

Abstract
In mammals an increase in glucose leads to block of ATP dependent potassium channels in pancreatic β cells leading to membrane depolarization. This leads to the repetitive firing of action potentials that increases calcium influx and triggers insulin granule exocytosis. Several important differences between species in this process suggest that a dedicated human-oriented approach is advantageous as extrapolating from rodent data may be misleading in several respects. We examined depolarization-induced spike activity in pancreatic human islet-attached β-cells employing whole-cell patch-clamp methods. We also reviewed the literature concerning regulation of insulin secretion by channel activity and constructed a data-based computer model of human β cell function. The model couples the Hodgkin-Huxley-type ionic equations to the equations describing intracellular Ca2+ homeostasis and insulin release. On the basis of this model we employed computational simulations to better understand the behavior of action potentials, calcium handling and insulin secretion in human β cells under a wide range of experimental conditions. This computational system approach provides a framework to analyze the mechanisms of human β cell insulin secretion.
Keywords: diabetes, electrophysiology, glucose, mathematical model, membrane potential, ranolazine, tetrodotoxin
Introduction
Raising external glucose in human and rodent pancreatic β-cells leads to the generation of intracellular intermediates (including ATP), which block ATP-sensitive K+ (KATP) channels. Closure of this key channel results in membrane depolarization, the onset of electrical activity and voltage-dependent Ca2+ entry. The resultant activation of Ca2+ channels and rise in cytosolic Ca2+ triggers a cascade of biochemical events involved in the control of insulin secretion (IS) and key cellular pathways.1-3 A schematic diagram of the consensus model for biochemical steps, Ca2+ handling, channels and mechanisms of IS regulation in β-cell is presented in Figure 1.
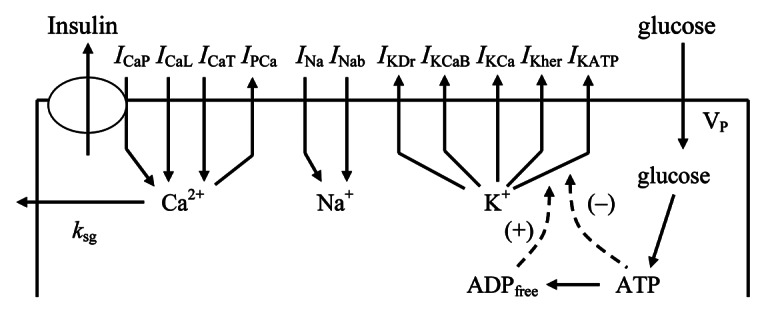
Figure 1. Schematic diagram of the ionic current, Ca2+ fluxes and exocytosis in human β-cell. Transmembrane currents are: ICaP is the voltage-gated P-type Ca2+ current, that is also responsible for insulin release (see text), ICaL is the voltage-gated L-type Ca2+ current, ICaT is the voltage-gated T-type Ca2+ current; IPCa, plasma membrane Ca2+ pump current; INa, voltage-gated Na+ current; INab, Na+ background current IKDr, delayed rectifier K+ current; IKCaB, Ca2+ and high voltage-activated K+ (BK channel) current; IKCa, Ca2+ activated K+ (SK channel) current; IKher is the human-ERG K+ channel current and IKATP, ATP-sensitive K+ channels current. Calcium enters the β-cells primarily through voltage-activated Ca2+ channels by diffusion along an inwardly directed electrochemical gradient. At the plasma membrane two processes are involved in transporting Ca2+ out of the cell; a Ca2+ pump and removal of Ca2+ sequestrated in insulin granules by exocytosis (coefficient ksq).
Over the physiological range of glucose concentrations β-cell electrical activity consists of spikes in plasma membrane potential (Vp). The electrical activity involves complex interactions between many different ion channels.1,3,4 This coordinated electrical activity is also necessary for the response of β-cells to secretagogues other than glucose that can act via modulation of activity of different ion channels.1,3,5 Changes in the action potential (AP) profile and the gating properties of the voltage-gated Ca2+ channels (VGCCs) can alter AP-driven Ca2+ influx. Interestingly, the sulfonylurea drugs developed for treatment of Type 2 diabetes modify the activity of KATP channels acting through PM depolarization.1,3,6 Defects in β-cell function have been shown to underlie both Type 2 and monogenic diabetes1,6,7 and this has increased the importance of understanding how β-cell electrical activity induced by glucose and other agonists is established and how it is altered in various pathological states.
The majority of investigations in pancreatic β-cell electrophysiology have been performed using rodent β-cells due to the difficulties in the isolation and culture of intact normal human islets.1,3,8 The mouse is also currently the most highly employed model for transgenic and molecular genetic studies of altered gene expression. However, while rodent and human islets show broad similarities, the expression and properties of certain ion channels found in human β-cells in particular are different from those expressed in mouse. In particular, human β-cells have active voltage-gated Na+ and T-type calcium currents that may be absent in mouse and have greater effects on electrophysiological properties in comparison with mouse β-cells.9-12 In general, these differences can underlie the species specific patterns of β-cell APs, voltage-gated Ca2+ influx and IS. Therefore, a direct comparison of β-cell electrical activity and the channel types between species is difficult. Given the importance of IS in diabetes and obesity, human β-cells require a special and detailed consideration.
Here, we examined the ionic mechanisms mediating depolarization-induced spike activity in pancreatic human β-cells, employing whole-cell patch-clamp methods on islet-attached cells. We also reviewed the literature on a regulation of IS by specific ion channel activities. To achieve a better understanding of human β-cell physiology and the possible roles of key ion channels we undertook an integrated consideration of data obtained on isolated human β-cells and islets. We used a computational systems biology approach with construction of a mathematical model of intracellular processes (see Appendix 1 and 2 in the Supplementary Material).
We have previously focused on experimental and theoretical studies of electrical activity, Ca2+ dynamics and IS in mouse pancreatic β-cells.13-17 Here we have studied and evaluated the role of different ion channels in human β-cells using the approaches we developed previously for β-cells.
The mathematical model for human β-cells was tested in various scenarios and the results of simulations were compared with the experimental data. The model gives a reasonable fit to important aspects of experimentally measured AP firing, Ca2+ dynamics and IS properties of human β-cells and provides a framework for analyzing the role of individual ionic currents and to determine their influence on insulin release. This allows also a comparison of the mechanisms of regulation of IS in rodent and human β-cells.
Materials and Methods
To illustrate the role of specific channels in glucose-stimulated APs, human islet-attached β-cell membrane potentials were recorded using the perforated-patch current-clamp configuration. Experimental methods were performed as previously described.14
Human islets were obtained from cadaveric donors, gifts from the University of Chicago Islet transplant program. All studies were approved by the University of Chicago Institutional Review Board.
Role of Ca2+ Channels and Cytoplasmic Ca2+ in Insulin Exocytosis
Insulin release is influenced by physiological β-cell electrical activity (for a review see refs.1,12). Capacitance measurements in rodent and human β-cells have shown that the insulin granule exocytosis is negligible at membrane potentials more negative than −20 mV but increases steeply at more depolarized voltages.2,10 This behavior may reflect a necessity for local calcium entry through high voltage-gated non-L-type Ca2+ channels because the process of release of primed granules may need a local increased calcium in specific microdomains (for a review see refs.2 and 10). Voltage-gated P/Q-type Ca2+ channels may serve in human β-cells as the main component of non-L-type Ca2+ channels that trigger depolarization-evoked exocytosis of the insulin–containing granules.2,10
Ca2+ enters into cells through voltage-gated Ca2+ channels and is removed by Ca2+ pumps and exchangers. Ca2+ handling can also include ER and mitochondrial sequestration. It is perhaps surprising that detailed data on [Ca2+]c in human islet β-cells is lacking. Our unpublished data suggest that human islet preparations are highly variable. Similarly, there is no data available for [Ca2+]c behavior in human β-cells during one spike. Therefore, our evaluations of [Ca2+]c are based on mouse experiments and our model for mouse β-cells13 (see Supplemental Material and section “Regulation of Ca2+ entry and bursting” for details). A correlation also exists between [Ca2+]c and insulin release at least in rodent islets from threshold to moderate levels of [Ca2+]c.18,19 These mechanisms were taken into account during simulations of insulin release in our mathematical model (see Supplemental Material, Appendix 1).
Mechanisms of Spike Generation in Human β-Cells
At resting state (low glucose levels) the rate of metabolism in β–cells is relatively low. KATP channels are mostly in the open state and maintain a hyperpolarized membrane potential. VDCCs are closed below their activation threshold. Under these conditions the influx of Ca2+ is small, [Ca2+]c is low and insulin secretion (IS) is at a low basal (constitutive) level. For convenience we define a “low glucose concentration” is approximately 2 mM (where IS is minimal), “intermediate glucose concentration” is 5–7 mM (where IS is intermediate) and “high glucose concentration” is above 10 mM (where IS is maximal) to compare our simulations with experimental data.
Available experiments
Spikes are induced in isolated human islets following increased extracellular glucose.9,10,12,20,21 A typical glucose-induced spike pattern for β-cells in isolated human islets is shown in Figure 2A. Spikes can also show complex behaviors as illustrated in Figure 2B.
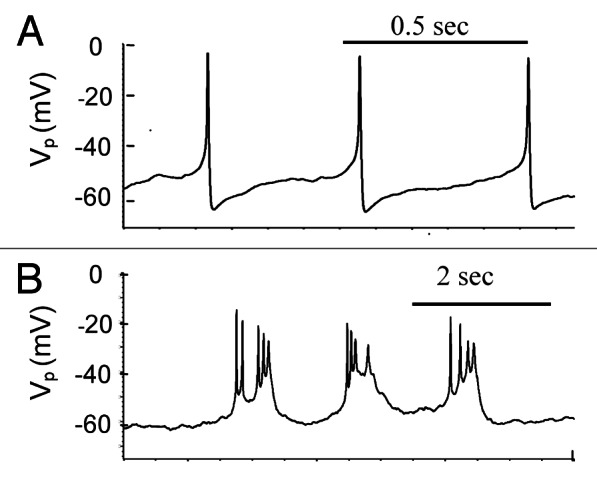
Figure 2. Electrical activity in human islets during treatment of 14 mM glucose. Examples of representative action potentials (AP). (A) Simple spikes. (B) Complex spikes. Experiments were performed as described in “Materials and Methods.”
Simulations and analysis
In our model KATP channels are substantially open at simulated low glucose concentrations (low ATP/ADP level) and mediate the hyperpolarized resting PM potential (left part of Figure 3). A background current that is carried mainly by Na+ (INab) is balanced at resting potential by KATP channels. With the parameter set given in Tables S1 and S2 and with high free [ADP]c (100 μM), that corresponds to the low glucose level, the model has a steady-state solution with a resting membrane potential of approximately -69 mV, the [Ca2+]c of just 123 nM (left part of Fig. 3) and relative IS of 0.1 relative unit, that is consistent with the data obtained for human and rodent β-cells at low glucose levels.2,13,18,22 It should be noted that [Ca2+]c at low glucose level depends significantly on activity of T-type Ca2+ channels in our model (see section “T-type Ca2+ channels” fordetails).
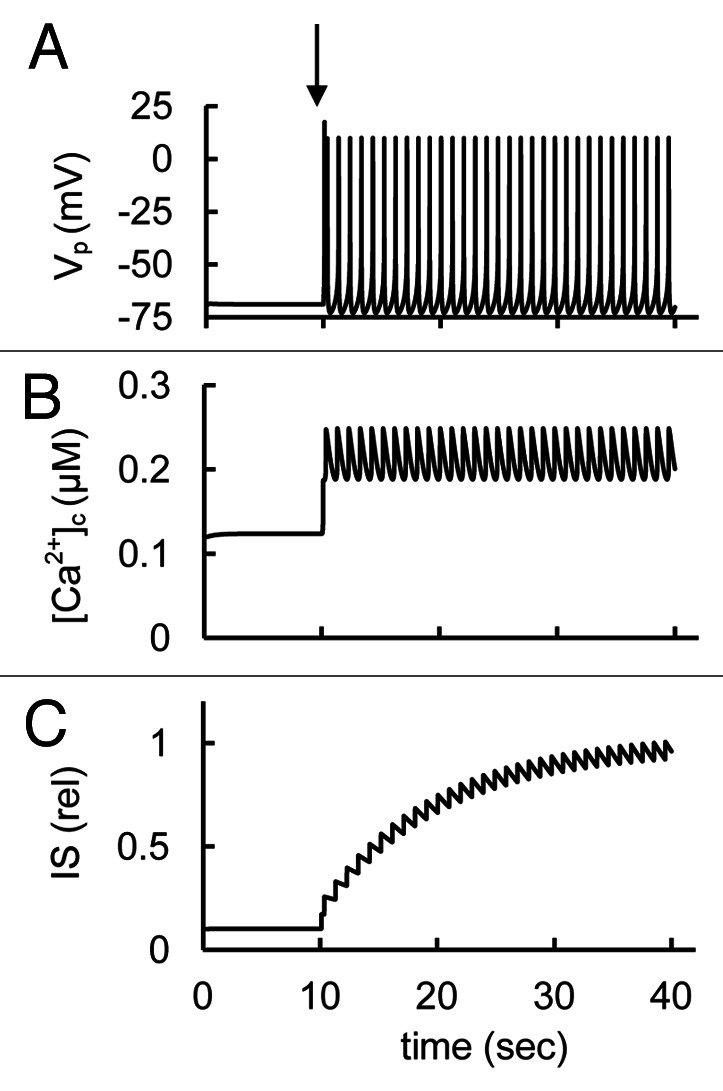
Figure 3. Modeling of spontaneous glucose-stimulated spikes, the changes of intracellular Ca2+ concentration and insulin secretion. (A) Action potentials (Vp); (B) Free cytoplasmic Ca2+ concentration ([Ca2+]c); (C) relative insulin secretion (IS). Glucose-induced spikes were simulated at a step decrease of the free ADP concentration at the arrow from a high to an intermediate value (from [ADPf]c = 100 µM to 15 µM; all other parameter were taken from the basal set of parameters (Tables S1 and S2). Corresponding changes in [Ca2+]c and IS were simulated using Eqs. Two and 7 in Appendix 1 in Supplemental Materials.
We simulated the effects of a glucose increase by decreasing free ADP concentration leading to increased [ATP]/[ADP], closing KATP channels (arrow in Fig. 3A). According to our simulation closing KATP channels leaves the background Na+ current (INab) unbalanced, leading to PM depolarization, spike generation and increased [Ca2+]c and IS. Narrow spikes with one maximum were simulated in this case. We were also able to simulate complex spikes when KATP, Kv, KCa or P-type Ca2+ channels were blocked in comparison with basal level (see discussion below).
The following scheme was suggested for the upstroke of human β-cell APs.1,9,10,12 (1) Activation of T-type Ca2+ current at low-voltage is likely to depolarize the membrane rapidly to voltage threshold for recruiting INa, ICaL and ICaP. (2) Recruitment of INa, at VP approaching –40 mV is likely to hasten the upstroke of the AP and underlies its overshoot, as well as to insure activation of voltage-dependent K+ currents (IKDr and IKCab) that produce rapid repolarization.
Possible mechanisms underlying the AP changes and the roles for specific currents can be better understood by considering the simulated voltage-dependent currents during one cycle of spontaneous activity in the ionic model (Fig. 4). Both Na+ and Ca2+ voltage dependent channels participate in the upstroke of the AP. The upstroke phase is clearly initially generated by voltage-dependent Na+ (INa) and T-type Ca2+ (ICaT) channels. When Na+ and Ca2+ enters the cell, the membrane is further depolarized and high-voltage-activated Ca2+ channels open. Then high-voltage-activated Ca2+ channels activate transiently. The AP repolarization phase includes activation of fast BK channels. After that the rapid delayed rectifier K+ channels also activate (IKDr current) leading to increased repolarizing current. The repolarization phase also includes the time-dependent inactivation of Na+ and Ca2+ channels (Fig. 4B). Other currents included in the model were relatively small during a spike and had an insignificant influence on AP shape (Fig. 4C). However, these currents can play a considerable role in total interspike current and influence spike frequency and height (see discussion below).
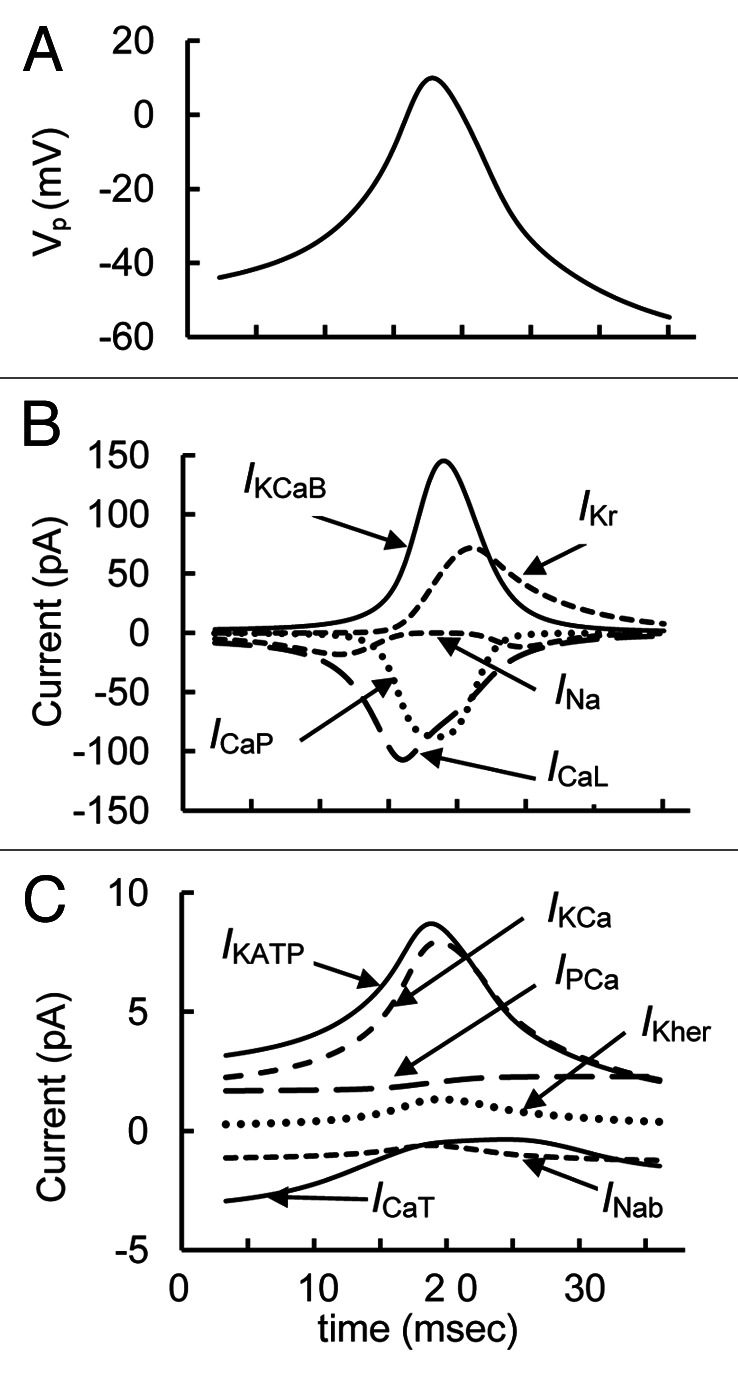
Figure 4. Detailed simulated curves of parameters during a single simple spike from Figure 3. (A) Action potential (Vp); (B) Principal currents for AP (ICaL, ICaP, IKCaB, IKDr, and INa) are represented for one characteristic spike. (C) Small currents (ICaT, IPCa, INab, IKCa, IKher and IKATP) are also represented for this interval.
K+ Currents
K+ channels are among the most important determinants of cellular excitability. Whole-cell patch clamp studies in human β-cells have identified several K+ current components.
KATP channels
The KATP channel is a key component of stimulus-secretion coupling in the pancreatic β-cells. Its hetero-octameric complex comprises four Kir6.2 subunits and four sulphonylurea receptor subunits. Kir6.2 is an inwardly rectifying K+ channel. KATP channels are subject to modulation by ATP/ADP ratio in both rodent and human β-cells. These channels and corresponding IKATP in human is generally similar to those observed in rodent β-cells (for a review see refs.12, 23 and 24). In our simulation closing KATP channels through increasing the ATP/ADP ratio leads to membrane depolarization and an increase in [Ca2+]c and IS at increased glucose levels (Fig. 3). This corresponds to the well-established idea that a block of KATP channels leads to increased IS in human β cells.12,23,24
Available experiments
Additional activation or inhibition of KATP channels can be obtained using KATP channel activators or blockers leading to PM potential changes. Freshly isolated human islets responded to tolbutamide (KATP channel blocker) with a sustained rise in [Ca2+]c after a glucose-induced initial [Ca2+]c increase.25 An increased IS rate was also found in response to tolbutamide after further increase in glucose,10 suggesting that we can use this as evidence of a tolbutamide-induced increase in [Ca2+]c and IS.
Simulations and analysis
Activation or inhibition of KATP channels can be simulated by changing the maximal conductance of KATP channels. Results of these simulations are shown for Vp, [Ca2+]c and IS changes (Fig. 5). Activation of KATP channels leads to decreased IS mainly as a consequence of decreased AP frequency (left part of Fig. 5). A broad complex spike train was evoked when we decreased KATP conductivity from basal level (right part of Fig. 5), determined using the default parameters from Tables S1 and S2. The changes in spike shape can be explained by the additional PM depolarization and activation of BK and KCa channels as a result of increased [Ca2+]c. The decrease in KATP channel conductance led to increased [Ca2+]c and IS, because the integrated Ca2+ influx was increased in this case. In general, increased IS and [Ca2+]c with decreased KATP channel conductance is consistent with the experimental findings. Increased IS may also have to do with other actions of sulfonylureas including binding to specific exchange protein activated by cAMP (EPAC).26
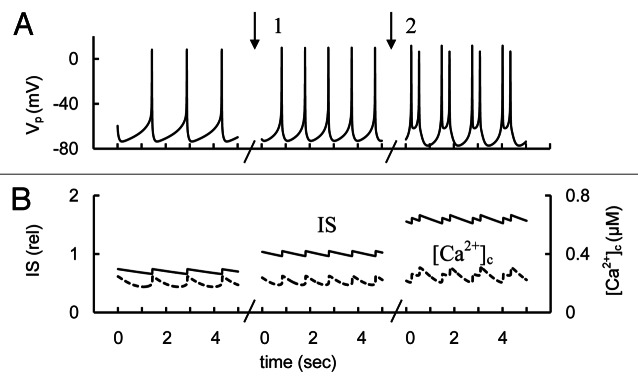
Figure 5. Simulated AP firing, [Ca2+]c and IS transients in response to activation or block of KATP channel conductance. (A) AP firing (Vp); (B) [Ca2+]c (– – –) and relative IS (——) during persistent spike activity (after transient changes). For simulation of KATP channel activation the maximal conductance (gmKATP, Eq. A41, Appendix 2 in Supplemental Material) was initially increased to 65 nS. Than gmKATP was decreased from 65 nS to 45 nS (basal level from Table S2) at arrow 1. For simulation of KATP channel block the gmKATP was decreased from 45 nS to 4 nS at arrow 2. Glucose induced APs were simulated as in Figure 3 up to the point of persistent spike activity. Several intervals in five second are represented for each case.
Transient component (BK channels)
One of the high voltage-gated K+ current in human β-cells is a transient component that activates rapidly upon membrane depolarization and depends on Ca2+-influx.10 This K+ current activates near -40 mV, inactivates rapidly through a voltage-dependent mechanism and peak amplitude exhibited a bell-shaped voltage-dependence with a maximum of about 40 mV.10 This current was blocked by iberiotoxin, a specific BK channel blocker and was suggested that it carried by large-conductance Ca2+ activated K+ channels (BK-channels) encoded by KCNMA1.10 Human β-cells express a relatively high level of the BK-type channels.10,14,27 Interestingly, recent studies indicate that BK-channels are also expressed in mouse pancreatic β-cells where the BK channel current can contribute to the outward current under specific experimental conditions.14,28
BK channels are activated by membrane depolarization and elevation of intracellular Ca2+ concentration.29,30 However, isolated BK channels are gated to open only at micromolar Ca2+ concentration.29 This contrasts with recorded levels of cytoplasmic β-cell [Ca2+]c, which are approximately 100 nM at low glucose levels and typically only double or triple following glucose stimulation, excluding microdomain effects. For this reason, BK channels should be closed at low [Ca2+]c according to data obtained for isolated channels. How can we, therefore, resolve the contradiction between experimental data for human β-cells and mouse islets which showed large BK currents10,14,28 with data obtained from isolated BK channels?
BK channel physiology has not been well characterized in intact pancreatic β-cells. However, for example in neurons, BK channels are colocalized or physically associated with voltage-dependent Ca2+ channels.29,31,32 Ca2+ concentrations near the Ca2+ source, termed the microdomain, can be much higher than in other parts of the cytoplasm30,33 to ensure the opening of colocalized BK channels. In this case BK channels open at less-positive membrane potentials.29-31 It was suggested that BK channels should open during the depolarizing phase of APs together with the opening of Ca2+ channels in β-cells, i.e., during a large Ca2+ influx a cell quickly increases Ca2+ concentration in microdomains.10,12 We therefore included in our model an increased sensitivity of BK channel currents to changes of [Ca2+]c than for isolated BK channels to simulate the effect of microdomain Ca2+ (Equations A28–A33, Appendix 2, Supplemental Material).
Available experiments
Blockade of BK channels using iberiotoxin, a specific BK channel blocker, increased significantly spike height in β-cells and enhanced IS by 70% in human islets (in the presence of tolbutamide).10
Simulations and analysis
It has been proposed that in human β-cells the large conductance (BK-type) Ca2+-activated K+-channels play a role in the AP repolarizing phase.10 Our theoretical consideration also confirms that the BK current could play a significant role in regulating β-cell electrical excitability by repolarization after fast Na+ channel and VGCCs activation (Fig. 4). We also simulated decreased BK channel activity by reducing their maximal conductance. In this case block of BK channels significantly increased the spike height and IS (Fig. 6). This occurs because IKCaB takes part in fast repolarization of the AP and the efficiency of this process decreased with decreased BK channel activity. This allows Vp to progress to higher potentials. Ca2+ influx through P/Q type VGCC and corresponding IS were also significantly increased in this case. [Ca2+]c was increased insignificantly. Thus, our simulation is consistent with the experimental findings.
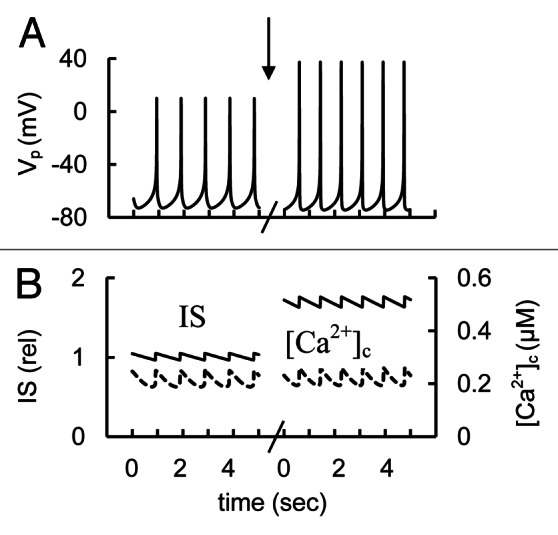
Figure 6. Simulation of K+ BK channel blocker application. (A) AP firing (Vp); (B) [Ca2+]c (– – –) and relative IS (——). The maximal conductance (gmkcab,Eq. A28, Appendix 2in Supplemental Material) was decreased from 25 nS (basal level from Table S2) to 0 nS at arrow. Glucose induced AP firing was simulated as in Figure 3.
Delayed rectifier K+ channels
Another high voltage-gated K+ (Kv) current in human β-cell is a relatively rapid delayed rectifier K+ current (IDKr).10,34 Expression of KV2.1 and KV2.2 proteins that are responsible for IDKr has been reported in human islets.10,35,36 This current is generally similar to those observed in rodent β-cells and insulinoma cells.1,3,37-39
Available experiments
KV2.1 and KV2.2 channels mediate the majority of the voltage-dependent outward K+ current in mouse and plays a key role in AP repolarization.3,13,14,40,41 However, these channels do not appear to play a prominent role in human β–cell spike generation in response to glucose since the KV2.1/2.2 blocker stromatoxin had only weak effects on electrical activity and IS in human β-cells.10
Simulations and analysis
Simulation of stromatoxin action at low glucose levels did not increase resting [Ca2+]c or IS significantly in our model (not shown). This can be explained by the very low conductivity of IKDr below -40 mV. For this reason, block of IKDr at simulated rest conditions for the β-cell where Vp is close to -70 mV cannot change the total current to depolarize the PM.
We also simulated experiments with decreased KDr channel conductance that corresponds to the effects of specific blockers of these channels (for example, stromatoxin) at high glucose levels (Fig. 7). We obtained an insignificant increase in spike height or IS to compare with simulated block of BK channels [see section “Transient component (BK channels)”] the even though in our model high voltage-activated delayed rectifier K+ channels make a significant contribution to the AP (see section “Mechanisms of spike generation in human β-cells”). The reason is that the main repolarizing current is IKCaB (Fig. 4) and the IKCaB increases sharply with increased PM potential in the Vp range close to 0 mV (Fig. S4). This compensates for a decreased KDr channels conductance even with an insignificant increase in Vp. Thus, our simulation is consistent with these experimental findings.
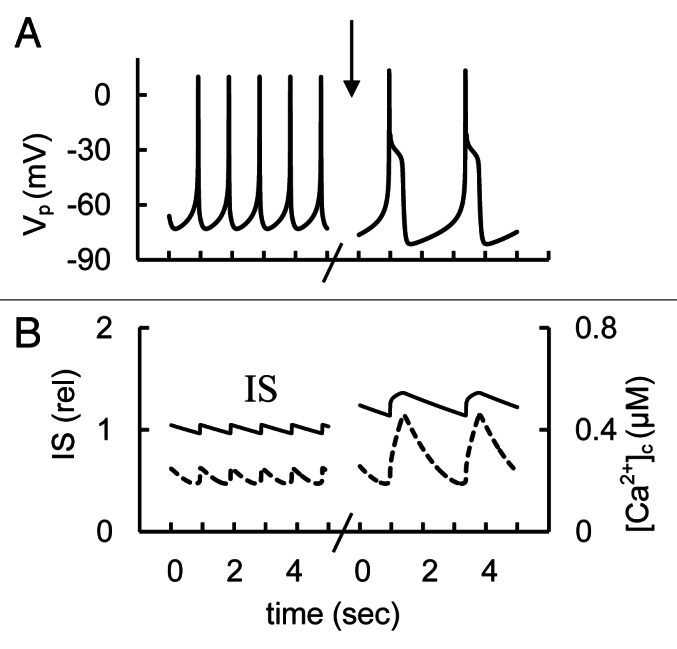
Figure 7. Modeling of glucose induced action potential firing, [Ca2+]c and IS during a simulation of rapid delayed rectified voltage-gated K+ channels blocking. (A) AP firing (Vp); (B) [Ca2+]c (– – –) and relative IS (——). For simulation of KDr channel blocking the maximal conductance (gmKr, Eq. A24, Appendix 2 in Supplemental Material) was decreased from 18 nS (basal level) to 0 nS at arrow. Glucose induced AP firing was simulated up to persistent spike activity as in Figure 3. Five second intervals are represented.
Thus the experimental data and our analysis show that although KV2.1 and KV2.2 channels may have modulatory effects on simple single spikes, these channels are not as crucial to spike generation in human β–cells as they are in mouse cells. Our analysis shows that contrary to mouse β-cell models13 IKDr was not able to provide the principal pacemaker agent for APs and contributes to a lesser extent compared with mouse to the AP repolarization phase. This reduced role is due to their slow activation, not fast enough to repolarize β-cells after the initial fast depolarization generated by Na+ channels and VGCCs. The activation of fast-activated BK channels is the most important repolarizing factor under these conditions (see Fig. 4).
HERG K+ current
Human-ERG (HERG) like-membrane currents have been recorded from human islet β-cells. Human islets also contain detectable levels of KCNH2 (HERG1).21,42
Available experiments
Firing frequency and IS were increased in the presence of selective blockers of HERG channels in human islet β-cells.21 However, no [Ca2+]c was measured in these experiments. Interestingly, inhibition of this type of K+ channel enhanced insulin secretion and temporarily increased [Ca2+]c in isolated mouse and Min6 cells.42
Simulations and analysis
We have found that HERG K+ channels are unlikely to take part in mechanisms that create the AP spikes (see section “Mechanisms of spike generation in human β-cells”). However, according to our simulations the changes in HERG channel conductance may change PM polarization such that AP behavior can be modified. Blocking HERG channel conductance can cause an increased spike frequency and IS (Fig. 8). This simulation shows that block of HERG K+ channels may indeed increase IS in human β-cells. However, this increase can depend on the level of HERG channel conductance and further studies are necessary to investigate the influence of β-cell HERG K+ channels on IS.
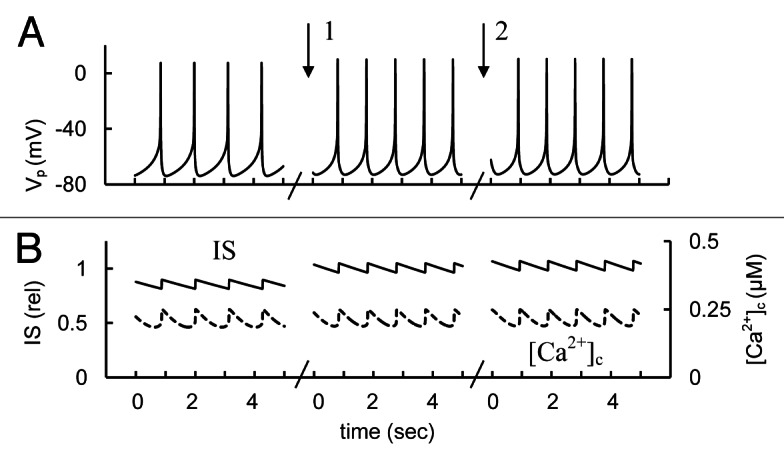
Figure 8. Simulation of changes of human ERG channel conductance. (A) AP firing (Vp); (B) [Ca2+]c (– – –) and relative IS (——). For simulation of HERG channel activation the maximal conductance (gmKhe, Eq. A34, Appendix 2 in Supplemental Material) was initially increased up to 1,000 nS. Then gmKhe was decreased from 1000 nS to 200 nS (basal level from Table S2) at arrow 1. For simulation of block of HERG channel the gmKhe was decreased from 200 pS to 20 nS at arrow 2. Glucose induced persistent AP firing was simulated as in Figure 3.
SK-type channel current
Small-conductance Ca2+ activated K+ channels (SK channels) and the corresponding current (IKCa) have been well defined in mouse islets.41,43 This current with voltage-independent gating variables was also found in human β-cells (SK-like current).10,14 Human islets express SK2 (KCNN2), SK3 (KCNN3) and SK4 (KCNN4).12
Available experiments
The role of SK channels in human islet β-cell electrical activity was studied during glucose stimulation with and without the SK3 channel blocker apamin. Two characteristic behaviors were found: (1) Block of SK channels following the addition of apamin led to a significantly increased spike frequency when glucose stimulated APs existed before adding apamin (Fig. 7C from ref. 14) and (2) AP firing was created when spike activity was suppressed before apamin addition (Fig. 7D from ref. 14). However, no IS or [Ca2+]c were measured in these experiments.
Simulations and analysis
We simulated block of SK channel activity by decreasing its maximal conductance. First, we modeled the effect of SK channel inhibition on spontaneous glucose-induced spike activity (Fig. 9A and B). In this case spike frequency was increased (if every peak is considered as a spike) in a manner consistent with the observations discussed above and simulated [Ca2+]c and IS were increased. Second, decreased SK channel conductance was simulated with increased [ADP]c to reproduce the decreased glucose concentration when spikes were abolished in our simulations (Fig. 9C and D). In this case an inhibition of SK channel conductance could generate spikes (consistent with experimental data) and significantly increases [Ca2+]c and IS.
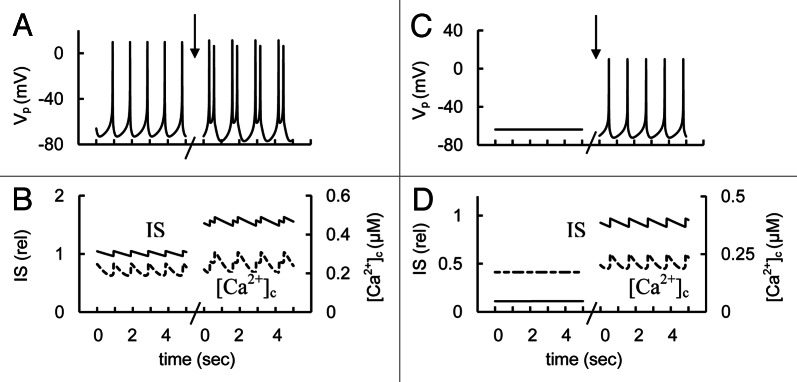
Figure 9. Simulated response to block of KCa channels. (A and C) AP firing (Vp). (B and D). [Ca2+]c (– – –) and relative IS (——). Effect of KCa blockers was simulated by reducing of the maximum conductance for KCa channels (gmKCa, Eq. A39, Appendix 2 in Supplemental Material) that was decreased from 150 pS (basal set from Table S2) to 20 pS at arrows. Other coefficients were as in Figure 3. (1). A and B represent the simulation at high glucose level as in Figure 3. C and D are the simulation at decreased glucose level ([ADP]f = 30 µM).
We have found that the SK-like current is unlikely to take part in creating the AP pacemaker mechanism (see section “Mechanisms of spike generation in human β-cells”). However, the changes in KCa channel conductance may change PM polarization such that AP behavior can be modified. Our calculations suggested the possibility that a block of SK-type channels can increase IS when AP firing occurs (Fig. 9A and B) and/or can lead to decreased threshold for glucose induced spike activity and corresponding increased [Ca2+]c and IS (Fig. 9C and D) when initial spike activity was suppressed with increased glucose. Note, in this simulation (Fig. 9C and D) the glucose level (simulated as [ADP]f changes) was higher than that used for low glucose but it was not enough to simulate spike activity. However, more research is needed to clearly understand the role of SK channels in insulin secretion.
Inwardly-rectifying K+ channels
Large inward K+ currents, that are unaffected by tolbutamide, were recorded from human β-cells.12 Expression of members of the inwardly rectifying K+ (Kir) channel family -Kir2.2 (KCNJ12), Kir2.3 (KCNJ4) and Kir4.2 (KNCJ15) was confirmed in human pancreatic islets.12,44
Available experiments
Inhibition of the Kir channels by Ba2+ may evoke membrane depolarization and stimulates AP firing in human β-cells and conversely, an increase in the inwardly rectifying current should reduce β-cell electrical activity.12
Simulations and analysis
Characteristics of Kir channels and their influence on electrophysiological properties have not been investigated in detail for human β-cells. For this reason, we were not able to directly include them in our modeling approach. However, changes in Kir may lead to the results that can be similar to the changes in KATP channel activity (because KATP channel is also an inwardly rectifying K+ channel, see section “KATP channels”.) although the magnitudes of the changes will be quite different, reflecting the larger current density of KATP channels. For example, our simulation of activated KATP channels has shown a decrease in spike frequency and IS. On the other hand, a blocking of conductivity of KATP channels led to PM depolarization and increase in IS (Fig. 5). Thus, these changes are consistent with behavior that was suggested at blocked and activated Kir channels in work.12
Interestingly, a polymorphism in KCNJ15 (the gene encoding Kir4.2) increases KCNJ15 mRNA levels and significantly increases risk for diabetes in an Asian population.44 These data lead to the conclusion that this gene variant may lower insulin secretion and increase diabetes risk via increased K+ conductance and reduced AP firing but direct evidence of this intriguing suggestion is lacking.
Na+ Currents
Voltage-gated Na+ currents (INa)
Two voltage-gated Na+ currents have been identified in rodent, dog and human β-cells.10,11,45,46 One of the Na+ currents is present in the mouse β-cell only when the β-cell is held at potentials below −100 mV before activation with a depolarizing step.47,48 As β-cells are at rest near −70 mV this current is most likely predominantly inactivated. However, human pancreatic β-cells are equipped with a large TTX-sensitive Na+ current that activates by depolarization to -30 mV and reaches maximal amplitude at 0 mV.9-11 Additional α subunits of the human β-cell Na+ current are likely encoded by SCN3A (Nav1.3)46 and SCN8A (Nav1.6) or SCN9A (Nav1.7).12 SCN1B (which encodes the β1 subunit) is expressed at higher level than is SCN3B (which encodes the β3 subunit).12 When β-cell APs fire at a threshold of about −40 mV, this Na+ current can play an important role in the upstroke (see Fig. 4).
Available experiments
Application of tetrodotoxin has a negligible effect on the electrical activity of mouse β-cells.48,49 By contrast, TTX has a large effect on the generation of APs in human β-cells decreasing the maxima of the spikes (Fig. 10, see also refs. 9 and 10). Insulin secretion elicited by glucose or tolbutamide was significantly reduced by TTX in human islets.9,10 However, [Ca2+]c dynamics was not measured. These differences have functional implications suggesting that the Na+ channels contribute little (if at all) to mouse β-cell electrical activity but can more strongly affect Na+, and thereby, Ca2+ entry in human β-cells.
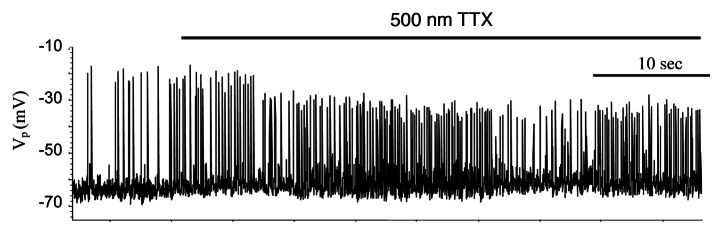
Figure 10. Effect of Na+ channel blocker tetrodotoxin (TTX) on spikes behavior in isolated human islets at 14 mM glucose. Representative examples of spikes. Experiments were performed as described in Sec. Two “Materials and methods.”
Simulations and analysis
We utilized the mathematical model to estimate the impact of Na+ channels on AP, [Ca2+]c and IS. Blockade of Na+ channels induced additional PM repolarization, reduced the AP peak voltage and IS (Fig. 11). Decreased spike amplitude and IS were roughly consistent with experimental data. Analysis of one cycle of AP spontaneous activity with blockaded Na+ channels (in Fig. 11, right part) shows that T- and L-type voltage dependent Ca2+ channels participate in the upstroke of the AP. The AP repolarization phase could include activation of HERG, KCa and BK K+ channels leading to increased repolarizing current (not shown).
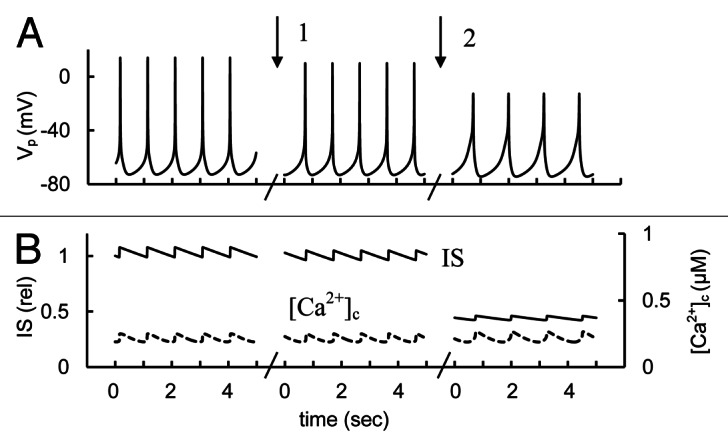
Figure 11. Simulated glucose induced spikes behavior, [Ca2+]c and IS changes at Na+ channel activation or blocking. (A) AP firing (Vp); (B) [Ca2+]c (– – –) and relative IS (——). For simulation of Na+ channel activation the maximal conductance (gmNa, Eq. A1, Appendix 2 in Supplemental Material) was initially increased to 20 nS, then gmNa was decreased from 20 nS to 10 nS (basal level from Table S2) at arrow 1. For simulation of block of Na+ channels the gmNa was decreased from 10 nS to 0.5 nS at arrow 2. Glucose induced APs were simulated as in Figure 3 up to persistent AP firing. Five second intervals are represented for every case.
In neurons, TTX-sensitive Na+ channels are critical for the development of the depolarizing phase of APs.50 However, experiments to date have generally reported that TTX application does not completely block AP firing in human β-cells (Fig. 10 and refs. 9 and 10). According to our simulation these results can be explained by participation of both Na+ and voltage-dependent Ca2+ channels in the AP upstroke (Fig. 4). These experiments and theoretical results argue against the idea that Na+ channels comprise the main generator potential for normal APs in human β-cells and show that VGCCs can be responsible for the AP upstroke during low or in the absence of Na+ channel activity. Activation of voltage-dependent Na+-channels is clearly not mandatory for all AP initiation. Mouse β-cells lack functional Na+ channels (likely due to steady-state inactivation) but are the classic preparation for studying islet APs. Typical spike patterns were obtained using mathematical models of APs without including Na+ channels.13,51 However, regulation of Na+ channel conductance can change the spike height and frequency and corresponding Ca2+ entry and IS in human β-cells (Figs. 10 and 11).
A shift of voltage-gated Na+ conductance and appearance of Na+ current can lead to increased Ca2+ influx in rodent pancreatic β-cells. For example, TsTx-V (scorpion venom α-toxins) and veratridine causes voltage dependent Na+ channels to stay open during a sustained membrane depolarization by decreasing inactivation.52 Using these agents it was possible to potentiate glucose-induced insulin release from isolated rat islets by enhancing β-cell membrane depolarization and increasing the relative duration of electrical activity during the active phase.53 Activation of Na+ channels by veratridine resulted in elevated [Ca2+]c level in isolated β-cells, cell clusters and islets from mouse.16,54 In general, increased flux through voltage–dependent Na+ channels during spikes may increase insulin secretion as reproduced in our simulations in Figure 11 if initial voltage-gated Na+ conductance was low.
Possible role of late (persistent) Na+ current
Ranolazine is novel antianginal drug, which may reduce the incidence of ischemia-related arrhythmias.55 Ranolazine is believed to have its effects via inhibition of the trans-cellular late (persistent) sodium current in cardiomyocytes.56 This drug is of particular interest because it has also been shown to improve glycemic control in patients with Type 2 diabetes, lowering average glycosylated hemoglobin by almost 1%.55
Available experiments
A recent study using isolated rat and human pancreatic islets suggest that ranolazine may increase insulin secretion in a glucose-dependent manner, although the mechanism remains uncertain.57 However, no electrophysiological activity or [Ca2+]c were measured in these experiments.
Simulations and analysis
We evaluated the possibility that a block of a late (persistent) Na+ current can lead to increased IS in human β-cells. A persistent sodium current could originate from a population of voltage-gated Na+ channels because some Na+ channel gating kinetics have incomplete inactivation of macroscopic currents for repolarization to subthreshold voltages.32,58 We modeled this by introducing a small voltage-independent coefficient in the equation for Na+ channels (kNar, Eq A1, Appendix 2 in Supplemental Material) that leads to a persistent Na+ current even at low PM potentials. (Note this coefficient was zero in other simulations.)
We decreased the coefficient kNar (Eq. A1, Appendix 2 in Supplemental Material) from an elevated level to simulate experiments with blocked persistent Na+ current. In this example a block of the persistent Na+ current increased the AP peaks and IS significantly despite decreased [Ca2+]c (Fig. 12). This result can be explained by increased P/Q type Ca2+ channel conductance, which is responsible for granule exocytosis (see section “P/Q-type Ca2+ channels” below) during high amplitude AP firing in comparison with L-type Ca2+ channels. This simulation shows that a block of a persistent inward Na+ current can increase IS. This mechanism can partially explain the mechanism of ranolazine action on pancreatic human β-cells.
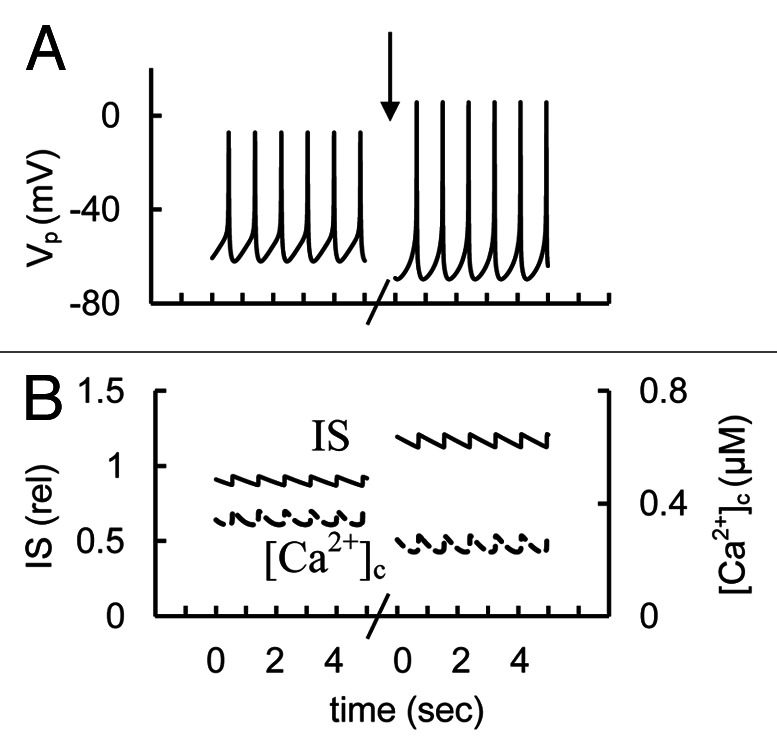
Figure 12. Simulated glucose induced AP firing, [Ca2+]c and relative IS at persistent (late) Na+ current changes. (A) AP firing (Vp); (B) [Ca2+]c (– – –) and relative IS (——). Initially the coefficient (kNar, Eq. A1, Appendix 2 in Supplemental Material) responsible for persistent Na+ current was 0.002 (basal level for this coefficient is zero in Table S2). For simulation of block of the persistent (late) Na+ currant this coefficient was decreased to 0.0006 at arrow. Glucose induced AP firing was simulated as in Figure 3. Five second intervals are represented.
However, a persistent Na+ current has not been well characterized in pancreatic β-cells. A small prolonged inward current was detected in human β-cells,9 however, the nature of this current and its relationship to the transient Na+ current underlying spikes and the influence of ranolazine on it remain to be clarified. According to our calculations this persistent Na+ current (with kNar as in Fig. 12) can be small (less than 1% of maximal amplitude of simulated INa current in Fig. S1A and C) to show a significant increase in IS following simulated block (see Fig. 12). This can make its experimental investigation difficult. For example, no persistent Na+ current was found in a related study.10 However, the experiments in this work may not have been performed with the required accuracy to find a small persistent Na+ current as they were not specifically sought. More studies (including electrophysiological investigations) are needed to clearly understand the role of the persistent (late) Na+ current in human β-cells and to determine whether ranolazine could have other mechanisms of action resulting in an improvement in glycemic control.
Based on the results of the experiments with rodent and human β-cells and our simulations we suggest that voltage-gated Na+ currents may be more generally important for IS in human than previously thought, as this re-analysis in mouse β-cell membrane polarization shows. Drugs that target voltage-gated Na+ channels or genetic variants in the channels genes may alter IS in humans.
Na+ background current
Closure of KATP-channels alone is not sufficient to cause membrane depolarization. Therefore, β-cells must be equipped with some sort of depolarizing inward current (see section “Mechanisms of spike generation in human β-cells”). However, the nature of the depolarizing current remains elusive.4,8 For example, this current in β-cells might result from an activation of cationic (principally Na+) Trp-channels12,59,60 or nonselective Na+ leak channels (NALCN) that can be activated in mouse β-cells by the binding of acetylcholine to the M3 receptor.61 The gene encoding NALCN channels may also be expressed in human islets.12 However, these currents have not been studied in human β-cells. In this article we used a model of some generalized inward Na+ background current (INab) (without specifying exactly which one) to simulate this depolarizing current in human β-cells (Eq. A45, Appendix 2, Supplemental Material).
Available experiments
In mouse β-cell line, acetylcholine causes plasma membrane depolarization by opening NALCN channels, a mechanism for resting Na+ permeability.62 However, no studies in human β-cells have directly examined this Na+ background current.
Simulations and analysis
We have found that Na+ background current is unlikely to take part in creating the AP pacemaker mechanism current (see section “Mechanisms of spike generation in human β-cells”).However, any change in inward voltage-independent Na+ background current may change the PM potential. According to proposal of Gilon and Rorsman61 acetylcholine activated NALCN can increase the firing of AP and corresponding IS when glucose is elevated. We attempted to simulate this proposal (Fig. 13).
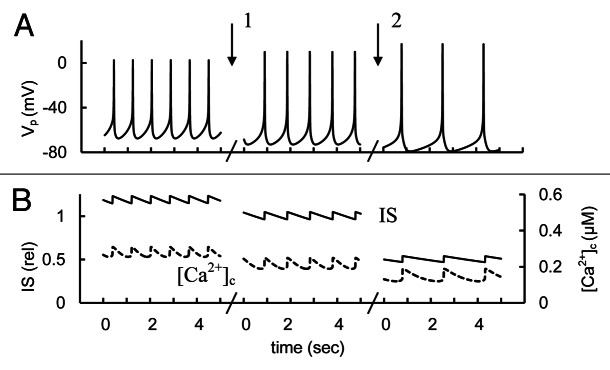
Figure 13. Simulated glucose induced AP firing, [Ca2+]c and relative IS at Na+ background current changes. (A) AP firing (Vp); (B) [Ca2+]c (– – –) and relative IS (——). For simulation of Na+ background current activation the maximal conductance (gbNa, Eq. A45, Appendix 2 in Supplemental Material) was initially increased up to 20 pS. Than gbNa was decreased from 20 pS to 10 pS (basal level from Table S2) at arrow 1. For simulation of block of Na+ background current the gbNa was decreased from 10 pS to 1 pS at arrow 2. Glucose induced persistent AP firing was simulated as in Figure 3. Five second intervals are represented for every case.
Simulated activation of the Na+ background current could increase spike frequency, [Ca2+]c and IS in our model (Fig. 13). Note, activation of a NALCN-type Na+ background current activity by increasing its maximal conductance may act similarly to KCa current block (see section “SK-type channel current”), because a similar PM depolarization takes place both by blocking voltage independent KCa channels and by activation of the Na+ background current. However, additional experiments are required to define the properties and regulation of this current and its influence on β-cells.
The Voltage-Gated Ca2+ Currents
The whole-cell Ca2+ current in mammalian β-cells flows principally through the voltage-gated Ca2+ channels.1,3,37 Whole-cell patch clamp studies in human islets initially identified two current components having different activation voltage thresholds: high- and low voltage-activated Ca2+ currents.9,10,63 Human β-cells are equipped mainly with L-(CaV1.3), P/Q- (CaV2.1) and T- (CaV3.2) other than R-type Ca2+ channels.10,12,64
L-type Ca2+ channels
The high-voltage-activated “L-type” Ca2+ current (ICaL) activates at potentials less than –40 mV and inactivates relatively slowly. In human β-cells this current was reported to be similar to those observed in rodent β-cells and insulinoma cells.10,37 Human islets express both α1C/Cav1.2 (CACNA1C) and α1D/Cav1.3 (CACNA1D) L-type Ca2+ channels.12
Available experiments
L-type Ca2+ channels blocker (isradipine) suppressed electrical activity induced by glucose or tolbutamide and decreased IS rate in human islets.10
Simulations and analysis
According to our analysis L-ype Ca2+ currents are largely responsible for the AP upstroke (Fig. 4). For this reason decreasing L–type Ca2+ channel conductance suppressed AP firing in our model. This suppression also results in a decreased Ca2+ influx, [Ca2+]c and IS (Fig. 14). Thus, our simulation is consistent with the experimental findings.
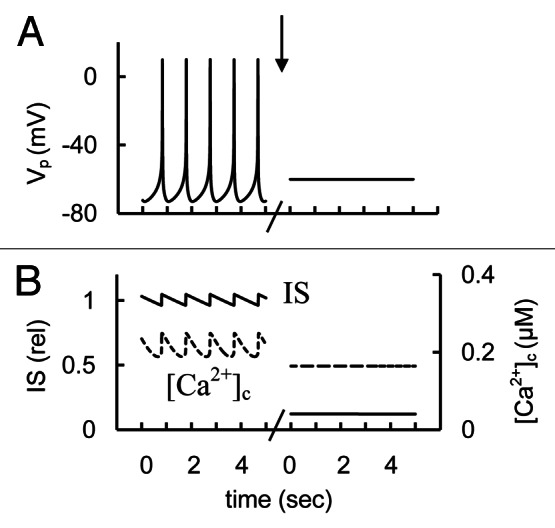
Figure 14. Simulated glucose induced AP spikes, [Ca2+]c and IS changes after simulation of L-type Ca2+ channel blockers application. (A) AP firing (Vp); (B) [Ca2+]c (– – –) and relative IS (——). The maximal conductance (gmCaL, Eq. A6, Appendix 2 in Supplemental Material) was decreased from 2.7 nS (basal level from Table S2) to 0.2 nS at arrow. Glucose induced persistent AP firing was simulated as in Figure 3. Five second intervals are represented.
P/Q-type Ca2+ channels
A high-voltage-activated P/Q-type Ca2+ current (ICaP) was also found in human β-cells that possess the fast activating and, apparently, only one slowly inactivating voltage-gated variable.2,10 Importantly, P/Q-type Ca2+ channels (encoded mainly by CACNA1A) may be opened at PM potentials that are above that for opening of L-type Ca2+ channels (see2,10) and we have modeled this peculiarity (see Fig. S3, Appendix 2 in Supplemental Material). P/Q-type Ca2+ channels may serve in human β-cells as the main component of non-L-type Ca2+ channels that trigger depolarization-evoked exocytosis of the insulin-containing granules2,10 (see also Sec. 3).
Available experiments
P/Q-type Ca2+ channel blocker (ω-agatoxin) significantly suppressed depolarization-evoked exocytosis in isolated human β-cells and inhibited insulin release in isolated human islets.2,10 However, no experimental measurements of the effects of P/Q-type Ca2+ channel blockers on [Ca2+]c have been reported.
Simulations and analysis
We simulated the action of a P/Q-type Ca2+ channel blocker by changing the maximal conductivity of these channels (Fig. 15). The results of these simulations showed that P/Q-type Ca2+ channel blockade does not fully eliminate spike activity, but can lead to complex spikes and increased [Ca2+]c. However P/Q-type Ca2+ channel blockade can significantly suppress IS. This may occur because in our model the high voltage-gated non-L-type Ca2+ channels have a more limited role on the AP upstroke and [Ca2+]c than L-type channels but they are primarily responsible for insulin release at high PM potential in response to AP firing. On the basis of specific experimental data (see references 2 and 10) and our computational analysis we suggest that block of P/Q-type Ca2+ channels in human β-cells may lead to suppressed insulin secretion even though the resulting change in electrophysiological behavior and cytoplasmic Ca2+ levels can be different for L- and P/Q-type Ca2+ channels.
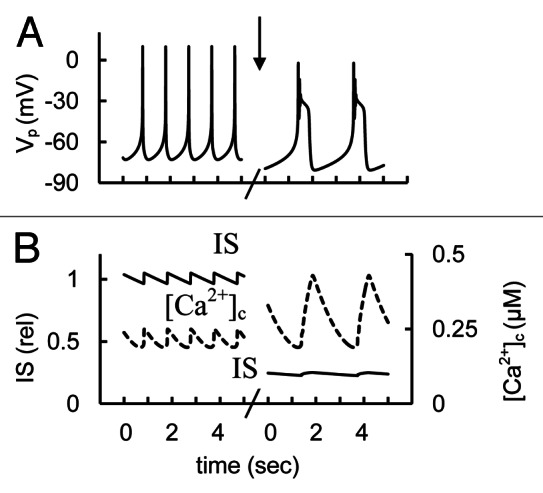
Figure 15. Simulated glucose induced AP spikes, [Ca2+]c and IS changes after of P-type Ca2+ channel blockers application. (A) AP firing (Vp); (B) [Ca2+]c (– – –) and relative IS (——). The maximal conductance (gmCaP, Eq. A14, Appendix 2) was decreased from 1.2 nS (basal level from Table S2) to 0.2 nS at arrow. Glucose induced AP firing was simulated as in Figure 3. Five second intervals are represented.
T-type Ca2+ channels
Low-voltage-activated “T-type” Ca2+ current (ICaT) and corresponding channels have been reported in human,10,11 rat, nonobese diabetic (or NOD) mouse β-cells, as well as in INS-1 and RINm5F cells.37,65,66 These channels activate near –70 mV and inactivate rapidly through a voltage-dependent mechanism.67 T-type Ca2+ channels in human β cells are likely encoded by CACNA1H (Cav3.2), which is expressed at much higher levels than are other T-type Ca2+ channel genes.10
Available experiments
T-type Ca2+ channel blocker (NNC) significantly suppressed electrical activity induced by glucose and tolbutamide in isolated human β-cells and inhibited insulin release in isolated human islets at 6 mM glucose.10 However, no experimental measurements of the effects of T-type Ca2+ channel blockers on [Ca2+]c have been reported. In rat insulinoma INS-1 cells activation of T-type Ca2+ channels increased IS and [Ca2+]c.68
Simulations and analysis
We simulated experiments with changed T-type Ca2+ channel conductance that corresponds to the effects of specific activators and blockers of these channels (Fig. 16). AP frequency and IS decreased in our model with decreased conductance of T-type Ca2+ channels from basal level. On other hand [Ca2+]c and IS increased with an increase of T-type Ca2+ channel conductance (left part of Fig. 16). This is consistent with experimental data.
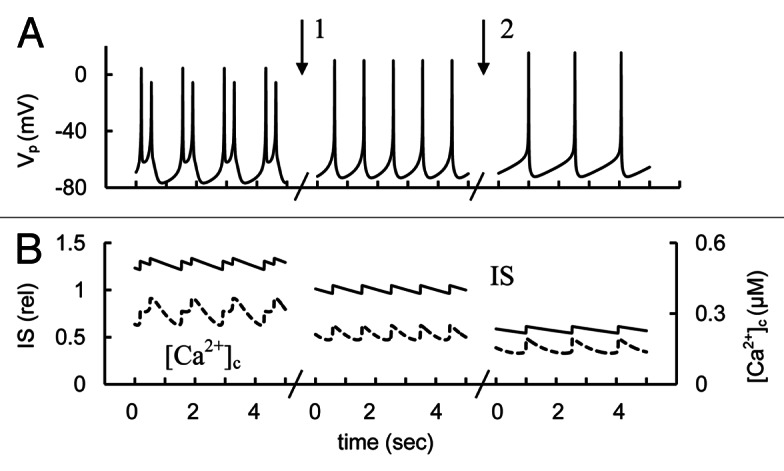
Figure 16. Simulated glucose induced spikes behavior, [Ca2+]c and IS changes at T-type Ca2+ channel activation or blocking. (A) AP firing (Vp); (B) [Ca2+]c (– – –) and relative IS (——). For simulation of T-type Ca2+ channel activation the maximal conductance (gmCaT, Eq. A19, Appendix 2 in Supplemental Material) was initially increased up to 500 pS. Than gmCaT was decreased from 500 pS to 250 pS (basal level from Table S2) at arrow 1. For simulation of block of T-type Ca2+ channel the gmCaT was decreased from 200 pS to 50 nS at arrow 2. Glucose induced persistent AP firing was simulated as in Figure 3.
The role of low-threshold T-type Ca2+ channels in islet physiology has been less well studied than L- and P/Q-types Ca2+ channels in part because these channels are not normally expressed in the mouse islet model. Progress in this field has also been hampered by the lack of selective T-type Ca2+ channel blockers.69 These factors restrict our possibility to analyze the role of low-threshold T-type Ca2+ channels. However, these channels may not be mandatory for AP firing because mouse β-cells have typical spike behavior even though they have only modest activity of T-type Ca2+ channels.
We have assigned only low conductivity for T-type Ca2+ channels in our model to compare with L- and P/Q- Ca2+ channels (see Figs. S2 and S3; Table S2). This suggestion was made because we found in simulations that increasing the maximal conductivity of T-type Ca2+ channels above the levels in Table S2 leads to significant increased Ca2+ cytoplasmic concentration in resting glucose conditions at low PM potential and to insignificant increase of [Ca2+]c with increase of glucose level from low to high that is unrealistic. The data that T-type Ca2+ current activation even under nonstimulatory conditions may raise basal [Ca2+]c in β-cells (NOD) mice65 supports the conclusion that increased activity of T-type Ca2+ channels can lead to increased [Ca2+]c in resting glucose conditions.
To resolve the discrepancy between significant activity of T-type Ca2+ channels in human β-cells that was found by Braun et al.10 and small [Ca2+]c at low glucose levels, we can suggest, for example, that low glucose levels lead to a decreased activity of T-type Ca2+ channels decreasing Ca2+ influx. This suggests that more research is needed to clearly understand the regulatory role of T-type Ca2+ channels in rodent and human β-cells.
Other Currents
Excitable cells (including pancreatic β-cells) have a great many other channels, pump and exchangers then we have analyzed here. For example, the Na+-K+ pump extrudes three Na+ ions from the cell in exchange for two K+ ions, generating a net outward, hyperpolarizing current, maintaining resting Vp. Currents provided by Na+-Ca2+ exchangers also contribute to Vp. Both these currents were included in our recent model for Ca2+ dynamics in β-cell.70
Genome wide association studies have pointed to an association of KCNQ1 with T2 diabetes.6,71 The voltage-gated potassium channel Kv7.1 (encoded by KCNQ1) is expressed in human islets. A polymorphism in KCNQ1 increases type 2 diabetes risk.72,73 Interestingly, block of Kv7.1 potassium current increased glucose induced IS in isolated rat islets, increasing AP firing and cytoplasmic free calcium.74
However, it is unclear whether these channels contribute to AP firing in human β-cells and so it was not directly included in our modeling and analysis. However, this model can be extended with reasonable facility by including the equations for new channels, pump and exchangers when additional experimental information will be obtained.
Regulation of Ca2+ Entry and Bursting
We modeled Ca2+ dynamics in these simulations. However, data are not yet available on the Ca2+ dynamics in one human β-cell during one spike that can facilitate a strong test of modeling. On the other hand, we have measured and analyzed cytoplasmic Ca2+ dynamics during single spikes in mouse β-cells.13,75 On the basis of these investigations we were able to construct a mathematical model of Ca2+ dynamics that fits reasonably well with experimental data.13 The main principles of regulation of Ca2+ fluxes through voltage-dependent Ca2+ channels and pumps appear to be similar for human and mouse β-cells. For this reason, we believe that the model we developed for Ca2+ dynamics in mouse β-cells will allow the simulation of fast Ca2+ dynamics in human β-cells. For example, we can suggest significant increase in [Ca2+]c during one complex spike (for example, see right part of Fig. 5 and left part of Fig. 7) since this was measured and simulated for mouse β-cells.13
Frequency, height and width of spikes can determine Ca2+ influx through VGCCs. We were able to simulate an increase in [Ca2+]c and IS even though the spike height was unchanged or decreased whereas spikes were broadened (see right parts of Figs. 5, 7 and 16). Consequently, decreased spike height cannot be assumed to lead to a decrease in [Ca2+]c or IS. Increased Ca2+ influx can also follow from broadening of spikes in mouse β-cells.13 Interestingly, in immortalized gonadotropin-releasing hormone-secreting neurons it was found that spike broadening can increase the integrated Ca2+ influx despite a decrease in spike height.76 This raises the question of whether the changes in spike broadening via channel regulation can lead to an appropriate increase in IS. This possibility has not yet been directly studied in human β-cells.
Glucose induced electrical activity in pancreatic mouse β-cells is thought to involve slow depolarizing waves that were called “bursting” in electrophysiological investigations.16,51,77 Bursts have a plateau from which APs rapidly fire with numerous spikes (that can also include complex spikes) during one burst and can last up to several minutes. Bursts are separated by quiescent period at potentials below the AP voltage threshold.16,51,77 Bursting processes have also been occasionally observed in human islets.12 It should be noted, the spikes with several maximums that we refer here as complex spikes are also denoted as “bursts” in some neurophysiological investigations and in the recent computational model of Pedersen.78 To avoid misinterpretation we use the term “burst” only as it was accepted for β-cells electrophysiological investigations.
Bursting in β-cells provides an effective mechanism for rapid changes in extracellular Ca2+ influx through VGCCs that leads to cytosolic [Ca2+]c and IS oscillations.16,25,77,79 Ca2+ oscillations have also been reported in human islets,12,22,25,80 and it seems that bursting activity is also characteristic of human islets. The mechanisms that generate and terminate spikes are responsible for the bursts of APs, Ca2+ and IS oscillations in mouse. Subsequent generation and termination of the bursts may be determined by small cyclic changes in some voltage-gated and/or voltage independent gating variable currents.13,16,79,81
There is no conventional model for bursting and [Ca2+]c oscillatory mechanisms even for mouse β-cells. However, several mechanisms were justified for β-cells (for review, see refs.16 and 82). For example, the ATP/ADP ratio in cytoplasm, calcium concentration in endoplasmic reticulum, Na+ concentration in cytoplasm and others can all be these pacemakers for oscillatory mechanisms. Slow changes of these components could gradually lead to a membrane repolarization down to the threshold potential for APs firing, shutting down the spikes and inactivating the VGCCs. Then Ca2+ influx decreases sharply, leading to decreased PM potential and a resting phase. Processes occur in opposite directions during a resting (repolarized) phase. However, the real pacemakers for bursting and [Ca2+]c oscillations in human β-cells were not investigated and further studies supported by improved mathematical models are necessary to define the mechanisms for possible spontaneous oscillatory phenomena in human β-cell physiology.
Mathematical Modeling of AP Firing in Pancreatic β-Cells
Chay and Keizer83 described the first Hodgkin-Huxley-type ionic model for mouse β-cells where spike generation was simulated as a result of an interaction between only two voltage-gated channels (Ca2+ and delayed-rectifier K+) and a leak current. Keizer and Magnus84 improved this model by including KATP channels, an approach also used in later models of spikes in pancreatic β-cells.
The interaction of the voltage-gated Ca2+ and delayed-rectifier K+ channels leads to spikes with one maximum.13,51,79,83 However, complex spikes were found in isolated mouse islets from Kv2.1 knockout mouse and similar spikes were obtained after application of specific Kv2.1 inhibitors, that blocked of the rapid delayed rectified current (IKDr).13,40 We were able to simulate the appearance of these complex spikes in our mathematical model of APs in mouse β-cells suggesting that the channels other than KDr can also play a prominent role in repolarization phase in mouse β-cells.13 These simulations shows that a transition from a spike with a single narrow maximum to a complex spike with several maxima can occur when, following an initial depolarization, the subsequent repolarizing signal is not strong enough to allow fast recovery of the membrane potential to a repolarized resting level. The final transition to the repolarizing level can occur with time after several small maxima when an additional repolarizing current increases.
The first Hodgkin-Huxley-type ionic model for AP firing, Ca2+ handling and IS in the human β-cell was recently developed78,85 which included models of voltage-gated Na+ and T-type Ca2+ channels. Complex spikes were also simulated by Pedersen78 following blockade of the delayed rectifier K+ channels. In this case the slowly activating K+ HERG current was employed as the repolarizing factor leading to final repolarization.
We used a mathematical modeling approach for a systematic review of function of channels in human β-cells. Several channels were added in the model for human β-cell to compare with our previous model for mouse13: BK channels, voltage-gated Na+ channels, P- and T-type Ca2+ channels. To compare with other models for human β-cell78,85 we also added the models of voltage-independent Ca2+ activated K+ current (IKCa) and plasma membrane Ca2+ pump current. We have performed also more detailed description of KATP, high L-type voltage-gated Ca2+ and Na+ currents and others changes in parameters and equations (see Supplementary Material). These differences lead to an improved description of human β-cell behavior. For example, the frequency for simple spikes in our model is about 1 spike per second that corresponds well to experimental data (see Fig. 2 and refs.9,10,20,21) instead of about 4 spike per sec,78 that corresponds better to the spike behavior in mouse islets (for example, see refs. 13, 51 and 83). In our review article we also evaluated the possible role of major channels that regulate AP firing and IS in human β-cells.
Several mathematical models have been developed to simulate bursting and [Ca2+]c oscillations in mouse β-cells on the basis of alternative mechanisms (for review, see Sec. Nine and refs. 16,79,82). The models developed were used to determine the mechanisms that generate and terminate spikes leading to bursting and Ca2 + oscillations in β-cells. Models describing spikes in human β-cells (included developed in this article) may be used for constructions of similar models for bursting, Ca2+ and IS oscillations in human islets.
Computational System Analysis in Electrophysiology of Human β-cells: Perspectives and Limitations
In general, an interpretation of the electrophysiological data and the effects of channel blockers and activators on electrical pacemaking is difficult because a change of any sufficiently large current potentially alters several aspects of the voltage trajectory and thus modifies activation of all other voltage-dependent currents. For this reason, using the mathematical modeling approach opens the way for better understanding of such complex interactions.
An important feature of the computational model of human β-cells is that we included all the major ionic currents fitted to recent experimental data in the context of expression data. We used the Hodgkin-Huxley formalism to describe the behavior of channels. This choice is consistent with our goal, to reconstruct the APs that result from ensemble currents through many individual channels. The key feature of the model is that it can be used to explore the putative mechanisms underlying AP spikes. We were able to show that the model we developed can reproduce a number of experimental observations ranging from voltage-clamp current traces of AP firing to Ca2+ dynamics and IS and can be used as the groundwork for in silico examination of the effects of agents that modulate insulin secretion via regulation of ion channels. As other channels are further characterized they can be readily incorporated into this basic model. This also creates opportunities for in silico studies of possible functional influence of different channels on human β–cell functional properties that it is difficult to measure at this time.
However, limitations in a simulation of the specific experiments are unavoidable because of the limited current knowledge of basic electrophysiological data, the considerable variation in experimental conditions, and the potentially deleterious effects procedures used in voltage-clamp experiments. Therefore it would be unreasonable to expect the model to accurately reproduce all previously observed experimental results.
Other limitations are tied to the mechanisms of insulin release. Insulin is secreted by exocytosis of large dense-core vesicles. Regulation of insulin–containing granule secretion in β-cells is sufficiently complex such that is not only a function of cytoplasmic Ca2+ concentration or calcium influx through different VGCCs located in the plasma membrane that we have included in the model. For example, cAMP pathway components, muscarinic pathways and other processes regulate exocytosis in β-cells.6,17 However, a consideration and modeling of the effect of these mechanisms on the rate of exocytosis of insulin–containing granules in human is outside the scope of this work.
Supplementary Material
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
Funding for this study was provided in part by the National Institute of Diabetes and Digestive and Kidney Diseases Grants DRTC P60DK020595, DK-48494, DK-063493, a Research Grant from the Keck foundation and the Blum-Kovler Foundation and the Kovler Diabetes Center.
Glossary
Abbreviations:
- AP
action potential
- BK
large-conductance Ca2+-activated K+-channels
- Cm
membrane capacitance
- [Ca2+]c
free cytoplasmic Ca2+
- HERG
human ERG K+ channels
- IbTX
iberiotoxin
- IS
insulin secretion
- KATP-channel
ATP-sensitive K+-channel
- KCa
calcium activated K+ channels, KDr, rapid delayed rectified voltage-gated K+ channels
- Kv
voltage-gated K+ channels
- SK
small conductance calcium activated K+ channels
- NALCN
nonselective Na+ leak channels
- PM
plasma membrane
- TTX
tetrodotoxin
- Vp
membrane potential
- VGCC
voltage-gated Ca2+ channel
Supplemental Material
Supplemental Material may be found here: http://www.landesbioscience.com/journals/islets/article/24166/
Footnotes
Previously published online: www.landesbioscience.com/journals/islets/article/24166
References
- 1.Jacobson DA, Philipson LH. Ion Channels and Insulin secretion. In: Seino S, Bell G, eds. Pancreatic Beta Cell in Health and Desease. Japan: Springer, 2008:91- 110. [Google Scholar]
- 2.Rorsman P, Braun M, Zhang Q. Regulation of calcium in pancreatic α- and β-cells in health and disease. Cell Calcium. 2012;51:300–8. doi: 10.1016/j.ceca.2011.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Drews G, Krippeit-Drews P, Düfer M. Electrophysiology of islet cells. Adv Exp Med Biol. 2010;654:115–63. doi: 10.1007/978-90-481-3271-3_7. [DOI] [PubMed] [Google Scholar]
- 4.Ashcroft FM, Rorsman P. Electrophysiology of the pancreatic beta-cell. Prog Biophys Mol Biol. 1989;54:87–143. doi: 10.1016/0079-6107(89)90013-8. [DOI] [PubMed] [Google Scholar]
- 5.Gilon P, Henquin JC. Mechanisms and physiological significance of the cholinergic control of pancreatic beta-cell function. Endocr Rev. 2001;22:565–604. doi: 10.1210/er.22.5.565. [DOI] [PubMed] [Google Scholar]
- 6.Rosengren AH, Braun M, Mahdi T, Andersson SA, Travers ME, Shigeto M, et al. Reduced insulin exocytosis in human pancreatic β-cells with gene variants linked to type 2 diabetes. Diabetes. 2012;61:1726–33. doi: 10.2337/db11-1516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Greeley SA, Tucker SE, Naylor RN, Bell GI, Philipson LH. Neonatal diabetes mellitus: a model for personalized medicine. Trends Endocrinol Metab. 2010;21:464–72. doi: 10.1016/j.tem.2010.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rorsman P, Eliasson L, Kanno T, Zhang Q, Gopel S. Electrophysiology of pancreatic β-cells in intact mouse islets of Langerhans. Prog Biophys Mol Biol. 2011;107:224–35. doi: 10.1016/j.pbiomolbio.2011.06.009. [DOI] [PubMed] [Google Scholar]
- 9.Barnett DW, Pressel DM, Misler S. Voltage-dependent Na+ and Ca2+ currents in human pancreatic islet beta-cells: evidence for roles in the generation of action potentials and insulin secretion. Pflugers Arch. 1995;431:272–82. doi: 10.1007/BF00410201. [DOI] [PubMed] [Google Scholar]
- 10.Braun M, Ramracheya R, Bengtsson M, Zhang Q, Karanauskaite J, Partridge C, et al. Voltage-gated ion channels in human pancreatic beta-cells: electrophysiological characterization and role in insulin secretion. Diabetes. 2008;57:1618–28. doi: 10.2337/db07-0991. [DOI] [PubMed] [Google Scholar]
- 11.Misler S, Barnett DW, Gillis KD, Pressel DM. Electrophysiology of stimulus-secretion coupling in human beta-cells. Diabetes. 1992;41:1221–8. doi: 10.2337/diabetes.41.10.1221. [DOI] [PubMed] [Google Scholar]
- 12.Rorsman P, Braun M. Regulation of insulin secretion in human pancreatic islets. Annu Rev Physiol. 2013;75:155–79. doi: 10.1146/annurev-physiol-030212-183754. [DOI] [PubMed] [Google Scholar]
- 13.Fridlyand LE, Jacobson DA, Kuznetsov A, Philipson LH. A model of action potentials and fast Ca2+ dynamics in pancreatic beta-cells. Biophys J. 2009;96:3126–39. doi: 10.1016/j.bpj.2009.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jacobson DA, Mendez F, Thompson M, Torres J, Cochet O, Philipson LH. Calcium-activated and voltage-gated potassium channels of the pancreatic islet impart distinct and complementary roles during secretagogue induced electrical responses. J Physiol. 2010;588:3525–37. doi: 10.1113/jphysiol.2010.190207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jacobson DA, Philipson LH. Action potentials and insulin secretion: new insights into the role of Kv channels. Diabetes Obes Metab. 2007;9(Suppl 2):89–98. doi: 10.1111/j.1463-1326.2007.00784.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fridlyand LE, Tamarina N, Philipson LH. Bursting and calcium oscillations in pancreatic beta-cells: specific pacemakers for specific mechanisms. Am J Physiol Endocrinol Metab. 2010;299:E517–32. doi: 10.1152/ajpendo.00177.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fridlyand LE, Philipson LH. Coupling of metabolic, second messenger pathways and insulin granule dynamics in pancreatic beta-cells: a computational analysis. Prog Biophys Mol Biol. 2011;107:293–303. doi: 10.1016/j.pbiomolbio.2011.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Henquin JC. Regulation of insulin secretion: a matter of phase control and amplitude modulation. Diabetologia. 2009;52:739–51. doi: 10.1007/s00125-009-1314-y. [DOI] [PubMed] [Google Scholar]
- 19.Smith PA, Duchen MR, Ashcroft FM. A fluorimetric and amperometric study of calcium and secretion in isolated mouse pancreatic beta-cells. Pflugers Arch. 1995;430:808–18. doi: 10.1007/BF00386180. [DOI] [PubMed] [Google Scholar]
- 20.Manning Fox JE, Gyulkhandanyan AV, Satin LS, Wheeler MB. Oscillatory membrane potential response to glucose in islet beta-cells: a comparison of islet-cell electrical activity in mouse and rat. Endocrinology. 2006;147:4655–63. doi: 10.1210/en.2006-0424. [DOI] [PubMed] [Google Scholar]
- 21.Rosati B, Marchetti P, Crociani O, Lecchi M, Lupi R, Arcangeli A, et al. Glucose- and arginine-induced insulin secretion by human pancreatic beta-cells: the role of HERG K(+) channels in firing and release. FASEB J. 2000;14:2601–10. doi: 10.1096/fj.00-0077com. [DOI] [PubMed] [Google Scholar]
- 22.Hellman B, Gylfe E, Bergsten P, Grapengiesser E, Lund PE, Berts A, et al. Glucose induces oscillatory Ca2+ signalling and insulin release in human pancreatic beta cells. Diabetologia. 1994;37(Suppl 2):S11–20. doi: 10.1007/BF00400821. [DOI] [PubMed] [Google Scholar]
- 23.Ashcroft FM. The Walter B. Cannon Physiology in Perspective Lecture, 2007. ATP-sensitive K+ channels and disease: from molecule to malady. Am J Physiol Endocrinol Metab. 2007;293:E880–9. doi: 10.1152/ajpendo.00348.2007. [DOI] [PubMed] [Google Scholar]
- 24.Clark R, Proks P. ATP-sensitive potassium channels in health and disease. Adv Exp Med Biol. 2010;654:165–92. doi: 10.1007/978-90-481-3271-3_8. [DOI] [PubMed] [Google Scholar]
- 25.Martín F, Soria B. Glucose-induced [Ca2+]i oscillations in single human pancreatic islets. Cell Calcium. 1996;20:409–14. doi: 10.1016/S0143-4160(96)90003-2. [DOI] [PubMed] [Google Scholar]
- 26.Seino S. Cell signalling in insulin secretion: the molecular targets of ATP, cAMP and sulfonylurea. Diabetologia. 2012;55:2096–108. doi: 10.1007/s00125-012-2562-9. [DOI] [PubMed] [Google Scholar]
- 27.Ferrer J, Wasson J, Salkoff L, Permutt MA. Cloning of human pancreatic islet large conductance Ca(2+)-activated K+ channel (hSlo) cDNAs: evidence for high levels of expression in pancreatic islets and identification of a flanking genetic marker. Diabetologia. 1996;39:891–8. doi: 10.1007/BF00403907. [DOI] [PubMed] [Google Scholar]
- 28.Houamed KM, Sweet IR, Satin LS. BK channels mediate a novel ionic mechanism that regulates glucose-dependent electrical activity and insulin secretion in mouse pancreatic β-cells. J Physiol. 2010;588:3511–23. doi: 10.1113/jphysiol.2009.184341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cui J, Yang H, Lee US. Molecular mechanisms of BK channel activation. Cell Mol Life Sci. 2009;66:852–75. doi: 10.1007/s00018-008-8609-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fakler B, Adelman JP. Control of K(Ca) channels by calcium nano/microdomains. Neuron. 2008;59:873–81. doi: 10.1016/j.neuron.2008.09.001. [DOI] [PubMed] [Google Scholar]
- 31.Berkefeld H, Sailer CA, Bildl W, Rohde V, Thumfart JO, Eble S, et al. BKCa-Cav channel complexes mediate rapid and localized Ca2+-activated K+ signaling. Science. 2006;314:615–20. doi: 10.1126/science.1132915. [DOI] [PubMed] [Google Scholar]
- 32.Bean BP. The action potential in mammalian central neurons. Nat Rev Neurosci. 2007;8:451–65. doi: 10.1038/nrn2148. [DOI] [PubMed] [Google Scholar]
- 33.Clapham DE. Calcium signaling. Cell. 2007;131:1047–58. doi: 10.1016/j.cell.2007.11.028. [DOI] [PubMed] [Google Scholar]
- 34.Dai XQ, Manning Fox JE, Chikvashvili D, Casimir M, Plummer G, Hajmrle C, et al. The voltage-dependent potassium channel subunit Kv2.1 regulates insulin secretion from rodent and human islets independently of its electrical function. Diabetologia. 2012;55:1709–20. doi: 10.1007/s00125-012-2512-6. [DOI] [PubMed] [Google Scholar]
- 35.MacDonald PE, Wang G, Tsuk S, Dodo C, Kang Y, Tang L, et al. Synaptosome-associated protein of 25 kilodaltons modulates Kv2.1 voltage-dependent K(+) channels in neuroendocrine islet beta-cells through an interaction with the channel N terminus. Mol Endocrinol. 2002;16:2452–61. doi: 10.1210/me.2002-0058. [DOI] [PubMed] [Google Scholar]
- 36.Yan L, Figueroa DJ, Austin CP, Liu Y, Bugianesi RM, Slaughter RS, et al. Expression of voltage-gated potassium channels in human and rhesus pancreatic islets. Diabetes. 2004;53:597–607. doi: 10.2337/diabetes.53.3.597. [DOI] [PubMed] [Google Scholar]
- 37.Yang SN, Berggren PO. Beta-cell CaV channel regulation in physiology and pathophysiology. Am J Physiol Endocrinol Metab. 2005;288:E16–28. doi: 10.1152/ajpendo.00042.2004. [DOI] [PubMed] [Google Scholar]
- 38.Philipson LH. Beta-cell ion channels: keys to endodermal excitability. Horm Metab Res. 1999;31:455–61. doi: 10.1055/s-2007-978774. [DOI] [PubMed] [Google Scholar]
- 39.Dai XQ, Manning Fox JE, Chikvashvili D, Casimir M, Plummer G, Hajmrle C, et al. The voltage-dependent potassium channel subunit Kv2.1 regulates insulin secretion from rodent and human islets independently of its electrical function. Diabetologia. 2012;55:1709–20. doi: 10.1007/s00125-012-2512-6. [DOI] [PubMed] [Google Scholar]
- 40.Jacobson DA, Weber CR, Bao S, Turk J, Philipson LH. Modulation of the pancreatic islet beta-cell-delayed rectifier potassium channel Kv2.1 by the polyunsaturated fatty acid arachidonate. J Biol Chem. 2007;282:7442–9. doi: 10.1074/jbc.M607858200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Smith PA, Bokvist K, Arkhammar P, Berggren PO, Rorsman P. Delayed rectifying and calcium-activated K+ channels and their significance for action potential repolarization in mouse pancreatic beta-cells. J Gen Physiol. 1990;95:1041–59. doi: 10.1085/jgp.95.6.1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hardy AB, Fox JE, Giglou PR, Wijesekara N, Bhattacharjee A, Sultan S, et al. Characterization of Erg K+ channels in alpha- and beta-cells of mouse and human islets. J Biol Chem. 2009;284:30441–52. doi: 10.1074/jbc.M109.040659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tamarina NA, Wang Y, Mariotto L, Kuznetsov A, Bond C, Adelman J, et al. Small-conductance calcium-activated K+ channels are expressed in pancreatic islets and regulate glucose responses. Diabetes. 2003;52:2000–6. doi: 10.2337/diabetes.52.8.2000. [DOI] [PubMed] [Google Scholar]
- 44.Okamoto K, Iwasaki N, Nishimura C, Doi K, Noiri E, Nakamura S, et al. Identification of KCNJ15 as a susceptibility gene in Asian patients with type 2 diabetes mellitus. Am J Hum Genet. 2010;86:54–64. doi: 10.1016/j.ajhg.2009.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hiriart M, Matteson DR. Na channels and two types of Ca channels in rat pancreatic B cells identified with the reverse hemolytic plaque assay. J Gen Physiol. 1988;91:617–39. doi: 10.1085/jgp.91.5.617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Philipson LH, Kusnetsov A, Larson T, Zeng Y, Westermark G. Human, rodent, and canine pancreatic beta-cells express a sodium channel alpha 1-subunit related to a fetal brain isoform. Diabetes. 1993;42:1372–7. doi: 10.2337/diabetes.42.9.1372. [DOI] [PubMed] [Google Scholar]
- 47.Göpel S, Kanno T, Barg S, Galvanovskis J, Rorsman P. Voltage-gated and resting membrane currents recorded from B-cells in intact mouse pancreatic islets. J Physiol. 1999;521:717–28. doi: 10.1111/j.1469-7793.1999.00717.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Plant TD. Na+ currents in cultured mouse pancreatic B-cells. Pflugers Arch. 1988;411:429–35. doi: 10.1007/BF00587723. [DOI] [PubMed] [Google Scholar]
- 49.Tarvin JT, Pace CS. Glucose-induced electrical activity in the pancreatic beta-cell: effect of veratridine. Am J Physiol. 1981;240:C127–34. doi: 10.1152/ajpcell.1981.240.3.C127. [DOI] [PubMed] [Google Scholar]
- 50.Hille B. Ion channels of exitable cells. Sunderland: Sinauer Associates, Inc 2001. [Google Scholar]
- 51.Sherman A. Contributions of modeling to understanding stimulus-secretion coupling in pancreatic beta-cells. Am J Physiol. 1996;271:E362–72. doi: 10.1152/ajpendo.1996.271.2.E362. [DOI] [PubMed] [Google Scholar]
- 52.Ulbricht W. Effects of veratridine on sodium currents and fluxes. Rev Physiol Biochem Pharmacol. 1998;133:1–54. doi: 10.1007/BFb0000612. [DOI] [PubMed] [Google Scholar]
- 53.Gonçalves AA, Toyama MH, Carneiro EM, Marangoni S, Arantes EC, Giglio JR, et al. Participation of Na(+) channels in the potentiation by Tityus serrulatus alpha-toxin TsTx-V of glucose-induced electrical activity and insulin secretion in rodent islet beta-cells. Toxicon. 2003;41:1039–45. doi: 10.1016/S0041-0101(03)00086-2. [DOI] [PubMed] [Google Scholar]
- 54.Eberhardson M, Grapengiesser E. Role of voltage-dependent Na+ channels for rhythmic Ca2+ signalling in glucose-stimulated mouse pancreatic beta-cells. Cell Signal. 1999;11:343–8. doi: 10.1016/S0898-6568(99)00002-9. [DOI] [PubMed] [Google Scholar]
- 55.Sossalla S, Maier LS. Role of ranolazine in angina, heart failure, arrhythmias, and diabetes. Pharmacol Ther. 2012;133:311–23. doi: 10.1016/j.pharmthera.2011.11.003. [DOI] [PubMed] [Google Scholar]
- 56.Antzelevitch C, Burashnikov A, Sicouri S, Belardinelli L. Electrophysiologic basis for the antiarrhythmic actions of ranolazine. Heart Rhythm. 2011;8:1281–90. doi: 10.1016/j.hrthm.2011.03.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ning Y, Zhen W, Fu Z, Jiang J, Liu D, Belardinelli L, et al. Ranolazine increases β-cell survival and improves glucose homeostasis in low-dose streptozotocin-induced diabetes in mice. J Pharmacol Exp Ther. 2011;337:50–8. doi: 10.1124/jpet.110.176396. [DOI] [PubMed] [Google Scholar]
- 58.Kiss T. Persistent Na-channels: origin and function. A review. Acta Biol Hung. 2008;59(Suppl):1–12. doi: 10.1556/ABiol.59.2008.Suppl.1. [DOI] [PubMed] [Google Scholar]
- 59.Colsoul B, Schraenen A, Lemaire K, Quintens R, Van Lommel L, Segal A, et al. Loss of high-frequency glucose-induced Ca2+ oscillations in pancreatic islets correlates with impaired glucose tolerance in Trpm5-/- mice. Proc Natl Acad Sci U S A. 2010;107:5208–13. doi: 10.1073/pnas.0913107107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Jacobson DA, Philipson LH. TRP channels of the pancreatic beta cell. Handb Exp Pharmacol 2007:409-24. [DOI] [PubMed] [Google Scholar]
- 61.Gilon P, Rorsman P. NALCN: a regulated leak channel. EMBO Rep. 2009;10:963–4. doi: 10.1038/embor.2009.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Swayne LA, Mezghrani A, Varrault A, Chemin J, Bertrand G, Dalle S, et al. The NALCN ion channel is activated by M3 muscarinic receptors in a pancreatic beta-cell line. EMBO Rep. 2009;10:873–80. doi: 10.1038/embor.2009.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Misler S, Dickey A, Barnett DW. Maintenance of stimulus-secretion coupling and single beta-cell function in cryopreserved-thawed human islets of Langerhans. Pflugers Arch. 2005;450:395–404. doi: 10.1007/s00424-005-1401-y. [DOI] [PubMed] [Google Scholar]
- 64.Sher E, Giovannini F, Codignola A, Passafaro M, Giorgi-Rossi P, Volsen S, et al. Voltage-operated calcium channel heterogeneity in pancreatic beta cells: physiopathological implications. J Bioenerg Biomembr. 2003;35:687–96. doi: 10.1023/B:JOBB.0000008032.49504.48. [DOI] [PubMed] [Google Scholar]
- 65.Wang L, Bhattacharjee A, Fu J, Li M. Abnormally expressed low-voltage-activated calcium channels in beta-cells from NOD mice and a related clonal cell line. Diabetes. 1996;45:1678–83. doi: 10.2337/diabetes.45.12.1678. [DOI] [PubMed] [Google Scholar]
- 66.Bhattacharjee A, Whitehurst RM, Jr., Zhang M, Wang L, Li M. T-type calcium channels facilitate insulin secretion by enhancing general excitability in the insulin-secreting beta-cell line, INS-1. Endocrinology. 1997;138:3735–40. doi: 10.1210/en.138.9.3735. [DOI] [PubMed] [Google Scholar]
- 67.Perez-Reyes E. Molecular physiology of low-voltage-activated t-type calcium channels. Physiol Rev. 2003;83:117–61. doi: 10.1152/physrev.00018.2002. [DOI] [PubMed] [Google Scholar]
- 68.Kim YH, Shim YJ, Shin YJ, Sul D, Lee E, Min BH. 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) induces calcium influx through T-type calcium channel and enhances lysosomal exocytosis and insulin secretion in INS-1 cells. Int J Toxicol. 2009;28:151–61. doi: 10.1177/1091581809336885. [DOI] [PubMed] [Google Scholar]
- 69.Catterall WA. Voltage-gated calcium channels. Cold Spring Harb Perspect Biol. 2011;3:a003947. doi: 10.1101/cshperspect.a003947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Fridlyand LE, Tamarina N, Philipson LH. Modeling of Ca2+ flux in pancreatic beta-cells: role of the plasma membrane and intracellular stores. Am J Physiol Endocrinol Metab. 2003;285:E138–54. doi: 10.1152/ajpendo.00194.2002. [DOI] [PubMed] [Google Scholar]
- 71.Lu S, Xie Y, Lin K, Li S, Zhou Y, Ma P, et al. Genome-wide association studies-derived susceptibility loci in type 2 diabetes: confirmation in a Chinese population. Clin Invest Med. 2012;35:E327. doi: 10.25011/cim.v35i5.18706. [DOI] [PubMed] [Google Scholar]
- 72.Unoki H, Takahashi A, Kawaguchi T, Hara K, Horikoshi M, Andersen G, et al. SNPs in KCNQ1 are associated with susceptibility to type 2 diabetes in East Asian and European populations. Nat Genet. 2008;40:1098–102. doi: 10.1038/ng.208. [DOI] [PubMed] [Google Scholar]
- 73.Yasuda K, Miyake K, Horikawa Y, Hara K, Osawa H, Furuta H, et al. Variants in KCNQ1 are associated with susceptibility to type 2 diabetes mellitus. Nat Genet. 2008;40:1092–7. doi: 10.1038/ng.207. [DOI] [PubMed] [Google Scholar]
- 74.Finol-Urdaneta RK, Remedi MS, Raasch W, Becker S, Clark RB, Strüver N, et al. Block of Kv1.7 potassium currents increases glucose-stimulated insulin secretion. EMBO Mol Med. 2012;4:424–34. doi: 10.1002/emmm.201200218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Jacobson DA, Kuznetsov A, Lopez JP, Kash S, Ammälä CE, Philipson LH. Kv2.1 ablation alters glucose-induced islet electrical activity, enhancing insulin secretion. Cell Metab. 2007;6:229–35. doi: 10.1016/j.cmet.2007.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Van Goor F, LeBeau AP, Krsmanovic LZ, Sherman A, Catt KJ, Stojilkovic SS. Amplitude-dependent spike-broadening and enhanced Ca(2+) signaling in GnRH-secreting neurons. Biophys J. 2000;79:1310–23. doi: 10.1016/S0006-3495(00)76384-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Beauvois MC, Merezak C, Jonas JC, Ravier MA, Henquin JC, Gilon P. Glucose-induced mixed [Ca2+]c oscillations in mouse beta-cells are controlled by the membrane potential and the SERCA3 Ca2+-ATPase of the endoplasmic reticulum. Am J Physiol Cell Physiol. 2006;290:C1503–11. doi: 10.1152/ajpcell.00400.2005. [DOI] [PubMed] [Google Scholar]
- 78.Pedersen MG. A biophysical model of electrical activity in human β-cells. Biophys J. 2010;99:3200–7. doi: 10.1016/j.bpj.2010.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Cha CY, Nakamura Y, Himeno Y, Wang J, Fujimoto S, Inagaki N, et al. Ionic mechanisms and Ca2+ dynamics underlying the glucose response of pancreatic β cells: a simulation study. J Gen Physiol. 2011;138:21–37. doi: 10.1085/jgp.201110611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Quesada I, Todorova MG, Alonso-Magdalena P, Beltrá M, Carneiro EM, Martin F, et al. Glucose induces opposite intracellular Ca2+ concentration oscillatory patterns in identified alpha- and beta-cells within intact human islets of Langerhans. Diabetes. 2006;55:2463–9. doi: 10.2337/db06-0272. [DOI] [PubMed] [Google Scholar]
- 81.Göpel SO, Kanno T, Barg S, Eliasson L, Galvanovskis J, Renström E, et al. Activation of Ca(2+)-dependent K(+) channels contributes to rhythmic firing of action potentials in mouse pancreatic beta cells. J Gen Physiol. 1999;114:759–70. doi: 10.1085/jgp.114.6.759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Bertram R, Sherman A, Satin LS. Electrical bursting, calcium oscillations, and synchronization of pancreatic islets. Adv Exp Med Biol. 2010;654:261–79. doi: 10.1007/978-90-481-3271-3_12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Chay TR, Keizer J. Minimal model for membrane oscillations in the pancreatic beta-cell. Biophys J. 1983;42:181–90. doi: 10.1016/S0006-3495(83)84384-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Keizer J, Magnus G. ATP-sensitive potassium channel and bursting in the pancreatic beta cell. A theoretical study. Biophys J. 1989;56:229–42. doi: 10.1016/S0006-3495(89)82669-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Pedersen MG, Cortese G, Eliasson L. Mathematical modeling and statistical analysis of calcium-regulated insulin granule exocytosis in β-cells from mice and humans. Prog Biophys Mol Biol. 2011;107:257–64. doi: 10.1016/j.pbiomolbio.2011.07.012. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.