

Abstract
Objective:
To test the hypothesis that inhibition of the Na-K-2Cl transporter with bumetanide will reduce the susceptibility to decreases in muscle force in a mouse model of hypokalemic periodic paralysis (HypoPP).
Methods:
In vitro contraction tests were performed on soleus muscle isolated from mice with knock-in missense mutations that result in HypoPP (sodium channel NaV1.4-R669H) or hyperkalemic periodic paralysis (HyperPP; sodium channel NaV1.4-M1592V).
Results:
Bumetanide prevented the development of weakness in 2 mM K+ and also restored force during an established attack of HypoPP. Stimulation of the Na-K-2Cl transporter via induction of hyperosmolality exacerbated the weakness seen in low K+ and was also prevented by bumetanide. Bumetanide was more efficacious than acetazolamide for preventing weakness in low K+ conditions. Decreases in force in HyperPP muscle exposed to 10 mM K+ were not prevented by treatment with bumetanide.
Conclusions:
The Na-K-2Cl inhibitor bumetanide was highly effective in preventing attacks of weakness in the NaV1.4-R669H mouse model of HypoPP and should be considered for management of patients with HypoPP due to sodium channel mutations. Dehydration may aggravate HypoPP by stimulating the Na-K-2Cl transporter.
Familial hypokalemic periodic paralysis (HypoPP) is a disorder of skeletal muscle excitability that presents with recurrent episodes of weakness, often triggered by exercise, stress, or carbohydrate-rich meals.1 The transient decrease in muscle excitability during an attack is caused by sustained depolarization of the resting potential,2 arising from mutations in voltage-gated calcium3,4 or sodium5,6 channels. Management of symptoms1 involves avoidance of activities that provoke attacks, potassium supplements, or prophylactic use of the carbonic anhydrase inhibitors acetazolamide (ACTZ)7 or dichlorphenamide.8 These interventions are moderately effective for some patients, but those with sodium channel mutations often receive no benefit or may even have worsening of symptoms.9,10
New insights into the mechanism of depolarization that produces an attack of HypoPP11–13 support a prior hypothesis that inhibition of the sodium–potassium–2-chloride transporter (Na-K-2Cl) may reduce susceptibility to episodes of weakness.14 Influx of K+, Na+, and 2 Cl− ions in each cycle of the Na-K-2Cl transporter carries no net current,15 but has a depolarizing influence on the resting potential by increasing myoplasmic chloride levels. The Cl− effect is substantial because of the high membrane conductance to Cl− in resting muscle. More importantly, the paradoxical depolarization of the resting potential from low levels of K+ was predicted to be less likely if myoplasmic Cl− does not increase via flux through the Na-K-2Cl transporter.14
In the present study, we tested whether inhibition of the Na-K-2Cl transporter by bumetanide administration decreases the susceptibility to low-K+–induced reductions of muscle force in our sodium channel (NaV1.4-R669H) knock-in mutant mouse model of HypoPP.16
METHODS
This study was approved by the UT Southwestern Medical Center Institutional Animal Care and Use Committee. Genetically engineered mice with targeted knock-in mutations of the NaV1.4 sodium channel were used as models for HypoPP (NaV1.4-R669H) and hyperkalemic periodic paralysis (HyperPP) (NaV1.4-M1592V), as previously described.16,17 For the NaV1.4-R669H mice, heterozygous mutant animals, R669H+/m, and homozygous R669Hm/m animals were available for study. Wild-type littermates (+/+) served as controls, and all mouse colonies were maintained in the 129/Sv strain. Mice were killed using isoflurane inhalation and cervical dislocation. All studies were performed on isolated soleus muscles, excised from the hind limb of mice.
Measurements of muscle force were obtained by direct field stimulation of the soleus muscle maintained at 37°C in an oxygenated organ bath (Myobath, World Precision Instruments Inc., Sarasota, FL). The organ bath contained curare (0.25 μM) to block neuromuscular transmission from distal branches of motor axons. Tetanic stimuli (80 mA, 1 ms pulses, 100 Hz, n = 40) were applied with a pair of parallel wire electrodes. The bath was continuously bubbled with 95% O2/5% CO2 and contained 118 mM NaCl, 4.75 mM KCl, 1.18 mM MgSO4, 2.54 mM CaCl2, 1.18 mM NaH2PO4, 24.8 mM NaHCO3, 10 mM glucose, and 0.02 U/mL insulin. Test solutions containing low (2 mM) or high (10 mM) KCl levels were prepared with commensurate changes in NaCl to maintain a constant concentration of total monovalent cations and of Cl−. The standard bath solution was 288 mOsm, and a hypertonic test solution was made by titration with sucrose (approximately 50 mM) to achieve a measured osmolality of 325 mOsm. Some experiments were performed in Cl-free conditions in which Cl− was replaced by methanesulfonate, and 9-anthracene carboxylic acid (9-AC, 100 μM) was added to block ClC-1 chloride channels. Solutions with bumetanide (75 μM) were prepared from a 60-mM stock solution in 100% ethanol, and those with ACTZ (100 μM) from a 1-M stock solution in a 1:1 mixture of 95% ethanol and dimethyl sulfoxide. Control experiments with vehicle alone had no effect on muscle force. Test solutions were prewarmed and applied by 8 exchanges of the bath volume over 1 minute. Data are displayed as the mean ± SEM.
RESULTS
Bumetanide prevented decreases in force in HypoPP muscle.
Stimulation with 40 pulses at 100 Hz produced a fused tetanic contraction, as shown for the soleus muscle from a heterozygous R669H+/m mouse (figure 1A). Peak force during tetanic stimulation applied every 2 minutes was stable (figure 1B, 0–30 minutes). Reduction of the bath K+ from 4.75 mM to 2 mM K+, however, caused a dramatic 90% decrease in force for homozygous R669Hm/m muscle and a 60% loss of force for heterozygous R669H+/m muscle which is homologous to the mutant gene dosage in human HypoPP (figure 1A, middle, figure 1B, 30–60 minutes). Wild-type (+/+) muscle had a much smaller reduction of 30%. Application of bumetanide (75 μM) in the continued presence of 2 mM K+ produced a large sustained recovery of force for R669H and WT muscle (figure 1A, right, and figure 1B, 60–90 minutes). When bumetanide was added coincident with the onset of the 2 mM K+ challenge (figure 1C), decreases in force were completely prevented for WT and R669H+/m muscle and only a mild 10% reduction occurred in homozygous R669Hm/m muscle at the end of a 30-minute exposure. Washout of bumetanide led to a precipitous decrease in force in R669Hm/m muscle, but force was maintained for WT and R669H+/m.
Figure 1. Bumetanide prevents reduction in force in low K+ conditions.

(A) Force recorded during a tetanic contraction of the soleus muscle from a heterozygous R669H+/m mouse in 4.75 mM K+ bath solution (left), 20 minutes after switching to 2 mM K+ (middle), and 12 minutes after application of bumetanide (BMT) (right). Stimulus (blue) is shown below each force response. (B) Peak tetanic force was measured every 2 minutes and normalized to the amplitude in the 4.75 mM K+ bath solution. Exposure to 2 mM K+ elicited 90% decrease in force for R669Hm/m muscle (black squares, n = 8), a 60% decrease in R669H+/m (gray squares, n = 7), and only a 30% decrease in WT (open squares, n = 8). The partial recovery in force between 50 and 60 minutes for R669Hm/m is due to spontaneous oscillations with cyclical recovery and weakness.16 Addition of 75 μM BMT produced a full recovery of tetanic force. (C) Application of BMT at the onset of a 2 mM K+ exposure prevents decreases in force. Washout of BMT caused a large reduction in force for R669Hm/m muscle.
Hyperosmolality exacerbated low-K+–induced decreases in force.
Prior studies using WT mouse muscle have shown that hypertonicity activates the Na-K-2Cl transporter, which in turn promotes solute influx to establish a transcellular osmotic balance. This sequence of events increases Cl− concentration of the myoplasm,18 resulting in a positive shift in the equilibrium potential for Cl (ECl) and depolarization of the fiber resting potential.14,19 A lower resting potential reduces fiber excitability and peak tetanic force, as seen for WT soleus exposed to a 325-mOsm bath (figure 2A, 30–60 minutes). A larger reduction in tetanic force was observed for heterozygous R669H+/m soleus muscle. More importantly, a superimposed low-K+ challenge triggered a profound decrease in muscle force (figure 2A, 60–90 minutes). These data show synergy between hyperosmolality and hypokalemia for eliciting attacks of weakness. Bumetanide is a potent blocker of the Na-K-2Cl transporter, and pretreatment with the drug prevented force reduction caused by both hyperosmolality and low K+ levels (figure 2B). The 15% increase in peak tetanic force produced by bumetanide in the standard bath (figure 2B, 15–30 minutes) was most likely caused by hyperpolarization and enhanced excitability under control conditions.
Figure 2. Hyperosmolality-induced decreases in force that were aggravated by low K+ conditions and prevented by bumetanide.
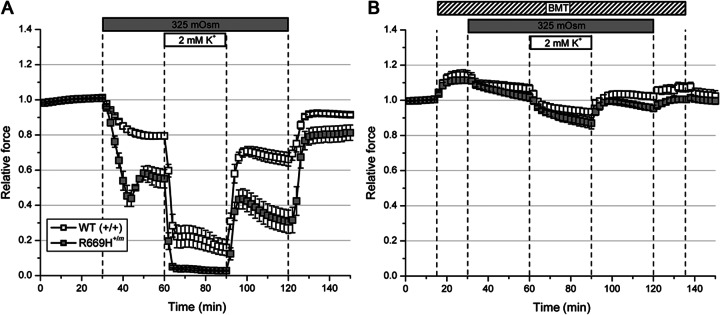
(A) Soleus from R669H+/m mice (n = 6) was more susceptible to reductions in force due to hyperosmolar challenge as compared to WT (n = 6). A concomitant reduction in K+ levels from 4.75 to 2 mM resulted in a complete loss of force for R669H+/m soleus. (B) Pretreatment with bumetanide (BMT) (75 μM) prevented decreases in force.
Protection from force reduction in a low-K+ environment by bumetanide was dependent on chloride.
The beneficial effect of bumetanide on the preservation of force in HypoPP muscle during a low-K+ challenge is thought to result from prevention of myoplasmic Cl− accumulation that results from inhibition of the Na-K-2Cl transporter.14 To test this hypothesis, we removed the influence of Cl− on the resting potential by using a Cl−-free bath solution and blocking efflux of myoplasmic Cl− through the ClC-1 chloride channel with the addition of 9-AC. These Cl−-free conditions enhanced muscle excitability which was discernible by 2 changes. First, the reduction of force was noticeably slowed at the end of a tetanic stimulation (figure 3A, inset) due to myotonia caused by the reduced Cl− conductance. Second, HypoPP muscle was much less susceptible to reductions in force in a low-K+ challenge. Exposure to 2 mM K+ induced only a 10% decrease in force in heterozygous R669H+/m soleus muscle (figure 3A), compared to a 60% loss in normal Cl− conditions (figure 1). For homozygous R669Hm/m muscle, a 2 mM K+ challenge caused an 80% reduction in force, but this was followed by large spontaneous fluctuations in peak force with transient recovery over 10 to 20 minutes (figure 3B). More importantly, application of bumetanide did not result in any recovery of force nor did it prevent subsequent fluctuations with profound decreases in force during these episodes in a low-K+ environment (figure 3, red squares). The lack of benefit from bumetanide in Cl−-free conditions is markedly different from our experience in a standard bath containing 128 mM Cl−; in the standard bath scenario, muscles isolated from more than 25 HypoPP mice always showed marked protection from weakness in the presence of bumetanide.
Figure 3. Beneficial effect of bumetanide was Cl− dependent.
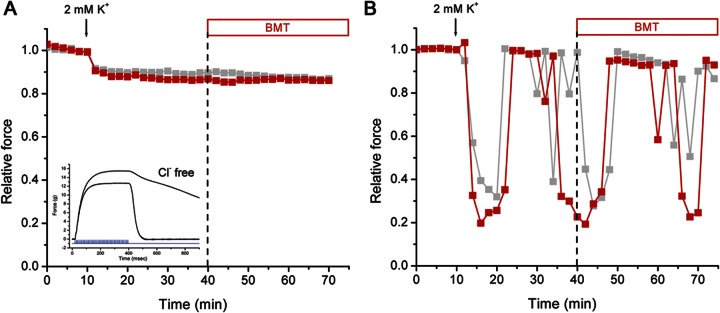
Tetanic contractions were measured in Cl−-free conditions. Peak forces recorded from the left and right soleus in separate tissue baths are superimposed. Both muscles were exposed to 2 mM K+ starting at 10 minutes, and only one preparation had bumetanide (BMT) (75 μM) applied at 40 minutes (red squares). (A) For soleus from a heterozygous R669H+/m mouse, the low-K+ challenge induced only a small decrease in force. Application of bumetanide did not produce recovery. Myotonia was evident from the slowed relaxation after a contraction (inset, contractions recorded before and after replacement of Cl− by methanesulfonate). (B) Low K+ triggered a large decrease in force for the soleus muscle from a homozygous R669Hm/m mouse. Spontaneous fluctuations in peak force occurred during continuous exposure to 2 mM K+, but the addition of BMT (40 minutes, red symbols) did not prevent subsequent transient decreases.
Bumetanide was superior to ACTZ in preventing low-K+–induced decrease in force for NaV1.4-HypoPP muscle.
The carbonic anhydrase inhibitor ACTZ is the most widely used prophylactic agent to reduce the frequency and severity of attacks of weakness in HypoPP.7,20 The efficacy of ACTZ in humans is lower, or its administration may even be deleterious, in cases of HypoPP caused by mutations of the NaV1.4 sodium channel compared to those with mutations in the CaV1.1 calcium channel.10,21 In the present study, there was only a modest benefit shown for pretreatment with ACTZ; low-K+–induced reductions in force were only 8% less than with no drug (p > 0.3, NS). In order to directly compare the efficacy of ACTZ to that of bumetanide at therapeutic levels, we performed a sequential test with 2 low-K+ challenges in the same muscle preparation: one using pretreatment with 100 μM ACTZ and the second with 0.5 μM bumetanide. ACTZ showed no significant protection against reductions in force for soleus muscle from R669H+/m females or males (figure 4). On the other hand, even after a preceding episode of weakness, a bath containing 0.5 μM bumetanide protected against force reductions in 2 mM K+. The beneficial effect of 0.5 μM bumetanide was consistently greater in muscle from females than males (p < 0.01, n = 3), and bumetanide was superior to no drug treatment for both sexes (females p < 0.0005, n = 3, males p < 0.01, n = 3).
Figure 4. Bumetanide was superior to acetazolamide in preventing decreases in force under low K+ conditions.

Sequential 2-mM K+ challenges were applied to soleus from heterozygous R669H+/m female (left) or male (right) mice. In each trial, a pair of muscles from the same animal was tested in separate tissue baths. The control had no drug applied (gray) while the other muscle was pretreated with acetazolamide (ACTZ) (100 μM) (red) in the first low-K+ challenge and bumetanide (BMT) (0.5 μM) before the second challenge. The modest improvement with ACTZ was not significantly different from untreated (p > 0.3, n = 6). BMT prevented decreases in force in females, and substantially reduced such decreases in males (p < 0.01, n = 3).
Bumetanide was not effective in preventing high-K+–induced decreases in force in HyperPP.
The mechanism by which bumetanide reduces susceptibility to an attack of HypoPP is most likely analogous to the reduced susceptibility of normal muscle to paradoxical depolarization in low K+; under such conditions, inhibition of the Na-K-2Cl transporter shifts the transition point of paradoxical depolarization to lower concentrations of K+.14 On the other hand, susceptibility to attacks of weakness in HyperPP occurs at high K+ levels, where an anomalous persistent inward current conducted by mutant Na+ channels produces marked depolarization of the resting potential.22 Because the mechanisms for aberrant depolarization of the resting potential are very different in HypoPP vs HyperPP, we predicted that bumetanide would not be effective in preventing anomalous depolarization and decreases in force in HyperPP muscle. We tested this hypothesis using a high-K+ challenge for soleus muscle isolated from NaV1.4-M1592V mice with HyperPP.17 Exposure to 10 mM K+ elicited a 35% decrease in tetanic force that was not affected by addition of 75 μM bumetanide (figure 5).
Figure 5. Bumetanide was not effective in preventing reductions in force in hyperkalemic periodic paralysis.
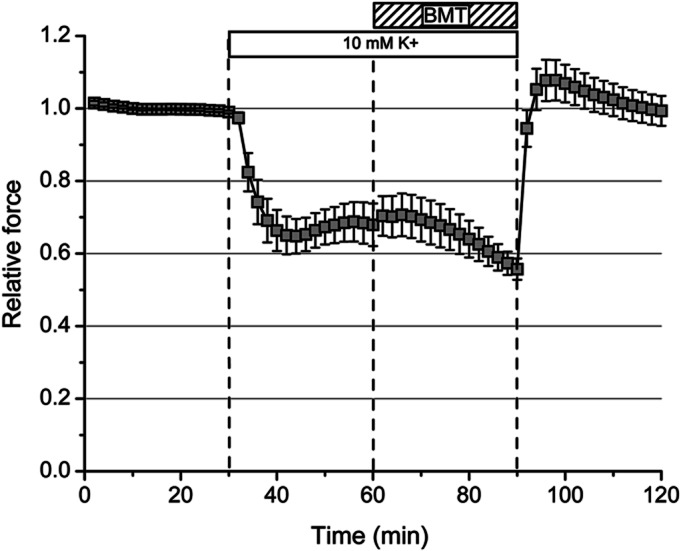
Peak tetanic force in soleus muscle of M1592V hyperkalemic periodic paralysis mice decreased by 35%, when challenged with 10 mM K+ (n = 6). Application of 75 μM bumetanide (BMT) did not produce a recovery, whereas return to 4.75 mM K+ resulted in complete recovery.
DISCUSSION
The rationale for testing whether bumetanide might be efficacious in reducing the severity of weakness in HypoPP was based on experimental studies demonstrating well-known but curious bistability of the resting potential for skeletal muscle in low K+conditions.14,18,23 When the extracellular K+ concentration is decreased from the normal 4 mM, as expected, the resting potential initially becomes more negative (hyperpolarized), due to the negative shift in the K+ Nernst potential (EK) caused by a steeper ion gradient [K+]in/[K+]out. At a very low range of about 1.5–2.0 mM K+, however, the resting potential may assume either of 2 possible values: the expected level of about −95 mV or a paradoxically depolarized value of about −60 mV. At even lower K+ concentrations, only the depolarized resting potential exists.
The resting potential in normal muscle is a few mV more positive than EK, primarily because myoplasmic Cl− accumulates to levels higher than would be attained by passive equilibrium.18 The mechanism for Cl− accumulation above equilibrium in muscle is influx via the Na-K-2Cl transporter. Inhibition of the transporter with bumetanide causes a reduction in myoplasmic Cl− levels and hyperpolarization of the resting potential.19 More importantly, inhibition of the Na-K-2Cl transporter also decreases the critical extracellular K+ concentration, leading to the bistable resting potential.14
These experimental observations in normal mammalian muscle fibers led to the suggestion that bumetanide might be effective in preventing attacks of transient weakness in HypoPP.14 The fundamental problem for muscle susceptible to attacks of weakness in HypoPP is that the critical K+ level for paradoxical depolarization is shifted and occurs in the lower end of the normal physiologic range (2.5–3.5 mM).2 This susceptibility occurs because of the leakage “gating pore” current in mutant NaV1.4 or CaV1.1 channels.11,12 Although there are no published studies or even anecdotal descriptions regarding the use of bumetanide in patients with periodic paralysis, the studies presented here using a mouse model of HypoPP caused by a missense mutation in NaV1.4 show that bumetanide was highly effective at preventing the reductions in force in low K+ conditions. Moreover, bumetanide application restored force in muscle that was profoundly weak due to prior exposure to 2 mM K+. The beneficial effect of bumetanide was Cl− dependent, consistent with the notion that the mechanism of action involved inhibition of the Na-K-2Cl transporter and not some off-target effect. Bumetanide was clearly superior to ACTZ, and may be the treatment of choice for the 20% of HypoPP patients with a NaV1.4 mutation.
Our in vitro studies using muscle from NaV.14 HypoPP mice demonstrate a proof of principle that bumetanide may be highly effective for both prevention and treatment of episodes of weakness in HypoPP. Chronic use of bumetanide to reduce attack severity and frequency in patients with HypoPP may be limited by increased urinary excretion of K+, but acute administration may still be effective for promoting recovery from an ongoing attack (figure 1B). Because the mechanism for destabilization of the resting potential under conditions of low K+ is similar for HypoPP resulting from mutations in NaV1.4 and CaV1.1, we predict bumetanide will be effective in patients with either channel mutation. In the present study, females showed a more favorable response than males (figure 4); however, a modest increase in drug concentration to 1.0 μM produced a comparable level of benefit in muscle isolated from either sex. Activation of the Na-K-2Cl transporter by hyperosmolality resulted in increased weakness (figure 3) and implies that patients with HypoPP should avoid treatments/activities that result in elevated osmolytes (e.g., hyperglycemia) or dehydration. Finally, because the mechanism for depolarization-induced loss of muscle excitability is fundamentally different in HyperPP22 and our mouse model of HyperPP showed no beneficial effect from even a high dose of the drug (75 μM), we predict that bumetanide will not be beneficial in patients with HyperPP.
GLOSSARY
- ACTZ
acetazolamide
- HyperPP
hyperkalemic periodic paralysis
- HypoPP
hypokalemic periodic paralysis
AUTHOR CONTRIBUTIONS
Dr. Wu performed the experiments, analyzed the data, and edited the manuscript. Dr. Mi developed the protocol and contributed to the data acquisition. Dr. Cannon conceived the study, performed the analysis, and wrote the manuscript.
STUDY FUNDING
Supported by NIAMS (AR 42703) of the NIH and the Muscular Dystrophy Association (RG 135835).
DISCLOSURE
The authors report no disclosures relevant to the manuscript. Go to Neurology.org for full disclosures.
REFERENCES
- 1.Venance SL, Cannon SC, Fialho D, et al. The primary periodic paralyses: diagnosis, pathogenesis and treatment. Brain 2006;129:8–17 [DOI] [PubMed] [Google Scholar]
- 2.Rüdel R, Lehmann-Horn F, Ricker K, Kuther G. Hypokalemic periodic paralysis: in vitro investigation of muscle fiber membrane parameters. Muscle Nerve 1984;7:110–120 [DOI] [PubMed] [Google Scholar]
- 3.Jurkat-Rott K, Lehmann-Horn F, Albaz A, et al. A calcium channel mutation causing hypokalemic periodic paralysis. Hum Mol Genet 1994;3:1415–1419 [DOI] [PubMed] [Google Scholar]
- 4.Ptacek LJ, Tawil R, Griggs RC, et al. Dihydropyridine receptor mutations cause hypokalemic periodic paralysis. Cell 1994;77:863–868 [DOI] [PubMed] [Google Scholar]
- 5.Bulman DE, Scoggan KA, van Oene MD, et al. A novel sodium channel mutation in a family with hypokalemic periodic paralysis. Neurology 1999;53:1932–1936 [DOI] [PubMed] [Google Scholar]
- 6.Matthews E, Labrum R, Sweeney MG, et al. Voltage sensor charge loss accounts for most cases of hypokalemic periodic paralysis. Neurology 2009;72:1544–1547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Resnick JS, Engle WK, Griggs RC, Stam AC. Acetazolamide prophylaxis in hypokalemic periodic paralysis. New Engl J Med 1968;278:582–586 [DOI] [PubMed] [Google Scholar]
- 8.Tawil R, McDermott MP, Brown R, Jr, et al. Randomized trials of dichlorphenamide in the periodic paralyses. Working Group on Periodic Paralysis. Ann Neurol 2000;47:46–53 [PubMed] [Google Scholar]
- 9.Bendahhou S, Cummins TR, Griggs RC, Fu YH, Ptacek LJ. Sodium channel inactivation defects are associated with acetazolamide-exacerbated hypokalemic periodic paralysis. Ann Neurol 2001;50:417–420 [DOI] [PubMed] [Google Scholar]
- 10.Sternberg D, Maisonobe T, Jurkat-Rott K, et al. Hypokalemic periodic paralysis type 2 caused by mutations at codon 672 in the muscle sodium channel gene SCN4A. Brain 2001;124:1091–1099 [DOI] [PubMed] [Google Scholar]
- 11.Struyk AF, Cannon SC. A Na+ channel mutation inked to hypokalemic periodic paralysis exposes a proton-selective gating pore. J Gen Physiol 2007;130:11–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sokolov S, Scheuer T, Catterall WA. Gating pore current in an inherited ion channelopathy. Nature 2007;446:76–78 [DOI] [PubMed] [Google Scholar]
- 13.Struyk AF, Cannon SC. Paradoxical depolarization of Ba2+- treated muscle exposed to low extracellular K+: insights into resting potential abnormalities in hypokalemic paralysis. Muscle Nerve 2008;37:326–337 [DOI] [PubMed] [Google Scholar]
- 14.Geukes Foppen RJ, van Mil HG, Siegenbeek van Heukelom J. Effects of chloride transport on bistable behaviour of the membrane potential in mouse skeletal muscle. J Physiol 2002;542:181–191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Russell JM. Sodium-potassium-chloride cotransport. Physiol Rev 2000;80:211–276 [DOI] [PubMed] [Google Scholar]
- 16.Wu F, Mi W, Burns DK, et al. A sodium channel knockin mutant (NaV1.4-R669H) mouse model of hypokalemic periodic paralysis. J Clin Invest 2011;121:4082–4094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hayward LJ, Kim JS, Lee MY, et al. Targeted mutation of mouse skeletal muscle sodium channel produces myotonia and potassium-sensitive weakness. J Clin Invest 2008;118:1437–1449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Aickin CC, Betz WJ, Harris GL. Intracellular chloride and the mechanism for its accumulation in rat lumbrical muscle. J Physiol 1989;411:437–455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.van Mil HG, Geukes Foppen RJ, Siegenbeek van Heukelom J. The influence of bumetanide on the membrane potential of mouse skeletal muscle cells in isotonic and hypertonic media. Br J Pharmacol 1997;120:39–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Griggs RC, Engel WK, Resnick JS. Acetazolamide treatment of hypokalemic periodic paralysis: prevention of attacks and improvement of persistent weakness. Ann Intern Med 1970;73:39–48 [DOI] [PubMed] [Google Scholar]
- 21.Matthews E, Portaro S, Ke Q, et al. Acetazolamide efficacy in hypokalemic periodic paralysis and the predictive role of genotype. Neurology 2011;77:1960–1964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cannon SC, Brown RH, Jr, Corey DP. Theoretical reconstruction of myotonia and paralysis caused by incomplete inactivation of sodium channels. Biophys J 1993;65:270–288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kao I, Gordon AM. Mechanism of insulin-induced paralysis of muscles from potassium-depleted rats. Science 1975;188:740–741 [DOI] [PubMed] [Google Scholar]