

Abstract
β-cell apoptosis is a significant contributor to β-cell dysfunction in diabetes and ER stress is among the factors that contributes to β-cell death. We previously identified that the Ca2+-independent phospholipase A2β (iPLA2β), which in islets is localized in β-cells, participates in ER stress-induced β-cell apoptosis. Here, direct assessment of iPLA2β role was made using β-cell-specific iPLA2β overexpressing (RIP-iPLA2β-Tg) and globally iPLA2β-deficient (iPLA2β-KO) mice. Islets from Tg, but not KO, express higher islet iPLA2β and neutral sphingomyelinase, decrease in sphingomyelins, and increase in ceramides, relative to WT group. ER stress induces iPLA2β, ER stress factors, loss of mitochondrial membrane potential (∆Ψ), caspase-3 activation, and β-cell apoptosis in the WT and these are all amplified in the Tg group. Surprisingly, β-cells apoptosis while reduced in the KO is higher than in the WT group. This, however, was not accompanied by greater caspase-3 activation but with larger loss of ∆Ψ, suggesting that iPLA2β deficiency impacts mitochondrial membrane integrity and causes apoptosis by a caspase-independent manner. Further, autophagy, as reflected by LC3-II accumulation, is increased in Tg and decreased in KO, relative to WT. Our findings suggest that (1) iPLA2β impacts upstream (UPR) and downstream (ceramide generation and mitochondrial) pathways in β-cells and (2) both over- or under-expression of iPLA2β is deleterious to the β-cells. Further, we present for the first time evidence for potential regulation of autophagy by iPLA2β in islet β-cells. These findings support the hypothesis that iPLA2β induction under stress, as in diabetes, is a key component to amplifying β-cell death processes.
Keywords: RIP-iPLA2β-Tg, apoptosis, autophagy, caspase-3, ceramides, iPLA2β-KO, iPLA2β, mitochondrial membrane potential, β-cell
Introduction
Both types 1 and 2 diabetes mellitus (T1DM and T2DM) are associated with β-cell dysfunction as a consequence of β-cell death due to apoptosis.1-3 Apoptosis can occur not only via extrinsic death receptor or intrinsic mitochondrial pathway but also due to ER stress.4 The ER, in addition to serving as a cellular Ca2+ store, is the site where secretory proteins are synthesized, assembled, folded, and post-translationally modified. Interruption of any of these functions can lead to production of malfolded proteins and their accumulation in the ER. When an imbalance between the load of client proteins on the ER and the ER’s ability to process the load occurs, it results in ER stress and the Unfolded Protein Response (UPR).5 Prolonged ER stress, however, can trigger apoptotic factors in the ER,6 activate the intrinsic apoptotic pathway,7 lead to downstream activation of caspase-3, a protease that is central to the execution of apoptosis,8 and induce apoptotic cell death.
Being a site for Ca2+ storage, the ER responds to various stimuli to release Ca2+ and is therefore extremely sensitive to changes in cellular homeostasis. As a secretory organelle, the β-cells are endowed with highly developed ER and therefore have a greater susceptibility to developing ER stress and several experimental (PERK−/−, Akita, and NOD.k iHEL) and clinical (Wolcott-Rallison and Wolfram syndromes) reports have linked ER stress-induced β-cell apoptosis to the development of diabetes mellitus (reviewed in ref. 9). However, underlying cellular mechanisms that contribute to this process remain to be elucidated.
Ongoing studies in our laboratory suggest that the Ca2+-independent phospholipase A2β (iPLA2β) participates in ER stress-induced β-cell apoptosis. The iPLA2β belongs to the family of PLA2s that hydrolyse the sn-2 substituent of membrane phospholipids to release a free fatty acid and a lysophospholipid.10 Since its initial descriptions in the heart and pancreatic islet β cells,11,12 iPLA2β activation is now thought to contribute to various biological processes in the CNS, skeletal muscle, bone, eye and vascular smooth muscle. Among the many described roles for iPLA2β include its involvement in lipid remodeling, cell proliferation and signal transduction (reviewed in refs. 9 and 13). An additional role, first described in human leukemic monocyte lymphoma U937 cells14 and subsequently in other non-β-cells, is its participation in apoptosis (reviewed in ref. 9).
A potential role for iPLA2β in β-cell apoptosis was initially gleaned from observations reported by the Polonsky group.15 They noted that decreases in MIN6 insulinoma cell survival following exposure to various chemical ER stressors were Ca2+-independent and due to generation of a bioactive metabolite of arachidonic acid. Subsequently, Kudo’s group reported that inhibition of iPLA2β, but not of cPLA2 or sPLA2, prevented Fas-stimulated death of human leukemic U937 cells.14 Our studies revealed that thapsigargin, which depletes ER Ca2+ stores by inhibiting sarco-endoplasmic reticulum Ca2+-ATPase (SERCA) Ca2+ pumps causing ER stress, promotes hydrolysis of arachidonic acid from pancreatic islet membrane phospholipids.16 β-Cell membranes are enriched in arachidonic-containing phospholipids,17 and accordingly, thapsigargin-induced accumulation in arachidonic acid was suppressed by a bromoenol lactone (BEL) suicide substrate inhibitor of the iPLA2β.16 These observations raised the possibility that iPLA2β is activated during ER stress in β-cells.
Subsequent studies in our laboratory demonstrated that iPLA2β participates in ER stress-induced β-cell apoptosis and that this process involves iPLA2β-mediated increase in neutral sphingomyelinase-catalyzed hydrolysis of sphingomyelins leading to accumulation of ceramides and mitochondrial decompensation. These observations raised the possibility that modulating the activity of iPLA2β can be beneficial to β-cell survival and presumably offer a means to ameliorate β-cell apoptosis associated with diabetes mellitus. However, while these observations strengthened a link between iPLA2β and β-cell apoptosis, they are limited in that they were demonstrated in insulinoma cells and relied on utilization of BEL to discern the involvement of iPLA2β. While BEL, at concentrations that do not impact cPLA2 or sPLA2 activities, is a recognized potent inhibitor of iPLA2, it has been reported to also have inhibitory effects on non-iPLA2 enzymes (reviewed in ref. 9).
In the present study, we therefore used mice in which the rat insulin 1 promoter (RIP) was used to overexpress iPLA2β specifically in the β-cells (RIP-iPLA2β-Tg)18 and global iPLA2β-null (iPLA2β-KO) mice19 to more directly assess the role of iPLA2β in β-cell apoptosis. Our findings reveal that altered expression of iPLA2β impacts β-cell survival and also provide the first evidence for iPLA2β-mediated regulation of autophagy in the β-cells.
Results
Genotyping
Breeding pairs obtained from Dr. John Turk (WUSM in St. Louis, MO) were used to generate RIP-iPLA2β-transgenic (Tg), iPLA2β-deficient (KO) and age-matched littermate wild-type (WT) colonies at UAB in Birmingham, AL, and their genotype was identified by PCR analyses, as detailed.18,19 The expected band sizes for WT and RIP-iPLA2β-Tg are a single band at 450 bp and 200 bp, respectively. The expected band sizes for iPLA2β+/+ (WT), iPLA2β-/+ (Het) and iPLA2β−/− (KO) are a single band at 1400 bp, two bands at 1400 bp and 400 bp, and a single band at 400 bp, respectively. As shown in Figure 1, our breeding protocol generated progeny that could be identified as WT or RIP-iPLA2β-Tg and WT, Het, or iPLA2β-KO mice.
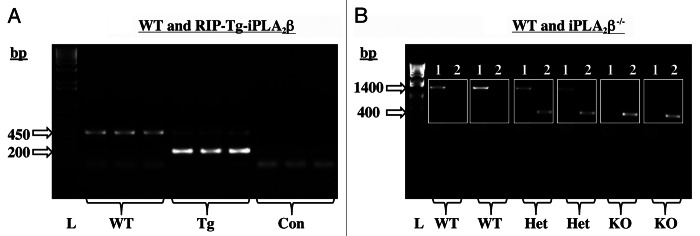
Figure 1. Mouse genotyping by PCR analyses. DNA was generated from tail clips and progeny were genotyped by PCR analyses. (A) WT and RIP-Tg-iPLA2β. Reactions were performed in the presence of two sets of primers and the expected bands for WT (450 bp) and RIP-Tg-iPLA2β (200 bp) in three mice each are presented (L, bp ladder and Con, control reactions without template). (B) WT and iPLA2β−/−. Reactions were performed in the presence of primers for the WT sequence (Set 1, lanes 1) or for the disrupted KO sequence (Set 2, lanes 2) for each mouse. The expected bands for WT (1400 bp), Hets (1400 and 200 bp) and KO (400 bp) in two mice each are presented (L, bp ladder).
Verification of RIP-iPLA2β-Tg and iPLA2β-KO models
To validate the genotyping results, iPLA2β expression in the progeny was assessed by iPLA2β message, activity, and protein expression analyses (Fig. 2). As seen in Figure 2A, iPLA2β mRNA is greater in the Tg islets and undetected in the KO islets, relative to corresponding WT islets. Enzymatic assays (Fig. 2B) revealed nearly 30-fold increase in catalytic activity in the Tg islets and a 50% decrease in KO islets, as compared with corresponding WT islets. Addition of ATP increased activity similarly in the WT and Tg groups but not in the KO group, relative to activity measured in the absence of ATP (Fold increase +ATP/−ATP: WT-Tg, 2.5 ± 0.6; Tg 2.1 ± 0.03; WT-KO, 3 ± 1.7; KO, 1.1 ± 0.5). Because ATP stimulation of activity is characteristic of iPLA2β, these findings suggest that the PLA2 activity in the WT and Tg groups is manifested by iPLA2β and that the low (near background) level of activity measured in the KO group is not. Immunofluorescence analyses in islet sections (Fig. 2C) confirmed higher iPLA2β expression in the Tg (Fig. 2C, left panels) and its absence in the KO (Fig. 2C, right panels) group, relative to WT groups. Further, the merging of iPLA2β fluorescence with insulin-containing cells confirms that the iPLA2β expression is localized within β-cells of pancreatic islets. Taken together, these findings confirm that iPLA2β expression is increased in islet β-cells from the RIP-iPLA2β-Tg-designated mice and is absent in the iPLA2β-KO-designated mice, relative to their corresponding age-matched WT littermates, and that they can be used to study the impact of differential iPLA2β expression on ER stress-induced apoptosis pathway in the β-cell.

Figure 2. Verification of RIP-iPLA2β-Tg and iPLA2β-KO models. Pancreatic islets were isolated from iPLA2β+/+ (WT), RIP-iPLA2β-transgenic (Tg) and iPLA2β-deficient (KO) mice and iPLA2β expression was assessed by the following methods: (A) PCR. Total islet RNA was prepared and cDNA generated for iPLA2β message analyses. (B). Enzymatic activity. Islet cytosol was prepared and iPLA2β enzymatic activity was assayed (n = 4–6) in 30 μg protein aliquots using [14C]-labeled PAPC substrate. (†Significantly different from corresponding WT group, p < 0.005 and *+ATP group significantly different from -ATP group, p < 0.05). (C) Immunofluorescence. Representative images of paraffin islet sections (8–10 μm) and co-stained for nuclei (DAPI), insulin (I) and iPLA2β (P).
Induction of ER stress-related factors
To assess susceptibility to ER stress, islets from WT, RIP-iPLA2β-Tg, and iPLA2β-KO mice were treated with thapsigargin for up to 48 h. Lysates were then prepared and resolved by SDS-PAGE to determine induction of ER stress factors CHOP, pPERK and GRP78. As seen in Figures 3A and 3B, all three factors increased within 24 h in the WT groups. In the RIP-iPLA2β-Tg group, the increases were amplified at both 24 and 48 h, relative to the corresponding WT group. In contrast, while increased expression of the factors was evident in the iPLA2β-KO group, it was either similar (CHOP) or lower (GRP78) than that observed in the corresponding WT group. However, pPERK while similar at 24 h, was still expressed at 48 h in the KO but not in the WT. These findings suggest that the RIP-iPLA2β-Tg islets have greater susceptible to developing ER stress than the iPLA2β-KO islets.

Figure 3. Thapsigargin-induced ER stress factors in WT, RIP-iPLA2β-Tg and iPLA2β-KO islets. Islets (300/condition) from WT, iPLA2β-Tg, and iPLA2β-KO mice were cultured O/N at 37°C under an atmosphere of 5%CO2/95% air and then treated with either vehicle (Con, DMSO) or with thapsigargin (T, 2 μM) for up to 48 h. Islet lysates were then prepared, resolved by SDS-PAGE, protein transferred to immunoblots and probed for ER stress factors. Immunoreactive bands were visualized by enhanced chemiluminescence. Each analysis was done a minimum of 3 times. (A) Immunoblotting analyses. Expression of CHOP, pPERK and GRP78 was determined using 30 μg protein aliquots. (B) Representative densitometry analyses. The ratios of the band intensity of protein of interest relative to loading control actin are presented. (*Significantly different from WT group, p < 0.05, n = 3/target.)
ER stress-induced activation of caspase-3
To further ascertain the impact of iPLA2β expression on β-cell apoptosis, cleavage (i.e., activation) of caspase-3 in islets was assessed in islets following treatment with thapsigargin. Immunoblotting analyses (Fig. 4A) revealed an increase in total caspase-3 in the WT and RIP-iPLA2β-Tg groups. In the Tg group, this was accompanied by an increase in cleaved caspase-3 (aC3) by 24 h. In contrast, total caspase-3 was unchanged and cleaved caspase-3 was undetectable in the KO group. Because immunoblotting was not sufficiently sensitive to reveal changes in cleaved caspase-3 in WT and KO groups, we performed two additional analyses. Immunofluorescence protocol allowed us to detect aC3 fluorescence in WT and KO islet sections but it was barely above background (Fig. 4B). In contrast, aC3 fluorescence was pronounced in the Tg islet and was found to be co-localized with insulin (I)-containing cells, as reflected by visualization of the merged yellow fluorescence. These findings suggest that ER stress promotes cleavage of caspase-3 and that this is amplified in RIP-iPLA2β-Tg islets and blunted in iPLA2β-deficient islets. Further evidence for this is provided in Figure 4C, which reveals amplified increase in aC3 activity in the RIP-iPLA2β-Tg islets, relative to WT and KO groups.

Figure 4. ER stress-induced activation of caspase-3 in WT, RIP-iPLA2β-Tg and iPLA2β-KO islets. Islets (200/condition) from WT, iPLA2β-Tg and iPLA2β-KO mice were cultured O/N at 37°C under an atmosphere of 5%CO2/95% air and then treated with either vehicle (DMSO) or with thapsigargin (Thaps, 2 μM) for up to 48 h and assessed for expression of cleaved (i.e., activated) caspase-3. (A) Immunoblotting analyses. Total and cleaved caspase-3 (ac3) expressions were determined using 30 μg aliquots of islet lysate. (B) aC3 Immunofluorescence. Representative paraffin islet sections (8–10 μm) co-stained for nuclei (DAPI), insulin (I) and aC3. (C) aC3 activity assay. Islet lysates were prepared and activity in 30 μg aliquot of protein was assayed using a colorimetric-based protocol. (*Significantly different from WT group, p < 0.05.)
Basal neutral sphingomyelinase (NSMase)
Earlier studies indicated that ceramides generated via NSMase-catalyzed hydrolysis of sphingomyelins was mediated by iPLA2β.20 We therefore hypothesized that islet NSMase expression is affected by iPLA2β expression levels. Total RNA was isolated and cDNA generated from islets from WT, RIP-iPLA2β-Tg and iPLA2β-KO mice and NSMase message expression was analyzed by qRT-PCR. As seen in Figure 5, NSMase mRNA was increased nearly 2-fold in the RIP-iPLA2β-Tg and significantly reduced in the iPLA2β-KO islets, relative to corresponding WT groups. These findings strengthen the notion that NSMase expression in pancreatic islets can be regulated by iPLA2β.
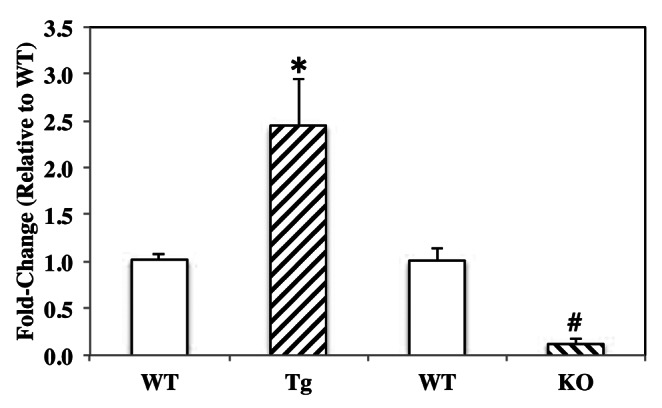
Figure 5. Basal neutral sphingomyelinase (NSMase) expression in WT, RIP-iPLA2β-Tg and iPLA2β-KO islets. Total RNA was isolated from islets from the three groups and cDNA generated for qRT-PCR analyses of NSMase message expression. (*#Significantly different from corresponding WT group, p < 0.05 and p < 0.005, respectively; n = 3).
Ceramide and sphingomyelin analyses by mass spectrometry
To corroborate findings in Figure 5, ESI/MS/MS analyses were used to determine abundances of ceramide and sphingomyelin molecular species in islets, as previously described.20-23 To compare the different genotypes, the abundance of each species relative to internal standard was determined and normalized to total phosphate. As observed in insulinoma cells20-23 and human islets,24 the fatty amide substituents of the major ceramide species (Fig. 6A) endogenous to rodent islets were found to be 16:0 (m/z 544), 18:0 (m/z 572), 20:0 (m/z 600), 22:0 (m/z 628), 24:1 (m/z 654) and 24:0 (m/z 656), and the major sphingomyelin species (Fig. 6C) endogenous to islets were found to be 16:0 (m/z 709), 18:0 (m/z 737), 22:0 (m/z 693), 24:1 (m/z 819) and 24:0 (m/z 821). Comparison of basal ceramide (Fig. 6B) and sphingomyelin (Fig. 6D) pools in islets revealed similar abundance of both in the KO group, whereas ceramides were increased nearly 3-fold and sphingomyelins decreased ca. 40% in the RIP-iPLA2β-Tg group, relative to corresponding WT groups. Following exposure of WT islets to thapsigargin, the pool of ceramides increased (180 ± 12%) and of sphingomyelins decreased (12 ± 11%), relative to vehicle-treated WT group. Treatment of RIP-iPLA2β-Tg group caused a further increase in ceramides (245 ± 30%) and decrease in sphingomyelins (42 ± 5%), relative to the corresponding pools in WT treated islets. In contrast, in the KO treated group the pool of ceramides was 109 ± 17% and of sphingomyelins 88 ± 14%, relative to corresponding pools in WT treated islets. These findings are consistent with iPLA2β-mediated accumulation of ceramides, in part, via hydrolysis of sphingomyelins.

Figure 6. Ceramide and sphingomyelin analyses by mass spectrometry. Islets were cultured O/N at 37°C under an atmosphere of 5%CO2/95% air and then prepared for ESI/MS/MS analyses. (A and C) Basal ceramide and sphingomyelin molecular species. Representative spectra obtained from WT mice illustrating molecular species of ceramides (A) and sphingomyelins (C) endogenous to islets. The labeled ions represent endogenous molecular species of ceramides and sphingomyelins. (B and D) Comparison of basal ceramide (CM) and sphingomyelin (SM) pools in RIP-iPLA2β-Tg and iPLA2β-KO islets. Abundance of each CM and SM molecular species, relative to internal standard was determined, normalized to total phosphate content, and the pool of ceramides and sphingomyelins in each group is presented as mean ± SEM (n = 3–5). (*Significantly different from WT, p < 0.050)
ER stress-induced islet cell apoptosis
We next determined the impact of differential expression of iPLA2β on ER stress-induced islet cell apoptosis. Following treatment of islets from WT, RIP-iPLA2β-Tg and iPLA2β-KO mice with thapsigargin, TUNEL analysis was used to visualize cells undergoing apoptosis. As seen in Figure 7A, vehicle treatment had minimal effect in all groups but TUNEL positivity increased in the islets following induction of ER stress. To facilitate quantitation of apoptotic cell number, the islets were dispersed and TUNEL fluorescence was quantitated by flow cytometry (Fig. 7B). Basal incidence of apoptosis was found to be similar among the groups. Following induction of ER stress, apoptosis was unchanged at 24 h but increased 3-fold at 48 h in both WT groups. In comparison, the fold increases were 5-fold and 22-fold greater at 24 h and 48 h, respectively, in the RIP-iPLA2β-Tg group. This reflected a nearly 5-fold higher incidence of apoptosis in the RIP-iPLA2β-Tg, relative to corresponding WT group. Further, ER stress induced iPLA2β protein in both the WT and RIP-iPLA2β-Tg islets (Fig. 7B, inset), with the increase occurring in the RIP-iPLA2β-Tg islets earlier than in the WT islets. In contrast, the fold increases were 3-fold and 11-fold greater at 24 h and 48 h, respectively, in the iPLA2β-KO group. Thus, while apoptosis was increased in the iPLA2β-KO, relative to WT group, it was 50% lower than in the RIP-iPLA2β-Tg group. These findings suggest that induction of ER stress in islets with thapsigargin promotes iPLA2β expression and apoptosis and that these effects are amplified in RIP-iPLA2β-Tg islets. In contrast, the islets deficient in iPLA2β exhibit a reduced susceptibility to ER stress-induced apoptosis.
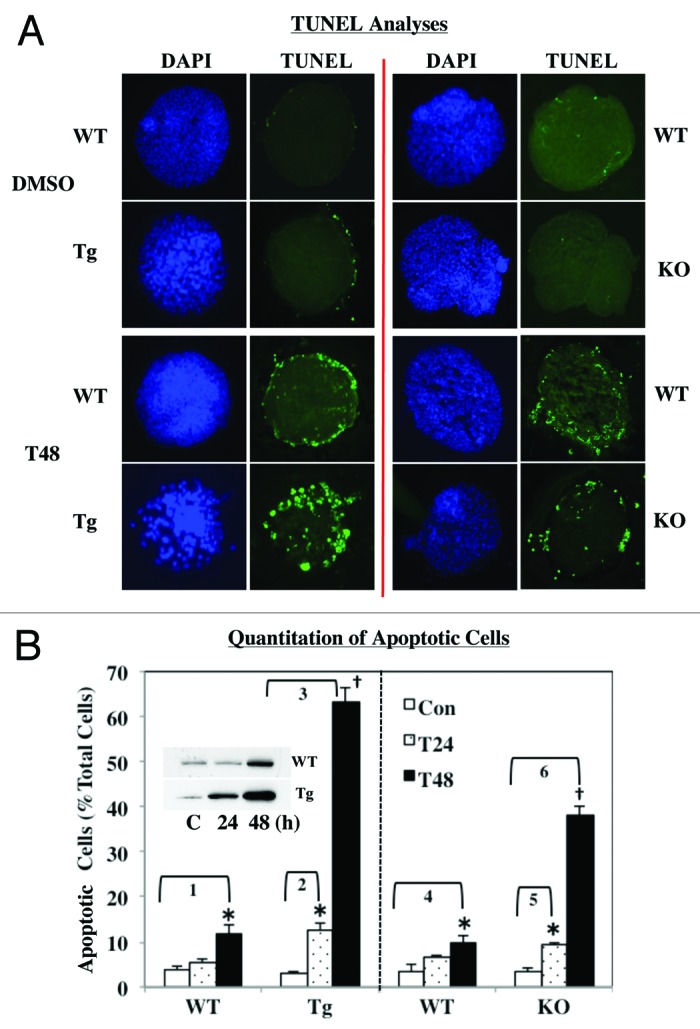
Figure 7. ER stress-induced apoptosis in WT, RIP-iPLA2β-Tg and iPLA2β-KO islets. Islets (200/condition) from WT, iPLA2β-Tg and iPLA2β-KO mice were cultured O/N at 37°C under an atmosphere of 5%CO2/95% air and then treated with either vehicle (Con, DMSO) or with thapsigargin (T, 2 μM) for up to 48 h. (A) TUNEL analyses. Intact islets were processed for TUNEL analyses to visualize cells undergoing apoptosis, as reflected by punctate green fluorescence. (B) Quantitation of apoptotic cell number. To assess the incidence of apoptosis, islets were dispersed into individual cells and flow cytometry protocol was used to determine the number of apoptotic cells, relative to total cell number in each preparation. The means ± SEM of the percentage of apoptotic cells in each group are presented. (*Significantly different from corresponding WT group and †significantly different from corresponding T24 and WT groups, p < 0.05, n = 3–5). Fold increases between groups are as follows: 1, 3.09 ± 0.50; 2, 4.50 ± 0.54; 3, 22.24 ± 1.18; 4, 2.73 ± 0.52; 5, 2.70 ± 0.19; and 6, 10.96 ± 0.64). Inset. Induction of iPLA2β during ER stress. Cytosol (30 μg WT and 10 μg iPLA2β-Tg) from thapsigargin-treated islets was resolved by SDS-PAGE, protein transferred to immunoblots and probed for iPLA2β.
ER stress-induced loss in mitochondrial membrane potential (∆Ψ) in islet β-cells
Loss of ∆Ψ is a hallmark of cellular apoptosis and ceramides are reported to participate in this process.21,22 We therefore assessed ∆Ψ in WT, RIP-iPLA2β-Tg and iPLA2β-KO groups following vehicle (DMSO) or thapsigargin treatment. To ensure that we were examining ∆Ψ only in β-cells, the islets were dispersed and FACS-purified to obtain a homogenous population of β-cells. The β-cells were then incubated with DiOC(6)3, a fluorochrome that accumulates in the mitochondria of healthy cells but is not retained in the mitochondria of compromised cells. As evidenced in Figure 8, the green DiOC(6)3, fluorescence is diminished in the WT β-cells following thapsigargin treatment, reflecting ∆Ψ loss in β-cell. In comparison, the loss in fluorescence is more dramatic in β-cells from the RIP-iPLA2β-Tg, indicative of significant loss in ∆Ψ in the RIP-iPLA2β-Tg, relative to WT group. Interestingly, the mitochondrial-associated fluorescence was also reduced in β-cells from the iPLA2β-KO, relative to corresponding WT group, suggesting that the loss of ∆Ψ in the iPLA2β-KO is greater than in the WT group. These findings suggest that iPLA2β expression (up or down) can impact islet β-cell mitochondrial integrity.
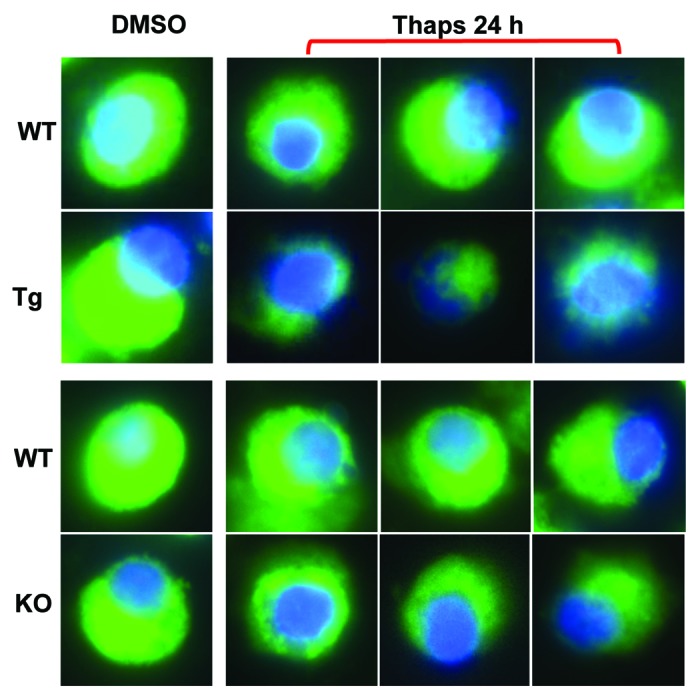
Figure 8. ER stress-induced loss in mitochondrial membrane potential (∆Ψ) in WT, RIP-iPLA2β-Tg and iPLA2β-KO β-cells. Islets were cultured O/N at 37°C under an atmosphere of 5%CO2/95% air and then treated with either vehicle (DMSO, control) or with thapsigargin (T, 2 μM) for 24 h. The islets were then dispersed and a FACS protocol was used to obtain a highly homogenous population of β-cells. The β-cells were loaded with blue nuclear (Hoechst) and green mitochondrial [DiOC(6)3] stains and examined by confocal microscopy. Merged images are shown in the panels.
Participation of iPLA2β in β-cell autophagy
Autophagy is an ongoing pro-survival process but under increased stress, dysregulation of autophagy can lead to cell death.25 As an initial response to ER stress, autophagy is activated to increase cell survival, however, prolonged stress can activate apoptotic processes and cause cell death.26 To determine if an autophagic response to ER stress is influenced by iPLA2β expression, autophagy was assessed in islets by monitoring conversion of LC3-I to LC3-II. Increased appearance of LC3-II is reflective of activation of the autophagic response in cells. As seen in Figure 9A and B, LC3-II is increased similarly in the WT groups. However, generation of LC3-II is markedly higher in the RIP-iPLA2β-Tg, relative to corresponding WT group (Fig. 9A). In contrast, the generation of LC3-II is reduced in the iPLA2β-KO, in comparison with corresponding WT group (Fig. 9B). This is reflected in the bar graphs, which illustrate higher LC3-II to loading control ratio in the RIP-iPLA2β-Tg and reduced ratio in the iPLA2β-KO, relative to corresponding WT groups.

Figure 9. ER stress-induced autophagy in WT, RIP-iPLA2β-Tg and iPLA2β-KO islets. Islets (500/condition) from WT, RIP-iPLA2β-Tg and iPLA2β-KO mice were cultured O/N at 37°C under an atmosphere of 5%CO2/95% air and then treated with either vehicle Control (D, DMSO) or with thapsigargin (T, 2 μM) for 24 or 48 h. (A and B) ER stress-induced autophagy. (C and D) Autophagy induction due to ER stress. The islets were co-treated with thapsigargin and PIs (200 μM leupeptin and 20 mM ammonium chloride) to inhibit autolysosome activation. In some experiments the islets were pretreated with GW4869 (G, 20 μM) for 30 min prior to thapsigargin addition. Ratios of LC3-II to loading control actin in WT and RIP-iPLA2β-Tg islets (A and C) and in WT and iPLA2β-KO islets (B and D), and representative insets of LC3 immunoblots are presented. (*Significantly different from WT, p < 0.05, n = 3–4/group.)
To determine whether the ER stress-induced autophagosome (LC3-II) accumulation is due to autophagy induction or to inhibition of downstream steps, a turnover assay was utilized. This assay is based on the observation that LC3-II is de-lipidated in the lysosome and inhibition of lysosomal activity allows for assessment of induction of autophagy.27 The islets were treated with protease inhibitors (PIs) ammonium chloride, which prevents lysosomal acidification, and leupeptin, which prevents lysosomal hydrolase activities.28 Flux was then determined by comparing the ratio of LC3-II (normalized to loading control) between control and treatment conditions. The fold-change in the ratio is expected to remain unchanged when autophagy initiation is unaffected by the experimental condition, whereas, the fold-change will increase when initiation is enhanced or decreased when initiation is inhibited.28 We find that addition of the PIs caused a modest decrease (Fig. 9C) or no change (Fig. 9D) in induction of autophagy in the WT groups and that induction is not significantly altered in either the RIP-iPLA2β-Tg (Fig. 9C) or iPLA2β-KO (Fig. 9D) groups, as compared with corresponding WT group. Because ceramides are recognized to participate in autophagy,29 islets were treated with the NSMase inhibitor GW4869 and while induction of autophagy was not altered in the WT group, it was modestly reduced in the RIP-iPLA2β-Tg group (Fig. 9C). These findings suggest that iPLA2β participates in β-cell autophagy and raises the possibility that it impacts the autophagy process beyond the initiation step, in part, via activation of NSMase.
Discussion
Emerging evidence suggests β-cell apoptosis is a prominent contributor to β-cell death in T1DM and T2DM3,30,31 making it important to understand the mechanisms underlying β-cell apoptosis if diabetes mellitus is to be prevented or delayed. Our studies with INS-1 cells,20,22,23 the Akita spontaneous ER stress model,21 and human pancreatic islet β-cells24 reveal that the Ca2+-independent phospholipase A2β (iPLA2β) participates in ER stress-induced β-cell apoptosis.9,22 These studies, however, are limited by several issues: (1) though insulinoma cell lines have contributed significantly to our understanding of β-cell biology, their functional properties are not identical to native pancreatic islet β-cells; (2) variability in human islet preparations, due to the nature of donor demise, isolation protocol at different procurement centers, and trauma related to shipment; and (3) reliance on BEL, a potent irreversible chemical inhibitor of iPLA2β but also noted for its inhibition of non-iPLA2β enzymes (reviewed in ref. 9), to discern the role of iPLA2β in the apoptotic process. Though knockdown of iPLA2β and NSMase in the insulinoma cells using siRNA protocols are supportive, they still leave open the question of whether iPLA2β expression in islet β-cells impacts their survival.
We therefore set out to assess the role of iPLA2β in ER stress-induced β-cell apoptosis by comparing responses in RIP-iPLA2β-Tg and iPLA2β-KO mouse islets. The RIP-iPLA2β-Tg mouse is a particularly valuable model because iPLA2β overexpression is restricted to only the islet β-cells. Our findings reveal that compared with the WT group, islet β-cells from RIP-iPLA2β-Tg mice express higher iPLA2β as expected, and exhibit increased susceptibility to outcomes due to thapsigargin-induced ER stress. In support of this, expression of ER stress factors (CHOP, GRP78 and pPERK) in the islets, and cleavage (i.e., activation) of caspase-3 and loss of membrane potential in the islet β-cells were all amplified in the RIP-iPLA2β-Tg group, when compared with age-matched WT littermates. As a consequence, ER stress-induced apoptosis of the β-cells was nearly 5-fold higher in the RIP-iPLA2β-Tg group than in the WT group.
As we previously linked iPLA2β-mediated ceramide generation via hydrolyses of sphingomyelins during ER stress with insulinoma cell apoptosis,20 we considered the possibility of this mechanism contributing to islet β-cell apoptosis observed in the present study. Surprisingly, basal abundance of ceramides was found to be increased and of sphingomyelins decreased in the RIP-iPLA2β-Tg, as compared with age-matched WT littermates. Consistent with this finding was an increase in basal NSMase message expression in the RIP-iPLA2β-Tg group. Following induction of stress with thapsigargin, the pool of ceramides was consistently higher (> 2-fold) and of sphingomyelin reduced (nearly 60%) in the RIP-iPLA2β-Tg group, relative to WT islets. However, these changes did not achieve statistical significance. In addition to day-to day experimental variability it is likely that the basal changes in the RIP-iPLA2β-Tg may have precluded further increases in ceramides and decreases in sphingomyelins. Alternatively, and a more likely possibility is that because the apoptotic process is amplified and occurs earlier in the RIP-iPLA2β-Tg islets, we were not able to capture the optimum time for analyses in the RIP-iPLA2β-Tg islets. Nevertheless, these findings suggest that increases in iPLA2β can lead to greater ceramide generation via sphingomyelin hydrolysis in islet β-cells.
It might be argued that if iPLA2β participates in the apoptosis process, a much more severe destruction of β-cell apoptosis should be evident under basal conditions as well. However, we did not find this to be the case as there were relatively small or no differences in outcomes (ER stress factors expression, mitochondrial membrane potential, activation of caspase-3, islet cell TUNEL positivity) measured in the WT and RIP-iPLA2β-Tg islets under basal conditions. The lack of basal changes in the presence of increased iPLA2β expression in β-cells is not entirely unexpected because with the exception of increased insulin secretion, neither profound morphological nor functional changes in the islet were reported in the initial characterization if the RIP-iPLA2β-Tg model.18 As noted in another model in which islet β-cell protein expression is modified,32 the β-cell appears to be resistant under basal conditions and only when stressed is the impact of the modification unmasked.
We also report for the first time induction of iPLA2β expression in a non-proliferative and native system (i.e., islet) during a biological process (i.e., ER stress). Intriguingly, iPLA2β is also induced in the RIP-iPLA2β-Tg islet during ER stress. These findings raise the possibility that in the presence of a stress stimulus, not only are processes to increase iPLA2β expression triggered, but that the potency of iPLA2β becomes unmasked. This may occur via manifestation of higher specific iPLA2β catalytic activity due to caspase-3 catalyzed cleavage of iPLA2β to a truncated and more active form14,23 or potential sequestration of iPLA2β in organelles that are sensitive to the stimulus.14,20,22,23 The present findings also suggest the potential importance of the induced iPLA2β in participating in the β-cell apoptotic process.
In contrast to the RIP-iPLA2β-Tg group, β-cells from the iPLA2β-KO mice are deficient in iPLA2β but as observed in the RIP-iPLA2β-Tg group, no significant basal abnormalities in the measured parameters are evident. While all outcomes in response to ER stress are significantly attenuated in the iPLA2β-deficient group, relative to the RIP-iPLA2β-Tg group, they varied in comparison with their age-matched WT group. For instance, with respect to ER stress factors there is no difference in CHOP expression, GRP78 is reduced, and increased pPERK expression is maintained at 48 h; activation of caspase-3 and ceramide and sphingomyelin pools (basal and following thapsigargin treatment) are similar; and basal NSMase is reduced. In spite of the apparent lack of differences between WT and iPLA2β-KO in various parameters, incidence of β-cell apoptosis in the iPLA2β-KO, though significantly lower than in the RIP-iPLA2β-Tg group, was higher than in age-matched WT group. It must be recognized that ER stress alone can induce β-cell apoptosis and the increases in ER stress factors and caspase-3 cleavage in the iPLA2β-deficient are evidence of that.
A question that arises therefore is what promotes the higher incidence of apoptosis in the iPLA2β-deficient group. This is even more paradoxical in view of our earlier demonstrations that chemical inhibition20,22,23 or siRNA-mediated knockdown (KD)21 of iPLA2β significantly attenuated insulinoma cell apoptosis. A salient difference between those and present studies is the duration of absence of an active iPLA2β enzyme. Both BEL inhibition (30 min prior to thapsigargin exposure) and siRNA-KD (2 d before thapsigargin exposure) of iPLA2β were acute protocols, in comparison with iPLA2β-deficiency for 3–4 mo in mice used in the present study. A potential explanation for the increased apoptosis in the iPLA2β-deficient group might be deduced from studies by Ma’s group. They reported that mitochondrial abnormalities and subsequent apoptosis promoted by staurosporine, which triggers the intrinsic mitochondrial apoptotic pathway,33 are prevented by expression of iPLA2β.34 They rationalized that this was due to availability of substrates normally provided by iPLA2β to facilitate repair of membrane phospholipids, in particular cardiolipins, which are susceptible to damage by ROS-mediated peroxidation. In support of this possibility, they found evidence of increased sensitivity of islets from iPLA2β-deficient mice to staurosporine-induced mitochondrial abnormalities and apoptosis.35 Consistent with their findings, in the present study we observed that islet β-cells from iPLA2β-deficient mice exhibit a greater loss in mitochondrial membrane potential than β-cells from age-matched WT littermates. However, as noted earlier the apoptosis was significantly greater than in the WT group, in spite of similar induction of caspase-3 cleavage. This raises the additional possibility that iPLA2β-deficiency triggers apoptosis via caspase-independent pathways, as was previously reported in non-β-cell systems.36
Whereas apoptosis is a well-studied process in β-cells, autophagy in β-cells has not received significant attention. Autophagy is a constitutively active process of cellular degradation in all cell types and is regarded as a generally protective process to prolong cell survival. However, it has been reported that β-cell death due to STZ, fatty acids, high fat diet and rapamycin is associated with increased autophagy in islet β-cells.37-42 Alternatively, decreases in autophagic turnover have also been shown to contribute to β-cell dysfunction and decrease β-cell survival in diabetes.43,44 For instance, β-cell-specific deletion of Atg7, an autophagy related gene essential for lipidation of LC3-I to form LC3-II during formation of the autophagosome, leads to islet degeneration, decreased glucose tolerance and reduced insulin secretion.45-47 Also noted were presence of abnormal, malformed and functionally defective β-cell mitochondria and a distended ER that presumably contributed to reduced insulin production, suggesting that basal autophagy is required during the maintenance of normal β-cell function.48 Further, feeding Atg7 knockout a high fat diet led to glucose intolerance, demonstrating the importance of autophagy during β-cell maintenance under stress.41 These reports illustrate a complex process of autophagy, which can be regulated at various steps that can potentially impact β-cell viability.
Germane to the present study, ER stress triggers the UPR to relieve the stress but prolonged ER stress in addition to activating intrinsic apoptosis pathway can upregulate autophagy as a means to clear mis-folded protein aggregates in the ER. It is being recognized that though autophagy is a pro-survival mechanism that it can also activate cell death processes.49 It is now suggested that UPR and autophagy are linked and share “functional duality,” where they exert cytoprotective activity under basal/unstressed states but are cytocidal in times of metabolic stress and cellular damage. In light of evidence linking ceramides with autophagy50 and our collective observations that ER stress induces ceramide generation via an iPLA2β-mediated mechanism, we addressed the possibility that iPLA2β expression modulates β-cell autophagy.
We found that ER stress promoted a greater increase in LC3-II in islets from RIP-iPLA2β-Tg mice, relative to islets from age-matched WT. In contrast, LC3-II generation was decreased in islets deficient in iPLA2β. Thapsigargin is thought to specifically block fusion of autophagosomes with lysosomes51 and we find that in the presence of inhibitors of autolysosomal activity, LC3-II accumulations in the RIP-iPLA2β-Tg and iPLA2β-KO are similar to their corresponding WT groups. Interestingly, inhibition of NSMase in the presence of thapsigargin had minimal effect in the WT islets but modestly reduced LC3-II flux in the Tg. These findings suggest that differential iPLA2β activation can impact β-cell autophagy and that the most likely point of effect is beyond the induction step. However, more detailed studies are needed to identify the precise location of iPLA2β impact on the autophagic response due to ER stress in the β-cells.
While this is the first demonstration of a link between the cytosol-associated iPLA2β and autophagy, it has been reported that mice deficient in the membrane-associated iPLA2γ had enlarged hippocampal mitochondrial and that their degeneration led to an increase in autophagy.52 They concluded that iPLA2γ-deficiency decreased mitochondrial membrane re-modeling, resulting in loss of membrane potential and subsequent mitochondrial dysfunction leading to cognitive dysfunction and increased autophagy in the hippocampus. These observations again illustrate the importance of normal expression of iPLA2β protein in maintaining cell survival.
The relevance of our findings is that biological processes is islet β-cells impacted by iPLA2β appear to be influenced by iPLA2β homeostasis, where both increased and decreased activity can have deleterious effects. For instance, clinical reports indicate that mutations in the PLA2G6 gene are associated with various CNS disorders.53-56 In contrast, increases in iPLA2β activity are associated with myocardial ischemia,57 cancer (reviewed in ref. 9) and inflammatory responses.58-60 Of importance to our findings, increases in iPLA2β activity (human neutrophils, and rodent myocardium and vascular smooth muscle)61-63 have been reported in T1DM. Further, gene expression analyses reveal increased expression of iPLA2β in peripheral blood mononuclear cells (PBMCs) from children with T1DM64 and interestingly, iPLA2β gene expression is induced in healthy PBMCs upon exposure to sera from recent diabetes patients.65 These observations are consistent with our hypothesis that increases in iPLA2β in the β-cells could contribute to β-cell dysfunction associated with diabetes.
In summary, our findings provide evidence for the involvement of iPLA2β in apoptosis and autophagy in β-cells due to ER stress. While our studies have focused on ER stress due to thapsigargin, to note is that many different stresses are associated with generation of mature (m) form of SREBP, which can translocate to the nucleus and induce expression of many gene products including that of PLA2G6, which encodes iPLA2β. The iPLA2β gene contains a sterol regulatory element, which can bind mSREBP to promote iPLA2β expression.24 Thus, we speculate that various stresses (ER, hyperglycemia, hyperlipidemia, oxidative, autoimmune responses)66-74 that are deleterious to the β-cell and associated with the evolution of diabetes potentially mediate their effects through activation of iPLA2β. In this regard, elevated glucose and cytokine levels both activate iPLA2β in pancreatic islets.75,76 Continued studies will lead to a better understanding of the underlying cellular mechanisms governing the role of iPLA2β in islet β-cells and lead to identification of specific loci that can be targeted for drug therapy.
Materials and Methods
Materials
Mice (RIP-iPLA2β-Tg and iPLA2β-KO breeding pairs), generously provided by Dr. John Turk (Washington University School of Medicine), were bred and maintained, according to the IACUC policies at the University of Alabama at Birmingham (UAB). Other materials are as follows: Rabbit polyclonal antibody to LC3 from Abcam (ab51520); SYBR Green PCR Kit from Applied Biosystems (4385612); brain and egg sphingomyelins, ceramide and other lipid standards from Avanti Polar Lipids; rabbit monoclonal phospho-PERK and cleaved caspase-3 antibodies from Cell Signaling Technology Inc. (3179 and 9664); Accumax from Innovative Cell Technologies (AM105); AccuPrime Taq DNA polymerase system, SuperScript III first-strand synthesis system for PT-PCR, guinea pig anti-insulin, DiOC6(3), and Hoechst Stain from Invitrogen (12339-016, 18080-051, 18-0067, D273 and H21491, respectively); SDS-PAGE supplies and Triton X-100 from BioRad (161-0156 and 161-0407); normal goat serum, Cy3-conjugated affinipure goat anti-rabbit IgG (H+L), and Dnk Anti-GPig IgG-FITC from Jackson Immuno Research Laboratories (005-000-121, 111-165-003, and 706-096-148, respectively); Immobilin-P PVDF membrane from Millipore Corp. (IPVH00010); Slow Fade® light antifade kit from Molecular Probes (S2828); mouse monoclonal antibody to LC3 from Nano Tools Antikörpertechnik GmbH and Co. (0231S0203); L-α-1-palmitoyl-2-arachidonyl [1-14C] phosphatidylcholine (PAPC), 58 mCi/mmol from Perkin Elmer (NEC765); RNeasy kit from Qiagen Inc. (74106); TUNEL kit from Roche Diagnostic Corporation (1684795); CHOP, GRP78, actin, tubulin, bovine anti-goat IgG-HRP and goat anti-rabbit IgG-HRP from Santa Cruz Biotech Inc. (sc-575, sc-1050, sc-7210, sc-8035, sc-2350, sc-2030, respectively); caspase-3 colorimetric assay kit ammonium chloride, leupeptin, NSMase inhibitor GW4869, and protease inhibitor cocktail from Sigma Chemical Co. (CASP3C, A0171, L9783, D1692 and P8340, respectively); and SuperSignal* West femto chemi-luminescent substrate from Thermo Scientific (34094). All other common reagents and salts were obtained from Sigma Chemical Company.
Generation of RIP-iPLA2β-Tg and iPLA2β−/− mice and genotyping by PCR analyses
Breeders obtained from WUSM were used to generate colonies of WT, RIP-iPLA2β-Tg, and iPLA2β−/− mice at UAB. Tg founders (TG1 line18) were mated with WT C57BL/6J mice (Jackson Laboratory) to generate RIP-iPLA2β-Tg mice and WT mice, and male and female iPLA2β+/− pairings were used to generate iPLA2β−/− (KO) and iPLA2β+/+ (WT) mice, as previously described.18,19 Progeny genotype was determined using PCR analyses and the following sense/antisense primers: WT and RIP-iPLA2β-Tg - Set 1, CCT CCG GAG AGC AGC GAT TAA AAG TGT CA/TAG AGC TTT GCC ACA TCA CAG GTC ATT CAG and Set 2, CTA GGC TCA GAC ATC ATG CTG GAC GAG GT/AAG ATC TCA GTG GTA TTT GTG AGC CAG GG were used together. Set 1 amplified the sequence in the internal control fatty acid- binding protein gene and the expected size of the product is 450 bp. Set 2 amplified the sequence that spans the junction of iPLA2β and globin cDNA in the TG construct and the expected size of the product is 200 bp. WT/ iPLA2β−/−—Set 1, AGC TTC AGG ATC TCA TGC CCA TC/CTC CGC TTC TCG TCC CTC ATG GA (expected size of the product for WT is 1400 bp) and Set 2, GGG GCC TCA GAC TGG GAA TC/Neo: TCG CCT TCT ATC GCC TTC TTG AC (expected size of the product for KO is 400 bp) were used separately.
Islet isolation
Mice (3–4 mo age) were killed by cervical dislocation, abdomen exposed, and pancreata isolated. The common bile duct was clamped at the duodenum-bile duct junction, and collagenase/Krebs-Ringer buffer (5 ml) was injected into the pancreas via the duct. Once the pancreas was completely distended, it was removed and placed in a scintillation vial with collagenase/Krebs-Ringer buffer (2.5 ml) and incubated in a 37°C water bath for 13 min. The vial was then vigorously shaken for 90 sec, followed by washing (3×) of the pancreas with Krebs-Ringer buffer containing 1 mM CaCl2 (50 ml). The pancreas was then resuspended in incomplete RPMI 1640 (without FBS or penicillin/streptomycin, 25 ml) and poured through a 70 μm cell strainer into a Petri dish. The cells were washed further with incomplete RPMI 1640 (75 ml). The cell strainer was then inverted and rinsed with complete RPMI 1640 (10% fetal bovine serum, 2 × penicillin/streptomycin, 25 ml) to collect the remaining islets in solution. The islets were hand-picked under a microscope, counted, and incubated overnight in media under an atmosphere of 5% CO2/95% air and 37°C and then used for the experiments described below.
Islet iPLA2β expression
To confirm that progeny islet expression of iPLA2β matched their genotype, total RNA was isolated from islets of each genotype using RNeasy kit (Qiagen Inc.). cDNA was then synthesized using SuperScript III kit (Invitrogen) for PCR analyses of iPLA2β message. PCR amplifications were performed using AccuPrime Taq DNA Polymerase System, and the sense/antisense primer sequences were ccgtatgaaggacgacctgt/cggtggcttcaggttaatgt. After verifying the genotypes, the RIP-iPLA2β-Tg and iPLA2β-KO mice were compared against corresponding age-matched littermate controls (WT) in subsequent studies.
iPLA2β activity assay
Cytosol fraction was prepared from islets and protein concentration was determined using Coomassie reagent. Ca2+-independent PLA2 catalytic activity in an aliquot of cytosolic protein (30 μg) was assayed under zero Ca2+ conditions (no added Ca2+ plus 10 mm EGTA) in the presence of 16:0/[14C]20:4 GPC (PAPC, 5 μM) as the substrate, and specific enzymatic activity was quantitated, as described.23 Because stimulation of activity by ATP is a characteristic property of iPLA2β,11 the activity assay was also performed in the presence of 10 mM ATP.
Immunoblotting analyses
Islets were harvested at various times following induction of ER stress, sonicated, homogenates analyzed by SDS-PAGE (8 or 15%), and proteins transferred onto Immobolin-P PVDF membranes and processed for immunoblotting analyses. The targeted proteins and (1° antibody concentrations) were as follows: CHOP (1:500), GRP78 (1:500), iPLA2 (T-14; 1:500), cleaved caspase-3 (1:1,000), LC3 (1:1,000), actin (1:500) and tubulin (1:1000). The secondary antibody concentration was 1:10,000. Immunoreactive bands were visualized by enhanced chemiluminescence.
In situ detection of DNA cleavage by TUNEL staining
Islets or dispersed islet cells were washed twice with ice-cold phosphate-buffered saline (PBS). Islets were then immobilized on slides by cytospin and fixed at room temperature (RT) with 4% paraformaldehyde (in PBS, pH 7.4, 1 h). Dispersed cells were directly fixed with 4% paraformaldehyde for 20 min. The islets or dispersed cells were then washed with PBS and incubated in permeabilization solution (0.1% Triton-X-100 in 0.1% sodium citrate in phosphate-buffered saline, 30 min, RT). The permeabilization solution was then removed, TUNEL reaction mixture (50 μl) added, and the cells were incubated (1 h, 37°C) in a humidified chamber. The islets were washed again with PBS and counterstained with 1 μg/ml DAPI (4',6'-diamidino-2-phenylindole) in PBS for 10 min to identify cellular nuclei. Incidence of apoptosis was assessed under a fluorescence microscope using a FITC filter. Dispersed cells and flow cytometry were used to determine the percentage of apoptotic cells in islets.
Cleaved caspase-3 activity
To obtain a quantitative measure of apoptosis in isolated islets, cleaved (activated) caspase-3 (aC3) activity was assayed according to Manufacturer’s instructions. Islets were treated, harvested, resuspended in 1× lysis buffer (100 μl/200 islets, 20 min), sonicated and centrifuged at 18,000 g for 20 min at 4°C. The supernatants were then collected for the activity assay, which is based on measurement of aC3-catalyzed generation of p-nitroaniline (pNA) from Ac-DEVD-pNA substrate. Ten μl of cell lysate or casp-3 positive control was incubated (3 h, 37°C, total volume of 1 ml) with 1 × assay buffer and the casp-3 substrate. The samples were then transferred to 1 ml quartz cuvettes and the absorbance of released pNA was read at 405 nm. Measured activity of cleaved caspase-3 was normalized to protein.
Immunostaining
Islets were fixed in 10% formalin and 5 μl of tissue marking dye, followed by the addition of 150 μl of low melting agarose. The mixture was spun down quickly to settle the islets at one surface of the agarose, which was then allowed to solidify. The islet-containing blocks were then processed, and paraffin sections (8–10 μm) were prepared for iPLA2β, aC3 and insulin staining. The sections were incubated overnight with primary antibodies (1:25), washed with PBS (4 × 30 min), incubated for 2–3 h with secondary antibodies (1:100 of Cy3 for insulin and Alexa Fluor 594 for iPLA2β or cleaved caspase-3) and washed with PBS (3×, 10 min each). DAPI stain (20–30 μl) was then added, and the sections were sealed with a coverslip using nail polish. Fluorescence was recorded using a Nikon Eclipse TE300 microscope and images were captured at 20× magnification.
Quantitative RT-PCR
To determine mRNA expression of neutral sphingomyelinase (NSMase), total RNA was isolated from islets using RNeasy kit. cDNA was then synthesized using SuperScript III kit (Invitrogen) and heat-inactivated (70°C for 15 min). A reaction without reverse transcriptase was performed to verify the absence of genomic DNA. PCR amplifications were performed using SYBR Green PCR kit in an ABI 7000 detection system (Applied Biosystems). The sense/antisense primer sets for NSMase and 18S were ccggatgcacactacttcagaa/ggattgggtgtctggagaaca and agtcctgccctttgtacaca/gatccgagggcctcactaaac, respectively.
Ceramide analyses by electrospray ionization (ESI)/MS/MS
Lipids were extracted from islets under acidic conditions, as described.22 Briefly, islets were harvested, gently pelleted and extraction buffer (chloroform/ methanol/2% acetic acid, 2/2/1.8; v/v/v) containing C8-ceramide (m/z 432) internal standard (IS, 500 ng), which is not an endogenous component of β-cell lipids, was added to the cellular pellet. After vigorous vortexing, the mixture was centrifuged (800 g) and the organic bottom layer was collected, concentrated to dryness under nitrogen and reconstituted in chloroform/methanol (1/4) containing 10 pmol/μl LiOH. To determine ceramide abundances, ESI/MS/MS standard curves were generated from a series of samples containing fixed amount of C8-ceramide standard and varied amounts of long-chain ceramide standards. The relative abundances of individual ceramide species, relative to the C8-ceramide IS, were measured by ESI/MS/MS scanning for constant neutral loss of 48, which reflects the elimination of formaldehyde and water from the [M + Li+] ion.23 This loss is characteristic of ceramide-Li+ adducts upon low energy collisionally-activated dissociation ESI/MS/MS. Lipid phosphorous measurements were used to normalize individual ceramide molecular species.
Sphingomyelin (SM) analyses by ESI/MS/MS
Lipid extracts prepared as above were used for the sphingomyelin analyses. Sphingomyelins are formed by reaction of a ceramide with CDP-choline, and similar to GPC lipids, they contain a phosphocholine as the polar head group. This feature of sphingomyelins facilitates identification of sphingomyelin molecular species by constant neutral loss scanning of trimethylamine ([M+ H]+ − N(CH3)3) or constant neutral loss of 59, as described.20 The prominent ions in the total ion current spectrum are those of the even mass PC molecular species, and these mask the odd mass sphingomyelin signals.20 Constant neutral loss of 59, however, facilitates emergence of signals for sphingomyelin species at odd m/z values, reflecting loss of nitrogen. Lipid extracts were prepared as above in the presence of a 14:0/14:0- GPC (m/z 684, 8 μg) IS, which is not an endogenous component of β-cell lipids, and analyzed by ESI/MS/MS. Sphingomyelins abundances in the samples was determined based on standard curves generated using commercially available brain and egg sphingomyelins with a known percentage of each fatty acid constituent and the internal standard as described.20 Lipid phosphorous measurements were used to normalize individual sphingomyelin molecular species.
Islet dispersion and cell sorting
Islets were collected and washed with PBS (2×) before being dispersed into single cells. Islets were resuspended in 1 ml of Accumax and incubated for 1 h at 37°C. Dispersed cells were then washed once and passed through a filter (35 μm pore size) to eliminate cell clumps. Sorting protocol was based on endogenous flavin adenine dinucleotide (FAD) auto-fluorescence in β-cells.77 High and low fluorescent cells were sorted on a fluorescence-activated cell sorter (FACS, BD Biosciences). The FAD content of the dispersed cells was analyzed at an excitation wavelength of 488 nm, and collected at 525 nm. The group of high autofluorescent cell population typically comprised 90–95% β-cells.
Assessment of mitochondrial membrane potential
β-cells obtained using the FACS protocol were washed with PBS (2×) at room temperature and then incubated with DiOC6(3) solution (175 nm) for 15 min under an atmosphere of 5% CO2/95% air (37°C). The Hoechst reagent (5 μg/ml) was then added to stain the nucleus. After 20 min, the cells were rinsed with PBS and mounted on slides, and then immediately examined using a confocal laser-scanning microscope (Zeiss) with a 488-nm argon laser and a 405-nm diode laser
Autophagy
Islets (500/condition) were treated with vehicle (DMSO) or thapsigargin (2 μM) in the absence or presence of leupeptin (200 μM) and ammonium chloride (20 mM), which inhibit autolysosomal activity. In some experiments, islets were pre-treated with the NSMase inhibitor GW4869 (20 μM) for 30 min prior to addition of thapsigargin. Autophagy was monitored by determining the ratio of LC3-II to loading control.
Statistical analyses
Data were converted to mean ± standard error of the means and the Students’ t-test was applied to determine significant differences between two samples (p < 0.05).
Acknowledgments
The authors would like to thank the expert technical assistance of Ms. Min Tan and Ms. Sheng Zhang, Washington University Diabetes Research and Training Center (DRTC) supported Morphology Core, UAB Comprehensive Diabetes Center Islet Biology Core, and the DRTC at UAB. This work was supported by grants from the National Institutes of Health (DK69455, DK34388, DK079626, P01-HL57278, P41-RR00954, P60-DK20579 and P30-DK56341), and the American Diabetes Association (S.R.).
Glossary
Abbreviations:
- BEL
bromoenol lactone suicide inhibitor of iPLA2β
- cPLA2
group IV cytosolic phospholipase A2
- DAPI
4',6'-diamidino-2-phenylindole
- ER
endoplasmic reticulum
- ESI
electrospray ionization
- GPC
glycerophosphocholine
- iPLA2β
β-isoform of group VIA calcium-independent phospholipase A2
- iPLA2β-KO
iPLA2β-deficient mice
- RIP-iPLA2β-Tg
transgenic mice in which iPLA2β is overexpressed specifically in pancreatic islet β-cells
- ∆Ψ
mitochondrial membrane potential
- MS
mass spectrometry
- NSMase
neutral sphingomyelinase
- PERK
ER-stress transducer pancreatic ER kinase
- PLA2
phospholipase A2
- SEM
standard error of the mean
- SERCA
sarcoendoplasmic reticulum Ca2+-ATPase
- SM
sphingomyelin
- T1DM and T2DM
type 1 and 2 diabetes mellitus
- UPR
unfolded protein response
- WT-KO and WT-Tg
age-matched control littermates of iPLA2β-KO and RIP-iPLA2β-Tg mice, respectively
Disclosure of Potential Conflicts of Interest
No conflicts of interest were disclosed.
Footnotes
Previously published online: www.landesbioscience.com/journals/islets/article/23758
References
- 1.Butler AE, Janson J, Bonner-Weir S, Ritzel R, Rizza RA, Butler PC. β-cell deficit and increased β-cell apoptosis in humans with type 2 diabetes. Diabetes. 2003;52:102–10. doi: 10.2337/diabetes.52.1.102. [DOI] [PubMed] [Google Scholar]
- 2.Bernard C, Berthault M-F, Saulnier C, Ktorza A. Neogenesis vs. apoptosis As main components of pancreatic β cell ass changes in glucose-infused normal and mildly diabetic adult rats. FASEB J. 1999;13:1195–205. doi: 10.1096/fasebj.13.10.1195. [DOI] [PubMed] [Google Scholar]
- 3.Butler AE, Janson J, Soeller WC, Butler PC. Increased β-cell apoptosis prevents adaptive increase in β-cell mass in mouse model of type 2 diabetes: evidence for role of islet amyloid formation rather than direct action of amyloid. Diabetes. 2003;52:2304–14. doi: 10.2337/diabetes.52.9.2304. [DOI] [PubMed] [Google Scholar]
- 4.Nakagawa T, Zhu H, Morishima N, Li E, Xu J, Yankner BA, et al. Caspase-12 mediates endoplasmic-reticulum-specific apoptosis and cytotoxicity by amyloid-beta. Nature. 2000;403:98–103. doi: 10.1038/47513. [DOI] [PubMed] [Google Scholar]
- 5.Ron D. Translational control in the endoplasmic reticulum stress response. J Clin Invest. 2002;110:1383–8. doi: 10.1172/JCI16784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mehmet H. Caspases find a new place to hide. Nature. 2000;403:29–30. doi: 10.1038/47377. [DOI] [PubMed] [Google Scholar]
- 7.Berridge MJ. The endoplasmic reticulum: a multifunctional signaling organelle. Cell Calcium. 2002;32:235–49. doi: 10.1016/S0143416002001823. [DOI] [PubMed] [Google Scholar]
- 8.Cohen GM. Caspases: the executioners of apoptosis. Biochem J. 1997;326:1–16. doi: 10.1042/bj3260001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lei X, Barbour SE, Ramanadham S. Group VIA Ca2+-independent phospholipase A2 (iPLA2β) and its role in β-cell programmed cell death. Biochimie. 2010;92:627–37. doi: 10.1016/j.biochi.2010.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gijón MA, Leslie CC. Phospholipases A2. Semin Cell Dev Biol. 1997;8:297–303. doi: 10.1006/scdb.1997.0151. [DOI] [PubMed] [Google Scholar]
- 11.Gross RW, Ramanadham S, Kruszka KK, Han X, Turk J. Rat and human pancreatic islet cells contain a calcium ion independent phospholipase A2 activity selective for hydrolysis of arachidonate which is stimulated by adenosine triphosphate and is specifically localized to islet beta-cells. Biochemistry. 1993;32:327–36. doi: 10.1021/bi00052a041. [DOI] [PubMed] [Google Scholar]
- 12.Wolf RA, Gross RW. Identification of neutral active phospholipase C which hydrolyzes choline glycerophospholipids and plasmalogen selective phospholipase A2 in canine myocardium. J Biol Chem. 1985;260:7295–303. [PubMed] [Google Scholar]
- 13.Balsinde J, Pérez R, Balboa MA. Calcium-independent phospholipase A2 and apoptosis. Biochim Biophys Acta. 2006;1761:1344–50. doi: 10.1016/j.bbalip.2006.07.013. [DOI] [PubMed] [Google Scholar]
- 14.Atsumi G-i, Tajima M, Hadano A, Nakatani Y, Murakami M, Kudo I. Fas-induced arachidonic acid release is mediated by Ca2+-independent phospholipase A2 but not cytosolic phospholipase A2, which undergoes proteolytic inactivation. J Biol Chem. 1998;273:13870–7. doi: 10.1074/jbc.273.22.13870. [DOI] [PubMed] [Google Scholar]
- 15.Zhou Y-P, Teng D, Dralyuk F, Ostrega D, Roe MW, Philipson L, et al. Apoptosis in insulin-secreting cells. Evidence for the role of intracellular Ca2+ stores and arachidonic acid metabolism. J Clin Invest. 1998;101:1623–32. doi: 10.1172/JCI1245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nowatzke W, Ramanadham S, Ma Z, Hsu FF, Bohrer A, Turk J. Mass spectrometric evidence that agents that cause loss of Ca2+ from intracellular compartments induce hydrolysis of arachidonic acid from pancreatic islet membrane phospholipids by a mechanism that does not require a rise in cytosolic Ca2+ concentration. Endocrinology. 1998;139:4073–85. doi: 10.1210/en.139.10.4073. [DOI] [PubMed] [Google Scholar]
- 17.Ramanadham S, Hsu FF, Bohrer A, Nowatzke W, Ma Z, Turk J. Electrospray ionization mass spectrometric analyses of phospholipids from rat and human pancreatic islets and subcellular membranes: comparison to other tissues and implications for membrane fusion in insulin exocytosis. Biochemistry. 1998;37:4553–67. doi: 10.1021/bi9722507. [DOI] [PubMed] [Google Scholar]
- 18.Bao S, Jacobson DA, Wohltmann M, Bohrer A, Jin W, Philipson LH, et al. Glucose homeostasis, insulin secretion, and islet phospholipids in mice that overexpress iPLA2β in pancreatic β-cells and in iPLA2β-null mice. Am J Physiol Endocrinol Metab. 2008;294:E217–29. doi: 10.1152/ajpendo.00474.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bao S, Miller DJ, Ma Z, Wohltmann M, Eng G, Ramanadham S, et al. Male mice that do not express group VIA phospholipase A2 produce spermatozoa with impaired motility and have greatly reduced fertility. J Biol Chem. 2004;279:38194–200. doi: 10.1074/jbc.M406489200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lei X, Zhang S, Bohrer A, Bao S, Song H, Ramanadham S. The group VIA calcium-independent phospholipase A2 participates in ER stress-induced INS-1 insulinoma cell apoptosis by promoting ceramide generation via hydrolysis of sphingomyelins by neutral sphingomyelinase. Biochemistry. 2007;46:10170–85. doi: 10.1021/bi700017z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lei X, Zhang S, Barbour SE, Bohrer A, Ford EL, Koizumi A, et al. Spontaneous development of endoplasmic reticulum stress that can lead to diabetes mellitus is associated with higher calcium-independent phospholipase A2 expression: a role for regulation by SREBP-1. J Biol Chem. 2010;285:6693–705. doi: 10.1074/jbc.M109.084293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lei X, Zhang S, Bohrer A, Ramanadham S. Calcium-independent phospholipase A2 (iPLA2 β)-mediated ceramide generation plays a key role in the cross-talk between the endoplasmic reticulum (ER) and mitochondria during ER stress-induced insulin-secreting cell apoptosis. J Biol Chem. 2008;283:34819–32. doi: 10.1074/jbc.M807409200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ramanadham S, Hsu FF, Zhang S, Jin C, Bohrer A, Song H, et al. Apoptosis of insulin-secreting cells induced by endoplasmic reticulum stress is amplified by overexpression of group VIA calcium-independent phospholipase A2 (iPLA2 β) and suppressed by inhibition of iPLA2 β. Biochemistry. 2004;43:918–30. doi: 10.1021/bi035536m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lei X, Zhang S, Bohrer A, Barbour SE, Ramanadham S. Role of calcium-independent phospholipase A(2)β in human pancreatic islet β-cell apoptosis. Am J Physiol Endocrinol Metab. 2012;303:E1386–95. doi: 10.1152/ajpendo.00234.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Choi KS. Autophagy and cancer. Exp Mol Med. 2012;44:109–20. doi: 10.3858/emm.2012.44.2.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schonthal AH. Targeting endoplasmic reticulum stress for cancer therapy. Front Biosci (Schol Ed) 2012;4:412–31. doi: 10.2741/s276. [Schol Ed] [DOI] [PubMed] [Google Scholar]
- 27.Mizushima N, Yoshimori T, Levine B. Methods in mammalian autophagy research. Cell. 2010;140:313–26. doi: 10.1016/j.cell.2010.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Klionsky DJ, Abdalla FC, Abeliovich H, Abraham RT, Acevedo-Arozena A, Adeli K, et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy. 2012;8:445–544. doi: 10.4161/auto.19496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tanida I. Autophagosome formation and molecular mechanism of autophagy. Antioxid Redox Signal. 2011;14:2201–14. doi: 10.1089/ars.2010.3482. [DOI] [PubMed] [Google Scholar]
- 30.Yoon KH, Ko SH, Cho JH, Lee JM, Ahn YB, Song KH, et al. Selective β-cell loss and α-cell expansion in patients with type 2 diabetes mellitus in Korea. J Clin Endocrinol Metab. 2003;88:2300–8. doi: 10.1210/jc.2002-020735. [DOI] [PubMed] [Google Scholar]
- 31.Oyadomari S, Takeda K, Takiguchi M, Gotoh T, Matsumoto M, Wada I, et al. Nitric oxide-induced apoptosis in pancreatic beta cells is mediated by the endoplasmic reticulum stress pathway. Proc Natl Acad Sci U S A. 2001;98:10845–50. doi: 10.1073/pnas.191207498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Remedi MS, Koster JC, Markova K, Seino S, Miki T, Patton BL, et al. Diet-induced glucose intolerance in mice with decreased beta-cell ATP-sensitive K+ channels. Diabetes. 2004;53:3159–67. doi: 10.2337/diabetes.53.12.3159. [DOI] [PubMed] [Google Scholar]
- 33.Yang J, Liu X, Bhalla K, Kim CN, Ibrado AM, Cai J, et al. Prevention of apoptosis by Bcl-2: release of cytochrome c from mitochondria blocked. Science. 1997;275:1129–32. doi: 10.1126/science.275.5303.1129. [DOI] [PubMed] [Google Scholar]
- 34.Seleznev K, Zhao C, Zhang XH, Song K, Ma ZA. Calcium-independent phospholipase A2 localizes in and protects mitochondria during apoptotic induction by staurosporine. J Biol Chem. 2006;281:22275–88. doi: 10.1074/jbc.M604330200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhao Z, Zhang X, Zhao C, Choi J, Shi J, Song K, et al. Protection of pancreatic beta-cells by group VIA phospholipase A(2)-mediated repair of mitochondrial membrane peroxidation. Endocrinology. 2010;151:3038–48. doi: 10.1210/en.2010-0016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Aoto M, Shinzawa K, Suzuki Y, Ohkubo N, Mitsuda N, Tsujimoto Y. Essential role of p38 MAPK in caspase-independent, iPLA(2)-dependent cell death under hypoxia/low glucose conditions. FEBS Lett. 2009;583:1611–8. doi: 10.1016/j.febslet.2009.04.028. [DOI] [PubMed] [Google Scholar]
- 37.Kaniuk NA, Kiraly M, Bates H, Vranic M, Volchuk A, Brumell JH. Ubiquitinated-protein aggregates form in pancreatic beta-cells during diabetes-induced oxidative stress and are regulated by autophagy. Diabetes. 2007;56:930–9. doi: 10.2337/db06-1160. [DOI] [PubMed] [Google Scholar]
- 38.Choi SE, Lee SM, Lee YJ, Li LJ, Lee SJ, Lee JH, et al. Protective role of autophagy in palmitate-induced INS-1 beta-cell death. Endocrinology. 2009;150:126–34. doi: 10.1210/en.2008-0483. [DOI] [PubMed] [Google Scholar]
- 39.Las G, Shirihai OS. The role of autophagy in β-cell lipotoxicity and type 2 diabetes. Diabetes Obes Metab. 2010;12(Suppl 2):15–9. doi: 10.1111/j.1463-1326.2010.01268.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Grasso D, Sacchetti ML, Bruno L, Lo Ré A, Iovanna JL, Gonzalez CD, et al. Autophagy and VMP1 expression are early cellular events in experimental diabetes. Pancreatology. 2009;9:81–8. doi: 10.1159/000178878. [DOI] [PubMed] [Google Scholar]
- 41.Ebato C, Uchida T, Arakawa M, Komatsu M, Ueno T, Komiya K, et al. Autophagy is important in islet homeostasis and compensatory increase of beta cell mass in response to high-fat diet. Cell Metab. 2008;8:325–32. doi: 10.1016/j.cmet.2008.08.009. [DOI] [PubMed] [Google Scholar]
- 42.Tanemura M, Ohmura Y, Deguchi T, Machida T, Tsukamoto R, Wada H, et al. Rapamycin causes upregulation of autophagy and impairs islets function both in vitro and in vivo. Am J Transplant. 2012;12:102–14. doi: 10.1111/j.1600-6143.2011.03771.x. [DOI] [PubMed] [Google Scholar]
- 43.Masini M, Lupi R, Bugliani M, Boggi U, Filipponi F, Masiello P, et al. A role for autophagy in β-cell life and death. Islets. 2009;1:157–9. doi: 10.4161/isl.1.2.9372. [DOI] [PubMed] [Google Scholar]
- 44.Las G, Serada SB, Wikstrom JD, Twig G, Shirihai OS. Fatty acids suppress autophagic turnover in β-cells. J Biol Chem. 2011;286:42534–44. doi: 10.1074/jbc.M111.242412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fujitani Y, Ebato C, Uchida T, Kawamori R, Watada H. β-cell autophagy: A novel mechanism regulating β-cell function and mass: Lessons from β-cell-specific Atg7-deficient mice. Islets. 2009;1:151–3. doi: 10.4161/isl.1.2.9057. [DOI] [PubMed] [Google Scholar]
- 46.Jung HS, Lee MS. Macroautophagy in homeostasis of pancreatic beta-cell. Autophagy. 2009;5:241–3. doi: 10.4161/auto.5.2.7518. [DOI] [PubMed] [Google Scholar]
- 47.Jung HS, Chung KW, Won Kim J, Kim J, Komatsu M, Tanaka K, et al. Loss of autophagy diminishes pancreatic beta cell mass and function with resultant hyperglycemia. Cell Metab. 2008;8:318–24. doi: 10.1016/j.cmet.2008.08.013. [DOI] [PubMed] [Google Scholar]
- 48.Jung HS, Lee MS. Role of autophagy in diabetes and mitochondria. Ann N Y Acad Sci. 2010;1201:79–83. doi: 10.1111/j.1749-6632.2010.05614.x. [DOI] [PubMed] [Google Scholar]
- 49.Levine B, Yuan J. Autophagy in cell death: an innocent convict? J Clin Invest. 2005;115:2679–88. doi: 10.1172/JCI26390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Scarlatti F, Bauvy C, Ventruti A, Sala G, Cluzeaud F, Vandewalle A, et al. Ceramide-mediated macroautophagy involves inhibition of protein kinase B and up-regulation of beclin 1. J Biol Chem. 2004;279:18384–91. doi: 10.1074/jbc.M313561200. [DOI] [PubMed] [Google Scholar]
- 51.Ganley IG, Wong PM, Jiang X. Thapsigargin distinguishes membrane fusion in the late stages of endocytosis and autophagy. Autophagy. 2011;7:1397–9. doi: 10.4161/auto.7.11.17651. [DOI] [PubMed] [Google Scholar]
- 52.Mancuso DJ, Kotzbauer P, Wozniak DF, Sims HF, Jenkins CM, Guan S, et al. Genetic ablation of calcium-independent phospholipase A2γ leads to alterations in hippocampal cardiolipin content and molecular species distribution, mitochondrial degeneration, autophagy, and cognitive dysfunction. J Biol Chem. 2009;284:35632–44. doi: 10.1074/jbc.M109.055194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Biancheri R, Rossi A, Alpigiani G, Filocamo M, Gandolfo C, Lorini R, et al. Cerebellar atrophy without cerebellar cortex hyperintensity in infantile neuroaxonal dystrophy (INAD) due to PLA2G6 mutation. Eur J Paediatr Neurol. 2007;11:175–7. doi: 10.1016/j.ejpn.2006.11.013. [DOI] [PubMed] [Google Scholar]
- 54.Morgan NV, Westaway SK, Morton JE, Gregory A, Gissen P, Sonek S, et al. PLA2G6, encoding a phospholipase A2, is mutated in neurodegenerative disorders with high brain iron. Nat Genet. 2006;38:752–4. doi: 10.1038/ng1826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Westaway SK, Gregory A, Hayflick SJ. Mutations in PLA2G6 and the riddle of Schindler disease. J Med Genet. 2007;44:e64. doi: 10.1136/jmg.2006.044966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Crompton D, Rehal PK, MacPherson L, Foster K, Lunt P, Hughes I, et al. Multiplex ligation-dependent probe amplification (MLPA) analysis is an effective tool for the detection of novel intragenic PLA2G6 mutations: implications for molecular diagnosis. Mol Genet Metab. 2010;100:207–12. doi: 10.1016/j.ymgme.2010.02.009. [DOI] [PubMed] [Google Scholar]
- 57.Hazen SL, Gross RW. Human myocardial cytosolic Ca(2+)-independent phospholipase A2 is modulated by ATP. Concordant ATP-induced alterations in enzyme kinetics and mechanism-based inhibition. Biochem J. 1991;280:581–7. doi: 10.1042/bj2800581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Strokin M, Sergeeva M, Reiser G. Proinflammatory treatment of astrocytes with lipopolysaccharide results in augmented Ca2+ signaling through increased expression of via phospholipase A2 (iPLA2) Am J Physiol Cell Physiol. 2011;300:C542–9. doi: 10.1152/ajpcell.00428.2010. [DOI] [PubMed] [Google Scholar]
- 59.Sharma J, Turk J, McHowat J. Endothelial cell prostaglandin I(2) and platelet-activating factor production are markedly attenuated in the calcium-independent phospholipase A(2)β knockout mouse. Biochemistry. 2010;49:5473–81. doi: 10.1021/bi100752u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Liu S, Xie Z, Zhao Q, Pang H, Turk J, Calderon L, et al. Smooth muscle-specific expression of calcium-independent phospholipase A2β (iPLA2β) participates in the initiation and early progression of vascular inflammation and neointima formation. J Biol Chem. 2012;287:24739–53. doi: 10.1074/jbc.M112.340216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ayilavarapu S, Kantarci A, Fredman G, Turkoglu O, Omori K, Liu H, et al. Diabetes-induced oxidative stress is mediated by Ca2+-independent phospholipase A2 in neutrophils. J Immunol. 2010;184:1507–15. doi: 10.4049/jimmunol.0901219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Rahnema P, Shimoni Y, Nygren A. Reduced conduction reserve in the diabetic rat heart: role of iPLA2 activation in the response to ischemia. Am J Physiol Heart Circ Physiol. 2011;300:H326–34. doi: 10.1152/ajpheart.00743.2010. [DOI] [PubMed] [Google Scholar]
- 63.Xie Z, Gong MC, Su W, Xie D, Turk J, Guo Z. Role of calcium-independent phospholipase A2β in high glucose-induced activation of RhoA, Rho kinase, and CPI-17 in cultured vascular smooth muscle cells and vascular smooth muscle hypercontractility in diabetic animals. J Biol Chem. 2010;285:8628–38. doi: 10.1074/jbc.M109.057711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kaizer EC, Glaser CL, Chaussabel D, Banchereau J, Pascual V, White PC. Gene expression in peripheral blood mononuclear cells from children with diabetes. J Clin Endocrinol Metab. 2007;92:3705–11. doi: 10.1210/jc.2007-0979. [DOI] [PubMed] [Google Scholar]
- 65.Wang X, Jia S, Geoffrey R, Alemzadeh R, Ghosh S, Hessner MJ. Identification of a molecular signature in human type 1 diabetes mellitus using serum and functional genomics. J Immunol. 2008;180:1929–37. doi: 10.4049/jimmunol.180.3.1929. [DOI] [PubMed] [Google Scholar]
- 66.Kaplan M, Aviram M, Hayek T. Oxidative stress and macrophage foam cell formation during diabetes mellitus-induced atherogenesis: role of insulin therapy. Pharmacol Ther. 2012;136:175–85. doi: 10.1016/j.pharmthera.2012.08.002. [DOI] [PubMed] [Google Scholar]
- 67.Akarte AS, Srinivasan BP, Gandhi S. Vildagliptin selectively ameliorates GLP-1, GLUT4, SREBP-1c mRNA levels and stimulates β-cell proliferation resulting in improved glucose homeostasis in rats with streptozotocin-induced diabetes. J Diabetes Complications. 2012;26:266–74. doi: 10.1016/j.jdiacomp.2012.03.013. [DOI] [PubMed] [Google Scholar]
- 68.Chin HJ, Fu YY, Ahn JM, Na KY, Kim YS, Kim S, et al. Omacor, n-3 polyunsaturated fatty acid, attenuated albuminuria and renal dysfunction with decrease of SREBP-1 expression and triglyceride amount in the kidney of type II diabetic animals. Nephrol Dial Transplant. 2010;25:1450–7. doi: 10.1093/ndt/gfp695. [DOI] [PubMed] [Google Scholar]
- 69.Heller JJ, Qiu J, Zhou L. Nuclear receptors take center stage in Th17 cell-mediated autoimmunity. J Clin Invest. 2011;121:519–21. doi: 10.1172/JCI45939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wang H, Kouri G, Wollheim CB. ER stress and SREBP-1 activation are implicated in beta-cell glucolipotoxicity. J Cell Sci. 2005;118:3905–15. doi: 10.1242/jcs.02513. [DOI] [PubMed] [Google Scholar]
- 71.Boslem E, MacIntosh G, Preston AM, Bartley C, Busch AK, Fuller M, et al. A lipidomic screen of palmitate-treated MIN6 β-cells links sphingolipid metabolites with endoplasmic reticulum (ER) stress and impaired protein trafficking. Biochem J. 2011;435:267–76. doi: 10.1042/BJ20101867. [DOI] [PubMed] [Google Scholar]
- 72.Chan JY, Cooney GJ, Biden TJ, Laybutt DR. Differential regulation of adaptive and apoptotic unfolded protein response signalling by cytokine-induced nitric oxide production in mouse pancreatic beta cells. Diabetologia. 2011;54:1766–76. doi: 10.1007/s00125-011-2139-z. [DOI] [PubMed] [Google Scholar]
- 73.Véret J, Coant N, Berdyshev EV, Skobeleva A, Therville N, Bailbé D, et al. Ceramide synthase 4 and de novo production of ceramides with specific N-acyl chain lengths are involved in glucolipotoxicity-induced apoptosis of INS-1 β-cells. Biochem J. 2011;438:177–89. doi: 10.1042/BJ20101386. [DOI] [PubMed] [Google Scholar]
- 74.Yano M, Watanabe K, Yamamoto T, Ikeda K, Senokuchi T, Lu M, et al. Mitochondrial dysfunction and increased reactive oxygen species impair insulin secretion in sphingomyelin synthase 1-null mice. J Biol Chem. 2011;286:3992–4002. doi: 10.1074/jbc.M110.179176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ma Z, Ramanadham S, Corbett JA, Bohrer A, Gross RW, McDaniel ML, et al. Interleukin-1 enhances pancreatic islet arachidonic acid 12-lipoxygenase product generation by increasing substrate availability through a nitric oxide-dependent mechanism. J Biol Chem. 1996;271:1029–42. doi: 10.1074/jbc.271.2.1029. [DOI] [PubMed] [Google Scholar]
- 76.Ramanadham S, Gross RW, Han X, Turk J. Inhibition of arachidonate release by secretagogue-stimulated pancreatic islets suppresses both insulin secretion and the rise in beta-cell cytosolic calcium ion concentration. Biochemistry. 1993;32:337–46. doi: 10.1021/bi00052a042. [DOI] [PubMed] [Google Scholar]
- 77.Smelt MJ, Faas MM, de Haan BJ, de Vos P. Pancreatic beta-cell purification by altering FAD and NAD(P)H metabolism. Exp Diab Res. 2008;2008:1–11. doi: 10.1155/2008/165360. [DOI] [PMC free article] [PubMed] [Google Scholar]