

Abstract
A novel approach to specifically target tumor cells for detection and treatment is the proposed use of hetero-multivalent ligands, which are designed to interact with, and non-covalently crosslink, multiple different cell surface receptors. Although enhanced binding has been shown for synthetic homo-multivalent ligands, proof of cross-linking requires the use of ligands with two or more different binding moieties. As proof-of-concept, we have examined the binding of synthetic hetero-bivalent ligands to cell lines that were engineered to co-express two different G-protein coupled human receptors, viz. the human melanocortin 4 receptor (hMC4R) expressed in combination with either the human delta-opioid receptor (hδOR) or the human cholecystokinin-2 receptor (hCCK2R). Expression levels of these receptors were characterized by time-resolved fluorescence saturation binding assays using Europium-labeled ligands; Eu-DPLCE, Eu-NDP-α-MSH and Eu-CCK8 for the δOR, MC4R and CCK2R, respectively. Heterobivalent ligands were synthesized to contain a MC4R agonist connected via chemical linkers to either a δOR or a CCK2R agonist. In both cell systems, the hetero-bivalent constructs bound with much higher affinity to cells expressing both receptors, compared to cells with single receptors or to cells where one of the receptors was competitively blocked. These results indicate that synthetic hetero-bivalent ligands can non-covalently crosslink two unrelated cell surface receptors, making feasible the targeting of receptor combinations. The in vitro cell models described herein will lead to the development of multivalent ligands for target combinations identified in human cancers.
Keywords: heterobivalent ligands, dual expression, G-Protein Coupled Receptors, binding assay, cell surface receptor, cancer targeted therapy
Introduction
A major goal of cancer therapy is to kill or inhibit cancer cells, while minimizing deleterious effects on normal cells. Molecular biology and pharmacogenomics have revealed critical molecular differences between normal and tumor cells. These differences may allow for the development of agents that can be specifically targeted to cancer to inhibit signaling pathways for cell replication, differentiation, and survival. Such anticancer targeted therapies include small-molecule tyrosine kinase inhibitors, antisense messenger RNA inhibitors, and antibodies, which are in various stages of clinical development (1-3).
Although these approaches have resulted in clinical benefit for selected cancer subtypes, there are a number of limitations to targeting only single genes or gene products (1, 4). An alternative approach examined here involves agents that are capable of delivering a payload directly and specifically to receptor targets that need not be overexpressed or essential for survival. We and others have proposed an approach (“molecular Velcro®”), wherein hetero-multivalent ligands are designed to target to cell surface receptor combinations that are unique to the target cell (5-7).
Multivalent ligands consist of multiple binding moieties (pharmacophores) that are tethered together via chemical linkers. It is well known that multivalent binding can lead to high avidity and specificity in binding (6, 8, 9). A wide spectrum of binding moieties can be used, including small peptide fragments, truncated versions of antibodies, and carbohydrate analogues (10-13). Although monoclonal antibodies (mAbs) have found success in the clinic, the high molecular weight of mAbs is a drawback to their multimerization (14, 15). Small peptides, such as those used in our current study, do not share this limitation (7, 16).
Multivalent ligands can be homo-multivalent, with multiple copies of the same ligand, or they can be hetero-multivalent, with different types of ligands targeted to different types of receptors. Previous work has shown that homo-multivalent ligands exhibit increased avidity or potency and that flexible linkers of 20-50 Å provide the greatest enhancement of binding affinities (6, 8, 13, 17-19). However, in addition to requiring overexpression of a single receptor, homo-multivalent constructs cannot unequivocally distinguish statistical proximity effects from the non-covalent crosslinking (clustering) of receptors which would be needed for hetero-multivalent interactions. Thus, demonstration of receptor non-covalent crosslinking requires the use of hetero-multivalent constructs.
To evaluate the binding of hetero-bivalent ligands to their corresponding receptors, it was necessary to construct and stringently characterize cell lines that expressed one, or both, of the target receptors. In the current proof-of-concept studies, three different G-protein-coupled receptors (GPCRs) were chosen as target gene products: the human delta-opioid receptor, δOR, the human melanocortin receptor subtype 4, MC4R; and the human cholecystokinin-2 receptor, CCK2R. These were co-expressed in combinations of MC4R + δOR and MC4R + CCK2R for testing of Deltorphin-MSH7 and MSH7-CCK6 heterobivalent structural constructs, respectively.
Here, CHO cell lines were engineered to transiently co-express the MC4R and δOR receptors and were characterized by lanthanide-based time-resolved fluorescence (TRF) saturation binding assay using Europium-labeled monomeric ligands; Eu-NDP-α-MSH and Eu-DPLCE, respectively. An Deltorphin II-MSH7 heterobivalent ligand was synthesized and binding affinity determined in cells expressing one or both receptors. In another system, stable co-expression of the MC4R and CCK2R receptors was successfully established in the Hek293 cell line. This engineered line and derivatives were tested for their ability to bind the corresponding monomeric ligands as well as a heterobivalent ligand containing both MSH7 and CCK6 pharmacophores. In both cell systems, we observed similar results demonstrating that heterobivalent constructs were bound to two different receptors with increased avidity.
These results demonstrate the feasibility of simultaneously targeting multiple receptors using heterobivalent ligands. Additionally, this study shows that cell lines can be constructed that are suitable for screening heterobivalent ligands in high-throughput mode. The methodology described and the dual receptor expression system will facilitate further development of novel ligands for targeting human cancers.
Materials and Methods
Cell Culture
The parental cell lines employed in the experiments were the CHO-K1 (ATCC, CRL-9618), Hek293 (ATCC, CRL-1573) cell lines. The MC4R stable transfected Hek293 cell line (Hek293/MC4R) was described previously (20). All cells were maintained at 37 °C and 5% CO2. All cell lines except for the CHO cells were maintained in Dulbecco's Modified Eagle's Medium (DMEM)/Ham's Nutrient Mixture F-12 supplemented with 10% fetal bovine serum (FBS). CHO cells were maintained in Ham's F-12 media supplemented with 10% FBS.
Ligand Synthesis
Europium labeled ligands (Eu-NDP-α-MSH, Eu-CCK8, Eu-DPLCE) and heterobivalent compounds DeltII-[PG]15-MSH7 and MSH7-Pego-[PG]6-Pego-CCK6 (Table 1) were prepared as previously described (20, 21) by solid-phase synthesis. Briefly, ligands were synthesized using a manual synthesizer (Torviq, Niles, MI) with Nα-Fmoc/tBu chemistry. Polyethylene glycol units (PEGO) were introduced by first adding diglycolic anhydride to the free Nα-terminal and then activating the free carboxylate as an imidazolide for attachment of 4,7,10-trioxa-1,13- tridecanediamine to form PEGO (13). DTPA chelator was attached to peptides on solid support using the HOBT ester method to decrease unwanted cross-linking and dimerization (21). DPLCE requires a freeNα- terminal to retain biological activity therefore the chelator was conjugated via the Aloc-protected ε-side chain of lysine. Ligands were cleaved from the resin using a TFA-scavenger cocktail and purified by HPLC. DPLCE was cyclized by air oxidation and then chelate ligands were labeled with Europium (III) chloride in neutral pH buffers. Excess Europium was removed by size exclusion chromatography (Sepak C18, Waters). Structures were characterized by Mass Spec (ESI, Termoquest, LCQ; MALDI-TOF, Brucker Reflex III) and quantitative HPLC.
Table 1.
List of compounds and their structures used in this study
| Compound | Structure |
|---|---|
| Eu-NDP-α-MSH |
1
Eu-(DTPA)-Ser-Tyr-Ser-Nle-Glu-His-DPhe-Arg-Trp-Gly-Lys-Pro- Val-NH2 |
| Eu-DPLCE | H-Tyr-c[DPen-Gly-Phe-Cys]-Phe-Lys(Eu-(DTPA))-NH2 |
| Eu-CCK8 | Eu-(DTPA)-Asp-Tyr-Nle-Gly-Trp-Nle-Asp-Phe-NH2 |
| Delt II-[PG]15-MSH7 | 2 H-Deltorphin II-[Pro-Gly]15-MSH7-NH2 |
| MSH7-Pego-[PG]6-Pego- CCK6 |
3 Ac-MSH7-PEGO-[Pro-Gly]6-PEGO-CCK6-NH2 |
| MSH7-Pego-[PG]3-K(Cy5)- Pego-CCK6 |
Ac-MSH7-PEGO-[Pro-Gly]3-Lys(Cy5)-PEGO-CCK6-NH2 |
Eu-(DTPA)

Deltorphin II MSH7
Tyr-DAla-Phe-Glu-Val-Val-Gly Ser-Nle-Glu-His-DPhe-Arg-Trp
PEGO
![]()
CCK6
Nle-Gly-Trp-Nle-Asp-Phe
The Cy5 labeled ligand (MSH7-pego-[PG]3-K(Cy5)-pego-CCK6) (Table 1) was synthesized by conjugation of Cy5 dye to the bivalent ligand using the lysine side chain. After incorporation of the first Pego linker, Nα-Fmoc-Nε- Mtt, lysine was incorporated into the sequence and then peptide synthesis completed. Peptide was cleaved from the resin and purified by preparative HPLC. Purified peptide was dissolved in DMSO and Cy5-NHS ester (Amersham Biosciences) was added. The reaction was monitored using analytical HPLC at 280nm. Labeled peptide was purified using size exclusion chromatography (C-18 Sep-Pak), lyophilized, and characterized using MALDI-MS.
Construction of Expression Vector(s) for MC4R, δOR and CCK2R
MC4R and δOR cDNA were cloned into the pBudCE4.1 vector (Invitrogen V532-20) which is a dual promoter vector capable of expressing two independent recombinant proteins. Using pcDNA3.1-MC4R as a template, full length MC4R was amplified using PCR primers with adapters containing Kpn I and Bgl II restriction enzyme sites. Primers were designed using the MC4R sequence (accession #: NM_005912): sense, 5−- TCA ATC GGT ACC ATG GTG AAC TCC- 3′; and antisense, 5′- GGT ACC AGA TCT GCT TAA TAT CTG CT-3′. A Kozak sequence (A/G NN ATG G) was also generated at the 5′ end of the MC4R PCR fragment. The fragment was digested with Kpn I / Bgl II and ligated into the Kpn I / Bgl II site between the EF-1 alpha promoter and BGH poly A of the pBudCE4.1 vector. The sequence of the recombinant plasmid was verified by sequencing both strands and named pBudCE-MC4R. The plasmid pcDNA3.1(+)/ Hygro containing full-length δOR cDNA (accession #: U07882) was provided by Henry Yamamura, and digested with Apa I and EcoR I, blunting the 5′- and 3′- protruding termini with T4 DNA polymerase, and subsequently subcloned into pBudCE4.1-MC4R, which was cut with Xba I, blunting the 5′ end with T4 DNA polymerase and dephosphorylated. This δOR cDNA insert was subcloned between the CMV promoter and SV40 poly A of pBudCE-MC4R. The resulting plasmid, termed pBudCE-MC4R-δOR was verified using restriction enzyme analysis and DNA sequencing.
For the CCK2R construct, a 1700 base pair fragment containing the full length CCK2 receptor gene (accession #: NM_176875) was PCR amplified using a panel of human lymph cDNA (Clontech, K1426-1) as a template. The sequences of the forward and reverse primers were: CAC CGG TCG ACC GGG GGC CAT GGA AGC TGC TAA AG and GCA GGT CGA CCC TTG TCA GAG, respectively. This purified PCR product was used as template DNA for a second PCR reaction using the following forward and reverse primers: CAC CAT GGA GCT GCT AAA GCT GAA CCG G and TCA GCC AGG GCC CAG TGT, respectively. These primers were designed with a CACC sequence at the 5′ end so that the PCR product could be directly cloned into the pcDNA3.1 TOPO vector (Invitrogen, K4900-01). Subsequently, the CCK2R gene was cut out of the pcDNA3.1 TOPO vector using HindIII and XhoI, and inserted into the pcDNA3.1/Zeo(+) vector at the HindIII and XhoI sites. The constructs mentioned above were verified via DNA sequencing.
Construction of Cell Lines Co-expressing MC4R and δOR (CHO/MC4R/δOR Cells)
Cell transfections were performed using FuGENE 6 transfection reagent (Roche, 1814-443). For establishment of cell lines transiently expressing both MC4R and δOR receptors, CHO cells were plated on Wallac B&W Isoplate TC (Wallac/PerkinElmer, 1450-583) 96-well plates at a density of 20,000 cells per well, allowed to adhere for 24 h, and then transfected with pBudCE-MC4R-δOR vector. Forty-eight hours post transfection, cells were tested for MC4R and δOR cell surface expression by MC4R and δOR binding assay using Eu-NDP-α-MSH and Eu-DPLCE, respectively, as described previously (20, 21).
Transient transfection efficiencies were determined by co-transfection of pBudCE-MC4R-δOR vector and green fluorescent protein (GFP) reporter constructs (Invitrogen, A-150228) in CHO cells. Cells were harvested 48 h post transfection and analyzed by fluorescence activated cell sorting (FACS) for GFP expression.
Construction of Stable Transfected Cell Lines (Hek293/CCK2R and Hek293/MC4R/CCK2R Cells)
Hek293 cells were transfected with the pcDNA3.1/Zeo(+)-CCK2R construct. After 48 hours, replacement media contained 0.1 mg/mL zeocin. Single stable transfectants were observed in 3-4 weeks. The CCK2R surface expression level for each clone was measured by ligand binding assay using Eu-CCK8. A stable dual expressing cell line was made by transfecting Hek293/MC4R cells with the CCK2R construct. After 48 hours, dual selection media included both 0.1 mg/mL zeocin (Invitrogen, 450430) and 0.4 mg/mL geneticin (Gibco, 11811-031). Clones were tested for ligand binding at both receptors using Eu-CCK8 and Eu-NDP-α-MSH.
In Cyto Lanthanide-based Time-resolved Fluorescence Binding Assays
Lanthanide-based binding assays were performed on whole CHO or Hek293 cells as previously described (20, 21). Saturation- and competitive-binding assays confirmed the functional expression of cell surface receptors using monomeric ligands. For saturation binding, increased concentration of labeled ligands Eu-NDP-α-MSH, Eu-DPLCE and Eu-CCK8 were used for binding to the MC4R, δOR, and CCK2R respectively. Non-specific binding was determined in the presence of 10 μM NDP-α-MSH, 10 μM Naloxone or 1 μM CCK8, respectively. The competitive binding assay was used to evaluate heterobivalent ligand binding on engineered cells, i.e. increasing amounts of the Delt II-[PG]15-MSH7 ligand competed with known amounts of Eu-labeled ligand (10 nM Eu-NDP-α-MSH or 10 nM Eu-DPLCE), or the MSH7-Pego-[PG]6-Pego-CCK6 ligand competed with 10 nM Eu- NDP-α-MSH or 0.1 nM Eu-CCK8.
For screening, one-point ligand binding assays were performed for each clone. Respectively, MC4R, δOR or CCK2R expression was determined by incubation with 10 nM Eu-NDP-α-MSH, 10 nM Eu-DPLCE, or 0.1 nM Eu-CCK8 in the presence or absence of unlabeled 10 μM NDP-α-MSH, 10 μM Naloxone or 1 μM CCK8. Specific binding was determined as the difference between values in the absence (total) and presence (nonspecific) of competing unlabeled ligand.
Immunocytochemistry
Cells were grown on coverslips to ∼70% confluence. The MC4R antibody (RDI, RTMC4Rabr) recognizes an epitope on the cell exterior. Hence, live cells were incubated with this antibody (10 μg/mL) for 5 min, washed in antibody-free buffer (2 × 5 min), and fixed with 2% paraformaldahyde. Prior to labeling with secondary antibody (anti-rabbit Alexa 488, Invitrogen, A-11008), fixed cells were incubated with 25mM glycine and permeabilized with 0.1% TritonX-100 (22). Antibodies for δOR (mouse, Neuromics, RA10101) and CCK2R (rabbit, Abcam Inc, AB13173) recognize intracellular epitopes. Thus, cells were incubated with these antibodies following permeabilization.
For dual labeling of MC4R and δOR, MC4R labeled coverslips were incubated sequentially with the primary antibody to δOR, washed, and then labeled with a Texas Red conjugated anti-mouse antibody (Jackson Labs).
Since the antibodies for MC4R and CCK2R are both raised in rabbit, when dual labeling cells for these receptors, an intermediate blocking strategy was required to assure specificity of secondary antibodies for each specific primary (23). MC4R labeled coverslips were incubated overnight with a 10X concentration of non-labeled anti-rabbit IgG to block free anti-rabbit IgG sites remaining on the MC4R specific polyclonal antibody, washed (3 × 5 min) in phosphate buffered saline and incubated with the CCK2R antibody for 60 minutes followed by washing and incubation with a secondary anti-rabbit IgG labeled with Alexa480 (Invitrogen) for 45 min at 25°C. All processed coverslips were mounted onto glass slides using a 50% glycerol:saline solution containing the antibleach agent paraphenylendiamine (0.1%). To assure 100% blocking of rabbit anti-MC4R sites, a control was included in all batches, wherein the primary CCK2R antibody was omitted prior to incubation with secondary antibody. Any Texas Red labeling on these coverslips indicated that the blocking step was ineffective and the entire batch was discarded.
Cell Fluorescence Imaging
An inverted Olympus IX70 microscope equipped with a 40x 1.4 NA ultrafluor objective, and a 100 W Hg lamp as the excitation source was used for cell imaging experiments. For imaging dual receptor expressing cells using the Cy5 tagged ligand (Table 1), a # 1 coverslip bearing live Hek293/MC4R/CCK2R cells was placed in a 37°C chamber mounted on the microscope stage. Cy5 fluorescence was excited with a 20 nm BP filter centered at 640 nm and emitted light collected through a 30 nm filter centered at 690 nm. A liquid cooled CCD camera (Photometrics CH-250) was used to acquire images.
Cytotoxicity of Monovalent and Bivalent Ligands
Hek293 cells expressing MC4R and/or CCK2R were plated at a density of 20,000 cells/well into 96-well dishes and allowed to attach overnight. Cells were treated with 1 nM or 1 μM of NDP-α-MSH, or CCK8 or MSH7-Pego-[PG]6-Pego-CCK6 for 24 or 48 hours. Viable cell count was determined with a celltiter-Glo luminescent cell viability assay (Promega; G7571). Cytotoxicity was quantified as the percentage of treated cell viability relative to untreated controls, thus a higher relative viability corresponds to lower toxicity.
Data Analysis
Saturation and competition binding data were analyzed by non-linear regression analysis using GraphPad Prism (GraphPad Software, San Diego, CA). Saturation binding data were fitted to a classical one site binding (hyperbola) equation and competitive binding data were fitted to a classical one site binding competition equation. For saturation binding assays, Kd values were determined after correction for non-specific binding as the concentration that yielded half-maximal binding. For competitive binding assays, the IC50 was determined after correction for nonspecific binding as the concentration of unlabeled ligand sufficient to compete off 50% of the labeled ligand.
Results
Creation and characterization of transient systems coexpressing MC4R and δOR
Although tremendous efforts were made to construct a cell line stably expressing both MC4R and δOR, this was not achieved (data not shown). However, the MC4R and δOR were observed to co-express well in CHO cells for a short period of time using the pBudCE-MC4R-δOR vector. Hence, a transient dual receptor-expressing system using the same cell line and same vector was characterized. Maximal surface expression of both receptors in CHO cells was determined by in cyto lanthanide-based time-resolved fluorescence binding assays to be 48 h post transfection (supplemental Figure S1). All subsequent binding assays were performed at this time point. Saturation-binding assays showed that Eu-DPLCE bound to the δOR with a Kd of 10.5 ± 2.6 nM and a Bmax of 24,000 ± 2,000 AFU (Figure 1A); and that Eu-NDP-α-MSH bound to the MC4R with a Kd of 5.6 ± 2.7 nM and a Bmax of 7,700 ± 1,400 AFU (Figure 1B).
Figure 1. MC4R and δOR saturation binding and competitive binding analysis.

A. Saturation binding of Eu-DPLCE ligand to δOR in CHO/MC4R/δOR cells. The curve shows δOR specific binding only (total-nonspecific). From these data, the Kd = 10.5 ± 2.6 nM and Bmax = 24,000 ± 2,000 AFU (R2 = 0.91). Each data point indicates the average of four samples, with error bars indicating the standard error of the mean. B. Saturation binding assay of Eu-NDP-α-MSH ligand to MC4R in CHO/MC4R/δOR cells. The curve shows MC4R specific binding only. From these data, the Kd = 5.6 ± 2.7 nM and Bmax = 7,700 ± 1,400 AFU (R2 = 0.83). C-D. Competitive binding to CHO cells co-expressing MC4R and δOR. C. Increasing concentrations of Naloxone were added in the presence of 10 nM Eu-DPLCE. From these data, the IC50 was 65 nM with R2 = 0.90. D. Increasing concentration of NDP-α-MSH were added to cells in the presence of 10 nM Eu- NDP-α-MSH. From these data, IC50 was 0.77 nM with R2 = 0.89.
To further investigate these dual expressing cells, competitive binding assays were performed (Figure 1C and 1D). Naloxone or NDP-α-MSH effectively displaced Eu-DPLCE or Eu-NDP-α-MSH with IC50 values of 65 nM (R2 = 0.90) or 0.77 nM (R2 = 0.89), respectively. Importantly, testing of heterobivalent ligand binding using this cell system required that blocking one receptor would not interfere with the affinity of the other receptor and vice-versa. To examine this, competitive binding studies of each monomeric ligand to its receptor were performed in the presence of excess competitor for the heterologous receptor. The IC50 value for NDP-α-MSH to displace Eu-NDP-α-MSH was 0.65 nM when δOR was blocked with Naloxone, which is similar to the IC50 value of 0.77 nM measured in the absence of naloxone (as shown in Figure 1D). Similarly, the IC50 of Naloxone to displace Eu-DPLCE from the δOR was 55 nM in the presence of excess NDP-α-MSH, compared to an IC50 value of 65 nM in the absence of NDP-α-MSH (Figure 1C). Hence, blocking the δOR did not interfere with the binding affinity of the MC4R and vice-versa.
Transfection efficiency was determined for this transient dual expression system using green fluorescence protein (GFP) because it can be quantified at the single-cell level by flow cytometry and the efficiency of target gene expression roughly corresponds with GFP expression (24-26). CHO cells were co-transfected with GFP and pBudCE-MC4R-δOR plasmid and the percentage of cells expressing GFP was measured 48 hours after transfection. Transfection efficiency was 33 ± 4 % (n = 9) which is similar to that typically achieved using lipid-based transfection reagents.
Expression of δOR and MC4R simultaneously in the same cells within the transfected population was also examined using immunocytochemistry (supplemental Figure S2). Only surface resident MC4 receptors were labeled and a peripheral distribution was clearly observed. Since the procedure for labeling the δOR allowed access to intracellular compartments, a significant amount of δOR was observed within the cell and determination of surface expression was less clear. But, overlap of signal from both antibodies at the cell periphery was observed within individual cells, and we conclude that the transfected cells are expressing both receptors types. Control samples without primary antibodies were devoid of signal (data not shown).
Construction and characterization of cell lines stably expressing both MC4R and CCK2R
A cell line that stably expressed both the MC4R and CCK2R (Hek293/MC4R/CCK2R) was constructed as described in the methods section. Immunocytochemistry was used to evaluate the pattern of expression in the derived lines. As shown in Figure 2A & 2B, and supplemental Figure S6, many cells express both receptors simultaneously. These cells were evaluated for surface expression levels by saturation binding assays. Saturation binding analysis showed that Eu-NDP-α-MSH bound to Hek293/MC4R/CCK2 cells with a Kd of 8.3 ± 1.9 nM and a Bmax of 732,000 ± 59,000 AFU, as shown in Figure 3A. This Kd value is consistent with our previous results using this ligand in MC4 single receptor-expressing cells (20). Similarly, saturation analysis of Eu-CCK binding to CCK2R was achieved with a Kd of 34.6 ± 3.9 nM, and a Bmax of 1,600,000 ± 83,000 AFU (Figure 3B). This Kd value was consistent with that observed in the cell line expressing the CCK2R alone, e.g. Kd of 38.6 ± 3.8 nM using this same Eu-CCK ligand.
Figure 2. Immuno-labeling of MC4R and CCK2R and the distribution of heterobivalent ligand labeling in MC4R and CCK2R expressing cells.

A & B. Images of Hek293 cells, that stably expressed both the CCK2R and MC4R, labeled with antibodies against the MC4R and CCK2R receptors. C & D. Images of cells incubated for 3 minutes with 0.8 nM of Cy5 labeled MSH-CCK ligand, then washed with ligand free media. The ligand distribution was determined immediately following the rinse (C), and 7 minutes thereafter (D). Scale bar = 20 μM for both images pairs.
Figure 3. MC4R and CCK2R saturation binding and Scatchard plot analysis.

A. Saturation curve of Eu-NDP-α-MSH obtained from the MC4R and CCK2 dual expression cell line. The figure shows total binding (■) and binding in the presence of 10 μM NDP-α-MSH (▲). From these data, the Kd = 8.3 ± 1.9 nM, and Bmax = 732,000 ± 59,000 AFU. Lines represent the computer modeled best fit of the data using GraphPad Prism software using the non-linear regression, one site-binding equation, with a R2 value of 0.81. Each data point indicates the average of four samples, with error bars indicating the standard error mean. B. Saturation curve of Eu-CCK obtained from the MC4R and CCK2 dual expression cell line. The figure shows total binding (■) and binding in the presence of 1 μM CCK8 (▲). From these data, the Kd = 34.6 ± 3.9 nM, and Bmax = 1,600,000 ± 83,000 AFU, with a R2 value of 0.96.
Determination of receptor numbers on cells
Standard curves for Eu-labeled ligands were generated by adding increasing amounts of Eu-NDP-α-MSH, Eu-DPLCE or Eu-CCK8 to quadruplicate wells of a 96-well plate. These were used to generate a relationship between fluorescence intensities and ligand concentrations (supplemental Figure S3). These calibration data allowed for the determination of the amount of ligand present at the Bmax obtained from the saturation binding data shown in Figures 1A & 1B and Figures 3A & 3B. For binding to the MC4 receptor on CHO/MC4R/δOR cells, the Bmax was 7,700 ± 1,400 AFU which corresponds to 0.76 ± 0.14 fmol/well. For δOR binding, the Bmax was 24,000 ± 2,000 AFU which corresponds to 4.30 ± 0.36 fmol/well. The average cell number per well was 75,000 ± 2,300 cells (n = 4). After correcting for transfection efficiency (33 ± 4 %), it was calculated that there were 18,000 ± 3,300 MC4R/cell and 100,000 ± 8,600 δOR/cell. Thus δOR outnumbered MC4R by a ratio of approximately 6:1.
Taking a similar approach, receptor numbers were determined in Hek293/MC4R/CCK2R cells. It was calculated that there were 1,100,000 ± 56,000 CCK2 receptors/cell and 640,000 ± 52,000 MC4R receptors/cell in this stably expressing line. Hence, the CCK2 receptors outnumbered MC4R by a ratio of approximately 2:1.
Evaluation of heterobivalent ligand binding
Ligand binding assays for heterovalent ligands are more complicated than for monovalent ligands, as binding to multiple different receptor types must be validated. For the purpose of testing heterobivalent constructs there are two ways that binding assays may be designed, as illustrated in Figure 4. In one type of experiment, heterodimers are tested on two different cell lines; one that expresses a single complementary receptor (Figure 4A1) and another that expresses both of the complementary receptors (Figure 4A2). This system was used to test the binding of the heterobivalent MSH-CCK ligand. Alternatively, heterodimeric binding can be assessed using a single cell line that expresses both complementary receptors in the presence (Figure 4B1) or absence (Figure 4B2) of receptor blockage using excess unlabeled ligand. This system was used to test the binding of the heterobivalent MSH-δOR ligand.
Figure 4. Evaluation of heterobivalent ligand binding.
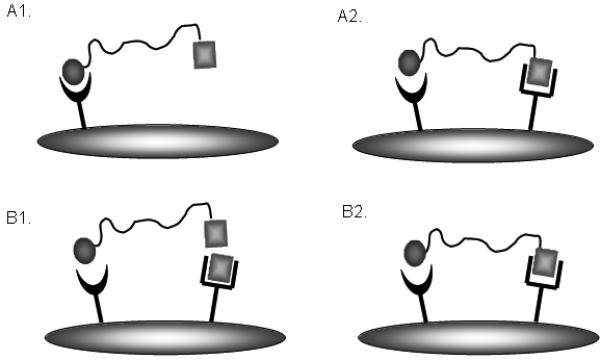
There are two different approaches by which heterobivalent binding can be evaluated. One approach uses two different cell types (Figure A1 & A2); one that expresses a single complimentary receptor and another that expresses both of the complimentary receptors, while another approach uses the same cell type but blocks binding at one receptor by addition of an agent (Figure B1 & B2).
Heterobivalent ligands were prepared to target the MC4R-δOR and MC4R-CCK2R combinations. DeltII-[PG]15-MSH7 was constructed and tested for binding to the MC4R and δOR receptors as described above. This ligand competed Eu-NDP-α-MSH with an IC50 value of 3.3 ± 1.8 nM and 159.6 ± 46.3 nM in the absence and presence of naloxone (a δOR antagonist), respectively (number of replicates, n = 5, p < 0.01). Figure 5A shows a representative binding curve in competition with Eu-NDP-α-MSH in the absence (dimer) and presence (monomer) of naloxone. The DeltII-[PG]15-MSH7 ligand competed with Eu-DPLCE with IC50 values of 230 ± 74 nM and 500 ± 90 nM in the absence and presence of excess NDP-a-MSH (n = 5, p > 0.05), respectively. From the hMC4R data, the heterodimer bound with much higher affinity when both complimentary receptors are available, compared to its binding when the δOR was blocked. In contrast, binding to the δOR did not appear to be affected by the availability of the second receptor (MC4R). These results were interpreted to indicate that the enhancement of divalent binding was only apparent at the less abundant of the two receptors. In other words, all of the MC4 receptors can be engaged in a heterobivalent complex whereas only 1/6 of the δOR can be thus occupied. The remaining 5/6 of the δOR receptors thus must bind monovalently. In all, these data are consistent with receptor crosslinking and heterobivalent interactions (supplemental Figure S4). Notably, a short linker ligand, DeltII-[PG]3-MSH7 (18-atom long linker) showed no significant increase of binding affinity between MC4R monovalent ( IC50 = 110 ± 9 nM) and bivalent (IC50 = 180 ± 30 nM) binding modes. The observation that smaller compounds show no enhancement suggests that the enhancement with longer linkers was not simply due to a local concentration (statistical) effect.
Figure 5. Representative competitive binding assay at the MC4R.

A. Increasing concentrations of the MSH-Deltorphin heterobivalent ligand were added to CHO/MC4R/δOR cells in the presence of 10 nM Eu-DTPA-NDP-α-MSH and the absence (dimer) and presence (monomer) of competing naloxone. For dimer binding (absence of naloxone), the IC50 = 3.2 nM with R2 = 0.83; for monomer binding (presence of naloxone), the IC50 = 134.0 nM with R2 = 0.93. B. Increasing concentrations of the MSH-CCK heterobivalent ligand were added to cells in the presence of 10 nM Eu-DTPA-NDP-α-MSH. For dimer binding, Hek293/CCK2R/MC4R cells were used and for monomer binding, Hek293/MC4R cells were used. For dimer binding, the IC50 = 4.5 nM with R2 = 0.88; for monomer binding, the IC50 = 349.5 nM with R2 = 0.96.
In the alternative system, an MSH-CCK heterodimer, MSH7-Pego-[PG]6-Pego-CCK6, was constructed. This ligand was tested using the competitive binding assay in the MC4R or CCK2R single receptor cell lines (i.e., “monomeric” binding) as well as the dual receptor MC4R + CCK2R cell line (i.e., “dimeric” binding), as outlined in Figures 4A1 & 4A2. This ligand competed with Eu-NDP-α-MSH as a monomer with an IC50 value of 251.4 ± 38.8 nM (n = 5) and as a dimer with an IC50 value of 3.1 ± 0.3 nM (n = 5, *P < 0.01); Thus, an 81-fold enhancement was observed. Figure 5B shows a representative competitive binding curve for dimeric and monomeric binding to the MC4R. For the CCK2 receptor, this ligand competed with Eu-CCK with an IC50 value of 190.0 ± 51.3 nM and 167.5 ± 20.2 nM for monomeric and dimeric binding, respectively (n = 5, not significant). These results were consistent with those presented above; i.e., that the less abundant receptor showed significant enhancement for divalent, compared to monovalent interactions, whereas the more abundant receptors did not.
Imaging of Cy5 labeled heterobivalent ligand binding and ligand-induced receptor internalization
Binding of the heterobivalent MSH-CCK ligand tagged with the fluorophore Cy5 to the dual CCK2R/MC4R expressing line was also evaluated at the cell level. Immediately following incubation with the ligand (15 sec w/ 0.8 nM), significant binding to the cell surface was observed. After 3 minutes the ligand was washed from the cell chamber, and receptor distribution was followed (Figure 2C). By 10 minutes, all ligand was observed to be in punctate structures assumed to be receptor capping zones (Figure 2D). To determine if the receptor/ligand complex was significantly internalized, CCK ligand (50 μM) was added to the chamber in an attempt to compete ligand from receptors remaining on the surface. No significant loss of ligand was observed with this strategy, indicating that the receptor/ligand complex had been substantially internalized.
Cytotoxicity of monovalent and bivalent ligands
Synthetic heterobivalent constructs are novel, and may have unanticipated effects on cell behavior, such as proliferation or survival. Viability was assayed in cells expressing MC4R or CCK2R or both following incubation with either monovalent ligands or bivalent ligands. None of the monovalent or bivalent ligands had a toxic effect on any of the cell lines at a 1 nM concentration. However, at 1 μM, there was reduced viability in dual receptor expressing cells following 24 and 48 h of treatment with either CCK or the heterobivalent ligands (supplemental Figure S5A-F).
Discussion
Expressing pairs of GPCRs in heterologous cell systems is a useful approach to investigate and characterize the binding of bivalent pharmacophores (27). Pairs of functionally-related GPCR subtypes have been co-overexpressed in the same cell, such as the β2- and β3 –adrenoceptors (28), and the CXC chemokine receptors, CXCR1 and CXCR2 (29). The current work employed hetero-expression of GPCRs as system for proof-of-concept studies to evaluate heterobivalent ligand binding.
In the current study, we readily developed a stable cell line expressing both the MC4R and the CCK2R receptor in the Hek293 cell line. It seems reasonable to speculate that MC4R and CCK2R receptors may have a functional relationship as both are involved in the control of meal size and food intake (30, 31). Although the other receptor pair MC4R and δOR, are expressed in the brain, they are unlikely expressed in the same cells, as their expression profiles are unique, and they are functionally unrelated. Establishing stable cell lines expressing both MC4R and δOR receptors was a particularly elusive challenge. Several different expression systems, including two C-terminal truncated receptors (32) and a pDisplay vector (33), were investigated in six cell lines with different biochemistries (34-36) without success. However, it was interesting to observe that one cell clone achieved high expression levels of both receptors detected by ligand binding assay, but this clone underwent a distinctive morphological change which led to cell death after 20 days. These observations have led us to surmise that cross-talk prevented co-expression of these heterologous receptors in the same cell. Numerous recent studies have indicated that cross-talk can occur between GPCR classes, which may give rise to synergistic or more complicated effects (37-40). For the receptor pair MC4R and δ-opioid, it is possible that there may be cross-talk between the receptors that results in a pro-apoptotic signal. Further experiments will be needed to confirm this molecular mechanism.
To test the dual expressing systems, heterobivalent ligands with MSH7-deltorphin and CCK6-MSH7 were synthesized by the solid phase method. The constructs tested were part of larger families of heterobivalent constructs that are described elsewhere (41). These were connected by proline-glycine repeats as the core of the linker unit (Table 1). In one case (MC4R-δOR) these were tested in the same cell line in the presence and absence of heterologous blockade, and in the other (CCK2R-MC4R) these were tested in cell lines that expressed either one or both receptors. It is interesting to observe that, in both cases, the receptor with lower abundance (MC4R) showed significant enhancement in the binding affinity when both receptors were available (dimer) compared to cases where only the single receptor was available (monomer), which indicated the heterobivalent ligands were engaged and bound to both receptors. The finding that no enhancement was observed when binding was determined at the higher abundance receptor is expected (supplemental Figure S4). After the lower abundance receptors are saturated by bivalent interactions, the remaining higher abundance receptors are only available to bind the ligands as monomers, which is weak binding. This finding is in agreement with the mathematical model that the absolute number of receptors and the ratio of receptor expression on the target cell is critical to achieving specificity (42).
Previous work from our laboratory using homobivalent ligands with relatively short linker lengths demonstrated enhancement by “statistical” binding, where affinity is increased simply via increased local concentration and not receptor “cross-linking” (13, 20, 21, 43). In order to discriminate receptor cross-linking effects from statistical effects, a series of MSH7-deltorphin heterobivalent ligands with variable linker lengths and rigidities were designed, synthesized and screened for binding affinities using a cell system that expressed both the MC4R and the δOR as described in this study (41). A construct with the optimal linker length for binding enhancement, viz., DeltII-[PG]15-MSH7 with a 90-atom linker, was used in the current study. Short linker constructs evidenced no enhancement. A similar series of, CCK6-MSH7 heterobivalent ligands were also designed, synthesized and screened. The optimally binding construct, viz. MSH7-Pego-[PG]6-Pego-CCK6 (76 atoms) was used herein and exhibited potent binding compared to compounds with shorter linkers, i.e. MSH-Pego-CCK6 (20 atoms) and MSH-Pego-Pego-CCK6 (40 atoms) which exhibited no significant binding enhancement between single and dual expressing cells (data not shown).
In addition to the efficient cell binding of these heterobivalent ligands, the internalization of these ligand-receptor complexes will be important for drug targeting, imaging and eventual delivery of therapies. Internalization of these heterobivalent constructs would amplify any potential imaging signal, and could equally allow for payloads to be delivered to the cell nuclei. In addition to delivering a therapeutic payload, it was equally important to assess whether these novel agents induced any toxicity of their own. Although cytotoxicity may often be a goal in cancer, these agents may also be useful to deliver positive therapies, such as gene-replacements to the nucleus (vide supra). Notably, the bivalent MSH7-CCK6 ligand used in this study showed no reduction in viability at either low (1 nM) or high doses (1 μM) in the non-targeted cells, yet there was toxicity in the target cells. This toxicity was also observed with CCK alone, suggesting that the effect was not specific to receptor crosslinking and was a product of engaging the CCK-R. This is an area that warrants further investigation.
In summary, we have developed a system to investigate the effects of the binding of heterobivalent ligands to two different cell surface receptors in target cells (i.e. those that co-express both receptors). Such an approach may be useful for the development of potent ligands for targeting physiologically relevant receptor combinations, especially in cancers, such as, Her2/Her3, Her2/Her4, etc. Using the in vitro cell systems described herein, developing the multivalent ligands for target combinations in pancreatic cancer (44) are underway.
Supplementary Material
Acknowledgements
This work was supported by research grants from the National Institutes of Health Grants CA123547 and CA95944 and Grant 06-006 from the Arizona Biomedical Research commission to RJG.
Grant Support: National Institute of Health Grants CA123547 and CA95944 and Grant 06-006 from the Arizona Biomedical Research Commission to RJG
Abbreviations
- hδOR
human delta-opioid receptor
- hMC4R
human melanocortin 4 receptor
- hCCK2R
human cholecystokinin-B receptor
- CHO
chinese hamster ovary
- Hek293
human embryonic kidney
- NDP-α-MSH
[Nle4, D-Phe7]-α-melanocyte stimulating hormone
- DPLCE
[D-Pen2,L-Cys5]Enkephalin
- CCK8
cholecystokinin-8
- MSH7
melanocyte stimulating hormone-7
- CCK6
cholecystokinin-6
- Deltorphin II
δ-opioid agonist
- Eu
europium
- DTPA
diethylenetriaminepentaacetic acid
- DMEM
Dulbecco's modified eagle Medium
- FBS
fetal bovine serum
- BSA
bovine serum albumin
- PEGO
polyethylene glycol
- GFP
green fluorescent protein
- FACS
fluorescent activated cell sorting
- AFU
average fluorescence units
- CMV
cytomegalovirus
- EF-α
elongation factor-1α
Footnotes
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed
Reference List
- 1.Finley RS. Overview of targeted therapies for cancer. Am J Health Syst Pharm. 2003;60:S4–10. doi: 10.1093/ajhp/60.suppl_9.S4. [DOI] [PubMed] [Google Scholar]
- 2.Lindsay MA. Target discovery. Nat Rev Drug Discov. 2003;2:831–8. doi: 10.1038/nrd1202. [DOI] [PubMed] [Google Scholar]
- 3.Ross JS, Schenkein DP, Pietrusko R, et al. Targeted therapies for cancer 2004. Am J Clin Pathol. 2004;122:598–609. doi: 10.1309/5CWP-U41A-FR1V-YM3F. [DOI] [PubMed] [Google Scholar]
- 4.Kaptain S, Tan LK, Chen B. Her-2/neu and breast cancer. Diagn Mol Pathol. 2001;10:139–52. doi: 10.1097/00019606-200109000-00001. [DOI] [PubMed] [Google Scholar]
- 5.Gillies RJ, Hruby VJ. Expression-driven reverse engineering of targeted imaging and therapeutic agents. Expert Opin Ther Targets. 2003;7:137–9. doi: 10.1517/14728222.7.2.137. [DOI] [PubMed] [Google Scholar]
- 6.Handl HL, Vagner J, Han H, Mash E, Hruby VJ, Gillies RJ. Hitting multiple targets with multimeric ligands. Expert Opin Ther Targets. 2004;8:565–86. doi: 10.1517/14728222.8.6.565. [DOI] [PubMed] [Google Scholar]
- 7.Shadidi M, Sioud M. Selective targeting of cancer cells using synthetic peptides. Drug Resist Updat. 2003;6:363–71. doi: 10.1016/j.drup.2003.11.002. [DOI] [PubMed] [Google Scholar]
- 8.Kiessling LL, Gestwicki JE, Strong LE. Synthetic multivalent ligands in the exploration of cell-surface interactions. Curr Opin Chem Biol. 2000;4:696–703. doi: 10.1016/s1367-5931(00)00153-8. [DOI] [PubMed] [Google Scholar]
- 9.Mammen M, Choi S-K, Whitesides GM. Polyvalent interactions in biological systems: implications for design and use of multivalent ligands and inhibitors. Angew Chem Int Ed. 1998;37:2754–94. doi: 10.1002/(SICI)1521-3773(19981102)37:20<2754::AID-ANIE2754>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 10.Dam TK, Roy R, Das SK, Oscarson S, Brewer CF. Binding of multivalent carbohydrates to concanavalin A and Dioclea grandiflora lectin. Thermodynamic analysis of the “multivalency effect”. J Biol Chem. 2000;275:14223–30. doi: 10.1074/jbc.275.19.14223. [DOI] [PubMed] [Google Scholar]
- 11.Pluckthun A, Pack P. New protein engineering approaches to multivalent and bispecific antibody fragments. Immunotechnology. 1997;3:83–105. doi: 10.1016/s1380-2933(97)00067-5. [DOI] [PubMed] [Google Scholar]
- 12.Todorovska A, Roovers RC, Dolezal O, Kortt AA, Hoogenboom HR, Hudson PJ. Design and application of diabodies, triabodies and tetrabodies for cancer targeting. J Immunol Methods. 2001;248:47–66. doi: 10.1016/s0022-1759(00)00342-2. [DOI] [PubMed] [Google Scholar]
- 13.Vagner J, Handl HL, Gillies RJ, Hruby VJ. Novel targeting strategy based on multimeric ligands for drug delivery and molecular imaging: homooligomers of alpha-MSH. Bioorg Med Chem Lett. 2004;14:211–5. doi: 10.1016/j.bmcl.2003.09.079. [DOI] [PubMed] [Google Scholar]
- 14.Schrama D, Reisfeld RA, Becker JC. Antibody targeted drugs as cancer therapeutics. Nat Rev Drug Discov. 2006;5:147–59. doi: 10.1038/nrd1957. [DOI] [PubMed] [Google Scholar]
- 15.Segota E, Bukowski RM. The promise of targeted therapy: cancer drugs become more specific. Cleve Clin J Med. 2004;71:551–60. doi: 10.3949/ccjm.71.7.551. [DOI] [PubMed] [Google Scholar]
- 16.Shadidi M, Sioud M. Selection of peptides for specific delivery of oligonucleotides into cancer cells. Methods Mol Biol. 2004;252:569–80. doi: 10.1385/1-59259-746-7:569. [DOI] [PubMed] [Google Scholar]
- 17.Gestwicki JE, Cairo CW, Strong LE, Oetjen KA, Kiessling LL. Influencing receptor-ligand binding mechanisms with multivalent ligand architecture. J Am Chem Soc. 2002;124:14922–33. doi: 10.1021/ja027184x. [DOI] [PubMed] [Google Scholar]
- 18.Sharma SD, Jiang J, Hadley ME, Bentley DL, Hruby VJ. Melanotropic peptide-conjugated beads for microscopic visualization and characterization of melanoma melanotropin receptors. Proc Natl Acad Sci U S A. 1996;93:13715–20. doi: 10.1073/pnas.93.24.13715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shrivastava A, von Wronski MA, Sato AK, et al. A distinct strategy to generate high-affinity peptide binders to receptor tyrosine kinases. Protein Eng Des Sel. 2005;18:417–24. doi: 10.1093/protein/gzi049. [DOI] [PubMed] [Google Scholar]
- 20.Handl HL, Vagner J, Yamamura HI, Hruby VJ, Gillies RJ. Lanthanide-based time-resolved fluorescence of in cyto ligand-receptor interactions. Anal Biochem. 2004;330:242–50. doi: 10.1016/j.ab.2004.04.012. [DOI] [PubMed] [Google Scholar]
- 21.Handl HL, Vagner J, Yamamura HI, Hruby VJ, Gillies RJ. Development of a lanthanide-based assay for detection of receptor-ligand interactions at the delta-opioid receptor. Anal Biochem. 2005;343:299–307. doi: 10.1016/j.ab.2005.05.040. [DOI] [PubMed] [Google Scholar]
- 22.Lynch RM, Fogarty KE, Fay FS. Modulation of hexokinase association with mitochondria analyzed with quantitative three-dimensional confocal microscopy. J Cell Biol. 1991;112:385–95. doi: 10.1083/jcb.112.3.385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lynch RM, Carrington W, Fogarty KE, Fay FS. Metabolic modulation of hexokinase association with mitochondria in living smooth muscle cells. Am J Physiol. 1996;270:C488–99. doi: 10.1152/ajpcell.1996.270.2.C488. [DOI] [PubMed] [Google Scholar]
- 24.Chen R, Greene EL, Collinsworth G, et al. Enrichment of transiently transfected mesangial cells by cell sorting after cotransfection with GFP. Am J Physiol. 1999;276:F777–85. doi: 10.1152/ajprenal.1999.276.5.F777. [DOI] [PubMed] [Google Scholar]
- 25.Davies A, Greene A, Lullau E, Abbott WM. Optimisation and evaluation of a high-throughput mammalian protein expression system. Protein Expr Purif. 2005;42:111–21. doi: 10.1016/j.pep.2005.03.012. [DOI] [PubMed] [Google Scholar]
- 26.Ducrest AL, Amacker M, Lingner J, Nabholz M. Detection of promoter activity by flow cytometric analysis of GFP reporter expression. Nucleic Acids Res. 2002;30:e65. doi: 10.1093/nar/gnf064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xie Z, Bhushan RG, Daniels DJ, Portoghese PS. Interaction of bivalent ligand KDN21 with heterodimeric delta-kappa opioid receptors in human embryonic kidney 293 cells. Mol Pharmacol. 2005;68:1079–86. doi: 10.1124/mol.105.012070. [DOI] [PubMed] [Google Scholar]
- 28.Breit A, Lagace M, Bouvier M. Hetero-oligomerization between beta2- and beta3-adrenergic receptors generates a beta-adrenergic signaling unit with distinct functional properties. J Biol Chem. 2004;279:28756–65. doi: 10.1074/jbc.M313310200. [DOI] [PubMed] [Google Scholar]
- 29.Wilson S, Wilkinson G, Milligan G. The CXCR1 and CXCR2 receptors form constitutive homo- and heterodimers selectively and with equal apparent affinities. J Biol Chem. 2005;280:28663–74. doi: 10.1074/jbc.M413475200. [DOI] [PubMed] [Google Scholar]
- 30.Sutton GM, Duos B, Patterson LM, Berthoud HR. Melanocortinergic modulation of cholecystokinin-induced suppression of feeding through extracellular signal-regulated kinase signaling in rat solitary nucleus. Endocrinology. 2005;146:3739–47. doi: 10.1210/en.2005-0562. [DOI] [PubMed] [Google Scholar]
- 31.Vaughan CH, Haskell-Luevano C, Andreasen A, Rowland NE. Effects of oral preload, CCK or bombesin administration on short term food intake of melanocortin 4-receptor knockout (MC4RKO) mice. Peptides. 2006;27:3226–33. doi: 10.1016/j.peptides.2006.08.002. [DOI] [PubMed] [Google Scholar]
- 32.Hirst RA, Smart D, Devi LA, Lambert DG. Effects of C-terminal truncation of the recombinant delta-opioid receptor on phospholipase C and adenylyl cyclase coupling. J Neurochem. 1998;70:2273–8. doi: 10.1046/j.1471-4159.1998.70062273.x. [DOI] [PubMed] [Google Scholar]
- 33.Chen L, Li G, Tang L, Wang J, Ge XR. The inhibition of lung cancer cell growth by intracellular immunization with LC-1 ScFv. Cell Res. 2002;12:47–54. doi: 10.1038/sj.cr.7290109. [DOI] [PubMed] [Google Scholar]
- 34.Bonander N, Hedfalk K, Larsson C, et al. Design of improved membrane protein production experiments: quantitation of the host response. Protein Sci. 2005;14:1729–40. doi: 10.1110/ps.051435705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Grisshammer R. Understanding recombinant expression of membrane proteins. Curr Opin Biotechnol. 2006;17:337–40. doi: 10.1016/j.copbio.2006.06.001. [DOI] [PubMed] [Google Scholar]
- 36.Tate CG, Grisshammer R. Heterologous expression of G-protein-coupled receptors. Trends Biotechnol. 1996;14:426–30. doi: 10.1016/0167-7799(96)10059-7. [DOI] [PubMed] [Google Scholar]
- 37.Breitwieser GE. G protein-coupled receptor oligomerization: implications for G protein activation and cell signaling. Circ Res. 2004;94:17–27. doi: 10.1161/01.RES.0000110420.68526.19. [DOI] [PubMed] [Google Scholar]
- 38.Hur EM, Kim KT. G protein-coupled receptor signalling and cross-talk: achieving rapidity and specificity. Cell Signal. 2002;14:397–405. doi: 10.1016/s0898-6568(01)00258-3. [DOI] [PubMed] [Google Scholar]
- 39.Maggio R, Novi F, Scarselli M, Corsini GU. The impact of G-protein-coupled receptor hetero-oligomerization on function and pharmacology. Febs J. 2005;272:2939–46. doi: 10.1111/j.1742-4658.2005.04729.x. [DOI] [PubMed] [Google Scholar]
- 40.Park PS, Filipek S, Wells JW, Palczewski K. Oligomerization of G protein-coupled receptors: past, present, and future. Biochemistry. 2004;43:15643–56. doi: 10.1021/bi047907k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vagner J, Xu L, Handl HL, et al. Heterobivalent ligands crosslink multiple cell-surface receptors: the human melanocortin-4 and delta-opioid receptors. Angew Chem Int Ed Engl. 2008;47:1685–8. doi: 10.1002/anie.200702770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Caplan MR, Rosca EV. Targeting drugs to combinations of receptors: a modeling analysis of potential specificity. Ann Biomed Eng. 2005;33:1113–24. doi: 10.1007/s10439-005-5779-1. [DOI] [PubMed] [Google Scholar]
- 43.Handl HL, Sankaranarayanan R, Josan JS, et al. Synthesis and evaluation of bivalent NDP-alpha-MSH(7) peptide ligands for binding to the human melanocortin receptor 4 (hMC4R) Bioconjug Chem. 2007;18:1101–9. doi: 10.1021/bc0603642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Balagurunathan Y, Morse DL, Hostetter G, et al. Gene expression profiling-based identification of cell-surface targets for developing multimeric ligands in pancreatic cancer. Mol Cancer Ther. 2008;7:3071–80. doi: 10.1158/1535-7163.MCT-08-0402. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.