

Abstract
Objective:
Acute encephalopathy with biphasic seizures and late reduced diffusion (AESD) is a childhood encephalopathy following severe febrile seizures, leaving neurologic sequelae in many patients. However, its pathogenesis remains unclear. In this study, we clarified that genetic variation in the adenosine A2A receptor (ADORA2A), whose activation is involved in excitotoxicity, may be a predisposing factor of AESD.
Methods:
We analyzed 4 ADORA2A single nucleotide polymorphisms in 85 patients with AESD. The mRNA expression in brain samples, mRNA and protein expression in lymphoblasts, as well as the production of cyclic adenosine monophosphate (cAMP) by lymphoblasts in response to adenosine were compared among ADORA2A diplotypes.
Results:
Four single nucleotide polymorphisms were completely linked, which resulted in 2 haplotypes, A and B. Haplotype A (C at rs2298383, T at rs5751876, deletion at rs35320474, and C at rs4822492) frequency in patients was significantly higher than in controls (p = 0.005). Homozygous haplotype A (AA diplotype) had a higher risk of developing AESD (odds ratio 2.32, 95% confidence interval 1.32–4.08; p = 0.003) via a recessive model. mRNA expression was significantly higher in AA than AB and BB diplotypes, both in the brain (p = 0.003 and 0.002, respectively) and lymphoblasts (p = 0.035 and 0.003, respectively). In lymphoblasts, ADORA2A protein expression (p = 0.024), as well as cellular cAMP production (p = 0.0006), was significantly higher in AA than BB diplotype.
Conclusions:
AA diplotype of ADORA2A is associated with AESD and may alter the intracellular adenosine/cAMP cascade, thereby promoting seizures and excitotoxic brain damage in patients.
During the course of acute febrile diseases, such as influenza and exanthema subitum, some children develop repetitive or prolonged seizures, followed by sustained impairment of consciousness. These conditions are collectively termed acute encephalopathy with inflammation-mediated status epilepticus (AEIMSE).1 Among AEIMSE, acute encephalopathy with biphasic seizures and late reduced diffusion (AESD)2 is the most common in Japan, affecting hundreds of children every year.3 Hemiconvulsion-hemiplegia syndrome, a condition encountered worldwide, often occurs during an infectious disease, and is regarded as a subgroup of AESD.4 AESD typically shows a biphasic clinical course, consisting of a prolonged febrile seizure on the first day and a cluster of complex partial seizures several days later (late seizure), each followed by postictal coma. Cranial MRI reveals high signal intensity lesions in the cerebral subcortical white matter on diffusion-weighted images, which appear around the occurrence of late seizure (figure 1).5,6 Excitotoxicity is considered to be the main pathologic mechanism of AESD.2,4 The genetic background of AESD remains to be elucidated. Recently, polymorphism of a gene encoding a mitochondrial enzyme, carnitine palmitoyltransferase II (CPT2), was identified as a genetic predisposition for AESD7; however, some patients with AESD have no such polymorphism, suggesting the involvement of genes other than CPT2.
Figure 1. Typical MRI findings of a patient with acute encephalopathy with biphasic seizures and late reduced diffusion.
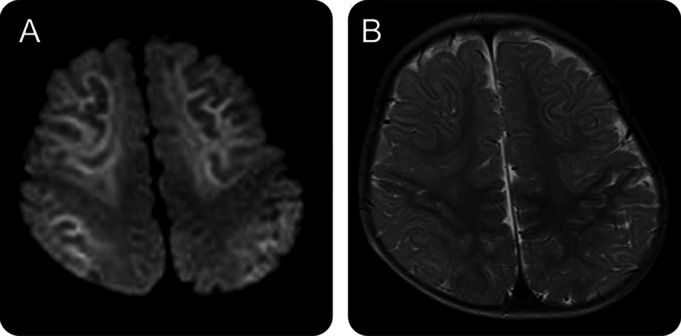
Magnetic resonance study of a 1-year-old boy on day 8 demonstrated lesions in the subcortical white matter that showed high signal intensity on diffusion-weighted (A) and T2-weighted (B) images. The lesions were prominent along the U-fibers with sparing of the peri-Rolandic region.
We hypothesized that the adenosine-mediated signal pathway is altered in AESD because theophylline, a nonselective adenosine receptor antagonist, aggravates AESD.4 To test this hypothesis, we studied the haplotype frequency of 4 single nucleotide polymorphisms (SNPs) located in the linkage disequilibrium block of the adenosine A2A receptor (ADORA2A) gene, and then examined the effects of ADORA2A diplotypes on their mRNA and protein expression, and those on cyclic adenosine monophosphate (cAMP) production in response to adenosine.
METHODS
Subjects.
We recruited patients with AESD from hospitals in Japan during 2008–2011 based on the diagnostic criteria.3 Eighty-five Japanese patients, 39 male and 46 female aged from 6 months to 10 years and 3 months (median, 1 year and 10 months), participated in this study. Detailed clinical data are shown in table e-1 on the Neurology® Web site at www.neurology.org. All patients had their first convulsion, mostly status epilepticus, within 24 hours from the onset of fever, followed by impairment of consciousness that improved on the second day in most cases. On the fourth to sixth day of illness, there was a recurrence of convulsions or a cluster of partial seizures, followed again by impairment of consciousness. Cranial MRI was normal on the first to second day of illness, but showed high signal intensity lesions in the cerebral subcortical white matter on the third to ninth day (figure 1). Pathogens of antecedent infections included human herpesvirus 6 (28 cases), influenza virus (5 cases), respiratory syncytial virus, rotavirus, adenovirus, mumps virus, and Mycoplasma pneumoniae.
Standard protocol approvals, registrations, and patient consents.
The procedures in this study were approved by the University of Tokyo Ethics Committee. Written informed consent was obtained from all guardians of participants in the study.
Controls.
We analyzed the ADORA2A genotypes of control subjects, consisting of 100 healthy Japanese adults, 50 men and 50 women, 20 to 69 years of age, using DNA extracted from Pharma SNP Consortium B cell lines obtained from the Human Science Research Resources Bank (Osaka, Japan). We searched the dbSNP database (http://www.ncbi.nlm.nih.gov/projects/SNP/) in the National Center for Biotechnology Information for the variation frequencies of ADORA2A SNPs and combined the data of 100 controls from the Pharma SNP Consortium and those of 84 Japanese in the National Center for Biotechnology Information dbSNP database.
Brain samples.
To examine ADORA2A gene expression levels in the brain, 100 human brain DNA and RNA samples were obtained from Stanley Medical Research Institute (SMRI) (Bethesda, MD). DNA and RNA were extracted from the occipital and anterior cingulate cortex, respectively. In this experiment, the ethnic background was Caucasian in the vast majority (at least 98 samples).
Lymphoblasts.
For expression studies and functional assays, we used 15 lymphoblast cell lines from control Japanese adults, obtained from control subjects at the University of Tokyo Hospital.
Procedures.
Peripheral blood samples were collected from the patients. Genomic DNA was extracted from the blood using standard protocols. All 5 exons of ADORA2A were PCR amplified with flanking intronic primers and standard PCR conditions (primer sequences are described in table e-2). PCR products of ADORA2A were sequenced on a 310 Genetic Analyzer, 3100 Genetic Analyzer, or 3130xl Genetic Analyzer (Life Technologies, Carlsbad, CA). To identify rs5751876 and rs2298383 SNPs, the PCR–restriction fragment length polymorphism method was adopted.8 For quantitative PCR, total RNA was isolated from control lymphoblasts using TRIzol reagent (Life Technologies) according to the manufacturer's protocol. Total RNA was reversely transcribed to cDNA by a Ready-To-Go You-Prime First-Stand Beads cDNA synthesis kit (GE Healthcare, Uppsala, Sweden) according to the manufacturer's protocol. Random Primer (Takara Bio, Otsu, Japan) was used. Gene expression was evaluated by the relative Quantification ABI PRISM 7000 Sequence Detection System (Life Technologies) with FastStart Universal SYBR Green Master [ROX] (Roche, Basel, Switzerland) reagent. The relative ADORA2A mRNA expression level was calculated using glucose-6-phosphate dehydrogenase (G6PDH) as the internal standard. Primer sequences of real-time PCR for ADORA2A and G6PDH are described in table e-3. Each value is shown as the mean value of 2 independent experiments in triplicate. For SMRI brain samples, genotyping and the gene expression study of ADORA2A were performed by the same methods as for AESD patient samples. Western blotting of the cell lysate from control lymphoblasts was performed by the standard protocol using a rabbit polyclonal antibody to human ADORA2A (Abcam, Cambridge, UK) at a dilution of 1:500. The relative ADORA2A protein expression level was calculated using β-actin as the internal standard. Each value is shown as the mean value of 3 independent experiments in duplicate. The cAMP concentration in lymphoblasts was measured after stimulation by adenosine (10 nM) and 8-cyclopentyl-1,3-dipropylxanthine (10 nM), a selective adenosine A1 receptor (ADORA1) antagonist, using the cAMP-Screen Direct System (Life Technologies) according to the manufacturer's protocol. Cellular cAMP levels were determined using SpectraMax Pro 5.3 software (Molecular Devices, Sunnyvale, CA). Each value is shown as the mean value of 2 independent experiments in triplicate.
Statistical analysis.
Differences in the demographic characteristics of the genotypes between patients (85 cases) and controls were assessed by Pearson χ2 test and Fisher exact test for categorical data. Goodness-of-fit to the Hardy-Weinberg equilibrium and differences in genotype and allele frequencies between AESD and control groups were examined by χ2 analysis. Significant differences were defined as p < 0.05 in conditional analysis. We estimated the odds ratio (OR) together with the 95% confidence interval (CI) for each allele haplotype frequency with AESD using Microsoft Office Excel 2010. Patients with AESD were compared with the controls under dominant, recessive, and additive models using a likelihood ratio χ2 test. These genetic models were also assessed using the Cochran-Armitage test for trend. The differences in mRNA and protein expression levels and cellular cAMP accumulation, expressed as the mean ± SEM, were calculated using analysis of variance followed by the Tukey-Kramer test in the case of multiple comparisons. p < 0.05 was considered a significant difference.
RESULTS
ADORA2A haplotype frequency.
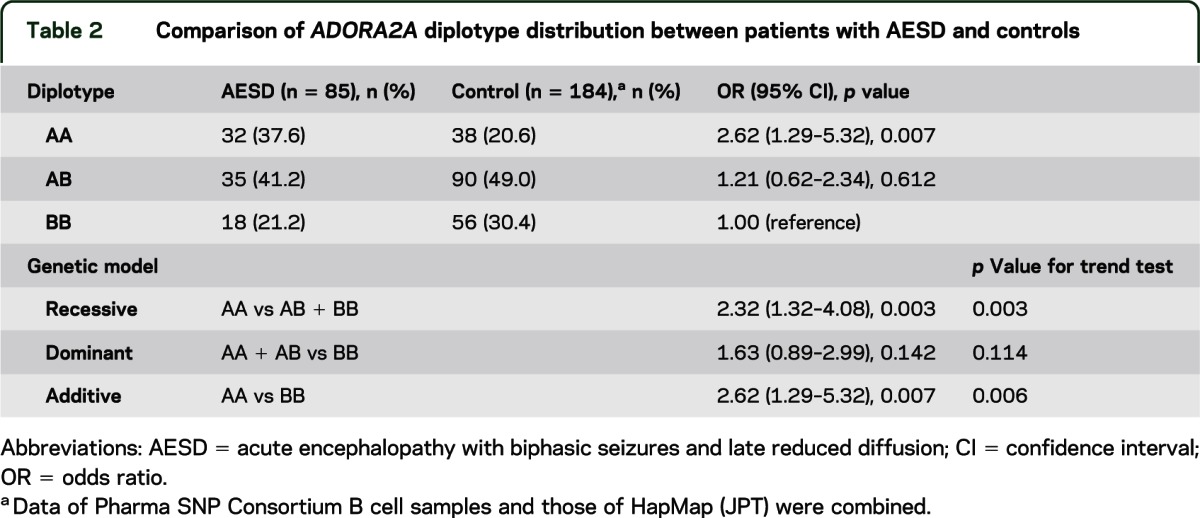
First, we analyzed the entire coding region of ADORA2A in patients with AESD and found no mutations. Second, we analyzed genetic variations of ADORA2A in patients with AESD and control subjects. Distribution of the ADORA2A polymorphisms in both AESD and controls met the Hardy-Weinberg equilibrium (p = 0.15 and 0.86, respectively). Four SNPs (figure e-1) in this gene, rs2298383, rs5751876, rs35320474, and rs4822492, had previously been reported to show complete linkage disequilibrium in 84 Japanese (human HapMap project, http://Apr2011.archive.ensemble.org). The present study also supported their complete linkage in both 85 AESD cases and 100 controls. Thus, there were only 2 haplotypes, haplotype A (C at rs2298383, T at rs5751876, deletion at rs35320474, and C at rs4822492) and haplotype B (T at rs2298383, C at rs5751876, T at rs35320474, and G at rs4822492). Table 1 shows haplotype frequency for the ADORA2A SNPs in AESD and control groups. Haplotype A was significantly more frequent in AESD than in controls (p = 0.005). The frequency of homozygous haplotype A (AA diplotype) in AESD and controls was 37.6% and 20.6%, respectively. There was a significant association between AA diplotype and increased risk of developing AESD for recessive model comparison (OR 2.32, 95% CI 1.32–4.08; p = 0.003) and additive model comparison (OR 2.62, 95% CI 1.29–5.32; p = 0.007), but not for the dominant model comparison (OR 1.63, 95% CI 0.89–2.99; p = 0.142) (table 2). The most significant p value was obtained under the recessive model using χ2 test, as well as Cochran-Armitage test for trend.
Table 1.
Comparison of ADORA2A haplotype frequency between patients with AESD and controlsa
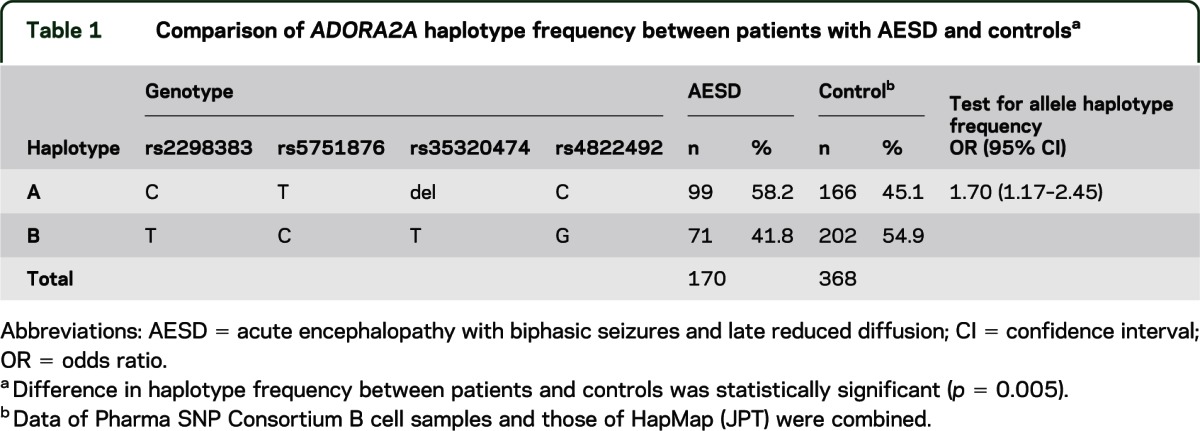
Table 2.
Comparison of ADORA2A diplotype distribution between patients with AESD and controls
ADORA2A mRNA expression in the brain.
Second, to evaluate the association of ADORA2A diplotypes with gene expression in the CNS tissue, we measured the amount of ADORA2A mRNA in SMRI samples after genotyping. Because the 4 SNPs were completely linked in 95 of 100 subjects (diplotype AA, 19 subjects; AB, 38 subjects; and BB, 38 subjects), we used these 95 samples. The relative expression level of ADORA2A mRNA (mean ± SEM) in AA, AB, and BB diplotypes was 0.246 ± 0.025, 0.179 ± 0.009, and 0.177 ± 0.009, respectively (figure 2). The expression level was 1.4-fold higher in the AA diplotype than in AB and BB, showing a significant difference (p = 0.003 and 0.002, respectively).
Figure 2. ADORA2A mRNA expression in the brain with different ADORA2A diplotypes.
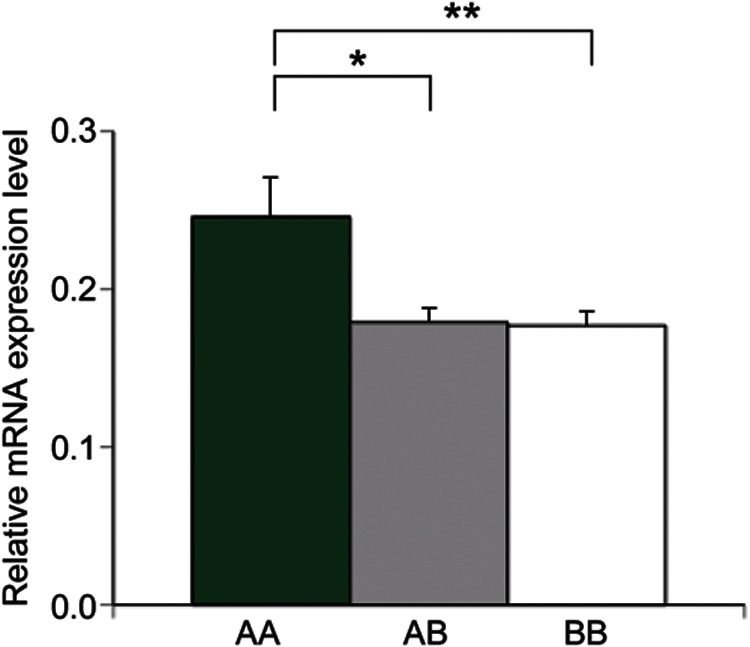
Relative ADORA2A mRNA expression level (ADORA2A/G6PDH) in the brain was higher in the AA diplotype (n = 19) than in AB (n = 38, *p = 0.003) and BB (n = 38, **p = 0.002).
ADORA2A mRNA and protein expression and production of cAMP in lymphoblasts.
ADORA2A is highly expressed in brain, heart, kidney, and lymphocytes.9,10 Because protein samples from the brain were unavailable, we used lymphoblast cell lines to determine the effect of ADORA2A diplotypes on ADORA2A protein expression. We again showed that the expression of ADORA2A mRNA in lymphoblasts with AA diplotype was higher than in those with AB and BB (figure 3A, p = 0.035 and 0.003, respectively). By Western blotting, the relative ADORA2A protein level (mean ± SEM) was evaluated as 0.611 ± 0.045, 0.439 ± 0.022, and 0.443 ± 0.044 for AA, AB, and BB, respectively (figure 3B). The protein expression was significantly higher in AA diplotype than in AB and BB (p = 0.021 and 0.024, respectively). Next, to elucidate the difference of intracellular signal transduction among 3 ADORA2A diplotypes, cAMP assay was performed. The cellular cAMP accumulation level (mean ± SEM) for AA, AB, and BB diplotypes was 2.016 ± 0.207, 1.421 ± 0.186, and 0.953 ± 0.118 pmol, respectively (figure 3C). As the number of haplotype A alleles increased, so did the adenosine-stimulated cAMP production. The cAMP level was significantly higher in the AA diplotype than in BB (p = 0.0006).
Figure 3. ADORA2A mRNA expression, ADORA2A protein expression, and cAMP production in lymphoblasts with different ADORA2A diplotypes.
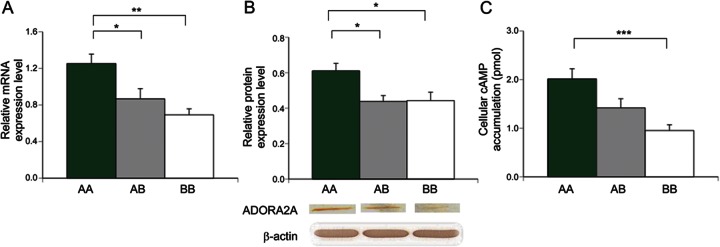
(A) Relative ADORA2A mRNA expression level (ADORA2A/G6PDH) is higher in AA diplotype than in AB (*p = 0.035) and BB (**p = 0.003) (n = 5 for each diplotype). (B) Relative ADORA2A protein expression level (ADORA2A/β-actin) was higher in AA than in AB (*p = 0.021) and BB (*p = 0.024) (n = 5 for each diplotype). Lower panel shows results of a representative Western blot, showing increasing band intensity with the number of haplotype A. (C) Cyclic adenosine monophosphate (cAMP) production in response to adenosine was higher in diplotype AA than in BB (***p = 0.0006) (n = 5 for each diplotype).
DISCUSSION
Previous studies have shown the complex roles of adenosine in the brain, deriving from the diversity of receptor subtypes. In the CNS, ADORA2A competes with ADORA1 in various neural functions. For synaptic transmission, ADORA2A enhances excitatory neurotransmitter release, whereas ADORA1 exerts an inhibitory effect.11 The role of adenosine as an endogenous anticonvulsant is mediated via ADORA1.12 Inhibition of ADORA1 function has been shown to cause status epilepticus.13 In a rat model of seizure kindling, ADORA1 in the hippocampal CA1 region reduces seizures, whereas ADORA2A promotes them.14 Adora2a knockout mice show a reduction of ethanol-induced seizures,15 whereas activation of ADORA2A renders rat pups susceptible to hyperthermia-induced seizures.16 Despite these findings, the association of ADORA2A variations with a seizure disorder has never been reported. They are known to be associated with anxiety induced by caffeine, an antagonist of ADORA1 and ADORA2A.17–19
The present study showed for the first time the association between an ADORA2A genetic variant and AESD, a typical syndrome of AEIMSE during early childhood. The results suggest that ADORA2A AA diplotype predisposes children to AESD by altering the intracellular adenosine/cAMP signal cascade.
We demonstrated that the frequency of ADORA2A AA diplotype was significantly higher in patients with AESD than in controls (table 2). These data show an apparent association between AA diplotype and AESD, although whether the recessive or additive model most accurately describes this association is unclear at this time. Haplotype A consists of 4 SNPs, rs2298383, rs5751876, rs35320474, and rs4822492, which show complete linkage disequilibrium with one another in Japanese. The rs2298383 SNP is located in a potential promoter region upstream of the recently identified exon variant,8 with a regulatory element predicted from alignment of human and other mammalian genes.20 Further evidence of its importance in gene expression regulation is provided by in silico analyses,21 which indicated the position of rs2298383 SNP within a triplex-forming oligonucleotide target sequence. The 35320474 SNP is located in the 3′ untranslated region including U-rich motifs. U-rich motifs are conserved across species and provide active sites for interaction with RNA-binding proteins. Thus, any of these SNPs may possibly alter the expression level of mRNA.
In fact, we found that the ADORA2A AA diplotype causes a high expression of ADORA2A mRNA in the brain and lymphoblasts, and a high expression of ADORA2A protein in lymphoblasts. Given its excitatory function, increased expression of ADORA2A may cause a functional imbalance between ADORA1 and ADORA2A, resulting in hyperexcitation of cerebral neurons.
In the present study, cellular cAMP accumulation in response to adenosine was high in lymphoblasts with ADORA2A AA diplotype. ADORA2A, together with coupled Gs proteins, activates adenylate cyclase and increases the cellular cAMP level. In this study, we observed high cellular cAMP in the AA diplotype, which supports our hypothesis that the signal cascade downstream of ADORA2A is excessively activated in AESD. cAMP promotes protein kinase A, which in turn enhances Ca2+ influx through the l-type Ca2+ channel in the basal ganglia, hippocampus, and striatum. Ca2+ then enhances glutamate efflux from the endoplasmic reticulum to the extracellular space, leading to excitotoxicity.22–25 An increase of extracellular glutamate in the brain lesion of AESD has recently been demonstrated by magnetic resonance spectrometry.5
Involvement of ADORA2A in the pathogenesis of AESD may have therapeutic implications. Experimental studies have previously shown that an ADORA2A antagonist, but not an ADORA1 agonist, can terminate or suppress seizures.26,27 Pharmacologic blockade or genetic disruption of ADORA2A may protect neurons from seizures by reducing glutamate release and excitotoxicity.27 Thus, ADORA2A antagonists are promising candidate drugs to ameliorate seizure-induced brain damage. Because this study showed alteration of the ADORA2A signal cascade in AESD, these drugs may also be particularly useful in the treatment of AESD. However, our data showed that 20% of patients with AESD have the BB diplotype, suggesting the involvement of factors other than ADORA2A in the etiology of AESD.
In conclusion, the present study demonstrated that polymorphisms of the ADORA2A, or AA diplotype, are risk factors of AESD, an acute encephalopathy with febrile status epilepticus. This diplotype showed a high ADORA2A expression level and high cAMP accumulation in response to adenosine, suggesting the involvement of the adenosine/cAMP signal cascade in the pathogenesis of AESD. Pharmacologic intervention in this pathway may improve the treatment of children with this devastating encephalopathy.
Supplementary Material
ACKNOWLEDGMENT
The authors thank the Stanley Medical Research Institute Brain Collection for providing the SMRI specimens, the contributors (listed on the Neurology® Web site at www.neurology.org) for providing patient samples and clinical records, and Ms. Kiyomi Noyama and Ms. Aya Shoda for technical assistance.
GLOSSARY
- ADORA1
adenosine A1 receptor
- ADORA2A
adenosine A2A receptor
- AEIMSE
acute encephalopathy with inflammation-mediated status epilepticus
- AESD
acute encephalopathy with biphasic seizures and late reduced diffusion
- cAMP
cyclic adenosine monophosphate
- CI
confidence interval
- CPT2
carnitine palmitoyltransferase II
- G6PDH
glucose-6-phosphate dehydrogenase
- OR
odds ratio
- SMRI
Stanley Medical Research Institute
- SNP
single nucleotide polymorphism
Footnotes
Supplemental data at www.neurology.org
AUTHOR CONTRIBUTIONS
M. Shinohara contributed to analysis and interpretation of the data. M. Saitoh contributed to design and conceptualization of the study, interpretation of the data, draft and revision of the manuscript for intellectual content. D. Nishizawa contributed to analysis and interpretation of the data. K. Ikeda contributed to design and conceptualization of the study, interpretation of the data, draft and revision of the manuscript for intellectual content. S. Hirose contributed to analysis and interpretation of the data. J. Takanashi, J. Takita, K. Kikuchi, M. Kubota, G. Yamanaka, T. Shiihara, A. Kumakura, M. Kikuchi, M. Toyoshima, T. Goto, and H. Yamanouchi contributed to interpretation of the data. M. Mizuguchi contributed to the design and conceptualization of the study, interpretation of the data, draft and revision of the manuscript for intellectual content.
STUDY FUNDING
No targeted funding reported.
DISCLOSURE
M. Shinohara reports no disclosures. M. Saitoh is funded by a Grant-in-Aid for Scientific Research from the Japan Society for the Promotion of Science No. 22591176. D. Nishizawa and K. Ikeda report no disclosures. S. Hirose is funded by a by a Grant-in-Aid for Nervous and Mental Disorder Grant 24-7 and H24-Nanji-Ippan-29 from the Ministry of Health, Labour and Welfare, Japan. J. Takanashi is funded by a Grant-in-Aid for research on Measures for Intractable Diseases H23-Nanji-Ippan-78 from the Ministry of Health, Labour and Welfare, Japan. J. Takita and K. Kikuchi report no disclosures. M. Kubota is funded by a Grant-in-Aid for research on Measures for Intractable Diseases H23-Nanji-Ippan-78 from the Ministry of Health, Labour and Welfare, Japan. G. Yamanaka, T. Shiihara, A. Kumakura, M. Kikuchi, M. Toyoshima, T. Goto, and H. Yamanouchi report no disclosures. M. Mizuguchi is funded by a Grant-in-Aid for Scientific Research from the Japan Society for the Promotion of Science No. 20390293 and by a Grant-in-Aid for research on Measures for Intractable Diseases H23-Nanji-Ippan-78 from the Ministry of Health, Labour and Welfare, Japan. Go to Neurology.org for full disclosures.
REFERENCES
- 1.Nabbout R, Vezzani A, Dulac O, Chiron C. Acute encephalopathy with inflammation-mediated status epilepticus. Lancet Neurol 2011;10:99–108 [DOI] [PubMed] [Google Scholar]
- 2.Takanashi J, Oba H, Barkovich AJ, et al. Diffusion MRI abnormalities after prolonged febrile seizures with encephalopathy. Neurology 2006;66:1304–1309 [DOI] [PubMed] [Google Scholar]
- 3.Hoshino A, Saitoh M, Oka A, et al. Epidemiology of acute encephalopathy in Japan, with emphasis on the association of viruses and syndromes. Brain Dev 2012;34:337–343 [DOI] [PubMed] [Google Scholar]
- 4.Mizuguchi M, Yannanouchi H, Ichiyama T, Shiomi M. Acute encephalopathy associated with influenza and other viral infections. Acta Neurol Scand 2007;115:45–56 [DOI] [PubMed] [Google Scholar]
- 5.Takanashi J, Tada H, Terada H, Barkovich AJ. Excitotoxicity in acute encephalopathy with biphasic seizures and late reduced diffusion. AJNR Am J Neuroradiol 2009;30:132–135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Takanashi J. Two newly proposed infectious encephalitis/encephalopathy syndromes. Brain Dev 2009;31:521–528 [DOI] [PubMed] [Google Scholar]
- 7.Shinohara M, Saitoh M, Takanashi J, et al. Carnitine palmitoyl transferase II polymorphism is associated with multiple syndromes of acute encephalopathy with various infectious diseases. Brain Dev 2011;33:512–517 [DOI] [PubMed] [Google Scholar]
- 8.Yu LQ, Frith MC, Suzuki Y, et al. Characterization of genomic organization of the adenosine A2A receptor gene by molecular and bioinformatics analyses. Brain Res 2004;1000:156–173 [DOI] [PubMed] [Google Scholar]
- 9.Olah ME, Stiles GL. Adenosine receptor subtypes: characterization and therapeutic regulation. Annu Rev Pharmacol Toxicol 1995;35:581–606 [DOI] [PubMed] [Google Scholar]
- 10.Huang S, Apasov S, Koshiba M, Sitkovsky M. Role of A2a extracellular adenosine receptor-mediated signaling in adenosine-mediated inhibition of T-cell activation and expansion. Blood 1997;90:1600–1610 [PubMed] [Google Scholar]
- 11.Fredholm BB, Chen JF, Cunha RA, Svenningsson P, Vaugeois JM. Adenosine and brain function. Int Rev Neurobiol 2005;63:191–270 [DOI] [PubMed] [Google Scholar]
- 12.Dragunow M. Adenosine: the brain’s natural anticonvulsant. Trends Pharmacol Sci 1986;7:128–130 [Google Scholar]
- 13.Avsar E, Empson RM. Adenosine acting via A1 receptors, controls the transition to status epilepticus-like behaviour in an in vitro model of epilepsy. Neuropharmacology 2004;47:427–437 [DOI] [PubMed] [Google Scholar]
- 14.Zeraati M, Mirnajafi-Zadeh J, Fathollahi Y, Namvar S, Rezvani ME. Adenosine A1 and A2A receptors of hippocampal CA1 region have opposite effects on piriform cortex kindled seizures in rats. Seizure 2006;15:41–48 [DOI] [PubMed] [Google Scholar]
- 15.El Yacoubi M, Ledent C, Parmentier M, et al. Absence of the adenosine A2A receptor or its chronic blockade decrease ethanol withdrawal-induced seizures in mice. Neuropharmacology 2001;40:424–432 [DOI] [PubMed] [Google Scholar]
- 16.Fukuda M, Suzuki Y, Hino H, Morimoto T, Ishii E. Activation of central adenosine A2A receptors lowers the seizure threshold of hyperthermia-induced seizure in childhood rats. Seizure 2011;20:156–159 [DOI] [PubMed] [Google Scholar]
- 17.Rogers PJ, Hohoff C, Heatherley SV, et al. Association of the anxiogenic and alerting effects of caffeine with ADORA2A and ADORA1 polymorphisms and habitual level of caffeine consumption. Neuropsychopharmacology 2010;35:1973–1983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Childs E, Hohoff C, Deckert J, Xu K, Badner J, de Wit H. Association between ADORA2A and DRD2 polymorphisms and caffeine-induced anxiety. Neuropsychopharmacology 2008;33:2791–2800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Alsene K, Deckert J, Sand P, de Wit H. Association between A2a receptor gene polymorphisms and caffeine-induced anxiety. Neuropsychopharmacology 2003;28:1694–1702 [DOI] [PubMed] [Google Scholar]
- 20.King DC, Taylor J, Elnitski L, Chiaromonte F, Miller W, Hardison RC. Evaluation of regulatory potential and conservation scores for detecting cis-regulatory modules in aligned mammalian genome sequences. Genome Res 2005;15:1051–1060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Conde L, Vaquerizas JM, Dopazo H, et al. PupaSuite: finding functional single nucleotide polymorphisms for large-scale genotyping purposes. Nucleic Acids Res 2006;34:W621–W625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Higley MJ, Sabatini BL. Competitive regulation of synaptic Ca2+ influx by D2 dopamine and A2A adenosine receptors. Nat Neurosci 2010;13:958–977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Skeberdis VA, Chevaleyre V, Lau CG, et al. Protein kinase A regulates calcium permeability of NMDA receptors. Nat Neurosci 2006;9:501–510 [DOI] [PubMed] [Google Scholar]
- 24.Cunha RA. Adenosine as a neuromodulator and as a homeostatic regulator in the nervous system: different roles, different sources and different receptors. Neurochem Int 2001;38:107–125 [DOI] [PubMed] [Google Scholar]
- 25.Fredholm BB, Chern Y, Franco R, Sitkovsky M. Aspects of the general biology of adenosine A2A signaling. Prog Neurobiol 2007;83:263–276 [DOI] [PubMed] [Google Scholar]
- 26.O’Regan MH, Simpson RE, Perkins LM, Phillis JW. The selective adenosine A2 receptor agonist CGS 21680 enhances excitatory transmitter amino acid release from the ischemic rat cerebral cortex. Neurosci Lett 1992;138:169–172 [DOI] [PubMed] [Google Scholar]
- 27.Jones PA, Smith RA, Stone TW. Protection against hippocampal kainate excitotoxicity by intracerebral administration of an adenosine A2A receptor antagonist. Brain Res 1998;800:328–335 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.