

Abstract
Since introduction into clinical practice over 60 years ago, aminoglycoside antibiotics remain important drugs in the treatment of bacterial infections, cystic fibrosis and tuberculosis. However, the ototoxic and nephrotoxic properties of these drugs are still a major clinical problem. Recent advances in molecular biology and biochemistry have begun to uncover the intracellular actions of aminoglycosides that lead to cytotoxicity. In this review, we discuss intracellular binding targets of aminoglycosides, highlighting specific aminoglycoside-binding proteins (HSP73, calreticulin and CLIMP-63) and their potential for triggering caspases and Bcl-2 signalling cascades that are involved in aminoglycoside-induced cytotoxicity. We also discuss potential strategies to reduce aminoglycoside cytotoxicity, which are necessary for greater bactericidal efficacy during aminoglycoside pharmacotherapy.
Introduction
Aminoglycoside antibiotics are among the most commonly-used antibiotics world-wide,1 and are highly effective in treating life-threatening Gram-negative bacterial infections, such as meningitis and bacterial sepsis in infants.2–4 In mammals, aminoglycosides are both nephrotoxic and ototoxic. Nephrotoxicity results in increased morbidity during and after treatment, and can cause acute kidney failure. After systemic delivery, aminoglycosides are primarily localized in epithelial cells lining the proximal tubules of the nephron. Distal tubule cells also take up aminoglycosides, but survive at drug concentrations that kill proximal tubule cells in vitro.5,6 In vivo, aminoglycosides disrupt distal tubule function by reversibly blocking luminal cation channels, leading to cation-wasting in urine.7,8 Renal cytotoxicity is reversible due to proximal tubule epithelial cell proliferation.9,10
Aminoglycoside-induced ototoxicity in mammals is frequently permanent as these drugs can kill inner ear sensory hair cells that cannot be spontaneously regenerated following hair cell death.11,12 Within the cochlea, aminoglycosides are preferentially localized in outer hair cells (OHCs) at the base of the cochlea, and hair cell death initially occurs in basal OHCs, and extends to inner hair cells (IHCs) and to more apical cochlear hair cells with increasing total dose.13,14 Aminoglycosides are also localized in stria vascularis and spiral ligament fibrocytes of the cochlear lateral wall, spiral ganglion neurons, and in supporting cells within the organ of Corti.15–18 Aminoglycosides also induce morphological changes in the stria vascularis, decreasing its volume and altering its structure.19,20
Several aminoglycosides are essential in clinical practice (Fig. 1). Gentamicin is administered systemically in intensive care units for prophylaxis in pre-term infants, and topically for major burn case. Tobramycin is preferentially used for treating Pseudomonas aeruginosa-induced pneumonia. Amikacin is most often used for treating severe, hospital-acquired infections with multidrug resistant Gram-negative bacteria. Gentamicin and tobramycin are thought to be preferentially vestibulotoxic, i.e. inducing hair cell death in the balance (vestibular) end-organs of the saccule, utricle, and the three ampullae of the semi-circular canals. Amikacin, neomycin and kanamycin are preferentially cochleotoxic.21 Clinically, gentamicin, tobramycin and amikacin are most frequently prescribed, while streptomycin remains important for treating tuberculosis, despite its severe ototoxicity.22
Fig. 1.

Structures of several aminoglycoside antibiotics. Chirality is indicated by the Natta projection method.
The bactericidal effects of aminoglycosides are largely due to inhibition and/or mis-translation during protein synthesis.23,24 In mammalian cells, aminoglycoside cytotoxicity occurs via several mechanisms (Fig. 2), including cytochrome c release from mitochondria, activation of caspase-9 and caspase-3, generation of toxic levels of reactive oxygen species (ROS), activation of c-Jun N-terminal kinases (JNKs), and protein cleavage by calpains.25–29 To exert these intracellular phenomena, aminoglycosides must first enter cells, typically by endocytosis or cation channel permeation.30–33 Endocytosis transports aminoglycosides to the endoplasmic reticulum (ER) and lysosomes. In lysosomes, aminoglycosides induce the release of cathepsins (lysosomal peptidases) and/or lysosomal rupture, either of which leads to cell death.32,34,35 Aminoglycoside permeation through non-selective cation channels into the cytosol can induce a wide range of drug–target interactions. In this review, we will focus on the intracellular binding targets of aminoglycosides, and their subsequent effect on physiological functions and downstream targets, with an emphasis on ototoxicity.
Fig. 2.
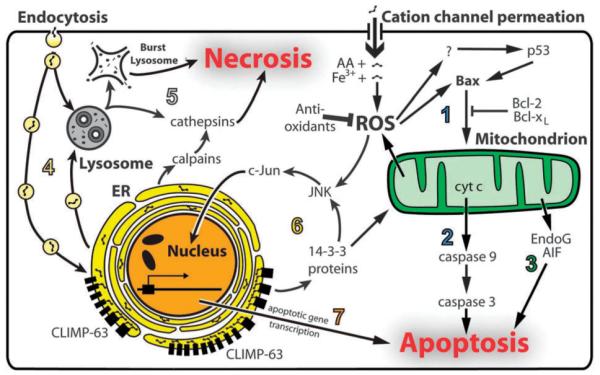
Cell death mechanisms induced by aminoglycosides. (1) Aminoglycosides can enter cells by permeating cation channels directly into the cytosol. Binding of aminoglycosides to iron generates ROS, with arachidonic acid (AA) acting as an electron donor. ROS activates Bax, which in turn translocates to mitochondrial membranes. (2) Cytochrome c (cyt c) is released from mitochondria through the mitochondrial transition pore formed by Bax-dependent mechanisms, activating caspase-9 and caspase-3, and leading to apoptosis. ROS are also released from mitochondria, further increasing cytosolic ROS levels. (3) In caspase-independent mechanisms, EndoG and AIF released from mitochondria also induce apoptosis. (4) Aminoglycosides can also be endocytosed and are trafficked to the ER and lysosome by vesicle transport mechanisms. (5) Aminoglycosides can induce lysosomal rupture, or the release of lysosomal cathepsins, either of which leads to necrosis. (6) Aminoglycosides within the lumen of the ER bind to CLIMP-63, inducing oligomerization that can activate 14-3-3 proteins, leading to mitochondrial apoptosis signaling and/or resulting in JNK activation and c-Jun translocation into nucleus. (7) The c-Jun transcription factor induces apoptotic gene transcription and subsequent apoptosis.
Aminoglycoside-binding molecules
Iron
It has been suggested that aminoglycosides bind to ferric iron (FeIII) and generate FeII–aminoglycoside complexes in the cytosol.36,37 This redox-active complex catalyzes the formation of reactive oxygen species (ROS) from molecular oxygen, using arachidonic acid as an electron donor.37,38 This can lead to excessive cytosolic production of ROS that induces apoptosis signalling cascades (Fig. 2). Toxic levels of ROS damage cells by triggering various cell death mechanisms, including caspase-dependent and independent apoptosis, and necrosis.39,40 Apoptotic signals also induce the release of mitochondrial ROS into the cytosol, further increasing cytosolic ROS levels.41–43 Animals that overexpress superoxide dismutase, a key anti-oxidative enzyme, are more resistant to aminoglycoside-induced ototoxicity compared to wild-type animals,44 supporting the role of ROS in aminoglycoside-induced ototoxicity. Although it is not clear how ROS induce multiple cell death signalling cascades, it is well-established that ROS activate c-Jun N-terminal kinases (JNK), which in turn induces apoptosis.45 Inhibition of the JNK pathway promotes acute hair cell survival during treatment with ototoxic levels of aminoglycosides.28,46,47
Phosphoinositides
Phosphoinositides are negatively-charged phospholipids located in the cytosolic face of eukaryotic cell membranes. Phosphoinositides are important second messengers in intracellular signal transduction pathways, and are a source of arachidonic acid.48 Aminoglycoside binding to phosphoinositides induces the release of arachidonic acid, which acts as an electron donor in FeII–aminoglycoside complex-mediated ROS formation, as discussed above.37,38
Interactions between the cationic aminoglycosides and phosphoinositides have long been considered a major component of aminoglycoside-induced ototoxicity.49 There is a good correlation between the decline of OHC receptor potential in response to acoustic stimulation during cochlear perfusion of aminoglycosides and the binding affinity of aminoglycosides to phosphoinositides.50 A deuterium-NMR study confirmed this correlation, and proposed that aminoglycosides sequester phosphoinositides and inhibit lysosomal phospholipase activity.51 This would increase cytoplasmic levels of drug-bound phosphoinositides,52–54 which are cytotoxic and may be a major cause of ototoxicity.55,56 A recent study postulated that aminoglycosides deplete cytoplasmic levels of free phosphoinositides that regulate KCNQ4 channel activity, resulting in the inhibition of potassium efflux necessary for cochlear sensory function and OHC survival.57,58
Phosphoinositides are ubiquitously expressed in mammalian cells and regulate a wide variety of ion channels.59,60 Because there is no tissue-specific expression of phosphoinositides, the cytotoxicity induced by the interactions between aminoglycosides and phosphoinositides is unlikely to be solely responsible for the selective susceptibility of kidney proximal tubule cells and inner ear mechanosensory hair cells.
RNA
It has been well established that aminoglycosides kill bacteria by binding to ribosomal 16S rRNA in the 30S subunit of the ribosome, inhibiting protein synthesis.23,61,62 In eukaryotes, mitochondria are thought to originate from bacteria,63 and mitochondrial ribosomes are highly similar to bacterial ribosomes compared to mammalian cytosolic ribosomes.64,65 The first analysis of familial aminoglycoside-induced deafness revealed a nucleotide 1555 A to G substitution (A1555G) in the mitochondrial 12S rRNA gene.66 This A1555G mutation makes the secondary structure of 12S rRNA more similar to the corresponding site in 16S rRNA of bacteria.66 Indeed, binding assays using RNA constructs demonstrated that the A1555G RNA analog binds to aminoglycosides with high affinity while the wild-type construct does not.67 Since the A1555G mutation also causes non-syndromic hearing loss in many families, factors other than aminoglycosides must also contribute to the deafness induced by this mutation.68 Additional mutations (C1494T, T1095C, T961) in the 12S rRNA also cause drug susceptibility.69–71 Biochemical studies show that these mutations also decrease mitochondrial protein translation efficacy.72 Therefore, aminoglycoside binding to mitochondrial 12S rRNA with these mutations could lead to high levels of inhibition or mistranslation of protein synthesis and subsequent cytotoxicity.73
One unexpected consequence of aminoglycoside binding to RNA has been its ability to readthrough premature termination codons (PTCs) by binding to the decoding site of 18S rRNA. Therapeutic approaches to promote readthrough of disease-causing PTCs have been developed. For example, in patients with cystic fibrosis (CF) resulting from PTC mutations in the CF transmembrane conductance regulator (CFTR), aminoglycoside treatment can lead to expression of full-length proteins.74,75 However, this readthrough efficacy is variable, depending on genes and mutations,76 and more understanding of the PTC readthrough mechanism is necessary.
Proteins
Although aminoglycosides bind to at least several proteins,77 it is not clear which proteins, when bound to aminoglycosides, become dysfunctional, and/or induce cytotoxicity in mammalian cells. Some aminoglycoside-binding proteins may sequester aminoglycosides and prevent the noxious intracellular effects of these drugs.
In kidney, a large glycoprotein called megalin binds to gentamicin at the apical membrane of proximal tubule cells, and delivers gentamicin to lysosomes following endocytosis, suggesting that megalin is involved in renal accumulation of aminoglycosides.78,79 However, a proximal tubule cell line LLC-PK1 and sensory hair cells in the inner ear do not express megalin, yet both take up aminoglycosides by endocytosis, and exhibit aminoglycoside-induced cytotoxicity.30,80,81
Recent studies using gentamicin affinity column chromatography identified HSP73 in porcine kidney,82 and calreticulin in bovine kidney83 as gentamicin-binding proteins (GBPs). HSP73 is a heat shock protein (HSP) that is constitutively expressed, and is not induced by cell stress. Constitutively-expressed HSPs function as molecular chaperones for newly-synthesized proteins in the ER, and these HSPs assist in protein folding and assembly, and in transporting proteins into subcellular organelles.84 Although the functional specificity of HSP73 is unknown, it is likely that HSP73 also functions as a chaperone protein. Since HSP73 is homogenously distributed throughout the kidney,85 it is unlikely to contribute to the difference in gentamicin susceptibility between proximal and distal tubule cells. There has been no report on HSP73 expression in the inner ear, although the ubiquitous expression of this protein elsewhere suggests a similar ubiquitous distribution in the cochlea. Interestingly, HSP70, another HSP protein, inhibits neomycin-induced hair cell death in mice.86 Unlike HSP73, HSP70 expression is induced by cellular stress, and protects the cell by stabilizing lysosomal membranes.87,88
Calreticulin is another chaperone protein localized in the ER.89 The ER may play an important role in aminoglycoside-induced cytotoxicity because endocytosed aminoglycosides are trafficked to the Golgi body and ER.32,90 Calreticulin binds to glycoproteins and assists in protein folding, quality control, and degradation.91 Calreticulin is expressed in both kidney proximal and distal tubules.82,142 Calreticulin is also expressed in the cochlea.92 We have immunolocalized calreticulin in the cytoplasm of marginal cells in the stria vascularis and in the stereociliary bundles of cochlear hair cells.142 These locations are exposed to high levels of aminoglycosides during trans-strial trafficking into endolymph and hair cell uptake of aminoglycosides (Fig. 3).30,31,93,94 The chaperone activities of both HSP73 and calreticulin are inhibited by gentamicin, and this inhibition could contribute to gentamicin-induced cytotoxicity.82,83
Fig. 3.
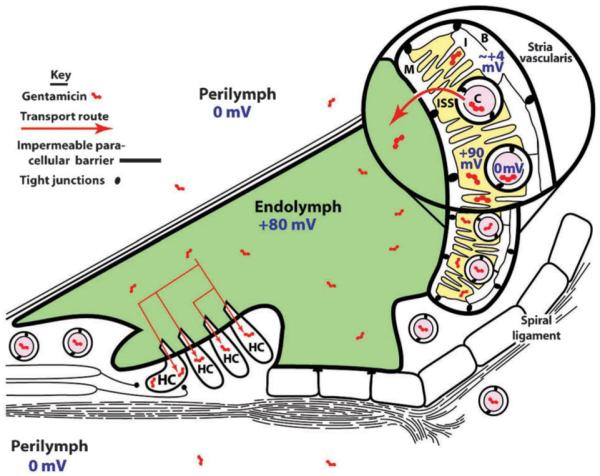
Schematic diagram of the cochlear duct cytoarchitecture and its electrophysiological environments. The stria vascularis, lining the spiral ligament on the inside lateral wall of the bony cochlear shell, contains basal (B) and marginal (M) cells connected together by tight junctions that form an impermeable paracellular barrier to solutes. Circulating aminoglycosides within strial capillaries (C) are preferentially transported through the strial blood–labyrinth barrier consisting of tight junction-coupled endothelial cells, into the intra-strial space (ISS). From there, aminoglycosides are trafficked through marginal cells into endolymph, and enter hair cells (HC) across their apical surfaces by endocytosis and non-selective cation channel permeation. The electrical potentials of various fluid compartments, separated by tight junction-coupled endothelial and epithelial cell barrier layers, are also indicated. Endolymph has a +80 mV, and hair cells have a resting potential of −60 to −75 mV, generating a considerable electrophoretic driving force across the apical endolymphatic membranes of hair cells.
The early GBP studies82,83 used CH- or CNBr-activated Sepharose 4B without a “spacer” molecule between gentamicin and Sepharose, which could cause steric hindrance between gentamicin and other GBPs. We employed a neutral 10-atom spacer between gentamicin and agarose, and identified another GBP, CLIMP-63, a protein that connects ER to the cytoskeleton.77 Notably, CLIMP-63 is heterogeneously expressed in kidney and cochlear cell lines. Many other GBPs were also observed in kidney cells, including calreticulin, but these did not show distinct differences in expression or localization between proximal and distal tubule epithelial cell lines in vitro.
Cell lines derived from the kidney proximal tubule and inner ear organ of Corti express significant amounts of CLIMP-63 dimers that are resistant to dithiothreitol (DTT) treatment. Gentamicin treatment increased CLIMP-63 dimerization. Knock-down of CLIMP-63 with siRNA transfection effectively reduced CLIMP-63 dimerization while retaining expression of the CLIMP-63, and cells were more resistant to gentamicin treatment. Although the nature of these DTT-resistant dimers of CLIMP-63 is unclear, these dimers could define the tissue selectivity of aminoglycoside-induced toxicity because they are not expressed in cells from the kidney distal tubule or other tissues.77 We have identified several 14-3-3 proteins as CLIMP-63-binding proteins, and that 14-3-3β is also involved in gentamicin-induced CLIMP-63-dependent cytotoxicity. Since 14-3-3 proteins have been implicated in various cell death and survival signaling pathways, it is possible that CLIMP-63 association with 14-3-3 proteins induces apoptosis. One possible pathway leads to JNKs or p38-MAPK-dependent apoptosis.95
One of the major questions in aminoglycoside-induced cytotoxicity is selective susceptibility of the inner ear hair cells and kidney proximal tubule cells. The conventional hypothesis for aminoglycoside susceptibility in these cell types is that these cells take up higher levels of aminoglycosides compared to other cells. Since aminoglycoside levels in the cytosol directly correlate with cytosolic ROS generation through iron-binding, this could be sufficient to explain the cell type-specific drug susceptibility. However, it remains to be explained why these cells retain high intracellular levels of aminoglycoside, while most cells are able to clear their cytosol of the drug.96 The ability of cells to clear aminoglycosides is also likely to be important in trafficking the drug across the tight junction-coupled endothelial and epithelial marginal cells in the cochlear blood–labyrinth barrier into the intra-strial space and endolymph, preventing or reducing cytotoxicity in these cochlear cell types (Fig. 3).
Based on the evidence that we have described of intracellular binding-targets of aminoglycosides, we speculate that aminoglycoside-binding proteins, like calreticulin, CLIMP-63, and possibly HSP73, contribute to the tissue selectivity of aminoglycoside-induced toxicity. Additionally in the cochlea, the positive endolymphatic potential and low Ca2+ level of endolymph bathing the hair cell apex favor a rapid influx of aminoglycosides through cation channels, such as mechano-transduction or TRPV4 channels, concentrating aminoglycosides in hair cells (Fig. 3).31,33
Downstream targets of aminoglycoside-induced cytotoxicity
Owing to recent advances in apoptosis research, especially related to cancer biology, we now have a better understanding of how aminoglycosides induce cell death signalling after initial drug–target interactions in the cytoplasm. Aminoglycosides induce phosphorylation of c-Jun in JNK signalling pathways that trigger hair cell death.28,97 Gentamicin treatment of rat cochlear explants also increases the binding activities of activating protein-1 (AP-1), a heterodimeric protein consisted of c-Jun and c-Fos family proteins.98 Caspases and Bcl-2 family proteins are essential components in apoptotic signalling. We will describe how these proteins are involved in aminoglycoside-induced apoptosis below.
Caspases
Among a dozen caspase family proteins identified, those that are activated by tumor necrosis factor receptors (TNFRs), including caspase-8, do not play a major role in aminoglycoside-induced ototoxicity.99 Activation of caspase-9, on the other hand, is induced by cytochrome c release from mitochondria into the cytosol, and has been detected in aminoglycoside-treated utricles, cochleae and kidney cells in vitro.26,100,101 Caspase-9 activates caspase-3, an “executioner” caspase, which cleaves anti-apoptotic proteins or inhibits deoxyribonucleases, to induce cell death.102,103 Aminoglycoside-treated hair cells showed enhanced levels of caspase-3 activity and increased cell death in vitro and in vivo.99,100,104 Direct infusion of the pan-caspase inhibitor z-VAD-fmk into the vestibule, or systemic administration of the inhibitor promoted hair cell survival after streptomycin treatment.105
In mice, chronic treatment with kanamycin induced hair cell death by caspase-independent pathway(s).35 While cytochrome c release, caspase-9, caspase-3 and JNK activation, and TUNEL staining were absent, endonuclease G (EndoG) translocation, calpain activation, and cathepsin D synthesis and activation were all observed after chronic treatment with kanamycin.35 This raises a question whether in vivo or clinical aminoglycoside administration induces ototoxicity by caspase-dependent apoptosis, by other caspase-independent apoptotic, by necrotic mechanisms, or by a combination of two or more of these mechanisms. One possible caspase-independent mechanism involves mitochondrial release of EndoG and apoptosis-inducing factor (AIF), while activation of calpain–cathepsin signalling cascades induces necrosis.106–108 Consistent with this, intracellular calcium levels are elevated by aminoglycoside treatment, and an increase in calcium levels can activate calpains, which leads to ototoxicity.29,109 Since these mechanisms have not been fully investigated, more studies using clinically-relevant experimental designs are required.
Bcl-2 family proteins
The cytoplasmic Bcl-2 family proteins consist of anti-apoptotic Bcl-2 and Bcl-xL and pro-apoptotic Bax and Bak, where Bcl-2 and Bcl-xL form heterodimers with Bax and Bak to inhibit apoptosis and maintain mitochondrial membrane integrity.110,111 When apoptotic signals overcome inhibition/protection by Bcl-2 and Bcl-xL, Bax translocates from the cytosol to mitochondria,112 releasing mitochondrial cytochrome c that in turn activates caspase-9.26 How Bax triggers release of cytochrome c is not well understood, but it likely involves the formation of the mitochondrial permeability transition pore.113,114
There have been numerous reports on Bcl-2 family proteins in the inner ear, and most early reports discuss their function in cochlear development. More recent evidence suggests that Bcl-2 protects against aminoglycoside-induced ototoxicity.115–117 Although little has been reported about the role of proapoptotic proteins, like Bax, in aminoglycoside-induced ototoxicity, studies on downstream targets suggest that they are involved. For example, loss of the mitochondrial membrane potential, an indication of Bax translocation to mitochondria, has been observed in the auditory hair cells after gentamicin treatment in vitro.25 In addition, cytochrome c release, also triggered by Bax activity, was detected in sensory hair cells treated with aminoglycosides.26,97,104 It is well-established that Bax can be up-regulated by the tumor suppressor p53 protein. The involvement of p53 in aminoglycoside-induced apoptosis is still unclear. However, a recent report suggested that non-transcriptional p53 activity is necessary for aminoglycoside-induced hair cell death (A. Coffin, personal communication).118 How p53 could be activated by aminoglycosides needs to be explored further.
Variation in aminoglycoside induction of toxicity
The utilization of zebrafish neuromast hair cells has provided an important tool to rapidly and reproducibly assess hair cell dose-response curves for different aminoglycosides. At a given dose, neomycin induced rapid hair cell death in some but not all hair cells, while gentamicin induced both acute hair cell death, and also continuing hair cell death over a longer period of time for a more complete ablation of neuromast hair cells.119 This effect was also seen with other aminoglycosides (kanamycin, streptomycin, tobramycin, amikacin) to varying degrees, implying that these drugs initiate a spectrum of cell death pathways.
Preclinical studies in aminoglycoside toxicity have also been confounded by the wide variation in rodent species susceptibility to aminoglycoside toxicity, with mice and rats being particularly resistant to aminoglycoside toxicity,120 compared to guinea pigs,121,122 for example. Furthermore, there is variation within a single species or strain to different types of aminoglycosides.120 In addition, there is individual variation within a study to the same dose of aminoglycosides that may correlate with the efficacy in trafficking aminoglycosides across the blood–labyrinth barrier.123 Nonetheless, with increasing availability of knockout and transgenic mice, a murine model for ototoxicity will be extremely advantageous to investigate specific mechanisms of drug trafficking in drug-induced cytotoxicity in the inner ear.120,124
The use of inner ear explants to study ototoxicity has increased in the last decade, and has both disadvantages and advantages. Obtaining inner ear sensory epithelial explants is a challenging procedure, subject to variation in the dissection procedure within experiments, and sensory cell death prior to ototoxic drug exposure.125 More importantly, explantation removes the unique three-dimensional electrochemical environment that bathes the mechano-sensitive hair cells (Fig. 3).126 However, inner ear explants provide high reproducibility in dose and duration of ototoxic drug exposure to hair cells, and avoid the experimental variability of animal studies due to species and inter-individual differences in the blood–labyrinth barrier. In addition, in vitro micro-Ussing chambers enable electrophysiological investigation of ion trafficking by the stria vascularis, a major function of the cochlear blood–labyrinth barrier.127,128
Otoprotection against aminoglycoside toxicity
One way to prevent ototoxicity may be the use of iron chelators, such as deferoxamine, to reduce ROS formation.120,129 Antioxidants also protect against ototoxicity by reducing ROS levels. Antioxidants that show otoprotective effects in animal models of aminoglycoside ototoxicity include: lipoic acid, d-methionine, and salicylate.130–132 In clinical studies in humans, aspirin, an antioxidant that is cheap and widely available, can ameliorate gentamicin-induced ototoxicity.133 Cellular enzymes with antioxidant properties, such as superoxide dismutases and glutathione S-transferase, could also be used to reduce ROS levels.134–136
Clinical interventions to interfere with aminoglycoside binding to phosphoinositides, RNA or proteins are far from established, partly because our understanding of these cytotoxicity mechanisms remains insufficient to devise a pharmacological strategy. However, in vitro studies and research on animal models are steadily accumulating evidence that support a few promising approaches to reduce aminoglycoside toxicity.
The first approach is to decrease cellular uptake of aminoglycosides in the cochlea or kidney by blocking drug entry into the cell via modulation of aminoglycoside-permissive ion channels (or transporters) at the cell membrane.33,137,143 This could be critical at the blood–labyrinth barrier, preventing aminoglycosides from entering the cochlea and hair cells.18 Another method would be to decrease intracellular accumulation by increasing cellular clearance of the drug. All cells initially take up aminoglycosides and most rapidly clear the drug.96 However, how the majority of cells clear aminoglycosides from their cytosol remains unknown. Up-regulating the intracellular expression of aminoglycoside-binding proteins to sequester these drugs and prevent their cytotoxicity may also be beneficial.
Another approach is to modulate cell death signalling by targeting specific proteins in cell death signaling, such as JNK or c-Jun. In order to block the functions of these proteins, pharmacological inhibitors or RNAi-based approaches may be useful, and this has been demonstrated in vitro.28,97 For example, l-carnitine, a naturally occurring neuroprotective agent, prevents expression of harakiri, a proapoptotic factor implicated in gentamicin-induced ototoxicity.138 Alternatively, increasing expression or efficacy of anti-apoptotic proteins such as Bcl-2 and Bcl-xL could promote hair cell survival. An apoptosis inhibitor protein survivin is a good candidate for this strategy.139 Similar approaches for modulating cell death signalling could also be used. These intracellular studies can be accelerated using zebrafish neuromast hair cells as a model system to identify potential intracellular otoprotectants.140,141 However, it remains to be determined whether hair cell survival extends beyond the initial assessment period, and that removal of these inhibitors of cell death signaling pathways has no adverse effects on hair cell survival.
Regardless of which approach ameliorates aminoglycoside toxicity, translation from in vitro studies and animal research to clinical medicine will be complex, and require extensive verification before clinical application. It will also be critical to determine how much reduction in eukaryotic toxicity in sensory receptors and kidney proximal tubules can occur while maintaining the drug’s inherent bactericidal properties. Any method that reduces both aminoglycoside ototoxicity and bactericidal efficacy will be unsuitable for clinical practice.
Insight, innovation, integration.
We present a review of aminoglycoside toxicity focusing on intracellular binding targets and downstream effects. A better understanding of the molecular mechanisms involved has recently become possible by innovative experimental approaches such as drug conjugation to agarose beads or fluorophores, and zebrafish hair cell death assays. Affinity chromatography or pull-down assays using gentamicin–agarose conjugates have identified several aminoglycoside-binding proteins. Zebrafish neuromast hair cells have become a primary model system to better understand intracellular pathways of cell death signaling induced by ototoxic drugs. Recent data using these methods give new insights into how aminoglycosides induce cytotoxicity. With increasing knowledge of intracellular drug binding targets and downstream effects, we can now begin to develop clinical strategies to reduce aminoglycoside-induced ototoxicity and nephrotoxicity.
Acknowledgements
We thank Dr Hongzhe Li for discussion on the manuscript. This work was supported by NIH NIDCD R03 DC009501 (TK) and R01 04555 (PSS).
Biographies
Takatoshi Karasawa received his PhD in Neuroscience in 2002 from Yale University, USA. Currently, he is Senior Research Associate at the Oregon Hearing Research Center, Oregon Health & Science University. His research interests include molecular mechanisms of ototoxicity induced by aminoglycosides and platinum-based anti-cancer drugs, identifying drug-binding proteins that are involved in ototoxicity.

Peter S. Steyger received his PhD in Cochlear Anatomy in 1991 from Keele University, UK. He is Associate Professor of Otolaryngology—Head & Neck Surgery at Oregon Health & Science University, and is also Scientific Director of the Deafness Research Foundation. The main focus of his research laboratory is to identify and then inhibit the molecular mechanisms that traffic systemically-administered ototoxic drugs across the blood–labyrinth barrier into the cochlear fluids and hair cells.

References
- 1.Forge A, Schacht J. Audiol. Neuro-Otol. 2000;5:3–22. doi: 10.1159/000013861. [DOI] [PubMed] [Google Scholar]
- 2.Edson RS, Terrell CL. Mayo Clin. Proc. 1999;74:519–528. doi: 10.4065/74.5.519. [DOI] [PubMed] [Google Scholar]
- 3.Grohskopf LA, Huskins WC, Sinkowitz-Cochran RL, Levine GL, Goldmann DA, Jarvis WR. Pediatr. Infect. Dis. J. 2005;24:766–773. doi: 10.1097/01.inf.0000178064.55193.1c. [DOI] [PubMed] [Google Scholar]
- 4.Twiss J, Byrnes C, Johnson R, Holland D. Int. J. Pharm. 2005;295:113–119. doi: 10.1016/j.ijpharm.2005.02.008. [DOI] [PubMed] [Google Scholar]
- 5.Bacon JA, Linseman DA, Raczniak TJ. Toxicol. in Vitro. 1990;4:384–388. doi: 10.1016/0887-2333(90)90085-8. [DOI] [PubMed] [Google Scholar]
- 6.Nonclercq D, Wrona S, Toubeau G, Zanen J, Heuson-Stiennon JA, Schaudies RP, Laurent G. Renal Failure. 1992;14:507–521. doi: 10.3109/08860229209047660. [DOI] [PubMed] [Google Scholar]
- 7.Quamme GA. Magnesium. 1986;5:248–272. [PubMed] [Google Scholar]
- 8.Kang HS, Kerstan D, Dai L, Ritchie G, Quamme GA. Can. J. Physiol. Pharmacol. 2000;78:595–602. [PubMed] [Google Scholar]
- 9.Nonclercq D, Toubeau G, Lambricht P, Heuson-Stiennon JA, Laurent G. Nephron. 1991;57:210–215. doi: 10.1159/000186253. [DOI] [PubMed] [Google Scholar]
- 10.Xie Y, Nishi S, Iguchi S, Imai N, Sakatsume M, Saito A, Ikegame M, Iino N, Shimada H, Ueno M, Kawashima H, Arakawa M, Gejyo F. Kidney Int. 2001;59:959–974. doi: 10.1046/j.1523-1755.2001.059003959.x. [DOI] [PubMed] [Google Scholar]
- 11.Forge A, Li L, Corwin JT, Nevill G. Science. 1993;259:1616–1619. doi: 10.1126/science.8456284. [DOI] [PubMed] [Google Scholar]
- 12.Warchol ME, Lambert PR, Goldstein BJ, Forge A, Corwin JT. Science. 1993;259:1619–1622. doi: 10.1126/science.8456285. [DOI] [PubMed] [Google Scholar]
- 13.Hiel H, Bennani H, Erre JP, Aurousseau C, Aran JM. Acta Oto-Laryngol. 1992;112:272–277. doi: 10.1080/00016489.1992.11665417. [DOI] [PubMed] [Google Scholar]
- 14.Hiel H, Schamel A, Erre JP, Hayashida T, Dulon D, Aran JM. Hear. Res. 1992;57:157–165. doi: 10.1016/0378-5955(92)90148-g. [DOI] [PubMed] [Google Scholar]
- 15.Imamura S, Adams JC. J. Assoc. Res. Oto-Laryngol. 2003;4:176–195. doi: 10.1007/s10162-002-2036-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kitahara T, Li HS, Balaban CD. Hear. Res. 2005;201:132–144. doi: 10.1016/j.heares.2004.09.007. [DOI] [PubMed] [Google Scholar]
- 17.Wang Q, Steyger PS. J. Assoc. Res. Oto-Laryngol. 2009;10:205–219. doi: 10.1007/s10162-009-0160-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dai CF, Steyger PS. Hear. Res. 2008;235:114–124. doi: 10.1016/j.heares.2007.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Forge A, Fradis M. Hear. Res. 1985;20:233–244. doi: 10.1016/0378-5955(85)90028-0. [DOI] [PubMed] [Google Scholar]
- 20.Kusunoki T, Cureoglu S, Schachern PA, Sampaio A, Fukushima H, Oktay MF, Paparella MM. Auris, Nasus, Larynx. 2004;31:383–388. doi: 10.1016/j.anl.2004.09.011. [DOI] [PubMed] [Google Scholar]
- 21.Rizzi MD, Hirose K. Current Opinion in Otolaryngology & Head and Neck Surgery. 2007;15:352–357. doi: 10.1097/MOO.0b013e3282ef772d. [DOI] [PubMed] [Google Scholar]
- 22.Durante-Mangoni E, Grammatikos A, Utili R, Falagas ME. Int. J. Antimicrob. Agents. 2009;33:201–205. doi: 10.1016/j.ijantimicag.2008.09.001. [DOI] [PubMed] [Google Scholar]
- 23.Fourmy D, Recht MI, Blanchard SC, Puglisi JD. Science. 1996;274:1367–1371. doi: 10.1126/science.274.5291.1367. [DOI] [PubMed] [Google Scholar]
- 24.Mingeot-Leclercq MP, Glupczynski Y, Tulkens PM. Antimicrob. Agents Chemother. 1999;43:727–737. doi: 10.1128/aac.43.4.727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dehne N, Rauen U, de Groot H, Lautermann J. Hear. Res. 2002;169:47–55. doi: 10.1016/s0378-5955(02)00338-6. [DOI] [PubMed] [Google Scholar]
- 26.Lee JE, Nakagawa T, Kim TS, Iguchi F, Endo T, Kita T, Murai N, Naito Y, Lee SH, Ito J. Acta Oto-Laryngol. 2004;551:69–74. doi: 10.1080/03655230310016799. [DOI] [PubMed] [Google Scholar]
- 27.Hirose K, Hockenbery DM, Rubel EW. Hear. Res. 1997;104:1–14. doi: 10.1016/s0378-5955(96)00169-4. [DOI] [PubMed] [Google Scholar]
- 28.Pirvola U, Xing-Qun L, Virkkala J, Saarma M, Murakata C, Camoratto AM, Walton KM, Ylikoski J. J. Neurosci. 2000;20:43–50. doi: 10.1523/JNEUROSCI.20-01-00043.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ding D, Stracher A, Salvi RJ. Hear. Res. 2002;164:115–126. doi: 10.1016/s0378-5955(01)00417-8. [DOI] [PubMed] [Google Scholar]
- 30.Hashino E, Shero M. Brain Res. 1995;704:135–140. doi: 10.1016/0006-8993(95)01198-6. [DOI] [PubMed] [Google Scholar]
- 31.Marcotti W, van Netten SM, Kros CJ. J. Physiol. 2005;567:505–521. doi: 10.1113/jphysiol.2005.085951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Steyger PS, Peters SL, Rehling J, Hordichok A, Dai CF. J. Assoc. Res. Oto-Laryngol. 2003;4:565–578. doi: 10.1007/s10162-003-4002-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Karasawa T, Wang Q, Fu Y, Cohen DM, Steyger PS. J. Cell Sci. 2008;121:2871–2879. doi: 10.1242/jcs.023705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hashino E, Shero M, Salvi RJ. Brain Res. 1997;777:75–85. doi: 10.1016/s0006-8993(97)00977-3. [DOI] [PubMed] [Google Scholar]
- 35.Jiang H, Sha SH, Forge A, Schacht J. Cell Death Differ. 2006;13:20–30. doi: 10.1038/sj.cdd.4401706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Priuska EM, Schacht J. Biochem. Pharmacol. 1995;50:1749–1752. doi: 10.1016/0006-2952(95)02160-4. [DOI] [PubMed] [Google Scholar]
- 37.Sha SH, Schacht J. Free Radical Biol. Med. 1999;26:341–347. doi: 10.1016/s0891-5849(98)00207-x. [DOI] [PubMed] [Google Scholar]
- 38.Lesniak W, Pecoraro VL, Schacht J. Chem. Res. Toxicol. 2005;18:357–364. doi: 10.1021/tx0496946. [DOI] [PubMed] [Google Scholar]
- 39.Genestra M. Cell. Signalling. 2007;19:1807–1819. doi: 10.1016/j.cellsig.2007.04.009. [DOI] [PubMed] [Google Scholar]
- 40.Chan PH. Ann. N. Y. Acad. Sci. 2005;1042:203–209. doi: 10.1196/annals.1338.022. [DOI] [PubMed] [Google Scholar]
- 41.Servais H, Ortiz A, Devuyst O, Denamur S, Tulkens PM, Mingeot-Leclercq MP. Apoptosis. 2008;13:11–32. doi: 10.1007/s10495-007-0151-z. [DOI] [PubMed] [Google Scholar]
- 42.Walker PD, Shah SV. Am. J. Physiol. 1987;253:C495–C499. doi: 10.1152/ajpcell.1987.253.4.C495. [DOI] [PubMed] [Google Scholar]
- 43.Murphy MP. Biochem. J. 2009;417:1–13. doi: 10.1042/BJ20081386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sha SH, Zajic G, Epstein CJ, Schacht J. Audiol. Neuro-Otol. 2001;6:117–123. doi: 10.1159/000046818. [DOI] [PubMed] [Google Scholar]
- 45.Davis RJ. Cell. Vol. 103. Cambridge, Mass.: 2000. pp. 239–252. [DOI] [PubMed] [Google Scholar]
- 46.Wang J, Van De Water TR, Bonny C, de Ribaupierre F, Puel JL, Zine A. J. Neurosci. 2003;23:8596–8607. doi: 10.1523/JNEUROSCI.23-24-08596.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Eshraghi AA, Wang J, Adil E, He J, Zine A, Bublik M, Bonny C, Puel JL, Balkany TJ, Van De Water TR. Hear. Res. 2007;226:168–177. doi: 10.1016/j.heares.2006.09.008. [DOI] [PubMed] [Google Scholar]
- 48.Nakashima S, Tohmatsu T, Shirato L, Takenaka A, Nozawa Y. Biochem. Biophys. Res. Commun. 1987;146:820–826. doi: 10.1016/0006-291x(87)90604-8. [DOI] [PubMed] [Google Scholar]
- 49.Schacht J. Archives of Oto-Rhino-Laryngology. 1979;224:129–134. doi: 10.1007/BF00455236. [DOI] [PubMed] [Google Scholar]
- 50.Lodhi S, Weiner ND, Mechigian I, Schacht J. Biochem. Pharmacol. 1980;29:597–601. doi: 10.1016/0006-2952(80)90382-2. [DOI] [PubMed] [Google Scholar]
- 51.Schanck A, Mingeot-Leclercq MP, Tulkens PM, Carrier D, Smith IC, Jarrell HC. Chem. Phys. Lipids. 1992;62:153–163. doi: 10.1016/0009-3084(92)90093-5. [DOI] [PubMed] [Google Scholar]
- 52.Knauss TC, Weinberg JM, Humes HD. Am. J. Physiol. 1983;244:F535–F546. doi: 10.1152/ajprenal.1983.244.5.F535. [DOI] [PubMed] [Google Scholar]
- 53.Ramsammy LS, Josepovitz C, Lane B, Kaloyanides GJ. Am. J. Physiol. 1989;256:C204–C213. doi: 10.1152/ajpcell.1989.256.1.C204. [DOI] [PubMed] [Google Scholar]
- 54.Sundin DP, Sandoval R, Molitoris BA. J. Am. Soc. Nephrol. 2001;12:114–123. doi: 10.1681/ASN.V121114. [DOI] [PubMed] [Google Scholar]
- 55.Beauchamp D, Laurent G, Maldague P, Abid S, Kishore BK, Tulkens PM. J. Pharmacol. Exp. Ther. 1990;255:858–866. [PubMed] [Google Scholar]
- 56.Kishore BK, Kallay Z, Lambricht P, Laurent G, Tulkens PM. J. Pharmacol. Exp. Ther. 1990;255:867–874. [PubMed] [Google Scholar]
- 57.Nouvian R, Ruel J, Wang J, Guitton MJ, Pujol R, Puel JL. Eur. J. Neurosci. 2003;17:2553–2562. doi: 10.1046/j.1460-9568.2003.02715.x. [DOI] [PubMed] [Google Scholar]
- 58.Leitner MG, Halaszovich CR, Oliver D. Mol. Pharmacol. 2011;79:51–60. doi: 10.1124/mol.110.068130. [DOI] [PubMed] [Google Scholar]
- 59.Logothetis DE, Petrou VI, Adney SK, Mahajan R. Pfluegers Arch. 2010;460:321–341. doi: 10.1007/s00424-010-0828-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hilgemann DW, Feng S, Nasuhoglu C. Science’s STKE. 2001;2001:re19. doi: 10.1126/stke.2001.111.re19. [DOI] [PubMed] [Google Scholar]
- 61.Moazed D, Noller HF. Nature. 1987;327:389–394. doi: 10.1038/327389a0. [DOI] [PubMed] [Google Scholar]
- 62.Purohit P, Stern S. Nature. 1994;370:659–662. doi: 10.1038/370659a0. [DOI] [PubMed] [Google Scholar]
- 63.Tommassen J. Microbiology. 2010;156:2587–2596. doi: 10.1099/mic.0.042689-0. [DOI] [PubMed] [Google Scholar]
- 64.Guan MX. Ann. N. Y. Acad. Sci. 2004;1011:259–271. doi: 10.1007/978-3-662-41088-2_25. [DOI] [PubMed] [Google Scholar]
- 65.Hutchin T, Haworth I, Higashi K, Fischel-Ghodsian N, Stoneking M, Saha N, Arnos C, Cortopassi G. Nucleic Acids Res. 1993;21:4174–4179. doi: 10.1093/nar/21.18.4174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Prezant TR, Agapian JV, Bohlman MC, Bu X, Oztas S, Qiu WQ, Arnos KS, Cortopassi GA, Jaber L, Rotter JI, et al. Nat. Genet. 1993;4:289–294. doi: 10.1038/ng0793-289. [DOI] [PubMed] [Google Scholar]
- 67.Hamasaki K, Rando RR. Biochemistry. 1997;36:12323–12328. doi: 10.1021/bi970962r. [DOI] [PubMed] [Google Scholar]
- 68.Estivill X, Govea N, Barcelo E, Badenas C, Romero E, Moral L, Scozzri R, D’Urbano L, Zeviani M, Torroni A. Am. J. Hum. Genet. 1998;62:27–35. doi: 10.1086/301676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zhao H, Li R, Wang Q, Yan Q, Deng JH, Han D, Bai Y, Young WY, Guan MX. Am. J. Hum. Genet. 2004;74:139–152. doi: 10.1086/381133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zhao L, Young WY, Li R, Wang Q, Qian Y, Guan MX. Biochem. Biophys. Res. Commun. 2004;325:1503–1508. doi: 10.1016/j.bbrc.2004.10.199. [DOI] [PubMed] [Google Scholar]
- 71.Li R, Xing G, Yan M, Cao X, Liu XZ, Bu X, Guan MX. Am. J. Med. Genet. 2004;124:113–117. doi: 10.1002/ajmg.a.20305. [DOI] [PubMed] [Google Scholar]
- 72.Guan MX, Fischel-Ghodsian N, Attardi G. Hum. Mol. Genet. 1996;5:963–971. doi: 10.1093/hmg/5.7.963. [DOI] [PubMed] [Google Scholar]
- 73.Hobbie SN, Akshay S, Kalapala SK, Bruell CM, Shcherbakov D, Bottger EC. Proc. Natl. Acad. Sci. U. S. A. 2008;105:20888–20893. doi: 10.1073/pnas.0811258106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wilschanski M, Famini C, Blau H, Rivlin J, Augarten A, Avital A, Kerem B, Kerem E. Am. J. Respir. Crit. Care Med. 2000;161:860–865. doi: 10.1164/ajrccm.161.3.9904116. [DOI] [PubMed] [Google Scholar]
- 75.Clancy JP, Bebok Z, Ruiz F, King C, Jones J, Walker L, Greer H, Hong J, Wing L, Macaluso M, Lyrene R, Sorscher EJ, Bedwell DM. Am. J. Respir. Crit. Care Med. 2001;163:1683–1692. doi: 10.1164/ajrccm.163.7.2004001. [DOI] [PubMed] [Google Scholar]
- 76.Linde L, Kerem B. Trends Genet. 2008;24:552–563. doi: 10.1016/j.tig.2008.08.010. [DOI] [PubMed] [Google Scholar]
- 77.Karasawa T, Wang Q, David LL, Steyger PS. Cell Death and Disease. 2010;1:e102. doi: 10.1038/cddis.2010.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Moestrup SK, Cui S, Vorum H, Bregengard C, Bjorn SE, Norris K, Gliemann J, Christensen EI. J. Clin. Invest. 1995;96:1404–1413. doi: 10.1172/JCI118176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Nagai J, Tanaka H, Nakanishi N, Murakami T, Takano M. Am. J. Physiol. Renal Physiol. 2001;281:F337–F344. doi: 10.1152/ajprenal.2001.281.2.F337. [DOI] [PubMed] [Google Scholar]
- 80.Girton RA, Sundin DP, Rosenberg ME. Am. J. Physiol. Renal Physiol. 2002;282:F703–F709. doi: 10.1152/ajprenal.00060.2001. [DOI] [PubMed] [Google Scholar]
- 81.Mizuta K, Saito A, Watanabe T, Nagura M, Arakawa M, Shimizu F, Hoshino T. Hear. Res. 1999;129:83–91. doi: 10.1016/s0378-5955(98)00221-4. [DOI] [PubMed] [Google Scholar]
- 82.Miyazaki T, Sagawa R, Honma T, Noguchi S, Harada T, Komatsuda A, Ohtani H, Wakui H, Sawada K, Otaka M, Watanabe S, Jikei M, Ogawa N, Hamada F, Itoh H. J. Biol. Chem. 2004;279:17295–17300. doi: 10.1074/jbc.M312217200. [DOI] [PubMed] [Google Scholar]
- 83.Horibe T, Matsui H, Tanaka M, Nagai H, Yamaguchi Y, Kato K, Kikuchi M. Biochem. Biophys. Res. Commun. 2004;323:281–287. doi: 10.1016/j.bbrc.2004.08.099. [DOI] [PubMed] [Google Scholar]
- 84.Hartl FU. Nature. 1996;381:571–579. doi: 10.1038/381571a0. [DOI] [PubMed] [Google Scholar]
- 85.Muller E, Neuhofer W, Ohno A, Rucker S, Thurau K, Beck FX. Pfluegers Arch. 1996;431:608–617. doi: 10.1007/BF02191910. [DOI] [PubMed] [Google Scholar]
- 86.Taleb M, Brandon CS, Lee FS, Lomax MI, Dillmann WH, Cunningham LL. J. Assoc. Res. Oto-Laryngol. 2008;9:277–289. doi: 10.1007/s10162-008-0122-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Nylandsted J, Gyrd-Hansen M, Danielewicz A, Fehrenbacher N, Lademann U, Hoyer-Hansen M, Weber E, Multhoff G, Rohde M, Jaattela M. J. Exp. Med. 2004;200:425–435. doi: 10.1084/jem.20040531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Kroemer G, Jaattela M. Nat. Rev. Cancer. 2005;5:886–897. doi: 10.1038/nrc1738. [DOI] [PubMed] [Google Scholar]
- 89.Fliegel L, Burns K, MacLennan DH, Reithmeier RA, Michalak M. J. Biol. Chem. 1989;264:21522–21528. [PubMed] [Google Scholar]
- 90.Sandoval RM, Molitoris BA. Am. J. Physiol.: Renal Physiol. 2004;286:F617–F624. doi: 10.1152/ajprenal.00130.2003. [DOI] [PubMed] [Google Scholar]
- 91.Williams DB. J. Cell Sci. 2006;119:615–623. doi: 10.1242/jcs.02856. [DOI] [PubMed] [Google Scholar]
- 92.Coling DE, Ding D, Young R, Lis M, Stofko E, Blumenthal KM, Salvi RJ. Hear. Res. 2007;226:140–156. doi: 10.1016/j.heares.2006.12.017. [DOI] [PubMed] [Google Scholar]
- 93.Hashino E, Shero M, Salvi RJ. Brain Res. 2000;887:90–97. doi: 10.1016/s0006-8993(00)02971-1. [DOI] [PubMed] [Google Scholar]
- 94.Gale JE, Marcotti W, Kennedy HJ, Kros CJ, Richardson GP. J. Neurosci. 2001;21:7013–7025. doi: 10.1523/JNEUROSCI.21-18-07013.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Zhou J, Shao Z, Kerkela R, Ichijo H, Muslin AJ, Pombo C, Force T. Mol. Cell. Biol. 2009;29:4167–4176. doi: 10.1128/MCB.01067-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Dai CF, Mangiardi D, Cotanche DA, Steyger PS. Hear. Res. 2006;213:64–78. doi: 10.1016/j.heares.2005.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Matsui JI, Gale JE, Warchol ME. J. Neurobiol. 2004;61:250–266. doi: 10.1002/neu.20054. [DOI] [PubMed] [Google Scholar]
- 98.Albinger-Hegyi A, Hegyi I, Nagy I, Bodmer M, Schmid S, Bodmer D. Neuroscience. 2006;137:971–980. doi: 10.1016/j.neuroscience.2005.10.010. [DOI] [PubMed] [Google Scholar]
- 99.Cunningham LL, Cheng AG, Rubel EW. J. Neurosci. 2002;22:8532–8540. doi: 10.1523/JNEUROSCI.22-19-08532.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Cheng AG, Cunningham LL, Rubel EW. J. Assoc. Res. Oto-Laryngol. 2003;4:91–105. doi: 10.1007/s10162-002-3016-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Servais H, Van Der Smissen P, Thirion G, Van der Essen G, Van Bambeke F, Tulkens PM, Mingeot-Leclercq MP. Toxicol. Appl. Pharmacol. 2005;206:321–333. doi: 10.1016/j.taap.2004.11.024. [DOI] [PubMed] [Google Scholar]
- 102.Lazebnik YA, Kaufmann SH, Desnoyers S, Poirier GG, Earnshaw WC. Nature. 1994;371:346–347. doi: 10.1038/371346a0. [DOI] [PubMed] [Google Scholar]
- 103.Kirsch DG, Doseff A, Chau BN, Lim DS, de Souza-Pinto NC, Hansford R, Kastan MB, Lazebnik YA, Hardwick JM. J. Biol. Chem. 1999;274:21155–21161. doi: 10.1074/jbc.274.30.21155. [DOI] [PubMed] [Google Scholar]
- 104.Mangiardi DA, McLaughlin-Williamson K, May KE, Messana EP, Mountain DC, Cotanche DA. J. Comp. Neurol. 2004;475:1–18. doi: 10.1002/cne.20129. [DOI] [PubMed] [Google Scholar]
- 105.Matsui JI, Haque A, Huss D, Messana EP, Alosi JA, Roberson DW, Cotanche DA, Dickman JD, Warchol ME. J. Neurosci. 2003;23:6111–6122. doi: 10.1523/JNEUROSCI.23-14-06111.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Tait SW, Green DR. Oncogene. 2008;27:6452–6461. doi: 10.1038/onc.2008.311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Lecain E, Omri B, Behar-Cohen F, Tran Ba Huy P, Crisanti P. Apoptosis. 2007;12:333–342. doi: 10.1007/s10495-006-0580-0. [DOI] [PubMed] [Google Scholar]
- 108.Golstein P, Kroemer G. Trends Biochem. Sci. 2007;32:37–43. doi: 10.1016/j.tibs.2006.11.001. [DOI] [PubMed] [Google Scholar]
- 109.Hirose K, Westrum LE, Stone JS, Zirpel L, Rubel EW. Ann. N. Y. Acad. Sci. 1999;884:389–409. doi: 10.1111/j.1749-6632.1999.tb08657.x. [DOI] [PubMed] [Google Scholar]
- 110.Kuwana T, Newmeyer DD. Curr. Opin. Cell Biol. 2003;15:691–699. doi: 10.1016/j.ceb.2003.10.004. [DOI] [PubMed] [Google Scholar]
- 111.Lindsten T, Zong WX, Thompson CB. Neuroscientist. 2005;11:10–15. doi: 10.1177/1073858404269267. [DOI] [PubMed] [Google Scholar]
- 112.Lalier L, Cartron PF, Juin P, Nedelkina S, Manon S, Bechinger B, Vallette FM. Apoptosis. 2007;12:887–896. doi: 10.1007/s10495-007-0749-1. [DOI] [PubMed] [Google Scholar]
- 113.Iverson SL, Orrenius S. Arch. Biochem. Biophys. 2004;423:37–46. doi: 10.1016/j.abb.2003.12.002. [DOI] [PubMed] [Google Scholar]
- 114.Scorrano L, Korsmeyer SJ. Biochem. Biophys. Res. Commun. 2003;304:437–444. doi: 10.1016/s0006-291x(03)00615-6. [DOI] [PubMed] [Google Scholar]
- 115.Cunningham LL, Matsui JI, Warchol ME, Rubel EW. J. Neurobiol. 2004;60:89–100. doi: 10.1002/neu.20006. [DOI] [PubMed] [Google Scholar]
- 116.Kashio A, Sakamoto T, Suzukawa K, Asoh S, Ohta S, Yamasoba T. J. Neurosci. Res. 2007;85:1403–1412. doi: 10.1002/jnr.21260. [DOI] [PubMed] [Google Scholar]
- 117.Pfannenstiel SC, Praetorius M, Plinkert PK, Brough DE, Staecker H. Audiol. Neuro-Otol. 2009;14:254–266. doi: 10.1159/000192953. [DOI] [PubMed] [Google Scholar]
- 118.Coffin A, Raible D, Rubel E. Association for Research in Otolaryngology Abstracts. 2011:#785. [Google Scholar]
- 119.Owens KN, Coffin AB, Hong LS, Bennett KO, Rubel EW, Raible DW. Hear. Res. 2009;253:32–41. doi: 10.1016/j.heares.2009.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Wu WJ, Sha SH, McLaren JD, Kawamoto K, Raphael Y, Schacht J. Hear. Res. 2001;158:165–178. doi: 10.1016/s0378-5955(01)00303-3. [DOI] [PubMed] [Google Scholar]
- 121.Lautermann J, McLaren J, Schacht J. Hear. Res. 1995;86:15–24. doi: 10.1016/0378-5955(95)00049-a. [DOI] [PubMed] [Google Scholar]
- 122.Dallos P, Harris D. J. Neurophysiol. 1978;41:365–383. doi: 10.1152/jn.1978.41.2.365. [DOI] [PubMed] [Google Scholar]
- 123.Ohtani I, Ohtsuki K, Aikawa T, Omata T, Ouchi J, Saito T. Acta Oto-Laryngol. 1982;94:413–419. doi: 10.3109/00016488209128929. [DOI] [PubMed] [Google Scholar]
- 124.Wu WJ, Sha SH, Schacht J. Audiol. Neuro-Otol. 2002;7:171–174. doi: 10.1159/000058305. [DOI] [PubMed] [Google Scholar]
- 125.Hordichok AJ, Steyger PS. Hear. Res. 2007;232:1–19. doi: 10.1016/j.heares.2007.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Nin F, Hibino H, Doi K, Suzuki T, Hisa Y, Kurachi Y. Proc. Natl. Acad. Sci. U. S. A. 2008;105:1751–1756. doi: 10.1073/pnas.0711463105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Sunose H, Liu J, Marcus DC. Hear. Res. 1997;114:107–116. doi: 10.1016/s0378-5955(97)00152-4. [DOI] [PubMed] [Google Scholar]
- 128.Wangemann P, Liu J, Marcus DC. Hear. Res. 1995;84:19–29. doi: 10.1016/0378-5955(95)00009-s. [DOI] [PubMed] [Google Scholar]
- 129.Mostafa BE, Tawfik S, Hefnawi NG, Hassan MA, Ismail FA. Acta Oto-Laryngol. 2007;127:234–239. doi: 10.1080/00016480600794495. [DOI] [PubMed] [Google Scholar]
- 130.Conlon BJ, Aran JM, Erre JP, Smith DW. Hear. Res. 1999;128:40–44. doi: 10.1016/s0378-5955(98)00195-6. [DOI] [PubMed] [Google Scholar]
- 131.Sha SH, Schacht J. Hear. Res. 2000;142:34–40. doi: 10.1016/s0378-5955(00)00003-4. [DOI] [PubMed] [Google Scholar]
- 132.Sha SH, Schacht J. Lab. Invest. 1999;79:807–813. [PubMed] [Google Scholar]
- 133.Sha SH, Qiu JH, Schacht J. N. Engl. J. Med. 2006;354:1856–1857. doi: 10.1056/NEJMc053428. [DOI] [PubMed] [Google Scholar]
- 134.McFadden SL, Ding D, Salvemini D, Salvi RJ. Toxicol. Appl. Pharmacol. 2003;186:46–54. doi: 10.1016/s0041-008x(02)00017-0. [DOI] [PubMed] [Google Scholar]
- 135.Nishida I, Takumida M. ORL J. Oto-Rhino-Laryngol. Relat. Spec. 1996;58:68–73. doi: 10.1159/000276801. [DOI] [PubMed] [Google Scholar]
- 136.Palodetto B, Postal M, Grignoli CR, Sartorato EL, Oliveira CA. Braz. J. Oto-Rhino-Laryngol. 2010;76:306–309. doi: 10.1590/S1808-86942010000300006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Myrdal SE, Steyger PS. Hear. Res. 2005;204:170–182. doi: 10.1016/j.heares.2005.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Kalinec GM, Fernandez-Zapico ME, Urrutia R, Esteban-Cruciani N, Chen S, Kalinec F. Proc. Natl. Acad. Sci. U. S. A. 2005;102:16019–16024. doi: 10.1073/pnas.0508053102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Knauer SK, Heinrich U-R, Bier C, Habtemichael N, Docter D, Helling K, Mann WJ, Stauber RH. Cell Death and Disease. 2010;1:e51. doi: 10.1038/cddis.2010.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Ou HC, Cunningham LL, Francis SP, Brandon CS, Simon JA, Raible DW, Rubel EW. J. Assoc. Res. Oto-Laryngol. 2009;10:191–203. doi: 10.1007/s10162-009-0158-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Ou HC, Santos F, Raible DW, Simon JA, Rubel EW. Drug Discovery Today. 2010;15:265–271. doi: 10.1016/j.drudis.2010.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Karasawa T, Wang Q, David LL, S P. Steyger, Toxicol. Sci. 2011 doi: 10.1093/toxsci/kfr196. (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Alharazneh A, Luk L, Huth M, Monfared A, Steyger PS, Cheng A, Ricci A. PLoS One. 2011 doi: 10.1371/journal.pone.0022347. (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]