

Abstract
Dysregulation of professional APC has been postulated as a major mechanism underlying Ag-specific T cell hyporesponsiveness in patients with patent filarial infection. To address the nature of this dysregulation, dendritic cells (DC) and macrophages (MΦ) generated from elutriated monocytes were exposed to live microfilariae (mf), the parasite stage that circulates in blood and is responsible for most immune dysregulation in filarial infections. DC exposed to mf for 24–96 h showed a marked increase in cell death and caspase-positive cells compared with unexposed DC, while mf exposure did not induce apoptosis in MΦ. Interestingly, 48 h exposure of DC to mf induced mRNA expression of the pro-apoptotic gene TRAIL and both mRNA and protein expression of TNF-α. mAb to TRAIL-R2, TNF-R1, or TNF-α partially reversed mf-induced cell death in DC, as did knocking down the receptor for TRAIL-R2 using small interfering RNA. Mf also induced gene expression of BH3-interacting domain death agonist (Bid) and protein expression of cytochrome c in DC; mf-induced cleavage of Bid could be shown to induce release of cytochrome c, leading to activation of caspase 9. Our data suggest that mf induce DC apoptosis in a TRAIL- and TNF-α-dependent fashion.
Keywords: Apoptosis, Dendritic cell, Parasitic-helminth
Lymphatic filariasis, a mosquito-borne disease that is a major cause of morbidity in tropical and subtropical regions of the world, is caused by infection with Wuchereria bancrofti, Brugia malayi, or Brugia timori. Patent infection, in large part, is clinically asymptomatic but is associated with the inability of T cells to proliferate or produce IFN-γ in response to parasite Ag (1). Dysregulation of professional APC—dendritic cells (DC) and macrophages (MΦ)—is one hypothesis felt to explain the lack of an Ag-specific T cell response. We have shown previously that microfilariae (mf) of B. malayi affect human DC in at least two ways: i) by interfering with their viability (2), and ii) by altering their function (2, 3). In addition, the infective stage (L3) of the parasite has been shown to alter the function of human Langerhans’ cells (LC) quite profoundly (4). Notably, these same filarial parasites, if injected into the peritoneal cavity of mice, induce suppressive nematode-elicited MΦ capable of blocking T cell proliferative responses (5). Moreover, data from another mouse system indicate that a phosphorylcholine-containing glycoprotein, ES-62, secreted by filarial nematode Acanthocheilonema viteae, induces maturation of DC2 with the capacity to induce Th2 responses (6) that may crossregulate Th1 responses.
Although both viral and bacterial infections are known to induce DC apoptosis (7), cell death or depletion of APC by filarial worms has been less understood. In addition to our previous report of B. malayi mf-mediated apoptotic cell death in human DC(2), there are additional studies indicating that filarial parasite proteins can cause cell death in lung epithelial cells (8) and that B. malayi mf can induce apoptosis in murine CD4+ T cells (9).
It has been documented that DC express surface receptors and ligands including those of the TNF family known to mediate apoptotic cell death (reviewed in 10). Binding of TNF-α or TRAIL to their receptors results in recruitment of several adaptor proteins and caspases that participate in both intrinsic and extrinsic pathways of apoptosis (11). The extrinsic pathway of apoptosis involves a subset of TNF receptors (TNFR) including DR3, Fas, TNF-R1, TRAIL-R1, and TRAIL-R2 that function as death receptors and, upon ligand engagement, results in activation of the initiator caspase, caspase 8, which in turn activates caspase 3 directly or indirectly through BH3-interacting domain death agonist (Bid) truncation (tBid), release of cytochrome c, and subsequent activation of caspase 9 through interaction with apoptotic protease-activating factor 1 (APAF1) (11–14). In contrast, intrinsic apoptotic stimuli—cellular stress or deprivation of growth factors—results in mitochondrial permeability directly, by inactivation of anti-apoptotic BCL-2 family members, or through activation of pro-apoptotic BCL-2 family members without activation of an initiator caspase (11).
We show that mf of B. malayi cause caspase-dependent apoptotic cell death in human monocyte-derived DC but not MΦ. We further show that the mechanism of this cell death involves upregulation of TRAIL and TNF-α in DC by mf, an upregulation that can be blocked by antibodies to TRAIL-R2, TNF-R1, or TNF-α. Finally, this apoptotic mf-induced cell death involves induction of Bid, translocation of tBid, and release of cytochrome c from mitochondria that leads to caspase-9 activation.
Materials and Methods
Mf preparation
Live B. malayi mf were provided by Drs. John McCall, Ray Kaplan, and Michael Dzimianski (University of Georgia, Athens, GA) as described previously (15) Briefly, mf were collected by peritoneal lavage of infected jirds and separated from peritoneal cells by Ficoll diatrizoate density centrifugation. mf were then washed repeatedly in RPMI with antibiotics and cultured overnight at 37°C in 5% CO2 before use.
In vitro generation of DC and MΦ
CD14+ peripheral blood-derived monocytes were isolated from leukopacks from healthy donors by counterflow centrifugal elutriation under protocols approved by the institutional review boards of both the National Institute of Allergy and Infectious Diseases and the Department of Transfusion Medicine of the National Institutes of Health for these studies. Fresh monocytes were cultured in 6-well tissue-culture plates at 2 to 3 × 106/mL (no. 3596 Costar; Fisher Scientific, Suwannee, GA) in complete RPMI 1640 (BioWhittaker, Walkersville, MD) supplemented with 20 mM glutamine (BioWhittaker), 2% heat-inactivated human AB serum (Gemini Bioproducts, Woodland, CA), 100 μg/mL penicillin, and 100 g/mL streptomycin (Biosource International, Rockville, MD). For generation of DCs, recombinant human (rh) IL-4 and recombinant human granulocyte-macrophage colony-stimulating factor (rhGM-CSF; PeproTech, Rocky Hill, NJ) were added to the culture at 50 ng/mL on days 1, 5, and 7 of culture. For generation of MΦs, rhM-CSF (PeproTech) was added at 1000 U/mL on the same days. Both cell types were exposed to live mf on day 7 at a final concentration of 50,000/well (per 1–2 × 106 DC or 0.5–1 × 106 MΦ). This number is equivalent to ~1000 mf/ml of blood (containing 0.02–0.04 DC). DC or MΦ were exposed to mf for 48 h, and then the cells were harvested at day 9 of culture with Versene/EDTA (Biofluids, Inc.), washed twice with PBS (without Ca++/Mg++), counted by trypan blue exclusion, and used for functional studies. DC harvested at day 8 were repeatedly shown to be CD1a+, HLA-DR+, CD86+, CD40+, CD3−, CD14−/lo, CD19−, and CD56− by flow cytometry with 98% purity (FACSCalibur; Becton Dickinson).
Cell death assay
Harvested cells were counted using trypan blue exclusion or propidium iodide (PI) (Sigma Chemical Corporation) staining followed by flow cytometry. Caspase positivity of the harvested cells was measured using 10 μM CaspACE ™ FITC-VAD-FMK In situ Marker (Promega). Z-VAD-FMK is a cell-permeable, irreversible inhibitor of caspases. Then Flow cytometry was performed to measure the activity of total caspases. To induce cell death in DC, at day 6 of DC culture, rTRAIL (SuperKiller trail; Axxora LLC) was added at 100 ng/ml alone or in the presence of isotope control or anti-TRAIL mAb (2E5; Axxora LLC) at a final concentration of 10 μg/ml. rTL1A (Axxora LLC) was added at 100 μg/ml in the presence and absence of 100 μg/ml cyclohexamide for 48 h. The cells were harvested and washed, and cell death was measured by PI staining.
mAb blocking experiments
DC were generated and at day 6 of culture were incubated either alone or with the following mAb at a final concentration of 10 μg/ml: anti-TRAIL (2E5), anti-TNF-α (TNF-D), anti-TRAIL-R2 (HS201), anti-TNF-R1 (H398), or the combination of antibodies for 2 h. Then mf at 50,000/well were added to the cultures for an additional 48 h, at which time point the cells were harvested and cell death measured using PI staining and flow cytometry. All antibodies were purchased from Axxora LLC.
RNA preparation
DC or MΦ were cultured in media alone or were exposed at day 6 to mf at 50,000/well (in a 6-well plate) for 48 h, after which the cells were harvested and total RNA prepared from independent donors using RNAEasy mini kit (Qiagen).
Real-time RT-PCR
RNA (1 μg) from DC or mf-exposed DC was used to generate cDNA and then assessed by multiplex TaqMan assays (Applied Biosystems). Briefly, random hexamers were used to prime RNA samples for reverse transcription using MultiScribe (Applied Biosystems) reverse transcriptase, after which PCR products for all the genes tested in this report, as well as an endogenous 18s ribosomal RNA control, were assessed in triplicate wells using TaqMan predeveloped assay reagents. The assay ID numbers from Applied Biosystems for the genes tested in this study is listed below:
| Gene name | Assay ID |
|---|---|
| TRAIL | Hs00234356_m1 |
| TNF-α | Hs00174128_m1 |
| TL1A | Hs00270802_s1 |
| DR3 | Hs00600930_g1 |
| BID | Hs00609632_m1 |
| BCL2 | Hs00153350_m1 |
| FADD | Hs00938709_m1 |
| TRADD | Hs00182558_m1 |
| TRAF2 | Hs00184186_m1 |
| FLIP | Hs00153439_ml |
| Caspase 8 | Hs00236278_m1 |
Relative transcript levels were determined by the formula:
where CT is the threshold cycle during the exponential phase of amplification. Average ΔCT above 24 indicates lack of expression of the gene. Real-time quantitative RT-PCR was performed on an ABI 7900HT system (Applied Biosystems).
Microarray analysis
Total RNA from 4 independent donors were pooled together to generate cRNA probes. Preparation of cRNA, hybridization, and scanning of the U95AV2 arrays were performed according to the manufacturer’s protocol (Affymetrix) and as described previously (16).
Microarray data processing
Microarray data were processed using the absolute expression analysis within the Affymetrix Microarray Suite. A target intensity of 500 was used as a scaling factor for all probes to establish present or absent calls for each transcript. Scaled data were analyzed using GeneSpring GX 7.3 software (Agilent Technologies, Inc.). DC samples were normalized to the corresponding media-only control. Normalized data were then imported and analyzed using Ingenuity Pathway Analysis software (Ingenuity Systems, Inc.). Pathway analysis revealed biologic pathways altered in DC following exposure to MF. Next, the raw expression values for experimental (mf exposed) and control (media only) were compared to identify those genes with the greatest difference. Those genes with the greatest difference were analyzed further by quantitative PCR. Those genes with multiple probe sets were analyzed in a probe specific manner.
The microarray data used in this study has been deposited in NCBI’s Gene Expression Omnibus (GEO) with the accession number GSE12787.
Flow cytometry
Staining of cells with antibodies was carried out according to standard protocols. PI was used to exclude nonviable cells from the analysis. DC (0.2–0.5 × 106) were harvested and washed with FACS media (HBSS) without phenol red and without Ca++/Mg++ (BioWhittaker) containing 0.2% human serum albumin (Sigma Chemical Corporation) and 0.2% sodium azide (Sigma Chemical Corporation). Cells were incubated with human gammaglobulin (Sigma Chemical Corporation) at 10 mg/ml for 10 minutes at 4°C to inhibit subsequent binding of mAb to FcR. Then cells were incubated with specific mAb conjugated with FITC or PE at saturating concentrations for 30 minutes at 4°C, washed twice with FACS media, and analyzed using a FACSCalibur (Becton Dickinson) and CellQuest software. All antibodies used were mouse anti-human mAb and consisted of the following: CD1a-PE (clone VIT6B; Caltag); CD11a-FITC (clone MEM25; Caltag); CD11b-FITC (clone CR3; Caltag); CD11c-PE (clone 3.9; Caltag); CD14-FITC (clone Tuk4; Caltag); CD40-FITC (clone 14G7; Caltag); CD54-FITC (ICAM-1) (clone MEM111; Caltag); CD58-FITC (clone IC3; PharMingen); CD80 (B7-1)-FITC (clone L307.4; PharMingen); CD86 (B7-2)-FITC (clone 2331; PharMingen); CD83-PE (clone HB15e, PharMingen); CD95 (Fas)-PE (clone DX2; Caltag); CD95L (FasL)-PE (clone Alf2.1; Caltag); HLA-A,B, C-FITC (clone G46-2.6; PharMingen); and HLA-DR-FITC (clone L243; PharMingen).
Cytokine assays
Detection of TNF-α was performed by Pierce Biotechnology using Searchlight™ proteome arrays. ELISA to detect human TL1A (PeproTech Inc.) was performed according to the manufacturer’s recommendation. All samples were tested in duplicate and results expressed as an average of duplicate samples ± error.
Immunoblot analysis
DC cultured in 6-well plates were exposed to mf at 50,000 mf/well for 48 h. At this time point, nonadherent cells were collected, lysis buffer (CHAPS buffer, catalog #7722; Cell Signaling Technology) was added to wells, and adherent cells were lysed by pipetting up and down. The collected nonadherent cells were then spun at 1200 rpm for 10 minutes and combined with the nonadherent cells of the respective well. Cell lysates were prepared according to the protocol to pellet the mitochondrial component from the cytosolic fraction. Then the cytosolic fraction was boiled for 5 minutes; 40 μl of protein were run in a 1.5 mm 18% Tris gel and transferred onto PVDF membranes. After blocking using 5% non-fat milk for 1 h for Bid, cytochrome c, or caspase 3 and overnight for tubulin, the membranes were incubated overnight at 4°C with anti-rabbit Bid (catalog # 2002; Cell Signaling Technology) or rabbit anti-cytochrome c (catalog # 4272; Cell Signaling Technology), or rabbit anti-caspase 9 (catalog # 9502; Cell Signaling Technology) or with mouse anti-tubulin (Sigma Chemical Corporation) for 2 h. After washing, the membranes were incubated with HRP-conjugated anti-rabbit IgG (Amersham Biosciences) at 1:10000 or anti-mouse IgG (Amersham Biosciences) at 1:10000) at room temperature for 2 h. For tubulin control, the membranes were stripped in stripping buffer (30% H2O2) for 30 minutes and re-probed with anti-α-tubulin (Sigma Chemical Corporation) antibody. Proteins were detected by chemiluminescence (Detection System; Cell Signaling Technology). Tubulin was used as an internal control because of low background detection and a m.w. distinct from the proteins of interest here. For positive control for all immunoblots, caspase 3 cell extracts (catalog # 9663; Cell Signaling Technology) were used.
ImageJ (http://rsb.info.nih.gov/ij/) was used to quantify intensity of bands in immunoblots.
Preparation of whole-cell lysates
DC exposed or unexposed to mf were harvested and whole-cell extracts prepared using a commercially available kit (Active Motif). Briefly, cells were washed in cold 1× PBS. After washing, cells were gently resuspended in complete lysis buffer and incubated on ice for 30 minutes. Then cells were centrifuged at 14,000×g for 20 minutes, and cell lysate supernatant was analyzed using the Bradford protein assay (Sigma Chemical Corporation) to measure the amount of protein.
Electron microscopy
DC and mf-exposed DC were harvested as described in the previous section. Cell pellets were then fixed in 2.5% glutaraldehyde and 4% paraformaldehyde in 0.1 M sodium cacodylate buffer (pH 7.2). Cell pellets were gently resuspended into a small volume of 2% NuSieve® low-melt agarose (Lonza Biologics), at 40°C and cooled to 4°C for 10 minutes. Cellular material was excised from agarose and processed as follows. Samples were washed twice for 30 minutes in 0.1 M sodium cacodylate pH 7.2, then post-fixed for 2 h in a mixture of 1% osmium tetroxide, 0.8% potassium ferrocyanide, and 0.1 M cacodylate buffer pH 7.2 (Ted Pella, Inc.). Samples were then washed for 30 minutes in cacodylate buffer and twice in water prior to staining for 1 h in 1% uranyl acetate in water. Following three more 30-minute washes in water, samples were dehydrated in an acetone series and embedded in araldite resin (Electron Microscopy Sciences). Sections were cut on a diamond knife and examined at 80 kV on a Hitachi H7500 transmission electron microscope. Digital images were captured using an XR-100 camera (Advanced Microscopy Techniques, Inc.).
Statistical analysis
The nonparametric Wilcoxon signed rank test was used throughout. All statistical analyses were performed with GraphPad Prism 4.0 (GraphPad Software, Inc.).
Results
mf induce caspase-dependent cell death in DC but not in MΦ
We have previously shown that the mf stage of B. malayi can induce apoptotic cell death in DC(2). To determine whether this mf-induced cell death is specific to DC and to investigate the mechanisms underlying this apoptotic cell death, we generated DC and MΦ from the same monocyte donors and compared the effect of mf on both cell types. After 48-h exposure to mf, DC showed a significant reduction in viable cells compared with unexposed DC or mf-exposed MΦ. These differences in cell death induction were detected by trypan blue exclusion (Fig. 1A; p = 0.0005) as well as by PI staining (Fig. 1B; P < 0.0001). Furthermore, longer exposure of MΦ to mf (up to 96 h) did not result in cell death induction in these cells (data not shown). Indeed we have shown in the past that live mf induce apoptosis in human monocyte-derived DC(2). In this report, we further assess apoptosis by using a FITC labeled pan-caspase inhibitor, Z-VAD-FMK (CaspACE ™ FITC-VAD-FMK In Situ Marker), which allows in situ labeling of activated caspases, and can be used as a quantitative tool for flow cytometric analysis of apoptosis. As shown in figure 1C, when cells were labeled with CaspACE ™ FITC-VAD-FMK In Situ Marker, there was a significant increase of caspase-positive cells with mf exposure only in DC and not in MΦ (Fig. 1C), providing additional evidence for caspase activation in mf-induced DC cell death (2). Furthermore, using electron microscopy, apoptotic morphology was detected in DC exposed to mf (Fig. 2). As seen, the majority of mf-unexposed DC had normal morphology (Fig. 2A) with intact organelles; however, exposure to mf resulted in chromatin condensation (Fig. 2B, both top and bottom cells), organelle degradation, loss of mitochondrial integrity (Fig. 2B, bottom cell) and cell shrinkage (Fig. 2B, both top and bottom cells).
FIGURE 1.

mf of B. malayi induce cell death in human DC and not MΦ. Cell viability represented A by trypan blue exclusion of dead cells and B by percentage of PI+ death cells. C, Percentage of caspase-positive cells as measured by flow cytometry using a FITC-labeled caspase inhibitor CaspACE ™ FITC-VAD-FMK In Situ Marker, in unexposed DC, unexposed MΦ, or DC and MΦ exposed to mf for 48 h. Results shown are dot plots with geometric mean of 16 (A, B) or 5 (C) independent donors.
FIGURE 2.
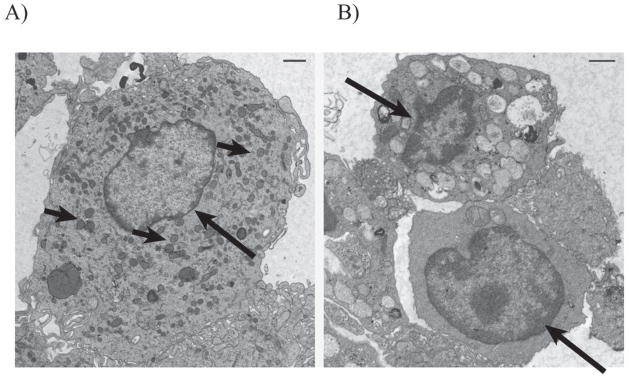
mf-exposed DC show apoptotic morphology. Electron microscopy was performed on A mf-unexposed DC or B DC exposed to mf for 48 h. B represents two cells: the top panel shows a cell with organelles intact; the bottom panel shows a cell with dissociated organelles. Long arrows, nucleoli; short arrows, mitochondria.
The mf upregulate gene expression of TRAIL, TL1A, and TNF-α
Having demonstrated that mf induce caspase-dependent cell death only in DC and not in MΦ from the same source, we assessed more globally the mechanisms involved in this mf-induced cell death utilizing microarray analysis. Therefore, RNA was prepared from unexposed DC and exposed to mf for 48 h. This RNA was labeled and hybridized to Illumina Sentrix bead chip microarrays (Illumina Inc.) and analyzed using GeneSpring GX 7.3 software (Agilent Technologies, Inc.) (Fig. 3 and GSE12787/Pending). As can be seen, mf induced expression of a large number of genes associated with apoptosis, including those in pro-apoptotic signaling (caspase 8, cytochrome c, and Bid). Further analysis using Ingenuity Pathway Analysis software (Ingenuity Systems, Inc.) revealed altered gene expression in two sets of pathways, one ligand/receptors, and two in downstream molecules involved in signaling events leading to apoptosis (see GSE12787/Pending). Moreover, upregulation of pro-apoptotic molecules was confined to ligands in death receptor signaling pathways (e.g., TRAIL and TNF-α) rather than their receptors, whose expression remained unchanged with an exception of TNF-R2 (Fig. 3), following exposure to mf.
FIGURE 3.

Expression of death receptor signaling genes by DC unexposed and exposed to mf. Gene expression of DC and DC exposed to mf for 48 h were assessed using Affymetrix U95AV2 microarrays. The raw expression values of apoptosis-related genes indicate a subset of these genes that is upregulated upon exposure to MF. Multiple lines for select genes reflect the presence of multiple probe sets present on the Affymetrix gene chip for a particular gene. Combined analysis of these data using GeneSpring GX and Ingenuity pathway analysis shows differential regulation of genes linked to death receptor signaling between DC and DC exposed to MF.
We confirmed these data by RT-PCR and show in Figure 4A that mf upregulated mRNA expression of TRAIL (P = 0.003; 10 of 13 donors had a fold increase ranging between 1.5 to 8.5), TNF-α (p = 0.03;), 5 of 6 had a fold increase ranging between 1.6 to 4.9), and TL1A (another member of the TNF family of ligands also known as VEG1; p = 0.01; ranging from 2 to 25 fold increase in all donors) only in DC and not in MΦ (data not shown). Notably, while we were unable to detect soluble TRAIL in the culture supernatant (data not shown), mf-exposed DC had a significant increase in the level of soluble TL1A (Fig. 4B) and TNF-α (Fig. 4B) in all donors tested.
FIGURE 4.

Pre-exposure to mf upregulates mRNA expression of TRAIL, TL1A, and TNF-α and induces production of soluble TL1A and TNF-α but downregulates mRNA expression of DR3 in DC. A, Results are expressed as 1/ΔCT for DC unexposed or mf-exposed (DC/mf). The higher the 1/average ΔCT, the higher the expression of the gene. Each line represents an independent donor. B, Data are expressed as ng/ml for DC unexposed or exposed to mf for 48 h (DC/mf). Each line represents an independent donor.
When the expression of the receptors for these ligands was examined, the mRNA levels of the TRAIL receptor TRAIL-R2 and of TNF receptor TNF-R1 were unchanged in mf-exposed DC (Fig. 4A). While the expression of TL1A receptor, DR3, was downregulated (Fig. 4A; p = 0.01; 7 of 10 donors had a fold decrease ranging from 1.4 to 8.5; and data not shown). TNFR2 in the microarray experiments (Fig. 3) was shown to be upregulated in mf-exposed DC. Furthermore, while cell surface expression of DR3 was slightly downregulated by mf (similar to mRNA expression), the expression of total TRAIL, membrane bound TRAIL, TRAIL-R2 and TNF-R1 remained unchanged after exposure to this parasite (data not shown).
TRAIL/TRAIL-R2 and TNF-α/TNF-RI pathways are involved in mf-induced DC cell death
To investigate whether upregulation of TRAIL, TNF-α, or TL1A directly translates to induction of cell death by mf (perhaps through binding of the ligands to the death receptors expressed on DC), we used blocking antibodies either to the ligands or to the receptors of these pathways. Because DC expressed both TRAIL-R1 and TRAIL-R2 on the cell surface (data not shown), we examined first whether these cells are susceptible to TRAIL-induced killing by exposing DC to recombinant TRAIL for 48 h and then measuring cell viability using PI staining (Fig. 5A). As shown, DC undergo TRAIL-dependent cell death that can be almost completely reversed using anti-TRAIL antibody; however, exposing DC to recombinant TL1A—a molecule shown originally to induce caspase activation (in the presence of cyclohexamide) in TF-1 cell line expressing DR3 (17)—did not induce cell death in DC (Fig. 5B). In concurrent studies, blocking the TL1A/DR3 pathway using a DR3-fusion protein did not reverse mf-induced DC cell death (data not shown), suggesting that this pathway is not the major pathway involved in killing of DC by mf. In contrast, blocking the TRAIL/TRAILR2 or the TNF-α/TNF-R1 pathways using neutralizing mAb to either the receptors or the ligands significantly reversed mf-induced cell death in DC. Since DC show a higher cell surface expression of TRAIL-R2 (data not shown) we used neutralizing antibody to TRAIL-R2 in our studies. While anti-TRAIL did not reverse mf-induced cell death, anti-TRAIL-R2 (P = 0.01), anti-TNF-α (P = 0.02), or anti-TNF-RI (P = 0.028), resulted in a significant increase in the percentage of viable cells in mf-exposed DC cultures (Fig. 6). It needs to be noted that neutralizing anti-TRAIL, anti-TRAIL-R1 or R2, or anti-TNF-α did not have a significant effect on the survival of untreated DC.
FIGURE 5.

DC are sensitive to TRAIL- (but not TL1A)–induced cell death. DC were cultured either alone or with A recombinant TRAIL in the presence of anti-TRAIL antibody or isotype control antibody for 48 h or B recombinant TL1A in the absence or presence of cyclohexamide. Cells were harvested, and viability of cells was measured by PI+ dead cells using flow cytometry.
FIGURE 6.

Anti-TRAIL-R2, anti-TNF-α, or anti-TNF-R1 reverse mf-induced cell death in DC. DC were exposed to live mf alone for 48 h or first incubated with the antibodies indicated in the graph for 2 h and then exposed to mf for 48 h. Cells were harvested, and cell viability was measured by PI staining of dead cells using flow cytometry. Results shown are the percentage of PI+ cells in DC/mf, or DC/mf pretreated with indicated mAb. Each line represents an independent donor.
We also confirmed our antibody blocking results utilizing siRNA in which we were able to achieve a 5-fold decrease in TRAIL-R2 mRNA expression as measured by RT-PCR that, in turn, resulted in an increase of 20% to 30% in viability of cells after exposure to mf (data not shown).
The mf induce the release of cytochrome c through tBid in DC
Because TRAIL or TNF-α can lead to apoptosis through caspase 8 activation (though either a mitochondria-dependent or independent pathway (18, 19) and because our microarray data suggested that Bid and cytochrome c (part of the mitochondria-dependent pathway) are upregulated in DC after exposure to mf, we assessed mf-induced expression of genes involved in TRAIL- or TNF-α-induced apoptotic pathway (FADD, TRADD, or TRAF2, Bcl2, TRAF2, FLIP, caspase 8). As seen in Figure 7A, mRNA expression of Bid was significantly induced in DC after 48-h exposure to mf (p = 0.04; eight of thirteen donors had an increase above 1.3 fold) compared with unexposed DC, as was caspase 8 (five of seven donors). Furthermore, 48 h of exposure of DC to mf resulted in a significant decrease in levels of tBid in cytoplasmic cell lysates (Fig. 7B; p = 0.05) compared with mf-unexposed DC. These data suggest that after exposure to mf, tBid has been translocated to the mitochondria, which in turn results in the release of cytochrome c. Therefore, we next measured release of cytochrome c in mf-exposed and unexposed DC. Notably, 48-h exposure of DC to mf resulted in a significant induction of cytochrome c protein in cytoplasmic lysates as compared with that seen in unexposed cells (Fig. 7C), suggesting that mf induction of cell death is through a mitochondria-dependent pathway. Furthermore, antibodies to TRAIL-R2, TNF-R1, or TNF-α diminished cytoplasmic cytochrome c in DC exposed to mf (Fig. 7C), suggesting that mf induction of cytochrome c release is mediated in this system through TNF-α and TRAIL binding to their appropriate receptors.
FIGURE 7.

Pre-exposure to mf results in upregulation of Bid mRNA expression but downregulation of cytoplasmic tBid protein. mf upregulate cytochrome c protein expression in DC in a TRAIL- and TNF-α-dependent manner and induce cleavage of 35-kDa and 17-kDa active caspase 9. A, Results are expressed as 1/average ΔCT in unexposed and mf-exposed DC (DC/mf) as individual lines representing each of 6 to 14 independent donors. The higher the 1/average ΔCT, the higher the expression of the gene. B, Total Bid (22 kDa) and tBid (15 kDa) in DC unexposed and exposed to mf for 48 h using western blot analysis in one representative of 12 different donors. Graph demonstrates the tBid:tubulin ratio for each of 12 donors. C, Cytochrome c (14 kDa) protein expression in DC unexposed and exposed to mf for 48 h in 1 representative of 11 different donors and in the absence or presence of anti-TRAIL-R2, anti-TRAIL-R1, or anti-TNF-α using immunoblot analysis. Graphs demonstrate the cytochrome c to tubulin ratio for each of 11 donors or of 4 donors. D, 47-kDa pro-caspase 9 was cleaved to 35-kDa and 17-kDa active fragments of DC exposed to mf for 48 h in one of three independent donors. For positive control in all immunoblots, caspase 3 cell extracts were used (see Materials and Method section).
The mf induce cleavage of 35-kDa and 17-kDa active caspase 9 in DC
Because active caspase 9 can mark cells undergoing apoptotic cell death through a mitochondria-dependent pathway, we examined expression of active caspase 9 in DC exposed to mf (Fig. 7D). As seen, mf induced protein expression of 47-kDa pro-caspase 9 in general and resulted in its cleavage to 35-kDa and 17-kDa active caspase 9 in DC.
Discussion
Programmed cell death, or apoptosis, is essential for the development, defense, and homeostasis of multicellular organisms and is an effective mechanism to remove cells that have encountered parasites. Induction of apoptosis in host T and B cells and MΦ by intracellular protozoan parasites has been shown to be one mechanism by which these organisms evade the host immune system (20). In other studies, a direct interaction between filarial proteins and lung epithelial cells was shown to result in epithelial cell death (8). Furthermore, it has been demonstrated that filarial sheath proteins can induce apoptosis in the human epithelial cell line HEp2 that can be reversed by bcl2 overexpression (21). In another study in a murine system, apoptosis of CD4+ T cells was shown to occur following injection of B. malayi mf (9). Recently, it has also been demonstrated that L3 B. malayi can induce apoptosis in human NK cells (22).
Thus, we have examined the mechanisms underlying filaria-induced cell death in DC and contrasted this to human MΦ. In so doing, we have demonstrated that this cell death is a caspase-dependent apoptotic process (Figs. 1 and 7) that occurs as a result of TNFR signaling and mitochondria-dependent pathway that involves Bid truncation, release of cytochrome c, and activation of caspase 9.
Our initial microarray analysis (Fig. 3; GSE12787/Pending) and corroborative quantitative RT-PCR suggested that TRAIL and TNF-α were involved in the extrinsic pathway of cell death (reviewed in 11), leading to mitochondria-dependent apoptosis. Notably, mRNA expression of the receptors for TRAIL-R2 and TNF-R1 remained unchanged, suggesting that gene alteration was at the level of ligands and not receptors. Furthermore, although we did not detect any soluble TRAIL, production of TNF-α was increased in these cells. Finally, confirmation that indeed both of these ligands are involved in mf-induced cell death came from our antibody-blocking experiments (which were also confirmed by siRNA; data not shown) showing that mf-induced cell death in DC was reversible using antibodies to TRAIL-R2, TNF-α, or TNF-R1 (Fig. 6). Because DC showed a higher cell surface expression of TRAIL-R2 than TRAIL-R1 (data not shown) we blocked TRAIL/TRAIL-R pathway using antibodies to TRAIL-R2. While anti-TRAIL-R2 significantly reversed DC cell death, blocking the ligand with anti-TRAIL neturalizing mAb at 10ug/ml failed to reverse cell death in these cells, suggesting that higher concentrations (or longer pre-incubation) of this antibody is needed. Furthermore, because TNF-R1 has been associated with cell death, our focus in the current study is on this particular receptor, although a role for TNF-R2 cannot be excluded in that blocking TNF-α was better than blocking TNF-R1 alone (see Fig. 6). Collectively, our data suggest an important role for both the TNF-α and TRAIL pathways in mf-induced DC cell death.
Upregulation of TNF ligands by intracellular parasites has been previously reported (22, 23). For example, mRNA upregulation of TRAIL and FAS in human epidermal keratinocytes upon exposure to Leishmania major has been demonstrated previously (23). Furthermore, upon infection with L. major, MΦ were shown to induce neutrophil apoptosis through a membrane-bound TNF (24). Thus, this work extends to extracellular parasites the ability of these parasites to regulate expression and production of TNF ligands and thereby induce cell death through TNF-α/TNF-R1 and TRAIL/TRAIL-R2 pathways of ligands/receptors.
Of interest, the apoptosis observed was not a result of general induction of TNF ligands in DC by mf, as another member of the TNF ligand superfamily, TL1A, highly upregulated by mf (both at the mRNA and the protein levels; Fig. 4) was not involved in mf-induced DC apoptosis. TL1A has been shown to be upregulated by TNF and IL-1α, and the interaction between TL1A and DR3 in cells expressing DR3 was shown to induce NF-κB and apoptosis (17). Whether DR3 downregulation by mf in DC is a result of ligand engagement or the reason that mf cannot induce apoptosis through induction of TL1A is not known. In addition, it has been documented that in MΦ-like cell types, interaction between TL1A and DR3 can result in induction of proinflammatory cytokines such as IL-8 (25). In our hands, mf highly upregulated production of IL-8 in DC(2); however, whether this production of IL-8 is TL1A dependent remains to be examined.
The extrinsic pathway of apoptosis can be induced through oligomerization of death receptors such as FAS, TNF-αR, DR3, TRAIL-R4, and TRAIL-R5 after engagement with their respective ligands. This oligomerization, in turn, results in recruitment of adaptor proteins and activation of caspase cascades. Initial activation of caspase 8 through the adaptor molecule FADD stimulates apoptosis in two ways: it can directly cleave and activate caspase 3 or, alternatively, it can cleave Bid, a pro-apoptotic Bcl2 family member. This cleaved (or truncated) bid (tBid) translocates to mitochondria, inducing cytochrome c release, sequentially activating caspase 9 and 3 and resulting in DNA fragmentation and cell death (11). We show that mf do not have any affect on mRNA expression of adapter molecules TRADD, FADD, or TRAF2. In contrast, however, exposure to this parasite significantly upregulates Bid gene expression (Fig. 7A) and results in a significant decrease in cytoplasmic tBid (Fig. 7B), suggesting translocation of tBid to mitochondria (12, 13). The upregulation of TNFR2 that we found by microarray (Fig. 3) may contribute to the TNF-induced apoptosis after mf exposure, as high levels of TNFR2 can skew TNFR1 signaling toward apoptosis (26). Furthermore, mf significantly induced the cytosolic release of cytochrome c (Fig. 7C), providing evidence that cell death is mitochondria dependent and involves activation of caspase 9.
A number of other infectious pathogens have been found to alter the machinery of apoptotic cell death (reviewed in 27, 28). For example, it has been shown that intracellular parasites such as Toxoplasma gondii are capable of inhibiting apoptosis of the host cell through direct inhibition of cytochrome c-induced caspase activation (29). In addition, other parasites such as L. major can prevent programmed cell death in infected MΦ through a repression of mitochondrial release of cytochrome c (30). Apoptosis of MΦ induced by Trichomonas vaginalis through phosphorylation of p38 mitogen-activated protein kinase that locates downstream of mitochondria-dependent caspase activation has also been reported (31). Furthermore, Plasmodium falciparum infection of RBC was found to involve caspase 9 activation through a mitochondrial pathway of cell death (32). Apoptosis of myeloid cells and lymphocytes during Listeria infection was also found to be partially dependent on TRAIL (33). Our work reveals that extracellular helminthes are also capable of manipulating the machinery of apoptosis through the action of two pro-apoptotic TNF-family ligands and their receptors.
We found that killing of human DC by B. malayi is both cell and parasite stage specific. In this report, we show that mf only kill DC and not MΦ from the same donors. Whether the differences between MΦ and DC reflect only intrinsic differences between the cell types or differences in tissue distribution (MΦ being commonly resident in tissues whereas DC are more mobile) awaits clarification. Also, the L3 infective stage of the parasite does not induce killing in DC or human LC (4)but can result in caspase-dependent apoptosis in human NK cells (34). We are in the process of performing proteomics on different stages of the parasite to get an insight in the differences they may have in triggering apoptosis in various cell types.
Our current model suggests that upregulation of TNF ligands by mf occurs either through direct interaction by certain worm ligands/receptors or (more likely) through soluble factors released by the parasites. Indeed, we have shown previously that DC cell death was also achieved by the excretory/secretory products from mf; however, the effect was less profound than when worms and cells were in physical contact (2). It is unlikely that this apoptotic cell death is the result of competition for nutrients by these extracellular worms, as other cell types (e.g., monocyte-derived MΦ [Fig. 1] or monocyte-derived pDC [Simon Metenou, personal communication]) do not die after being exposed to mf.
Why do mf parasites kill host DC? Are they killing DC for their own survival, or is this a mechanism by which the host avoids further inflammation and pathology during infection? Or, as we have suggested previously, is this a generalized mechanism by which mf deplete APC, leading to chronically poor Ag-specific T cell function? This Ag-induced loss of T cell function has been observed in vitro (2, 3), in patient cells ex vivo (1), and in murine systems of filarial infections at the time of patency (1, 35). One potential outcome of this increased cell death of DC induced by mf may involve clearance of dead cells through phagocytosis or other mechanisms. It is known that under normal conditions, apoptotic cells are rapidly cleared by phagocytes (36) and MΦ and subsets of DC are responsible for this phagocytosis (reviewed in 37–40). Therefore, a sudden increase in cell death may delay the normal process of phagocytosis of apoptotic cells, which in turn could result in other implications for patients with circulating mf—a hypothesis that needs to be investigated.
Thus, our studies reveal a mechanism for induction of apoptosis by extracellular helminths in DC. This mechanism involves signaling through TRAIL-R2 and TNFR1, activating Bid protein and resulting in cytochrome c release from mitochondria, which leads to activation of caspase 9. It is the depletion of these professional APC, however, that may provide a strong inhibitory signal leading to the Ag-specific T cell hyporesponsiveness seen in chronically filaria-infected individuals.
Acknowledgments
We thank Dr. Helen Sabzevari for critical reading of the manuscript, discussions, and useful advice; Dr. Rami Najjar from Cell Signaling Technologies for technical advice on immunoblots; and Victor Barcelona for help with the figures. We also thank NIAID intramural editor Brenda Rae Marshall for assistance.
Abbreviations used in this paper
- APAF1
apoptotic protease-activating factor 1
- Bid
BH3-interacting domain death agonist
- DC
dendritic cell
- LC
Langerhans’ cell
- MΦ
macrophage
- mf
live microfilariae
- PI
propidium iodide
- siRNA
Small interfering RNA
- tBid
Bid truncation
- TNFR
TNF receptor
Footnotes
This work was supported by the Intramural Research Program of the Division of Intramural Research, National Institute of Allergy and Infectious Diseases, National Institutes of Health.
Disclosures
The authors have no financial conflict of interest.
Because R.T.S., P.G.V., L.M., F.M., D.Chien, D.D., R.M.S., and T.B.N. are government employees and this is a government work, the work is in the public domain in the United States. Notwithstanding any other agreements, the NIH reserves the right to provide the work to PubMedCentral for display and use by the public, and PubMedCentral may tag or modify the work consistent with its customary practices. You can establish rights outside of the U.S. subject to a government use license.
References
- 1.Maizels RM, Balic A, Gomez-Escobar N, Nair M, Taylor MD, Allen JE. Helminth parasites--masters of regulation. Immunol Rev. 2004;201:89–116. doi: 10.1111/j.0105-2896.2004.00191.x. [DOI] [PubMed] [Google Scholar]
- 2.Semnani RT, Liu AY, Sabzevari H, Kubofcik J, Zhou J, Gilden JK, Nutman TB. Brugia malayi microfilariae induce cell death in human dendritic cells, inhibit their ability to make IL-12 and IL-10, and reduce their capacity to activate CD4+ T cells. J Immunol. 2003;171:1950–1960. doi: 10.4049/jimmunol.171.4.1950. [DOI] [PubMed] [Google Scholar]
- 3.Semnani RT, Sabzevari H, Iyer R, Nutman TB. Filarial antigens impair the function of human dendritic cells during differentiation. Infect Immun. 2001;69:5813–5822. doi: 10.1128/IAI.69.9.5813-5822.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Semnani RT, Law M, Kubofcik J, Nutman TB. Filaria-induced immune evasion: suppression by the infective stage of Brugia malayi at the earliest host-parasite interface. J Immunol. 2004;172:6229–6238. doi: 10.4049/jimmunol.172.10.6229. [DOI] [PubMed] [Google Scholar]
- 5.MacDonald AS, Maizels RM, Lawrence RA, Dransfield I, Allen JE. Requirement for in vivo production of IL-4, but not IL-10, in the induction of proliferative suppression by filarial parasites. J Immunol. 1998;160:1304–1312. [PubMed] [Google Scholar]
- 6.Whelan M, Harnett MM, Houston KM, Patel V, Harnett W, Rigley KP. A filarial nematode-secreted product signals dendritic cells to acquire a phenotype that drives development of Th2 cells. J Immunol. 2000;164:6453–6460. doi: 10.4049/jimmunol.164.12.6453. [DOI] [PubMed] [Google Scholar]
- 7.Bhardwaj N. Interactions of viruses with dendritic cells: a double-edged sword. J Exp Med. 1997;186:795–799. doi: 10.1084/jem.186.6.795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Maya A, Jayaraman K, Balakrishnan A. Necrosis of lung epithelial cells by filarial parasitic protein via an early induction of c-H-ras and TNF-α expression. Cell Biol Int. 1997;21:273–280. doi: 10.1006/cbir.1997.0139. [DOI] [PubMed] [Google Scholar]
- 9.Jenson JS, O’Connor R, Osborne J, Devaney E. Infection with Brugia microfilariae induces apoptosis of CD4+ T lymphocytes: a mechanism of immune unresponsiveness in filariasis. Eur J Immunol. 2002;32:858–867. doi: 10.1002/1521-4141(200203)32:3<858::AID-IMMU858>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 10.Matsue H, Takashima A. Apoptosis in dendritic cell biology. J Dermatol Sci. 1999;20:159–171. doi: 10.1016/s0923-1811(98)00078-4. [DOI] [PubMed] [Google Scholar]
- 11.Siegel RM. Caspases at the crossroads of immune-cell life and death. Nat Rev Immunol. 2006;6:308–317. doi: 10.1038/nri1809. [DOI] [PubMed] [Google Scholar]
- 12.Li H, Zhu H, Xu CJ, Yuan J. Cleavage of BID by caspase 8 mediates the mitochondrial damage in the Fas pathway of apoptosis. Cell. 1998;94:491–501. doi: 10.1016/s0092-8674(00)81590-1. [DOI] [PubMed] [Google Scholar]
- 13.Luo X, Budihardjo I, Zou H, Slaughter C, Wang X. Bid, a Bcl2 interacting protein, mediates cytochrome c release from mitochondria in response to activation of cell surface death receptors. Cell. 1998;94:481–490. doi: 10.1016/s0092-8674(00)81589-5. [DOI] [PubMed] [Google Scholar]
- 14.Gross A, Yin XM, Wang K, Wei MC, Jockel J, Milliman C, Erdjument-Bromage H, Tempst P, Korsmeyer SJ. Caspase cleaved BID targets mitochondria and is required for cytochrome c release, while BCL-XL prevents this release but not tumor necrosis factor-R1/Fas death. J Biol Chem. 1999;274:1156–1163. doi: 10.1074/jbc.274.2.1156. [DOI] [PubMed] [Google Scholar]
- 15.Gusmao RD, Stanley AM, Ottesen EA. Brugia pahangi: immunologic evaluation of the differential susceptibility of filarial infection in inbred Lewis rats. Exp Parasitol. 1981;52:147–159. doi: 10.1016/0014-4894(81)90070-9. [DOI] [PubMed] [Google Scholar]
- 16.Semnani RT, Keiser PB, Coulibaly YI, Keita F, Diallo AA, Traore D, Diallo DA, Doumbo OK, Traore SF, Kubofcik J, Klion AD, Nutman TB. Filaria-induced monocyte dysfunction and its reversal following treatment. Infect Immun. 2006;74:4409–4417. doi: 10.1128/IAI.01106-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Migone TS, Zhang J, Luo X, Zhuang L, Chen C, Hu B, Hong JS, Perry JW, Chen SF, Zhou JX, Cho YH, Ullrich S, Kanakaraj P, Carrell J, Boyd E, Olsen HS, Hu G, Pukac L, Liu D, Ni J, Kim S, Gentz R, Feng P, Moore PA, Ruben SM, Wei P. TL1A is a TNF-like ligand for DR3 and TR6/DcR3 and functions as a T cell costimulator. Immunity. 2002;16:479–492. doi: 10.1016/s1074-7613(02)00283-2. [DOI] [PubMed] [Google Scholar]
- 18.Sheridan JP, Marsters SA, Pitti RM, Gurney A, Skubatch M, Baldwin D, Ramakrishnan L, Gray CL, Baker K, Wood WI, Goddard AD, Godowski P, Ashkenazi A. Control of TRAIL-induced apoptosis by a family of signaling and decoy receptors. Science. 1997;277:818–821. doi: 10.1126/science.277.5327.818. [DOI] [PubMed] [Google Scholar]
- 19.Marsters SA, Sheridan JP, Pitti RM, Huang A, Skubatch M, Baldwin D, Yuan J, Gurney A, Goddard AD, Godowski P, Ashkenazi A. A novel receptor for Apo2L/TRAIL contains a truncated death domain. Curr Biol. 1997;7:1003–1006. doi: 10.1016/s0960-9822(06)00422-2. [DOI] [PubMed] [Google Scholar]
- 20.Ribeiro-Gomes FL, Silva MT, Dosreis GA. Neutrophils, apoptosis and phagocytic clearance: an innate sequence of cellular responses regulating intramacrophagic parasite infections. Parasitology. 2006;132(Suppl):S61–68. doi: 10.1017/S0031182006000862. [DOI] [PubMed] [Google Scholar]
- 21.Krishnamoorthy B, Renner W, Balakrishnan A. Apoptosis induced by filarial parasitic sheath protein in HEp 2 cell lines blocked by ectopic expression of bcl 2. Cell Biol Int. 1998;22:483–492. doi: 10.1006/cbir.1998.0294. [DOI] [PubMed] [Google Scholar]
- 22.Babu S, Blauvelt CP, Nutman TB. Filarial parasites induce NK cell activation, type 1 and type 2 cytokine secretion, and subsequent apoptotic cell death. J Immunol. 2007;179:2445–2456. doi: 10.4049/jimmunol.179.4.2445. [DOI] [PubMed] [Google Scholar]
- 23.Eidsmo L, Fluur C, Rethi B, Eriksson Ygberg S, Ruffin N, de Milito A, Akuffo H, Chiodi F. FasL and TRAIL induce epidermal apoptosis and skin ulceration upon exposure toLeishmania major. Am J Pathol. 2007;170:227–239. doi: 10.2353/ajpath.2007.060068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Allenbach C, Zufferey C, Perez C, Launois P, Mueller C, Tacchini-Cottier F. Macrophages induce neutrophil apoptosis through membrane TNF, a process amplified byLeishmania major. J Immunol. 2006;176:6656–6664. doi: 10.4049/jimmunol.176.11.6656. [DOI] [PubMed] [Google Scholar]
- 25.Kang YJ, Kim WJ, Bae HU, Kim DI, Park YB, Park JE, Kwon BS, Lee WJ. Involvement of TL1A and DR3 in induction of pro-inflammatory cytokines and matrix metalloproteinase-9 in atherogenesis. Cytokine. 2005;29:229–235. doi: 10.1016/j.cyto.2004.12.001. [DOI] [PubMed] [Google Scholar]
- 26.Chan FK, Lenardo MJ. A crucial role for p80 TNF-R2 in amplifying p60 TNF-R1 apoptosis signals in T lymphocytes. Eur J Immunol. 2000;30:652–660. doi: 10.1002/1521-4141(200002)30:2<652::AID-IMMU652>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 27.James ER, Green DR. Manipulation of apoptosis in the host-parasite interaction. Trends Parasitol. 2004;20:280–287. doi: 10.1016/j.pt.2004.04.004. [DOI] [PubMed] [Google Scholar]
- 28.Bruchhaus I, Roeder T, Rennenberg A, Heussler VT. Protozoan parasites: programmed cell death as a mechanism of parasitism. Trends Parasitol. 2007;23:376–383. doi: 10.1016/j.pt.2007.06.004. [DOI] [PubMed] [Google Scholar]
- 29.Keller P, Schaumburg F, Fischer SF, Hacker G, Gross U, Luder CG. Direct inhibition of cytochrome c-induced caspase activation in vitro by Toxoplasma gondii reveals novel mechanisms of interference with host cell apoptosis. FEMS Microbiol Lett. 2006;258:312–319. doi: 10.1111/j.1574-6968.2006.00241.x. [DOI] [PubMed] [Google Scholar]
- 30.Akarid K, Arnoult D, Micic-Polianski J, Sif J, Estaquier J, Ameisen JC. Leishmania major-mediated prevention of programmed cell death induction in infected macrophages is associated with the repression of mitochondrial release of cytochrome c. J Leukoc Biol. 2004;76:95–103. doi: 10.1189/jlb.1001877. [DOI] [PubMed] [Google Scholar]
- 31.Chang JH, Kim SK, Choi IH, Lee SK, Morio T, Chang EJ. Apoptosis of macrophages induced by Trichomonas vaginalis through the phosphorylation of p38 mitogen-activated protein kinase that locates at downstream of mitochondria-dependent caspase activation. Int J Biochem Cell Biol. 38:638–647. doi: 10.1016/j.biocel.2005.11.005. [DOI] [PubMed] [Google Scholar]
- 32.Pino P, Vouldoukis I, Kolb JP, Mahmoudi N, Desportes-Livage I, Bricaire F, Danis M, Dugas B, Mazier D. Plasmodium falciparum--infected erythrocyte adhesion induces caspase activation and apoptosis in human endothelial cells. J Infect Dis. 2003;187:1283–1290. doi: 10.1086/373992. [DOI] [PubMed] [Google Scholar]
- 33.Zheng SJ, Jiang J, Shen H, Chen YH. Reduced apoptosis and ameliorated listeriosis in TRAIL-null mice. J Immunol. 2004;173:5652–5658. doi: 10.4049/jimmunol.173.9.5652. [DOI] [PubMed] [Google Scholar]
- 34.Chen XM, Levine SA, Splinter PL, Tietz PS, Ganong AL, Jobin C, Gores GJ, Paya CV, LaRusso NF. Cryptosporidium parvum activates nuclear factor κB in biliary epithelia preventing epithelial cell apoptosis. Gastroenterology. 2001;120:1774–1783. doi: 10.1053/gast.2001.24850. [DOI] [PubMed] [Google Scholar]
- 35.Allen JE, Maizels RM. Th1–Th2: reliable paradigm or dangerous dogma? Immunol Today. 1997;18:387–392. doi: 10.1016/s0167-5699(97)01102-x. [DOI] [PubMed] [Google Scholar]
- 36.van Nieuwenhuijze AE, van Lopik T, Smeenk RJ, Aarden LA. Time between onset of apoptosis and release of nucleosomes from apoptotic cells: putative implications for systemic lupus erythematosus. Ann Rheum Dis. 2003;62:10–14. doi: 10.1136/ard.62.1.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rock KL, I, York A, Saric T, Goldberg AL. Protein degradation and the generation of MHC class I-presented peptides. Adv Immunol. 2002;80:1–70. doi: 10.1016/s0065-2776(02)80012-8. [DOI] [PubMed] [Google Scholar]
- 38.York IA, Goldberg AL, Mo XY, Rock KL. Proteolysis and class I major histocompatibility complex antigen presentation. Immunol Rev. 1999;172:49–66. doi: 10.1111/j.1600-065x.1999.tb01355.x. [DOI] [PubMed] [Google Scholar]
- 39.Albert ML, Sauter B, Bhardwaj N. Dendritic cells acquire antigen from apoptotic cells and induce class I-restricted CTLs. Nature. 1998;392:86–89. doi: 10.1038/32183. [DOI] [PubMed] [Google Scholar]
- 40.Steinman RM, Turley S, Mellman I, Inaba K. The induction of tolerance by dendritic cells that have captured apoptotic cells. J Exp Med. 2000;191:411–416. doi: 10.1084/jem.191.3.411. [DOI] [PMC free article] [PubMed] [Google Scholar]