the “pick the winner” strategy as an alternative to prioritize the treatment arms [29, 42]. In our study, it was designed to evaluate the efficacy of each of the two treatment arms. This study design has several features, such as randomization to reduce bias and to allow a greater degree of comparability, as well as to allow for termination of an ineffective treatment arm earlier.
There are several major populations of MDSC currently defined in cancer patients. Two of them, Lin− HLA-DR− CD33+ and CD14− CD11b+CD33+, were increased in patients with lung cancer. We previously showed that these populations are indeed immune suppressive MDSC [32]. It is important to point out that more than 60 % of MDSC are lost during freezing/thawing of the samples (data not shown). This may explain relatively small proportion of MDSC detected in this study. This was consistent with recent reports demonstrating that granulocytic (PMN) population of MDSC is cryosensitive [43, 44]. Despite these obvious limitations, the proportion of MDSC in frozen samples of peripheral blood from cancer patients showed correlation with clinical stage of the diseases, tumor progression, and response to immune therapy [13, 18, 27]. We used a short course of ATRA treatment that demonstrated the depletion of MDSC in a previous clinical trial in patients with renal cell carcinoma [27]. In the current trial, a significant reduction in MDSC was observed only in patients on arm C, which supported the hypothesis that ATRA indeed depleted these cells. No effect on the presence of DC or Treg was seen. This was not unexpected, since in animal studies only continuous long-term treatment with ATRA resulted in increased Tregs [45–47], whereas in this trial, three short (3-day each) courses of the drug were administered. P53-specific immune responses were not detected in any patient from arm A, or after first-line chemotherapy in arms B and C. Only 20 % of patients treated with vaccine alone developed immune response to vaccination. This was substantially lower than 57.1 % observed in our previous trial with the same vaccine [20]. The main differences between these two trials were in the selection of patients. The majority of patients in the previous trial were previously treated, having a relatively long history of the disease, whereas in the current randomized trial, only newly diagnosed patients were enrolled. As a result, in the previous trial, there was a bias toward patients with slow progression of disease and thus perhaps more responsive to the therapy. In the current trial, the addition of ATRA increased the immune responses to vaccination to almost 42 % indicating that MDSC depletion indeed may improve the immune response to vaccination. Importantly, our results also present the first indication that such improvement may translate into clinical benefits. The response rate to the second-line chemotherapy in patients from arm C was higher than in patients from arm B although this result is preliminary.
Although ATRA as a single agent is not effective in lung cancer, it was reported that in combination with chemotherapy, it might improve responses and survival of patients with non-small cell lung cancer [48]. However, in our trial, ATRA was given only for 3 days at the time of each vaccination. Chemotherapy was started at least 3–4 weeks after last vaccination (and ATRA administration). Therefore, ATRA could not directly affect the response of tumors to chemotherapy.
Thus, our study, for the first time, demonstrates that MDSC depletion in cancer patients may improve the antitumor immune response to vaccination and provides direct rationale to use this approach to augment cancer immune therapy.
Acknowledgments
This work was supported by NIH SPORE grant P50 CA119997 to Scott Antonia and Dmitry Gabrilovich and partially supported by Cell Therapy Core at H. Lee Moffitt Cancer Center. We thank Dr. Palucka (Baylor Institute of Immunology) for help with protocol evaluating intracellular cytokine production by T cells.
Conflict of interests
Authors declare no conflict of interest.
Footnotes
The authors Cristina Iclozan, Scott Antonia, and Alberto Chiappori contributed equally to this work.
References
- 1.Mellman I, Coukos G, Dranoff G. Cancer immunotherapy comes of age. Nature. 2011;480:480–489. doi: 10.1038/nature10673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Restifo NP, Dudley ME, Rosenberg SA. Adoptive immunotherapy for cancer: harnessing the T cell response. Nat Rev Immunol. 2012;12:269–281. doi: 10.1038/nri3191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pardoll D, Drake C. Immunotherapy earns its spot in the ranks of cancer therapy. J Exp Med. 2012;209:201–209. doi: 10.1084/jem.20112275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rabinovich GA, Gabrilovich D, Sotomayor EM. Immunosuppressive strategies that are mediated by tumor cells. Annu Rev Immunol. 2007;25:267–296. doi: 10.1146/annurev.immunol.25.022106.141609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zou W, Chen L. Inhibitory B7-family molecules in the tumour microenvironment. Nat Rev Immunol. 2008;8:467–477. doi: 10.1038/nri2326. [DOI] [PubMed] [Google Scholar]
- 6.Munn DH, Mellor AL. Indoleamine 2,3-dioxygenase and tumor-induced tolerance. J Clin Invest. 2007;117:1147–1154. doi: 10.1172/JCI31178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol. 2009;9:162–174. doi: 10.1038/nri2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gabrilovich DI, Ostrand-Rosenberg S, Bronte V. Coordinated regulation of myeloid cells by tumours. Nat Rev Immunol. 2012;12:253–268. doi: 10.1038/nri3175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Peranzoni E, Zilio S, Marigo I, Dolcetti L, Zanovello P, Mandruzzato S, Bronte V. Myeloid-derived suppressor cell heterogeneity and subset definition. Curr Opin Immunol. 2010;22:238–244. doi: 10.1016/j.coi.2010.01.021. [DOI] [PubMed] [Google Scholar]
- 10.Ostrand-Rosenberg S, Sinha P. Myeloid-derived suppressor cells: linking inflammation and cancer. J Immunol. 2009;182:4499–4506. doi: 10.4049/jimmunol.0802740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Filipazzi P, Valenti R, Huber V, et al. Identification of a new subset of myeloid suppressor cells in peripheral blood of melanoma patients with modulation by a granulocyte-macrophage colony-stimulation factor-based antitumor vaccine. J Clin Oncol. 2007;25:2546–2553. doi: 10.1200/JCO.2006.08.5829. [DOI] [PubMed] [Google Scholar]
- 12.Poschke I, Mougiakakos D, Hansson J, Masucci GV, Kiessling R. Immature immunosuppressive CD14 + HLA-DR-/low cells in melanoma patients are Stat3hi and overexpress CD80, CD83, and DC-sign. Cancer Res. 2010;70:4335–4345. doi: 10.1158/0008-5472.CAN-09-3767. [DOI] [PubMed] [Google Scholar]
- 13.Diaz-Montero CM, Salem ML, Nishimura MI, Garrett-Mayer E, Cole DJ, Montero AJ. Increased circulating myeloid-derived suppressor cells correlate with clinical cancer stage, metastatic tumor burden, and doxorubicin-cyclophosphamide chemotherapy. Cancer Immunol Immunother. 2009;58:49–59. doi: 10.1007/s00262-008-0523-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Montero AJ, Diaz-Montero CM, Deutsch YE, et al. Phase 2 study of neoadjuvant treatment with NOV-002 in combination with doxorubicin and cyclophosphamide followed by docetaxel in patients with HER-2 negative clinical stage II-IIIc breast cancer. Breast Cancer Res Treat. 2012;132:215–223. doi: 10.1007/s10549-011-1889-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yuan XK, Zhao XK, Xia YC, Zhu X, Xiao P. Increased circulating immunosuppressive CD14(+)HLA-DR(−low) cells correlate with clinical cancer stage and pathological grade in patients with bladder carcinoma. J Int Med Res. 2011;39:1381–1391. doi: 10.1177/147323001103900424. [DOI] [PubMed] [Google Scholar]
- 16.Eruslanov E, Neuberger M, Daurkin I, et al. Circulating and tumor-infiltrating myeloid cell subsets in patients with bladder cancer. Int J Cancer. 2012;130:1109–1119. doi: 10.1002/ijc.26123. [DOI] [PubMed] [Google Scholar]
- 17.Goedegebuure P, Mitchem JB, Porembka MR, Tan MC, Belt BA, Wang-Gillam A, Gillanders WE, Hawkins WG, Linehan DC. Myeloid-derived suppressor cells: general characteristics and relevance to clinical management of pancreatic cancer. Curr Cancer Drug Targets. 2011;11:734–751. doi: 10.2174/156800911796191024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Walter S, Weinschenk T, Stenzl A, et al. Multipeptide immune response to cancer vaccine IMA901 after single-dose cyclophosphamide associates with longer patient survival. Nat Med. 2012;18:1254–1265. doi: 10.1038/nm.2883. [DOI] [PubMed] [Google Scholar]
- 19.Nikitina EY, Clark JI, van Beynen J, Chada S, Virmani AK, Carbone DP, Gabrilovich DI. Dendritic cells transduced with full-length wild-type p53 generate antitumor cytotoxic T lymphocytes from peripheral blood of cancer patients. Clin Cancer Res. 2001;7:127–135. [PubMed] [Google Scholar]
- 20.Antonia SJ, Mirza N, Fricke I, et al. Combination of p53 cancer vaccine with chemotherapy in patients with extensive stage small cell lung cancer. Clin Cancer Res. 2006;12:878–887. doi: 10.1158/1078-0432.CCR-05-2013. [DOI] [PubMed] [Google Scholar]
- 21.Kusmartsev S, Cheng F, Yu B, Nefedova Y, Sotomayor E, Lush R, Gabrilovich DI. All-trans-retinoic acid eliminates immature myeloid cells from tumor-bearing mice and improves the effect of vaccination. Cancer Res. 2003;63:4441–4449. [PubMed] [Google Scholar]
- 22.Gabrilovich DI, Velders M, Sotomayor E, Kast WM. Mechanism of immune dysfunction in cancer mediated by immature Gr-1 + myeloid cells. J. Immunol. 2001;166:5398–5406. doi: 10.4049/jimmunol.166.9.5398. [DOI] [PubMed] [Google Scholar]
- 23.Kusmartsev S, Su Z, Heiser A, Dannull J, Eruslanov E, Kubler H, Yancey D, Dahm P, Vieweg J. Reversal of myeloid cell-mediated immunosuppression in patients with metastatic renal cell carcinoma. Clin Cancer Res. 2008;14:8270–8278. doi: 10.1158/1078-0432.CCR-08-0165. [DOI] [PubMed] [Google Scholar]
- 24.Weiss T, Vitacolonna M, Zoller M. The efficacy of an IL-1alpha vaccine depends on IL-1RI availability and concomitant myeloid-derived suppressor cell reduction. J Immunother. 2009;32:552–564. doi: 10.1097/CJI.0b013e31819b7b9e. [DOI] [PubMed] [Google Scholar]
- 25.Lee JM, Seo JH, Kim YJ, Kim YS, Ko HJ, Kang CY. The restoration of myeloid-derived suppressor cells as functional antigen-presenting cells by NKT cell help and all-trans-retinoic acid treatment. Int J Cancer. 2012;131:741–751. doi: 10.1002/ijc.26411. [DOI] [PubMed] [Google Scholar]
- 26.Song X, Guo W, Cui J, Qian X, Yi L, Chang M, Cai Q, Zhao Q. A tritherapy combination of a fusion protein vaccine with immune-modulating doses of sequential chemotherapies in an optimized regimen completely eradicates large tumors in mice. Int J Cancer. 2011;128:1129–1138. doi: 10.1002/ijc.25451. [DOI] [PubMed] [Google Scholar]
- 27.Mirza N, Fishman M, Fricke I, Dunn M, Neuger AM, Frost TJ, Lush RM, Antonia S, Gabrilovich DI. All-trans-retinoic acid improves differentiation of myeloid cells and immune response in cancer patients. Cancer Res. 2006;66:9299–9307. doi: 10.1158/0008-5472.CAN-06-1690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Simon R. Optimal two-stage designs for phase II clinical trials. Control Clin Trials. 1989;10:1–10. doi: 10.1016/0197-2456(89)90015-9. [DOI] [PubMed] [Google Scholar]
- 29.Wieand HS. Randomized phase II trials: what does randomization gain? J Clin Oncol. 2005;23:1794–1795. doi: 10.1200/JCO.2005.10.956. [DOI] [PubMed] [Google Scholar]
- 30.Rocha Lima C, Chiappori A. Treatment of relapsed small-cell lung cancer: a focus on the evolving role of topotecan. Lung Cancer. 2003;40:229–236. doi: 10.1016/S0169-5002(03)00039-4. [DOI] [PubMed] [Google Scholar]
- 31.Nagaraj S, Gabrilovich DI. Myeloid-derived suppressor cells in human cancer. Cancer J (Sudbury Mass) 2010;16:348–353. doi: 10.1097/PPO.0b013e3181eb3358. [DOI] [PubMed] [Google Scholar]
- 32.Nagaraj S, Youn J, Weber H, et al. Anti-inflammatory triterpenoid blocks immune suppressive function of myeloid-derived suppressor cells and improves immune response in cancer. Clin Cancer Res. 2010;16:1812–1823. doi: 10.1158/1078-0432.CCR-09-3272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Almand B, Clark JI, Nikitina E, van Beynen J, English NR, Knight SC, Carbone DP, Gabrilovich DI. Increased production of immature myeloid cells in cancer patients: a mechanism of immunosuppression in cancer. J Immunol. 2001;166:678–689. doi: 10.4049/jimmunol.166.1.678. [DOI] [PubMed] [Google Scholar]
- 34.Solito S, Falisi E, Diaz-Montero CM, et al. A human promyelocytic-like population is responsible for the immune suppression mediated by myeloid-derived suppressor cells. Blood. 2011;118:2254–2265. doi: 10.1182/blood-2010-12-325753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gudas LJ, Wagner JA. Retinoids regulate stem cell differentiation. J Cell Physiol. 2011;226:322–330. doi: 10.1002/jcp.22417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nefedova Y, Fishman M, Sherman S, Wang X, Beg AA, Gabrilovich DI. Mechanism of all-trans retinoic acid effect on tumor-associated myeloid-derived suppressor cells. Cancer Res. 2007;67:11021–11028. doi: 10.1158/0008-5472.CAN-07-2593. [DOI] [PubMed] [Google Scholar]
- 37.Gervais A, Leveque J, Bouet-Toussaint F, Burtin F, Lesimple T, Sulpice L, Patard JJ, Genetet N, Catros-Quemener V. Dendritic cells are defective in breast cancer patients: a potential role for polyamine in this immunodeficiency. Breast Cancer Res. 2005;7:R326–R335. doi: 10.1186/bcr1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mohty M, Morbelli S, Isnardon D, Sainty D, Arnoulet C, Gaugler B, Olive D. All-trans retinoic acid skews monocyte differentiation into interleukin-12-secreting dendritic-like cells. Br J Haematol. 2003;122:829–836. doi: 10.1046/j.1365-2141.2003.04489.x. [DOI] [PubMed] [Google Scholar]
- 39.Zitvogel L, Kepp O, Kroemer G. Immune parameters affecting the efficacy of chemotherapeutic regimens. Nat Rev Clin Oncol. 2011;8:151–160. doi: 10.1038/nrclinonc.2010.223. [DOI] [PubMed] [Google Scholar]
- 40.Gabrilovich DI. Combination of chemotherapy and immunotherapy for cancer: a paradigm revisited. The Lancet Oncol. 2007;8:2–3. doi: 10.1016/S1470-2045(06)70985-8. [DOI] [PubMed] [Google Scholar]
- 41.Ramakrishnan R, Gabrilovich DI. Mechanism of synergistic effect of chemotherapy and immunotherapy of cancer. Cancer Immunol Immunother. 2011;60:419–423. doi: 10.1007/s00262-010-0930-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dark GG, Calvert AH, Grimshaw R, et al. Randomized trial of two intravenous schedules of the topoisomerase I inhibitor liposomal lurtotecan in women with relapsed epithelial ovarian cancer: a trial of the national cancer institute of Canada clinical trials group. J Clin Oncol. 2005;23:1859–1866. doi: 10.1200/JCO.2005.02.028. [DOI] [PubMed] [Google Scholar]
- 43.Trellakis S, Bruderek K, Hütte J, Elian M, Hoffmann TK, Lang S, Brandau S (2012) Granulocytic myeloid-derived suppressor cells are cryosensitive and their frequency does not correlate with serum concentrations of colony-stimulating factors in head and neck cancer. Innate Immun. http://ini.sagepub.com/content/early/2012/11/16/1753425912463618.full.pdf+html [DOI] [PubMed]
- 44.Kotsakis A, Harasymczuk M, Schilling B, Georgoulias V, Argiris A, Whiteside TL. Myeloid-derived suppressor cell measurements in fresh and cryopreserved blood samples. J Immunol Methods. 2012;381:14–22. doi: 10.1016/j.jim.2012.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Golovina TN, Mikheeva T, Brusko TM, Blazar BR, Bluestone JA, Riley JL. Retinoic acid and rapamycin differentially affect and synergistically promote the ex vivo expansion of natural human T regulatory cells. PLoS ONE. 2011;6:e15868. doi: 10.1371/journal.pone.0015868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Van YH, Lee WH, Ortiz S, Lee MH, Qin HJ, Liu CP. All-trans retinoic acid inhibits type 1 diabetes by T regulatory (Treg)-dependent suppression of interferon-gamma-producing T-cells without affecting Th17 cells. Diabetes. 2009;58:146–155. doi: 10.2337/db08-1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kwok SK, Park MK, Cho ML, Oh HJ, Park EM, Lee DG, Lee J, Kim HY, Park SH. Retinoic acid attenuates rheumatoid inflammation in mice. J Immunol. 2012;189:1062–1071. doi: 10.4049/jimmunol.1102706. [DOI] [PubMed] [Google Scholar]
- 48.Arrieta O, Gonzalez-De la Rosa CH, Arechaga-Ocampo E, et al. Randomized phase II trial of All-trans-retinoic acid with chemotherapy based on paclitaxel and cisplatin as first-line treatment in patients with advanced non-small-cell lung cancer. J Clin Oncol. 2010;28:3463–3471. doi: 10.1200/JCO.2009.26.6452. [DOI] [PubMed] [Google Scholar]