

Abstract
Objective:
To assess eyes with neuromyelitis optica (NMO) for morphologic retinal abnormalities utilizing high-definition optical coherence tomography (OCT) imaging.
Methods:
In this cross-sectional study, 39 patients with NMO spectrum disorders and 39 age- and sex-matched healthy controls underwent spectral-domain OCT and visual function testing.
Results:
Microcystic macular edema (MME) of the inner nuclear layer (INL) was identified in 10 of 39 patients (26%) and was exclusively found in eyes with a history of optic neuritis (ON). MME eyes had lower high- and low-contrast letter-acuity scores (100%: p = 0.002; 2.5%: p = 0.002; 1.25%: p = 0.004), lower peripapillary retinal nerve fiber layer (RNFL) thickness (p = 0.04), lower macular RNFL thickness (p = 0.004), lower ganglion cell layer + inner plexiform layer (GCIP) thickness (p = 0.007), higher INL thickness (p < 0.001), and a greater number of ON episodes (p = 0.008) relative to non-MME eyes with a history of ON. After adjusting for history of multiple ON episodes, these findings remained significant for macular-RNFL thickness (p = 0.03), INL thickness (p < 0.001), and 100% and 2.5% contrast letter-acuity scores (p = 0.008 and p = 0.03, respectively). NMO spectrum eyes without ON history had lower macular RNFL thickness (p = 0.003), GCIP thickness (p = 0.002), outer nuclear layer thickness (p = 0.02), and low-contrast letter-acuity scores (2.5%: p = 0.03; 1.25%: p = 0.002) compared to healthy controls.
Conclusions:
We have identified a pattern of retinal morphologic abnormalities in NMO that is associated with severe retinal axonal and neuronal loss and corresponding visual disability. MME may contribute to poor visual outcomes following NMO-associated ON or alternatively represent a marker of ON severity. Additionally, our results support that subclinical involvement of the anterior visual pathway may occur in NMO spectrum disorders.
Neuromyelitis optica (NMO) is an inflammatory disorder of the CNS, the cardinal manifestations of which are optic neuritis (ON) and longitudinally extensive transverse myelitis (LETM). Autoantibodies (NMO–immunoglobulin G [IgG]) targeting aquaporin-4 are found in the sera of the majority of patients with NMO.1,2
NMO-associated ON is characterized by poor visual outcomes, often resulting in blindness.3,4 Studies utilizing optical coherence tomography (OCT) have identified profound retinal axonal and neuronal loss in NMO-ON eyes, primarily thought to represent sequelae of optic nerve injury.5–11 However, abnormalities of the retinal vasculature have also been identified in vivo following NMO-associated ON, suggesting that direct retinal vascular injury may also play a role.9 Additionally, subclinical involvement of the visual pathway has been suggested to occur in NMO-spectrum disorders, but data are conflicting.5–8,10,11
OCT studies in NMO have primarily utilized older, time-domain OCT,5–9 and studies employing modern, high-definition, spectral-domain OCT have focused solely on quantitative measures.10,11 Spectral-domain OCT renders high-resolution images (3–5 μm) and enables accurate visualization of retinal morphologic abnormalities. Utilizing spectral-domain OCT, we identified retinal abnormalities in the eyes of patients with NMO, namely microcystic macular edema (MME) of the inner nuclear layer (INL), an entity recently reported in a subset of patients with multiple sclerosis (MS).12,13 We proceeded to evaluate a cohort of patients with NMO-spectrum disorders for MME and other retinal abnormalities with OCT, and to examine associations with quantitative OCT measures, visual dysfunction, and ambulatory disability. Additionally, as a secondary objective, we sought to determine if subclinical retinal axonal and neuronal loss occurs in NMO-spectrum disorders.
METHODS
Standard protocol approvals, registrations, and patient consents.
Johns Hopkins University institutional review board approval was obtained for the study protocol and written informed consent was obtained from all participants prior to study enrollment.
Study participants and clinical data.
Patients with NMO-spectrum disorders14 were recruited from the Johns Hopkins Neuromyelitis Optica, Transverse Myelitis and MS Clinics, by unselected convenience sampling, and met a diagnosis of “definite NMO,” as defined by the revised 2006 diagnostic criteria by Wingerchuk et al.,15 or were NMO-IgG seropositive (Mayo Medical Laboratories or Athena Diagnostics) with a history of ON or LETM. Healthy controls (HCs) were recruited from among patients' families and Johns Hopkins University staff.
Subjects with diabetes, history of ocular surgery/trauma, glaucoma, or other ophthalmologic disorders were excluded from the study. Additionally, eyes within 3 months of acute ON were excluded from quantitative analyses, to minimize the effect of ON-related edema on OCT measurements.10,16
History of ON (including number of ON episodes per eye) was determined by patient self-report and confirmed by review of medical records. Mobility status was also recorded (unassisted, unilateral assistance, bilateral assistance, or uses wheelchair) by the treating physician.
Optical coherence tomography.
Retinal imaging was performed with spectral-domain OCT (Cirrus HD-OCT, Model 4000, Software version 5.0; Carl Zeiss Meditec, Dublin, CA), as described in detail elsewhere.17 Briefly, peripapillary and macular data scans were obtained with the Optic Disc Cube 200 × 200 and Macular Cube 512 × 128 protocols, respectively. Scans with signal strength <7/10, or with artifact, were excluded from the study. For eyes with extremely poor visual function (unable to fixate), OCT scans were acquired with external fixation of the fellow eye.
All acquired macular cube scans were qualitatively assessed for MME, as well as other abnormalities of the retina or vitreoretinal interface,18 by 2 reviewers masked to clinical status (E.S.S. and S.S.). MME was defined as cystic, lacunar areas of hyporeflectivity with clear boundaries, evident on at least 2 contiguous B-scans, or visible in a comparable region on at least 2 separate acquisitions. The reviewers agreed in all cases of MME. All scans designated as fulfilling MME criteria, as well as scans designated as demonstrating other morphologic abnormalities, were also reviewed and verified by a retinal specialist, masked to clinical status (Q.D.N.).
Macular cube scans were segmented utilizing an automated 3D segmentation algorithm, as described in detail elsewhere.10,17 Briefly, the segmentation software yields the thicknesses of the following retinal layers: 1) macular–retinal nerve fiber layer (RNFL); 2) ganglion cell layer + inner plexiform layer (GCIP); 3) INL + outer plexiform layer; 4) outer nuclear layer (ONL; including inner and outer photoreceptor segments). This segmentation protocol has been shown to be highly reproducible in MS and HCs (inter-rater intraclass correlation coefficients: 0.91–0.99 for all layers).17
Visual function.
Monocular visual acuity was assessed using standardized retroilluminated eye charts (Precision Vision, La Salle, IL). High-contrast (100%) Early Treatment Diabetic Retinopathy Study and low-contrast (2.5% and 1.25%) Sloan Letter charts were used. The total number of correct letters identified on each chart was recorded to determine letter-acuity scores for each contrast level (maximum score of 70 letters).
Statistical methods.
Statistical analyses were performed with Stata version 11 (StataCorp, College Station, TX). The Shapiro-Wilk test was used to evaluate the normality of distributions. Comparisons between groups were performed with the Student t test (for age; normally distributed), the Wilcoxon rank sum test (for number of ON episodes and time elapsed from initial ON episode; these variables were not normally distributed), and the χ2 test (for ON history, sex, mobility status, and NMO-IgG seropositivity). OCT measures and letter-acuity scores were compared between groups utilizing multivariate mixed-effects linear regression models including age and sex, and accounting for within-subject intereye correlations. Comparisons of MME and non-MME eyes with a history of ON were performed additionally adjusting for a history of multiple ON episodes, utilizing either the number of ON episodes or a binary indicator of single vs multiple episodes of ON. Results did not differ between these 2 methods, although the latter approach is more appropriate, since the number of ON episodes was not associated linearly with quantitative OCT measures and letter-acuity scores, and because this approach minimizes the effect of outlying values. Statistical significance was defined as p < 0.05.
RESULTS
Study population.
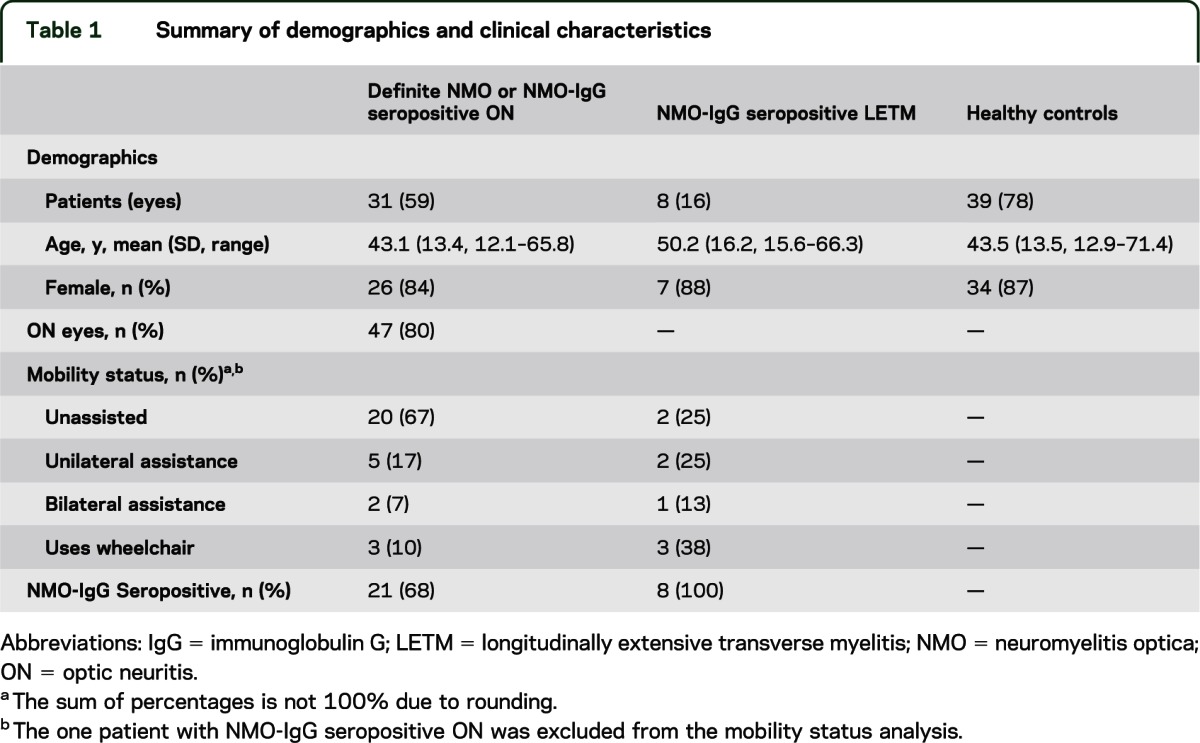
Thirty-nine patients with NMO-spectrum disorders (30 definite NMO, 1 NMO-IgG seropositive ON, and 8 NMO-IgG seropositive LETM) and 39 age- and sex-matched HCs participated in the study (table 1). OCT scan acquisition failed in 3 NMO eyes with ON history, because fixation was not possible due to poor visual function (external fixation of the fellow eye was also attempted but scan quality did not meet inclusion criteria). Two eyes were scanned within 3 months of acute ON and were reviewed qualitatively (no retinal abnormalities were noted), but were excluded from OCT measure and visual function analyses. None of the patients had a history of uveitis. Two patients with definite NMO had concomitant systemic rheumatologic disease (1 patient had a diagnosis of systemic lupus erythematosus [SLE], and 1 patient had a diagnosis of both SLE and Sjögren syndrome).
Table 1.
Summary of demographics and clinical characteristics
Characteristics of patients and eyes exhibiting microcystic macular edema.
MME (figure 1) was identified in 10 patients (14 eyes; MME was bilateral in 4 patients), all of whom had a diagnosis of definite NMO (26% of the entire NMO-spectrum cohort, 33% of the definite NMO patients). One patient with unilateral MME had a concomitant diagnosis of SLE and one patient with bilateral MME presented 2 months after the study visit with unilateral acute anterior uveitis. In general, the microcysts exhibited a patchy distribution throughout the macula, and were predominantly localized to the INL, although in one eye the microcysts extended to the ONL (figure 1). An epiretinal membrane, without associated vitreomacular traction, was identified in one eye with MME. Ophthalmologic examinations of 5 patients with MME (8 MME eyes) were performed at the Wilmer Eye Institute, including slit-lamp examination and funduscopy, and were unremarkable for abnormalities other than optic atrophy and disc pallor. Therefore, it appears that routine ophthalmologic examination is insensitive for detection of MME. No retinal abnormalities were found in HC eyes on OCT.
Figure 1. Microcystic macular edema of the inner nuclear layer identified by spectral-domain optical coherence tomography.

Images from 5 different eyes with microcystic macular edema (MME) of 5 individual patients. (A, B) Images from the same eye, traversing the foveal dip (A) and the inferior macula (B). MME is predominantly localized to the inner nuclear layer (INL) (red arrows), but extension to the outer nuclear layer is also noted (A; gray arrow). Vessel artifacts (black arrows) are demonstrated for comparison. (C, D) Images from the same eye, traversing the superior edge of the foveal dip (C) and the superior macula (D). MME of the INL is visible (red arrows). Vessel artifacts (black arrows) are demonstrated for comparison. (E, F) Images from the same eye are contiguous B-scans from the same macular cube scan (spaced ∼47 μm apart). MME of the INL (red arrows) is visible in the same area on both scans. (G, H) Images from 2 different patients demonstrate diffuse (G) and localized (H) MME of the INL (red arrows).
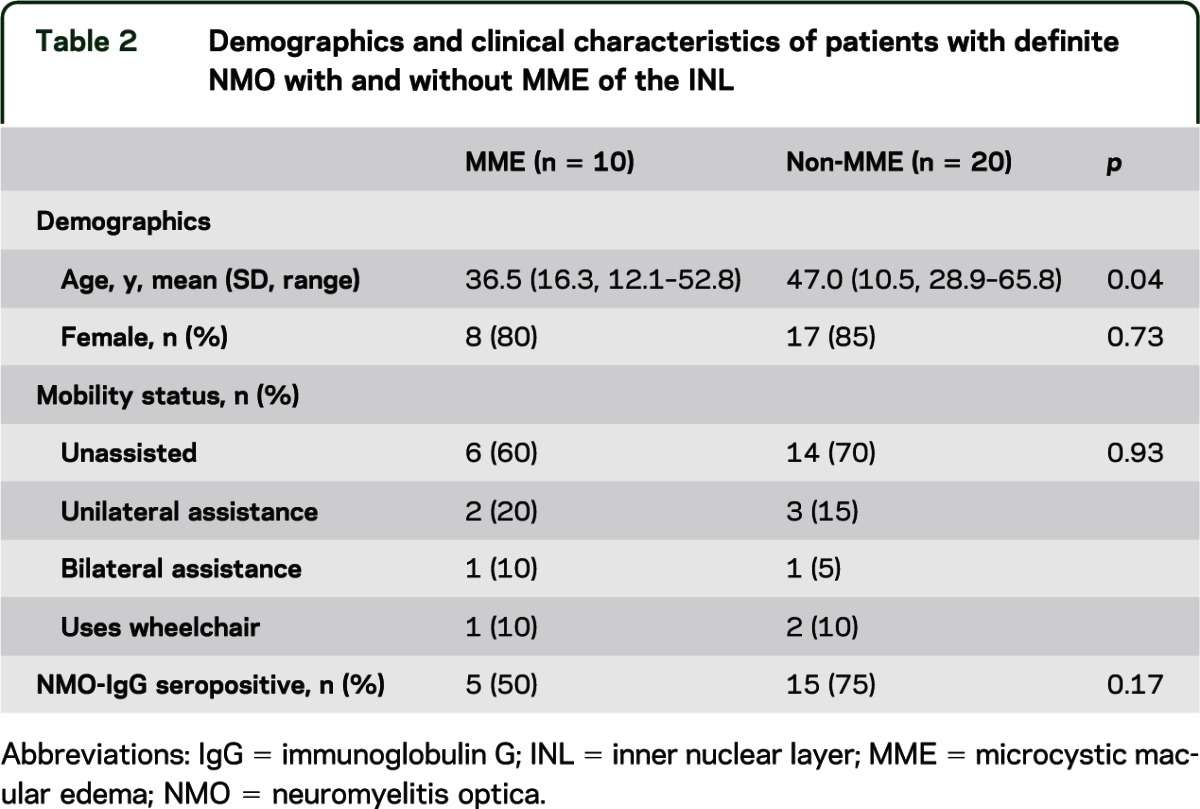
All eyes with MME had a history of ON and the prevalence of ON history was greater than among non-MME eyes of patients with definite NMO or NMO-IgG seropositive ON (100% vs 73%; p = 0.03). MME was present in 14 of the 47 eyes with ON history (30%). Sex, NMO-IgG seropositivity, and mobility status did not differ between patients with definite NMO with and without MME, but the age of MME patients was lower (table 2).
Table 2.
Demographics and clinical characteristics of patients with definite NMO with and without MME of the INL
MME eyes had lower high- and low-contrast letter-acuity scores (100%: p = 0.002; 2.5%: p = 0.002; 1.25%: p = 0.004), lower peripapillary RNFL thickness (p = 0.04), lower macular RNFL thickness (p = 0.004), lower GCIP thickness (p = 0.007), higher INL thickness (p < 0.001), and a greater number of previous ON episodes (p = 0.008) relative to non-MME eyes with a history of ON (table 3, figure 2). After adjusting for history of multiple ON episodes, these findings remained significant for macular-RNFL thickness (p = 0.03), INL thickness (p < 0.001), and 100% and 2.5% contrast letter-acuity scores (p = 0.008 and p = 0.03, respectively). Furthermore, after additionally adjusting letter-acuity score comparisons for peripapillary RNFL thickness, MME was no longer associated with decreased low-contrast letter-acuity (2.5%: p = 0.71; 1.25%: p = 0.69) and trended to significance for high-contrast letter-acuity (p = 0.08; adjusted mean difference = −13 letters). Time elapsed from the initial ON episode to the time of study did not differ between the eyes with and without MME (p = 0.56).
Table 3.
Comparisons of OCT measures and letter-acuity scores between groups
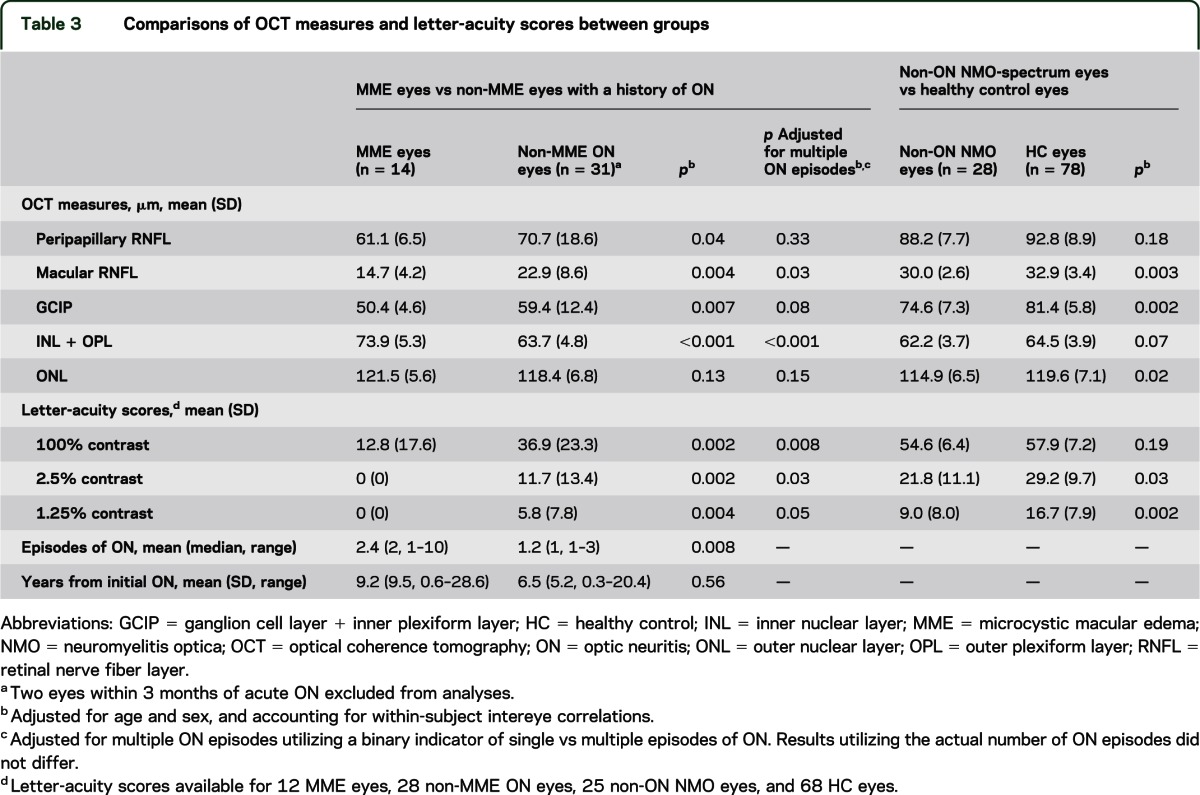
Figure 2. Boxplots of retinal layer thicknesses.

The p values were calculated using mixed-effects linear regression models including age and sex, and accounting for within-subject intereye correlations. GCIP = ganglion cell layer + inner plexiform layer; INL = inner nuclear layer; MME = microcystic macular edema; ON = optic neuritis; RNFL = retinal nerve fiber layer.
Comparisons of OCT and visual function measures between healthy controls and NMO-spectrum eyes without a history of ON.
Eyes without ON history of patients with NMO-spectrum disorders (unaffected eyes of patients with a history of ON and eyes of patients with NMO-IgG seropositive LETM) had lower macular-RNFL thickness (p = 0.003), lower GCIP thickness (p = 0.002), lower ONL thickness (p = 0.02), and lower low-contrast letter-acuity scores (2.5%: p = 0.03; 1.25%: p = 0.002), relative to HC eyes (table 3, figure 2).
DISCUSSION
In this study, MME of the INL was found to be common in NMO and was restricted to eyes with a history of ON. MME eyes exhibited greater decreases in RNFL (axonal) and GCIP (neuronal) thicknesses, and had experienced a greater number of previous ON episodes, relative to non-MME eyes affected by NMO-associated ON. Importantly, MME was associated with visual dysfunction, independently of the occurrence of multiple ON episodes, suggesting that MME may directly contribute to poor visual outcomes following ON in NMO, or alternatively represent a marker of ON severity. Furthermore, after adjusting for peripapillary RNFL thickness, MME was associated with decreased letter-acuity scores at high contrast (although this was not statistically significant, most probably due to sample size issues), adding further evidence that MME may possibly contribute to poor visual outcomes independently of optic nerve damage. Additionally, our results support that subclinical involvement of the anterior visual pathway may occur in NMO-spectrum disorders, as evidenced by decreased low-contrast letter-acuity scores and quantitative OCT abnormalities detected in NMO-spectrum eyes without a history of symptomatic ON.6,10,11
Recently, MME of the INL was reported to occur in a subset of patients with MS (∼5%), was more common in eyes with a history of ON, and was associated with visual and ambulatory disability.12,13 Our study's results demonstrate that the prevalence of MME in definite NMO may be much higher than in MS (33% vs ∼5%). Moreover, MME in NMO appears to be more closely linked with ON, with MME in our study being exclusively identified in eyes with a history of symptomatic ON. However, an important caveat is that the high prevalence of ON in NMO may hinder our ability to draw definitive conclusions regarding the absoluteness of this relationship, and it is possible that evaluating a larger NMO cohort may lead to the detection of MME in non-ON eyes.
Macular edema may result from multiple potential etiologies, including diabetes, retinal vein occlusion, uveitis, advanced vitreomacular traction, and intraocular surgery.19 Compromise of the blood–retinal barrier is a major pathophysiologic mechanism underlying the development of macular edema. The plexiform layers that form the boundaries of the INL act as diffusion barriers, favoring cyst formation in the INL when leakage occurs from the inner retinal vascular plexuses.20 Fluid accumulation is thought to occur primarily in the extracellular compartment; however, swelling of the Müller glial cells (the cell bodies of which are located in the INL) has also been suggested to contribute significantly, with cysts being formed by swollen and dying Müller cells.19
The pathophysiologic mechanism underlying the development of MME in NMO is unclear, but it is plausible that a primary retinal inflammatory process is operative, leading to breakdown of the blood–retinal barrier. In a study of eyes with macular edema, 25% of eyes with diffuse fluorescein leakage, a marker of breakdown of the blood–retinal barrier, were found to have microcysts present in the INL on OCT.21 Abnormalities of the retinal vasculature have been observed in vivo in NMO,9 and pathologic studies have identified prominent vascular hyalinization and perivascular inflammatory cell infiltration and immunoglobulin deposition in NMO lesions of the spinal cord and optic nerves/chiasm.22 Interestingly, aquaporin-4 is highly expressed in the retina by Müller glial cells and astrocytes, especially in end-feet membranes facing blood vessels.23 Additionally, aquaporin-4 deletion in a murine model results in a decreased capability of the Müller cells to withstand osmotic stress, and also induces retinal inflammation.24 Therefore, an immune response targeting aquaporin-4 in the retina is a possible explanation of our findings. The fact that NMO-IgG seropositivity was not more common in patients demonstrating MME does not mitigate this hypothesis, since a larger sample size may be necessary to detect such a relationship. Also, improvements in assay sensitivity have led to the detection of antibodies targeting aquaporin-4 in sera of patients considered to have seronegative disease.25 Alternatively, other antigens may be the target of the presumed autoimmune process, a hypothesis consistent with the frequent detection of other serum autoantibodies in NMO, and the overlap of NMO with systemic rheumatologic diseases, of which uveitis is a prominent manifestation.26,27 Furthermore, it is possible that, in the absence of comprehensive ophthalmologic evaluation, episodes of uveitis in patients with NMO may be unrecognized and misdiagnosed as ON. Interestingly, one of the patients demonstrating bilateral MME presented shortly after the study visit with acute anterior uveitis.
Another pathobiologic explanation for the development of the microcystic pathology characterized in our study is that such morphologic changes represent the consequence of a retrograde process extending to the INL, secondary to optic nerve injury and ganglion cell loss. This may either relate purely to transsynaptic degeneration of cells within the INL or a dysregulated glial inflammatory response induced secondary to retinal ganglion cell death due to retrograde degeneration, or a combination of both. Indeed, 2 pathologic studies have reported cystic changes in the INL following lesions of the pregeniculate visual pathway in animals and humans,28,29 but studies investigating retinal changes following optic nerve lesions have failed to demonstrate that retrograde transsynaptic degeneration occurs in the INL.30–33 Glial cell activation in response to neuronal injury is a tightly regulated response that may occur early, and exert variable effects depending on the underlying molecular signals.34–36 Additionally, in response to optic nerve injury, retinal microglial cells become activated, proliferate, and phagocytose cellular debris from degenerating retinal ganglion cells.34,37 This activation of the retinal microglia is not restricted to the ganglion cell layer, but has been shown to also occur in the microglial cells located in the inner INL and inner plexiform layer.37 OCT studies evaluating other optic neuropathies for the presence of INL pathology are important to exclude or confirm a transsynaptic etiology of our findings, although it is possible that OCT may lack the sensitivity to detect subtle changes in conditions associated with less severe optic nerve damage.38–40
Additionally, our investigations have identified decreased thicknesses of the macular-RNFL, GCIP, and ONL, as well as low-contrast visual loss, in NMO-spectrum eyes without a history of symptomatic acute ON. Although the pathoetiology of these abnormalities remains to be determined, a plausible hypothesis is that these changes relate to subclinical optic neuropathy or to other processes similar to those outlined above. Importantly, peripapillary RNFL thickness was not significantly lower in non-ON eyes relative to healthy controls, in contrast to the macular-RNFL and GCIP thicknesses. This indicates that macular segmentation may enable more sensitive detection of retinal axonal and neuronal loss, and that the use of the peripapillary RNFL (a gross measure of global retinal axonal integrity) in previous reports examining “unaffected” eyes of NMO-spectrum patients may account for the conflicting results across studies.5–8,11
Our current study bears a number of limitations that warrant discussion. Comprehensive ophthalmologic evaluations of the study subjects were not performed. Importantly, though, only patients without a history of ophthalmologic disorders were recruited to this study and routine ophthalmologic evaluations were performed for the majority of MME eyes. Moreover, OCT scans were assessed for other retinal pathology (apart from MME) and the vitreoretinal interface was examined in order to exclude the possibility of vitreomacular traction,18 which may potentially lead to cystic retinal changes. Therefore, the risk that confounding ophthalmologic disorders may account for the high prevalence of MME in NMO is extremely unlikely. The cross-sectional nature of our study did not allow us to draw conclusions regarding the evolution of MME over time in NMO, or to determine the temporal relationship of MME development and acute NMO-associated ON. In order to address these issues, longitudinal studies incorporating detailed ophthalmologic assessments, including fluorescein angiography, are warranted and may help elucidate the mechanisms of development of MME in NMO. Testing for autoantibodies targeting aquaporin-4 was not performed with the same assay for all patients and antibody titers were not available, thus limiting our ability to detect a hypothesized association of anti-AQP4 antibodies and MME. Finally, our study is limited to cases of NMO, and the findings or pathogenesis may differ in other conditions associated with MME.
We have identified a pattern of morphologic abnormalities that is common in NMO and is associated with severe retinal axonal and neuronal loss, and visual disability (thereby coupling functional consequences with changes in retinal architecture). It is plausible that these findings may be due to a primary retinal process occurring in conjunction with ON or a transsynaptic process occurring secondary to optic nerve injury, and the identified retinal pathology may directly account for poor visual outcomes following NMO-associated ON, or serve as a marker of the severity of axonal and neuronal injury. Further studies are warranted to investigate the mechanisms underlying this retinal pathology, its potential implications for the prognosis and treatment of ON in NMO, to determine if therapeutic interventions to control the macular edema may help improve visual function, and to examine associations of MME with the overall clinical course of NMO.
GLOSSARY
- GCIP
ganglion cell layer + inner plexiform layer
- HC
healthy control
- IgG
immunoglobulin G
- INL
inner nuclear layer
- LETM
longitudinally extensive transverse myelitis
- MME
microcystic macular edema
- MS
multiple sclerosis
- NMO
neuromyelitis optica
- OCT
optical coherence tomography
- ON
optic neuritis
- ONL
outer nuclear layer
- RNFL
retinal nerve fiber layer
- SLE
systemic lupus erythematosus
AUTHOR CONTRIBUTIONS
Conceptualization of the study/study concept and design: E.S. Sotirchos, S. Saidha, Q.D. Nguyen, M. Levy, P.A. Calabresi. Acquisition of data: E.S. Sotirchos, S. Saidha, G. Byraiah, M.A. Mealy. Analysis and interpretation of the data: E.S. Sotirchos, S. Saidha, C.M. Crainiceanu, Q.D. Nguyen, P.A. Calabresi. Drafting/revising the manuscript: E.S. Sotirchos, S. Saidha, G. Byraiah, M.A. Mealy, M.A. Ibrahim, Y.J. Sepah, S.D. Newsome, J.N. Ratchford, E.M. Frohman, L.J. Balcer, C.M. Crainiceanu, Q.D. Nguyen, M. Levy, P.A. Calabresi. Critical revision of the manuscript for important intellectual content: E.S. Sotirchos, S. Saidha, G. Byraiah, M.A. Mealy, M.A. Ibrahim, Y.J. Sepah, S.D. Newsome, J.N. Ratchford, E.M. Frohman, L.J. Balcer, C.M. Crainiceanu, Q.D. Nguyen, M. Levy, P.A. Calabresi. Study supervision: Q.D. Nguyen, M. Levy, P.A. Calabresi.
STUDY FUNDING
National Multiple Sclerosis Society (TR 3760-A-3 to P.A.C. and RG 4212-A-4 to L.J.B. subcontracted to P.A.C.), National Eye Institute (R01 EY 014993 and R01 EY 019473 to L.J.B. subcontracted to P.A.C.), and Braxton Debbie Angela Dillon and Skip (DADS) Donor Advisor Fund (to P.A.C. and E.M.F.).
DISCLOSURE
E.S. Sotirchos reports no disclosures. S. Saidha has received consulting fees from MedicalLogix for the development of continuing medical education programs in neurology, and has received educational grant support from TEVA Neuroscience. G. Byraiah reports no disclosures. M.A. Mealy has received honoraria from EMD Serono and MS Association of America, consulting fees from Novartis and Teva Neuroscience, and academic research support from Guthy Jackson Charitable Foundation. M.A. Ibrahim and Y.J. Sepah report no disclosures. S.D. Newsome has received consulting fees from Biogen Idec and Novartis, and speaker honoraria from Biogen Idec and Teva Neuroscience. J.N. Ratchford has consulted for Bristol-Myers Squibb and Diogenix, received a speaking honorarium from TEVA, and receives research funding for clinical trials from Novartis and Biogen Idec. E.M. Frohman has received speaker honoraria from Biogen Idec, Novartis, Teva, Acorda, and Athena. He has also received consulting fees from Biogen Idec, Novartis, Teva, Acorda, Genzyme, and Abbott Laboratories. L.J. Balcer has received speaking and consulting honoraria from Biogen Idec, Bayer, and Novartis. C.M. Crainiceanu has received consulting fees from Merck and On-X. Q.D. Nguyen serves on the Scientific Advisory Board for Heidelberg Engineering, Inc. The Johns Hopkins University has received research support from Heidelberg to conduct studies; however, Heidelberg has had no inputs into the design, conduct, or analyses of the studies. M. Levy has received commercial research support and honoraria from ApoPharma Inc, travel funding from Amplimmune, academic research support from Guthy Jackson Charitable Foundation, book royalties from Lippincott, and legal fees for expert witness services. P.A. Calabresi has provided consultation services to Novartis, Teva, Biogen Idec, Vertex, Vaccinex, and Genentech, and has received grant support from EMD-Serono, Teva, Biogen Idec, Genentech, Bayer, Abbott, and Vertex. Go to Neurology.org for full disclosures.
REFERENCES
- 1.Lennon VA, Wingerchuk DM, Kryzer TJ, et al. A serum autoantibody marker of neuromyelitis optica: distinction from multiple sclerosis. Lancet 2004;364:2106–2112 [DOI] [PubMed] [Google Scholar]
- 2.Lennon VA, Kryzer TJ, Pittock SJ, Verkman AS, Hinson SR. IgG marker of optic-spinal multiple sclerosis binds to the aquaporin-4 water channel. J Exp Med 2005;202:473–477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wingerchuk DM, Hogancamp WF, O'Brien PC, Weinshenker BG. The clinical course of neuromyelitis optica (Devic's syndrome). Neurology 1999;53:1107–1114 [DOI] [PubMed] [Google Scholar]
- 4.Merle H, Olindo S, Bonnan M, et al. Natural history of the visual impairment of relapsing neuromyelitis optica. Ophthalmology 2007;114:810–815 [DOI] [PubMed] [Google Scholar]
- 5.de Seze J, Blanc F, Jeanjean L, et al. Optical coherence tomography in neuromyelitis optica. Arch Neurol 2008;65:920–923 [DOI] [PubMed] [Google Scholar]
- 6.Merle H, Olindo S, Donnio A, Richer R, Smadja D, Cabre P. Retinal peripapillary nerve fiber layer thickness in neuromyelitis optica. Invest Ophthalmol Vis Sci 2008;49:4412–4417 [DOI] [PubMed] [Google Scholar]
- 7.Naismith RT, Tutlam NT, Xu J, et al. Optical coherence tomography differs in neuromyelitis optica compared with multiple sclerosis. Neurology 2009;72:1077–1082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ratchford JN, Quigg ME, Conger A, et al. Optical coherence tomography helps differentiate neuromyelitis optica and MS optic neuropathies. Neurology 2009;73:302–308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Green AJ, Cree BA. Distinctive retinal nerve fibre layer and vascular changes in neuromyelitis optica following optic neuritis. J Neurol Neurosurg Psychiatry 2009;80:1002–1005 [DOI] [PubMed] [Google Scholar]
- 10.Syc SB, Saidha S, Newsome SD, et al. Optical coherence tomography segmentation reveals ganglion cell layer pathology after optic neuritis. Brain 2012;135:521–533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Monteiro ML, Fernandes DB, Apostolos-Pereira SL, Callegaro D. Quantification of retinal neural loss in patients with neuromyelitis optica and multiple sclerosis with or without optic neuritis using optical coherence tomography. Invest Ophthalmol Vis Sci 2012;53:3959–3966 [DOI] [PubMed] [Google Scholar]
- 12.Gelfand JM, Nolan R, Schwartz DM, Graves J, Green AJ. Microcystic macular oedema in multiple sclerosis is associated with disease severity. Brain 2012;135:1786–1793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Saidha S, Sotirchos ES, Ibrahim MA, et al. Microcystic macular oedema, thickness of the inner nuclear layer of the retina, and disease characteristics in multiple sclerosis: a retrospective study. Lancet Neurol 2012;11:963–972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wingerchuk DM, Lennon VA, Lucchinetti CF, Pittock SJ, Weinshenker BG. The spectrum of neuromyelitis optica. Lancet Neurol 2007;6:805–815 [DOI] [PubMed] [Google Scholar]
- 15.Wingerchuk DM, Lennon VA, Pittock SJ, Lucchinetti CF, Weinshenker BG. Revised diagnostic criteria for neuromyelitis optica. Neurology 2006;66:1485–1489 [DOI] [PubMed] [Google Scholar]
- 16.Henderson AP, Altmann DR, Trip AS, et al. A serial study of retinal changes following optic neuritis with sample size estimates for acute neuroprotection trials. Brain 2010;133:2592–2602 [DOI] [PubMed] [Google Scholar]
- 17.Saidha S, Syc SB, Ibrahim MA, et al. Primary retinal pathology in multiple sclerosis as detected by optical coherence tomography. Brain 2011;134:518–533 [DOI] [PubMed] [Google Scholar]
- 18.Mirza RG, Johnson MW, Jampol LM. Optical coherence tomography use in evaluation of the vitreoretinal interface: a review. Surv Ophthalmol 2007;52:397–421 [DOI] [PubMed] [Google Scholar]
- 19.Bringmann A, Reichenbach A, Wiedemann P. Pathomechanisms of cystoid macular edema. Ophthalmic Res 2004;36:241–249 [DOI] [PubMed] [Google Scholar]
- 20.Antcliff RJ, Hussain AA, Marshall J. Hydraulic conductivity of fixed retinal tissue after sequential excimer laser ablation: barriers limiting fluid distribution and implications for cystoid macular edema. Arch Ophthalmol 2001;119:539–544 [DOI] [PubMed] [Google Scholar]
- 21.Brar M, Yuson R, Kozak I, et al. Correlation between morphologic features on spectral-domain optical coherence tomography and angiographic leakage patterns in macular edema. Retina 2010;30:383–389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lucchinetti CF, Mandler RN, McGavern D, et al. A role for humoral mechanisms in the pathogenesis of Devic's neuromyelitis optica. Brain 2002;125:1450–1461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hamann S, Zeuthen T, La Cour M, et al. Aquaporins in complex tissues: distribution of aquaporins 1-5 in human and rat eye. Am J Physiol 1998;274:C1332–C1345 [DOI] [PubMed] [Google Scholar]
- 24.Pannicke T, Wurm A, Iandiev I, et al. Deletion of aquaporin-4 renders retinal glial cells more susceptible to osmotic stress. J Neurosci Res 2010;88:2877–2888 [DOI] [PubMed] [Google Scholar]
- 25.Waters PJ, McKeon A, Leite MI, et al. Serologic diagnosis of NMO: a multicenter comparison of aquaporin-4-IgG assays. Neurology 2012;78:665–671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wingerchuk DM. Evidence for humoral autoimmunity in neuromyelitis optica. Neurol Res 2006;28:348–353 [DOI] [PubMed] [Google Scholar]
- 27.Wingerchuk DM, Weinshenker BG. The emerging relationship between neuromyelitis optica and systemic rheumatologic autoimmune disease. Mult Scler 2012;18:5–10 [DOI] [PubMed] [Google Scholar]
- 28.Van Buren JM. Trans-synaptic retrograde degeneration in the visual system of primates. J Neurol Neurosurg Psychiatry 1963;26:402–409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gills JP, Wadsworth JAC. Retrograde transsynaptic degeneration of the inner nuclear layer of the retina. Invest Ophthalmol Vis Sci 1967;6:437–448 [Google Scholar]
- 30.Hollander H, Bisti S, Maffei L, Hebel R. Electroretinographic responses and retrograde changes of retinal morphology after intracranial optic nerve section: a quantitative analysis in the cat. Exp Brain Res 1984;55:483–493 [DOI] [PubMed] [Google Scholar]
- 31.Darby JE, Carr RA, Beazley LD. Retinal ganglion cell death during regeneration of the frog optic nerve is not accompanied by appreciable cell loss from the inner nuclear layer. Anat Embryol 1990;182:487–492 [DOI] [PubMed] [Google Scholar]
- 32.Williams RR, Cusato K, Raven MA, Reese BE. Organization of the inner retina following early elimination of the retinal ganglion cell population: effects on cell numbers and stratification patterns. Vis Neurosci 2001;18:233–244 [DOI] [PubMed] [Google Scholar]
- 33.Sriram P, Graham SL, Wang C, Yiannikas C, Garrick R, Klistorner A. Transsynaptic retinal degeneration in optic neuropathies: optical coherence tomography study. Invest Ophthalmol Vis Sci 2012;53:1271–1275 [DOI] [PubMed] [Google Scholar]
- 34.Thanos S, Pavlidis C, Mey J, Thiel HJ. Specific transcellular staining of microglia in the adult rat after traumatic degeneration of carbocyanine-filled retinal ganglion cells. Exp Eye Res 1992;55:101–117 [DOI] [PubMed] [Google Scholar]
- 35.Zhang S, Wang H, Lu Q, et al. Detection of early neuron degeneration and accompanying glial responses in the visual pathway in a rat model of acute intraocular hypertension. Brain Res 2009;1303:131–143 [DOI] [PubMed] [Google Scholar]
- 36.Logan MA, Hackett R, Doherty J, Sheehan A, Speese SD, Freeman MR. Negative regulation of glial engulfment activity by Draper terminates glial responses to axon injury. Nat Neurosci 2012;15:722–730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Garcia-Valenzuela E, Sharma SC, Pina AL. Multilayered retinal microglial response to optic nerve transection in rats. Mol Vis 2005;11:225–231 [PubMed] [Google Scholar]
- 38.Abegg M, Zinkernagel M, Wolf S. Microcystic macular degeneration from optic neuropathy. Brain 2012;135:e255. [DOI] [PubMed] [Google Scholar]
- 39.Balk LJ, Killestein J, Polman CH, Uitdehaag BMJ, Petzold A. Microcystic macular oedema confirmed, but not specific for multiple sclerosis. Brain 2012;135:e226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gelfand JM, Green AJ. Reply: Microcystic macular degeneration from optic neuropathy and Reply: microcystic macular oedema confirmed, but not specific for multiple sclerosis. Brain 2012;135:e227. [DOI] [PMC free article] [PubMed] [Google Scholar]