

SUMMARY
HSP70i and other stress proteins have been used in anti-tumor vaccines. This begs the question whether HSP70i plays a unique role in immune activation. We vaccinated inducible HSP70i (Hsp70-1) knockout mice and wild-type animals with optimized TRP-1, a highly immunogenic melanosomal target molecule. We were unable to induce robust and lasting depigmentation in the Hsp70-1 knockout mice, and in vivo cytolytic assays revealed a lack of CTL activity. Absence of T cell infiltration to the skin and maintenance of hair follicle melanocytes was observed. By contrast, depigmentation proceeded without interruption in mice lacking a tissue specific constitutive isoform of HSP70 (Hsp70-2) vaccinated with TRP-2. Next, we demonstrated that HSP70i was necessary and sufficient to accelerate depigmentation in vitiligo-prone Pmel-1 mice, accompanied by lasting phenotypic changes in dendritic cell subpopulations. In summary, these studies assign a unique function to HSP70i in vitiligo, and identify HSP70i as targetable entity for treatment.
Keywords: vitiligo, autoimmunity, HSP70, knockout mouse
INTRODUCTION
Vitiligo is a T cell mediated autoimmune disease of the skin. Patients present with progressive depigmentation that advances at an average rate of 1% bodily surface area a year (Cedercreutz et al, 2010). Genetic predisposition has been assigned to candidate genes including MHC class I and II, PTPN22 and IL2RA, supporting the autoimmune etiology of the disease (Jin et al, 2010). Approximately 25% more women than men develop vitiligo (Cedercreutz et al, 2010). At this time, there is no definitive evidence to support an association between vitiligo and a particular ethnic background. However, the progressively depigmenting skin is clearly more apparent for patients with a darker skin tone. Also, the social implications of disease are particularly devastating in countries where leprosy is a considerable health problem, as the appearance of skin lesions can bare similarities among both patient groups, and patients with vitiligo are mistakenly identified as individuals expressing a contagious disease (Millington and Levell, 2007).
Aside from a hereditary component, vitiligo pathogenesis involves environmental factors that contribute to precipitating the disease. Among these, exposure to skin bleaching phenols has been particularly well studied (Boissy and Manga, 2004). It is of great interest to identify the molecular connection between environmental precipitating factors and the autoimmune response that follows, as such studies can serve to identify candidate target molecules to develop novel and effective treatment strategies for the disease. There is a lack of effective therapeutics available for vitiligo, in part because halting disease progression and re-establishing pigmentation are conceptually different phases of the disease that may have to be addressed by separate means (Le Poole and Luiten, 2008).
Establishing pigmentation in patients with active disease is likewise problematic. An important step forward in developing vitiligo therapeutics is thus to recognize that progressive disease must first be effectively brought to a halt. Understanding of mechanisms at work in progressive disease will be important. In this respect, heat shock and glucose regulated proteins are prime candidates to connect stress to the skin with the autoimmune response to follow. In particular gp96 and HSPs 60, 70 and 90 have been implicated in immune cell activation (Multhoff, 2006). Stress proteins support immune reactivity by activating dendritic cells to more efficiently phagocytize, process and present antigens (Murshid et al, 2008). This concept has been elaborately exploited in the design of anti-tumor vaccines for melanoma, renal cell cancer and less immunogenic tumor types (Tosti et al, 2009).
Among heat shock proteins, inducible HSP70 (HSP70i) stands out as it can be secreted by live cells (Asea, 2007). Secretion of HSP70i by live cells would thus indicate that even in the absence of cell death, an immune response can be triggered to proteins and peptides derived from the cells under stress, chaperoned by HSP70i. This has guided an interest in defining the involvement of HSP70i in autoimmune vitiligo. Interestingly, heat shock proteins have been implicated in other autoimmune diseases as well, including rheumatoid arthritis, and other skin disorders such as psoriasis (Millar and Ohashi, 2007).
After observing consistent differential expression of HSP70i in non-lesional and vitiligo skin from 3 patients (Le Poole and Luiten, 2008), the concept was tested that expression of inducible HSP70 is a major contributor to the development of vitiligo in mice. Accelerated depigmentation was associated with enhanced cytotoxic T lymphocyte (CTL) responses to the vaccinated antigen, supporting that depigmentation occurs in a T cell mediated fashion in a vaccine based model of vitiligo (Denman et al, 2008). Also, melanocyte-directed overexpression of HSP70 can enhance anti-tumor responses to melanoma (Sanchez-Perez et al, 2006).
Whereas HSP70i can induce autoimmune depigmentation, given the shared chaperone and immune activating functions, the same is likely to hold true for other stress proteins as well. The question then remains, whether a single stress protein can be held responsible for mediating progressive disease, or whether there is redundancy in this mechanism. The HSP70 family of proteins is extensive, with estimates of up to 17 genes in human (Kabani et al, 2008) and 3 in mouse (Snoek et al, 1993). The canonical HSP70 isoforms are functionally redundant, with the main differences in their spatio-temporal expression (Kabani et al, 2008). The constitutively expressed isoforms are considered important for cellular housekeeping, whereas the inducible isoforms protect the cell from stressful conditions (Kabani et al, 2008; Daugaard et al, 2007).
What remains to be understood is whether HSP70i is critically important to vitiligo development. If the stress protein is redundant, we expect that in the absence of HSP70i another stress protein can mediate depigmentation. This issue requires resolution before considering HSP70i depletion in the treatment of vitiligo. Here we addressed whether HSP70i is solely responsible in vitiligo pathogenesis using different animal models. Initially, HSP70 knockout mice were vaccinated with DNA encoding melanocyte specific target antigens and assessed for progressive depigmentation, melanocyte loss, T cell infiltration and CTL activity. These experiments were performed in Hsp70-1 knockout animals lacking inducible HSP70 (Hunt et al, 2004), and repeated with Hsp70-2 knockout animals that lack a constitutively expressed, tissue-specific isoform of the protein important for spermatogenesis (E.M. Eddy, 1998). Next, a vitiligo-prone model was used to analyze whether HSP70i alone is sufficient to support progressive depigmentation. This model established by the group of N. Restifo at NIH carries a gp100 reactive T cell receptor transgene that is expressed by the majority of circulating T cells (Overwijk et al, 2003). Untreated animals develop minor depigmentation by approximately 9 months of age. Finally, blocking of HSP70i induced DC activation by antibodies was assessed using bone marrow derived dendritic cells. Taken together, these experiments provide a solid means to identify HSP70i as a potential target for immunotherapy of vitiligo.
RESULTS
HSP70i is uniquely required for autoimmune depigmentation
To evaluate redundancy of the involvement of HSP70i in depigmentation, wild-type C57BL/6 and inducible hsp70 knockout (Hsp70-1) mice were gene gun vaccinated with antigenic mouse Tyrp1ee (optimized TRP-1) encoding plasmid, an antigen more potent than TRP-2 to ensure that even the most subtle depigmentation responses may be detectable (Guevara-Patino et al, 2006). Gene-gun vaccinations involve coating gold particles with gene encoding plasmids which are then rapidly fired into the skin via pressurized helium. Four weeks after the final vaccination, we observed that depigmentation was virtually absent in the Hsp70-1 knockout mice (Figure 1A), and confirmed this with image analysis (Figure 1B). By contrast, 16.9% depigmentation was detected in wild-type mice at the vaccination site. Including the human HSP70i-encoding plasmid in the vaccine is insufficient to restore the depigmentation process in Hsp70-1 knockout mice, and resulted in 23.9% depigmentation in wild-type mice at the vaccination site (Figure 1A, B). We also followed depigmentation in Hsp70-2 knockout mice gene gun vaccinated with DNA encoding human TRP-2 (Supplementary Figure 1A, B). The HSP70-2 mice lack tissue specific expression of a constitutive isoform of hsp70 involved in spermatogenesis. Here we observed no differences in depigmentation compared to wild-type C57BL/6 mice, with both sets of animals displaying approximately 15% depigmentation at the vaccination site after four weeks (Supplementary Figure 1B). Taken together, the data indicate a critical and non-redundant role for HSP70i in inducing a depigmentation response.
Figure 1.
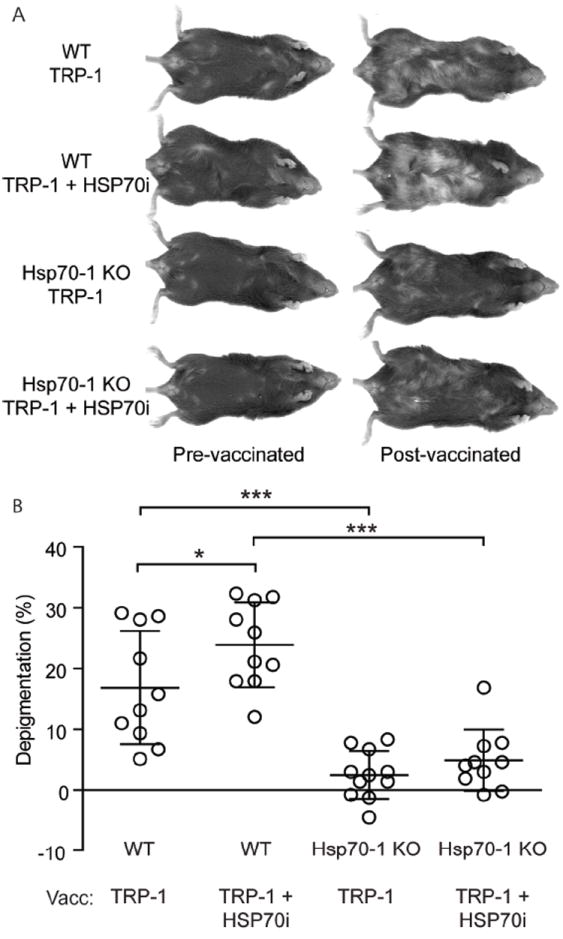
Inducible Hsp70 knockout mice are resistant to depigmentation after TRP-1 vaccination. (A) Wild-type (WT) and Hsp70-1 knockout (Hsp70-1 KO) mice were gene gun vaccinated five times every six days with DNA encoding either 6 μg of optimized TRP-1 or 3 μg each of optimized TRP-1 and HSP70i and imaged 4 weeks after the final vaccination. (B) Image analysis indicates that wild-type mice depigment significantly more compared to the Hsp70-1 KO mice after either vaccination (vacc), indicating that expression of HSP70i is required for efficient induction of autoimmunity. Data are presented as means ± SEM. (*P < 0.05; ***P < 0.001; n = 10 per group.)
HSP70i increases a cytotoxic response towards melanocyte antigens
To determine whether HSP70i mediates the immune activation of CTLs, wild-type C57BL/6 and Hsp70-1 knockout mice vaccinated with a combination of optimized TRP-1 and HSP70i encoding plasmids were boosted by two additional vaccinations three days apart, and six days later were assessed for in vivo cytotoxicity towards the encoded antigen as well as reactivity towards human TRP-2 (Figure 2A). Gene gun vaccinated wild-type mice displayed killing of 20.6% more splenocytes pulsed with a peptide derived from optimized TRP-1 compared to Hsp70-1 knockout mice (Figure 2B), indicating that CTL activation had taken place primarily in mice capable of expressing HSP70i. The data also reveal cytotoxicity towards approximately 25% of TRP-2 peptide-pulsed splenocytes indicating that significant epitope spreading had occurred (Figure 2B). By contrast, there were no differences in cytotoxicity towards peptide-pulsed splenocytes from wild-type and Hsp70-2 knockout mice vaccinated with the TRP-2 encoding plasmid (Supplementary Figure 1C). This is unlike the Hsp70-1 knockout mice which displayed less killing towards the antigen. It should be noted that mice vaccinated with optimized TRP-1 display 2-3 fold increased cytotoxicity towards the derivative peptide as compared to TRP-2 vaccinated animals, and in general achieve greater levels of depigmentation as compared to mice vaccinated with TRP-2 (Figure1; Figure 2). Together, these data further demonstrate the unique requirement for inducible HSP70 expression in the skin in precipitating autoimmune vitiligo.
Figure 2.
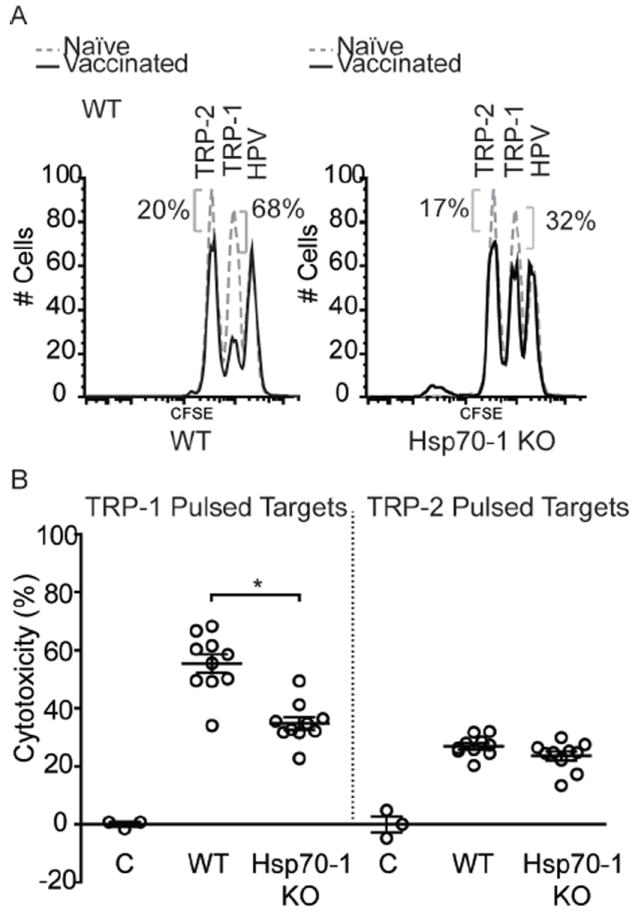
CTL killing towards melanoctye antigens is reduced in Hsp70-1 knockout mice. (A) For in vivo cytotoxicity assays, mice from Figure 1 were challenged with splenocytes pulsed with immunodominant peptides from TRP-1 or TRP-2, or irrelevant control peptides plus differing concentrations of CFSE. Spleens were harvested 18 hours after for analysis of CTL activity by FACS. Data from individual wild-type (WT) and Hsp70-1 KO mice are shown. (B) 20.6% more cytotoxicity was observed in wild-type mice (55.4%) towards TRP-1 pulsed splenocytes than in Hsp70-1 KO mice (34.8%). Approximately 25% cytotoxicity towards TRP-2-pulsed splenocytes is indicative of epitope spreading. Peptide pulsed splenocytes from control naïve mice are depicted as (C). Data are presented as means ± SEM. (*P < 0.05; n = 10 per group.)
HSP70i is associated with melanocyte loss and T cell infiltration in actively depigmenting skin
To further support that depigmentation following HSP70i-induced skin immune reactivity reflects a vitiligo phenotype, we evaluated local differences in immune reactivity in response to HSP70i. We probed skin from wild-type C57BL/6 and Hsp70-1 knockout mice one week following booster gene gun vaccinations using antibodies against the pan T lymphocyte marker CD3 and evaluated the density and location of the stained cells (Figure 3A). The vaccinated skin of wild-type mice contained 58.1% more T cells than Hsp70-1 knockout mice (Figure 3B). Further analysis revealed that the vast majority (>95%) of skin-infiltrating CD3+ cells co-express CD8 in both wild-type and Hsp70-1 knockout mice (Supplementary Figure 2). Although rare, a few CD3+/CD4+ double positive cells were detected, most CD4 expression as found primarily in the dermis could be assigned to a CD3-, non-T cell population (Supplementary Figure 2). Consistent with the depigmentation data, immunodetection of the melanocyte antigen TRP-1 revealed a 2.8 fold reduction in melanocyte-containing hair follicles in wild-type mice compared to Hsp70-1 knockout mice (figure 3C, D), supporting the loss of melanocytes in the latter that was not observed in mice lacking Hsp70i. Taken together, the immunohistology data establish the development of vaccine induced vitiligo in the skin of depigmenting wild-type animals.
Figure 3.
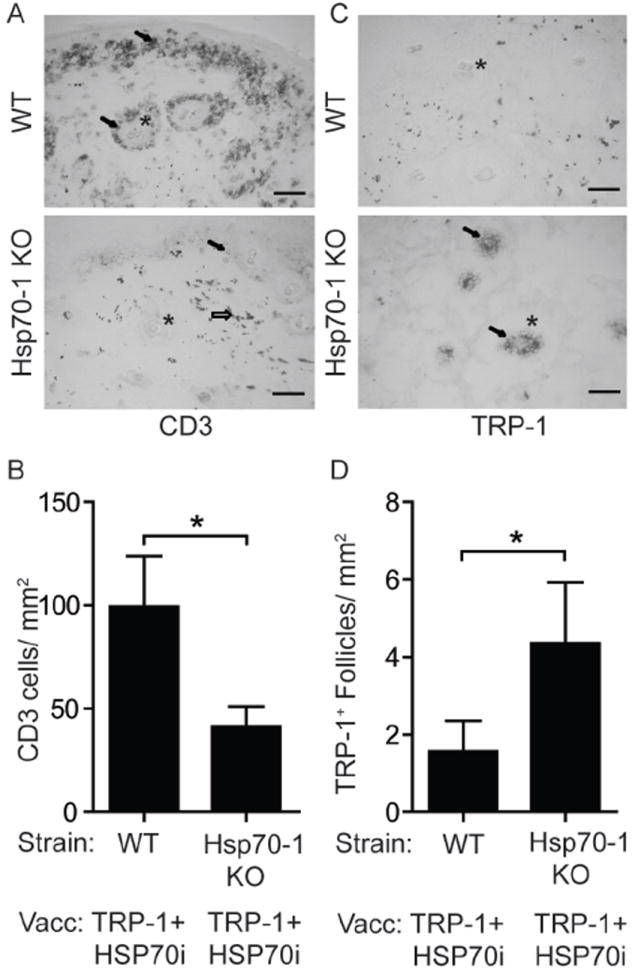
Immunohistology indicates inducible HSP70 is necessary for T cell-mediated loss of melanocytes. Treatment of mice engaged in this experiment is described under Figure 1. (A) Image of skin near the dermo-epidermal junction from mice one week after the final booster gene gun vaccination. CD3+ T cells (arrows) are more abundant near hair follicles (*) of wild-type (WT) than of Hsp70-1 KO mice. Gold particles can also be observed (open arrow). (B) Quantification of T cell infiltration. (C) Image of skin showing more melanocyte-containing hair follicles in vaccinated Hsp70-1 knockout mice compared to wild-type mice. TRP-1 expressing melanocytes (arrows) are shown within hair follicles (*) in an Hsp70-1 KO mouse. (D) Quantification of melanocyte-containing follicles. Depigmentation coincides with loss of melanocytes and T cell infiltration only in wild-type mice. Scale equals 50 μm. Data are presented as means ± SEM. (*P < 0.05; n = 10 per group.)
HSP70i is sufficient to support depigmentation in a spontaneous model of autoimmune vitiligo
We next tested whether HSP70i alone is sufficient to establish depigmentation in animals predisposed to developing autoimmune depigmentation, akin to human vitiligo. The T cell receptor (TCR) transgenic mouse strain Pmel-1 and C57BL/6 mice were gene gun vaccinated with either HSP70i encoding plasmid or empty vector control DNA. Six months after the final vaccination, Pmel-1 mice approximately 9 months of age vaccinated with HSP70i encoding DNA displayed markedly increased depigmentation (3.7 fold ventral; 2.8 fold dorsal) compared to mice vaccinated with control DNA (Figure 4A, B). Pmel-1 mice display subtle depigmentation even after vaccinating with empty vector DNA alone, with 18% and 25% depigmentation ventral and dorsal respectively (Figure4 A, B). This is similar to depigmentation in animals of that age which had not been vaccinated (Supplementary Figure 3). As expected, no depigmentation was observed in wild-type mice gene gun vaccinated with any plasmid (Figure 4A, B). This data set implies that elevated HSP70i expression alone is sufficient to augment the depigmentation process in animals prone to vitiligo development.
Figure 4.
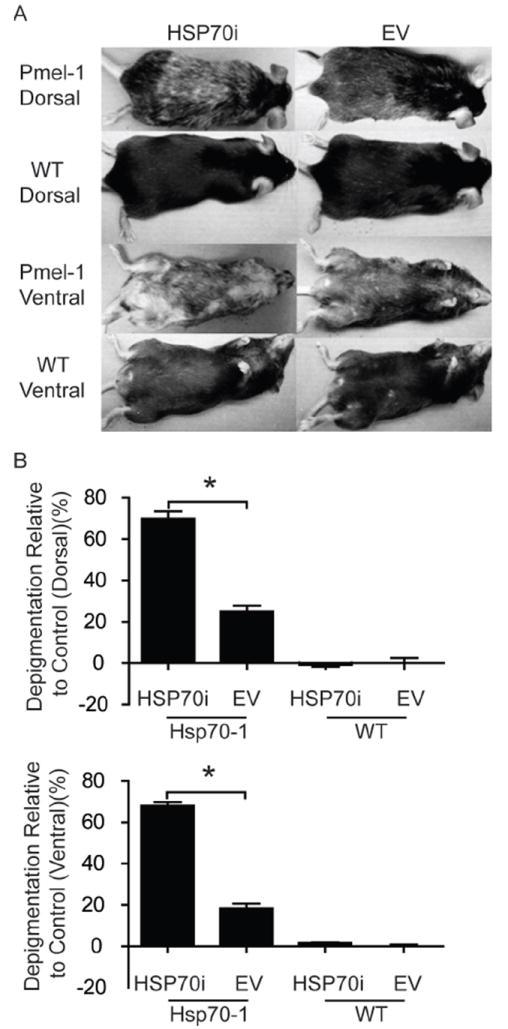
HSP70i accelerates depigmentation in vitiligo-prone mice. (A) Wild-type (WT) and Pmel-1 mice were vaccinated three times every 7 days with 4 μg of either HSP70i encoding or empty vector (EV) control DNA. Depicted are ventral and dorsal images of representative vaccinated animals six months after the final gene gun vaccination. Depigmentation in wild-type mice occurred within and distal to the site of the gene gun vaccination (B) Upon quantification, Pmel-1 mice receiving HSP70i-encoding DNA displayed significantly more depigmentation compared to control vaccinations and vaccinations in wild-type mice, confirming that focal overexpression of HSP70i is sufficient to induce vitiligo in disease-prone animals. Data are presented as means ± SEM. (**P < 0.01; ***P < 0.001; n = 3 per group.)
HSP70i provides a durable shift of the dendritic cell population towards an inflammatory subset
We next tested whether HSP70i has lasting effects on monocyte-derivative cell populations. Leukocyte phenotypic profiles were obtained from splenocytes of 9 month old mice euthanized 6 months after the final vaccination. The mouse splenocytes were stained for lymphocyte markers CD3, CD8, and Thy1.2, and the non-lymphocyte markers CD11b, CD11c, and F4/80. No sustained differences in the abundance of lymphocyte subpopulations were found, yet we observed marked differences in the abundance of monocyte-derived subpopulations discriminated based on CD11b and CD11c expression levels, shown as R1 through R3 (Figure 5A). The R1 population is characterized by the expression of CD11bint/hiCD11clo, typical for macrophages. Intriguingly, we show that the population R1 remained markedly suppressed for several months after vaccination with HSP70i (Figure 5B). By contrast, the dendritic cell population R2 (CD11bintCD11cint), representative of pro-inflammatory dendritic cells, showed an opposite trend (Figure 5B). These data demonstrate that HSP70i can drive the relative abundance of dendritic cell subpopulations with inflammatory versus immunosuppressive potential, associated with autoimmune depigmentation.
Figure 5.
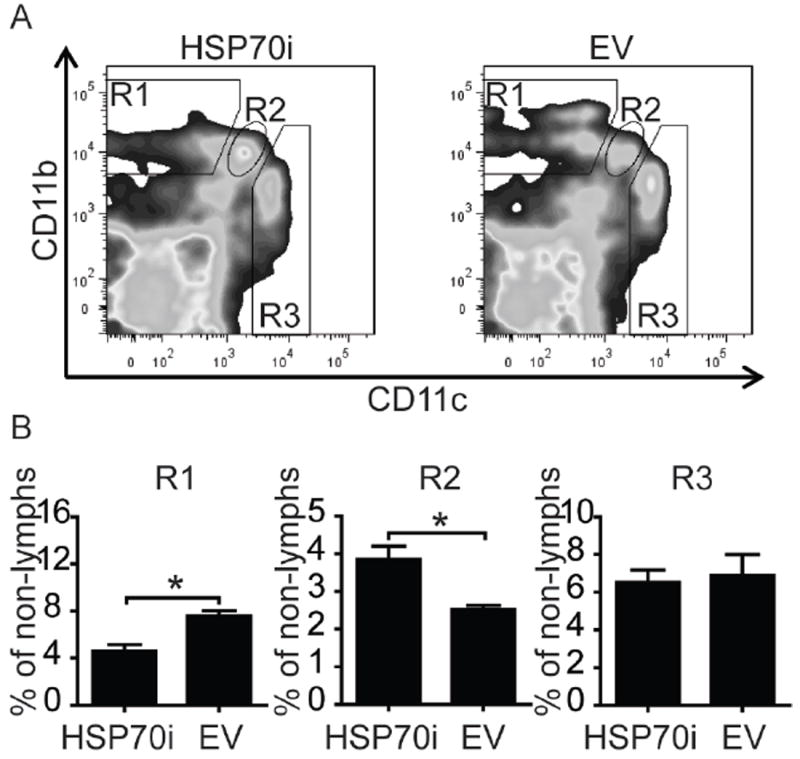
HSP70i has prolonged effects on peripheral leukocyte composition. Splenocytes obtained from Pmel-1 mice nine months after gene gun vaccination with HSP70i or empty vector DNA were stained to discriminate leukocyte subpopulations, and staining was quantified by flow cytometry. (A) Three distinct populations of cells were observed after gating for CD11b and CD11c cells in the non-lymphocyte population, with high (hi), low (lo) or intermediate (int) levels of expression. Macrophages are observed in the R1 population as CD11bint/hiCD11clo cells, while dendritic cells are amongst the R2 and R3 populations as CD11bintCD11cint and CD11bloCD11chi cells, respectively. (B) Quantification of CD11b and CD11c-expressing cells determined differential expression of these markers among leukocytes after vaccination with HSP70i encoding or empty vector plasmids. The macrophage population (CD11bint/hiCD11clo) shown in R1 displays decreased quantitative differences in response to HSP70i. By contrast, the inflammatory dendritic cell population shown in R2 (CD11bintCD11cint) shows an opposite trend. These results demonstrate the differential effects HSP70i on monocyte derivative leukocyte populations. Student’s t-test *P < 0.05, n=3.
Blocking HSP70i inhibits dendritic cells activation in vitro
Based on the FACS profiles suggesting that HSP70i induced sustained changes in dendritic cell populations, we assessed whether blocking the HSP70i protein could prevent dendritic cell activation. Mouse bone marrow monocytes were magnetically sorted and driven to a dendritic cell phenotype by addition of GM-CSF and IL-4 cytokines. After one week, HSP70i with or without blocking antibodies were added. To prevent the HSP70i antibody from inducing Fc receptor mediated dendritic cell stimulation itself, Fc receptor blocking antibodies were included. LPS and control media were used as controls. Twenty-four hours later, the dendritic cells were stained for the activation markers CD11c, CD80, CD83, CD86, and MHCII and analyzed by flow cytometry (Supplementary Figure 4; Figure 6). Gating on the CD11c+ population, it was observed that the entire population expressed CD80, CD86, and MHCII, with marked increases in expression attributed to the addition of HSP70i and LPS (Figure 6A). By contrast, addition of HSP70i antibodies markedly decreased activation (Figure 6A). Staining for CD83 revealed expressing and non-expressing cells (Figure 6A), with increases noted in the CD83+ population after the addition of LPS or HSP70i, and decreased activation with HSP70i antibodies (Figure 6B). Together, these data confirm that the HSP70i protein directly activates dendritic cells, while strongly suggesting that antibodies to HSP70i may be useful in preventing dendritic cell activation in vitiligo.
Figure 6.
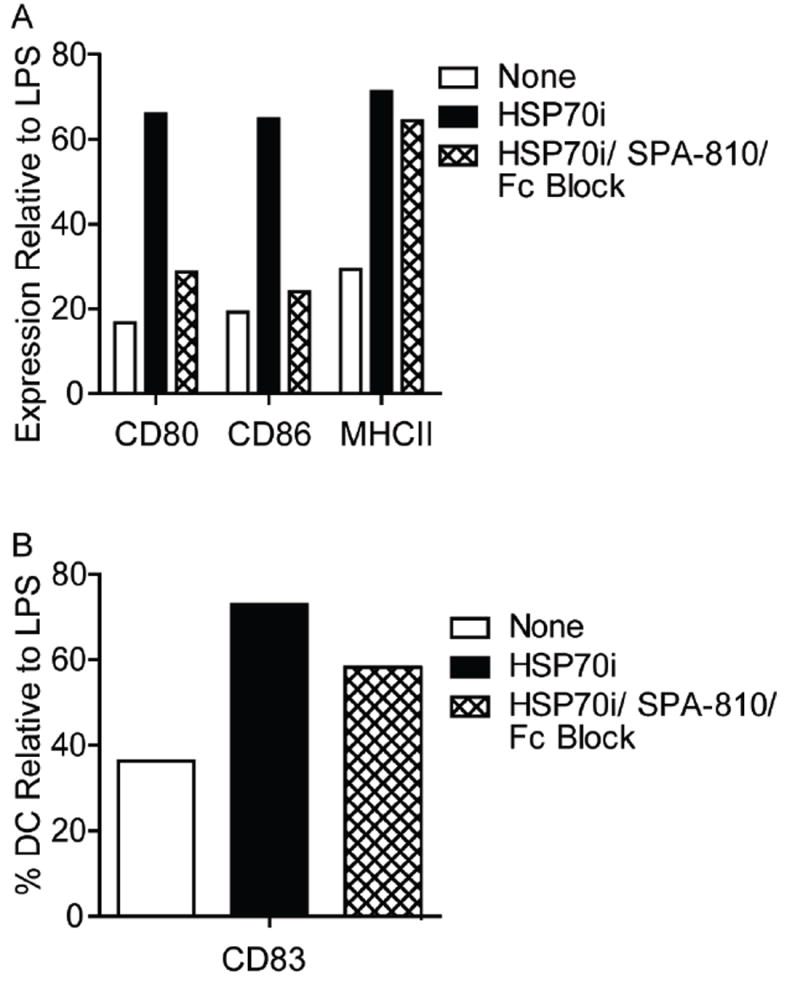
Antibodies to HSP70i interfere with dendritic cell activation in vitro. Dendritic cells derived from mouse bone marrow monocytes were grown in culture for 7 days followed by the addition of either 1 μg/ml of HSP70i, or a combination of HSP70i and Fc receptor blocking antibodies. LPS and control media were used as controls. Twenty-four hours later the cells were stained for the dendritic cell maturation markers CD11c, CD80, CD83, CD86, or MHCII and analyzed by FACS. Subsequent gating was performed on the CD11c+ cells. (A) Mean fluorescent intensity (MFI) indicates higher expression levels of CD80, CD86, and MHCII after addition of HSP70i as compared to control media, whereas addition of blocking antibodies was able to reduce expression levels close to baseline. (B) A higher percentage of dendritic cells expressing CD83+ were detected after addition of HSP70i as compared to control media, whereas blocking HSP70i reduced the cell number close to baseline. All data is represented as % stimulation compared to LPS. These results demonstrate the stimulatory effects of HSP70i on dendritic cells which can be augmented by blocking antibodies.
DISCUSSION
Our current data provide support to the notion that inducible HSP70 is a critical component in mediating autoimmune reactivity to melanocytes in vitiligo-prone animals. Previously, we have shown that overexpression of the heat shock protein, together with melanocyte-specific target antigen TRP-2 of human origin, could serve to induce progressive depigmentation in mice (Denman et al, 2008). Such depigmentation was originally restricted to the vaccination site, but would later diffusely spread to other sites as described (Denman et al, 2008). Melanocytes were not detected in follicles giving rise to depigmented hair, which is similar to the actual loss of melanocytes (rather than reduced melanization by existing melanocytes) observed in human vitiligo skin (Le Poole et al, 1993b). The vaccine combining TRP-2 and HSP70i was effectively used to generate an inducible animal model of the disease, useful to study immune effector responses involved in vitiligo. At that time we did not address the unique or redundant role of HSP70i versus other stress proteins, or discuss whether antigens other than TRP-2 can likewise serve to direct immune responses towards melanocytes.
As emphasized by the current data, the role of TRP-2 as a target antigen is not unique; we can replace this antigen by others expressed within the melanosomal compartment to likewise introduce progressive, CTL mediated responses to melanocytes. This is in concordance with the literature where the application of anti-melanoma vaccines targeting tyrosinase or TRP-2 was likewise associated with depigmentation of the pelage (Chapman and Wolchok, 2002). Depigmentation in response to DNA vaccines encoding human gp100 was mentioned previously as well (Chapman and Wolchok, 2002). There is however a difference in the type and extent of immune activation elicited by individual antigens. In fact, minimal humoral immunogenicity has been assigned to TRP-1, whereas TRP-2 can drive CTL responses (Avogadri et al, 2010). To elicit robust immune responses to vaccines, the immunogenicity of naturally occurring TRP-1 has been adjusted in its optimized version Tyrp-ee, generating peptides with enhanced binding to MHC and neopeptides revealed by deglycosylation (Guevara-Patino et al, 2006). Indeed in the current studies, optimized TRP-1 resulted in greater levels of depigmentation, as well as a 2-3 fold increase in cytotoxicity as compared to vaccination with TRP-2 in C57BL/6 wild-type mice. Also following optimized TRP-1 vaccination, responses were observed to TRP-2 derivative peptide SVYDFFWL. This peptide shares homology to the TRP-1 protein spanning the first 8 amino acids. As vaccination with native TRP-1 does not induce anti-TRP-2 responses, this phenomenon is most likely due to epitope spreading, with T cells targeting heteroclytic epitopes killing melanocytes which subsequently results in responses to TRP-2 as well. At this time however, we cannot rule out that reactivity to TRP-2 is rather a consequence of cross reactivity, where optimized TRP-1 has altered its processing in such a way that the homologous TRP-1 derivative peptide directly elicits a response to TRP-2.
The use of Tyrp-ee, eliciting optimal responses to melanocytes was important for the greatest possible odds of detecting vitiligo in vaccinated animals in the absence of inducible HSP70. Even with this optimized antigen, however, consistent depigmentation was not observed in animals lacking hsp70i, the inducible form of hsp70. In contrast, depigmentation was uninhibited in mice lacking hsp70-2, a tissue-specific isoform of hsp70 constitutively expressed in testes, even in response to vaccination with suboptimal antigen TRP-2. These findings suggest that HSP70 isoforms expressed outside the skin are less important to vitiligo development.
It is important to note that the progressive loss of melanocytes and T cell infiltration to the skin are considered hallmarks of vitiligo, and such hallmark events were likewise observed in our depigmenting mice (Badri et al, 1993; Ongenae et al, 2003). Enhanced T cell infiltration was inversely related to the loss of melanocytes within the wild-type mice, which can be attributed to increases in self-reactive CTLs. Meanwhile enhanced T cell infiltration or reduced melanocyte numbers were not observed in animals lacking expression of inducible HSP70. Removing isoform Hsp70-2 however still resulted in melanocyte death. Taken together our data demonstrate a critical role reserved for inducible HSP70 in supporting depigmentation.
Regarding potential redundancy of HSP70i, anti-melanoma vaccines are commonly applied in the absence of adjuvant heat shock proteins. This led us to consider that stress to the skin involved with the vaccine application process may be sufficient to enhance heat shock protein expression in the skin, serving as a natural adjuvant to enhance autoimmunity. Stress induced depigmentation is conclusively supported by reports of depigmentation occurring at the site of injection in mice previously vaccinated against melanocyte antigen TRP-2 (Lane et al, 2004). The question addressed here was whether there is a unique role reserved for HSP70i in autoimmune depigmentation in vitiligo. As other heat shock proteins including HSP27 and HSP110 have likewise been implicated as adjuvants in anti-melanoma vaccines, a non-redundant role for HSP70i would appear unusual (Srivastava, 2002). Our preliminary data in fact showed that in contrast to HSP60, HSP27 was differentially expressed in vitiligo lesional skin (not shown). This implies that HSP27 may play a role similar to HSP70, where patchy expression of the heat shock protein was noted in lesional, but not in non-lesional skin (Kroll et al, 2004). Also, gp96 has been implicated in autoimmune disease (Han et al, 2010), and it has been hypothesized that it is secreted in response to stress or cell death (Li et al, 2002). Investigators have modified gp96 to allow it to be secreted; however, there are no published data available supporting natural secretion of gp96, and this finding remains exclusively reserved for HSP70i (Strbo and Podack, 2008). In contrast to HSP70i, which is generally located in the cytoplasm or nucleus of the cell, gp96 is expressed in the endoplasmic reticulum (Li et al, 2002). The difference in subcellular location and function suggest that in the absence of HSP70i, gp96 cannot simply compensate for a loss of HSP70i. The current data indeed support that compensation does not occur, which is best explained by the different circumstances that call for overexpression of either stress protein, as well as its differential role in folding and chaperoning newly synthesized versus resident cellular proteins (Srivastava, 2002).
The current data definitively support a non-redundant role for HSP70i in progressive depigmentation, as we were unable to confer significant depigmentation upon mice that lack expression of the inducible isoform of HSP70. Although HSP70i was included in the vaccine, focal expression does not restore depigmentation, suggesting that systemic expression of HSP70i is required to drive biologically relevant responses to melanocytes.
We next chose to determine whether HSP70i alone was sufficient to define depigmentation in a model genetically prone to develop vitiligo. In this regard, the recent development of relevant T cell receptor transgenic mouse models now provides an opportunity to study and modulate effector responses in vitiligo. TCR transgenic animals with the majority of T cells reactive to a TRP-1 derived peptide presented in the context of MHC class II were engaged in experiments to provide a first line of evidence that a newly discovered subset of helper T cells, the Th17 cell, provides a driving force behind immune reactivity to melanocyte differentiation antigens in vitiligo (Muranski et al, 2008). This novel finding has yet to be translated to the human setting, where an infiltrate of CD4 cells may likewise be composed primarily of Th17 cells, as Th17 have now been implicated in several autoimmune settings (Hu et al, 2011). The TRP-1 reactive, TCR transgenic model is maintained in cappuccino mice, lacking any expression of the target antigen, as TRP-1 reactive T cells are otherwise clonally deleted (Muranski et al, 2008). Interestingly, clonal deletion does not occur in a setting of MHC class I restricted TCRs. The FH mouse expressing TCR transgenic CD8 T cells reactive with mouse tyrosinase is therefore the first reported spontaneous model of vitiligo, with depigmentation developing by 4 weeks of age (Gregg et al, 2010). This model has been pivotal to identify the central role for IFN-γ in depigmentation (Gregg et al, 2010). Due to the rapid development of vitiligo, this model is however less amenable to prophylactic measures that may prevent depigmentation. On the other hand, the Pmel-1 mouse expresses a gp100-reactive transgenic TCR on the majority of circulating CD8+ T cells, but mice are generally not maintained to the point where spontaneous depigmentation is observed (Overwijk et al, 2003). We found that by 9 months of age, minimal yet measurable depigmentation will occur. Delayed depigmentation can possibly be explained by a relatively lesser affinity of this TCR for its antigen, allowing T cell to circulate without causing damage to melanocytes. It is also possible that the cognitive peptide is less immunogenic or has reduced binding affinity to MHC. Whatever the cause, the Pmel-1 mouse has provided us with the currently exploited opportunity to study precipitating factors in vitiligo, as we were able to demonstrate that HSP70i is a critical player in defining the depigmentation process.
Earlier it was reported that CD11bintCD11cint dendritic cells induce IL-17-producing T cells associated with autoimmunity, which fits with our observations that this dendritic cell population (shown as R2 in Figure 5) remains increased at 6 months after vaccination with wild-type HSP70i (Denning et al, 2007). Further, Denning et al. (2007) demonstrated that the CD11b+F4/80+CD11clo macrophage population induced Foxp3+ regulatory T cells (Tregs). Although we recognize the preliminary nature of our observations, we currently demonstrate that the same subset of Treg-inducing macrophages is downregulated by HSP70i. Thus, HSP70i may contribute to vitiligo through the inhibition of Tregs via reduced macrophage activity, and through supporting Th17-mediated autoimmunity by inflammatory dendritic cells. It appears then that HSP70i may be acting on two fronts, by activating CTLs via dendritic cell activation, and simultaneously downregulating Treg activity.
Taken together, these data suggest that stress signals to the skin are funneled through inducible HSP70 to activate dendritic cells in the immediate environment of the stressful event. Thus, HSP70i may be regarded as a targetable entity, important for the development of new treatment strategies in vitiligo. When considering this opportunity, current knowledge about heat shock protein mediated dendritic cell activation can be used for guidance (Basu et al, 2000; Kuppner et al, 2001). Under stress, secreted inducible HSP70 serves as an adjuvant in vitiligo. Indeed, HSP70 has been previously shown to break T cell tolerance towards self antigens as mediated by dendritic cells (Millar et al, 2003). Thus, if dendritic cell activation is mediated primarily by HSP70i, such activation may be prevented by preventing the stress protein from binding dendritic cells.
Several stress protein receptors have been identified on the surface of immature dendritic cells. Such receptors include TLR4, TLR2, CD14, CD91, and CD40 (Chen et al, 2009; Asea et al, 2002; Asea et al, 2000). Preventing binding of HSP70i by blocking its receptors through antibodies is not likely to meet with success, as antibody binding will have dendritic cell-activating consequences itself, and may have cytolytic effects on cells other than the pivotal dendritic cell population. Thus we are currently focusing attention on HSP70i itself and attempting to prevent its binding to dendritic cells. Here we demonstrate that antibodies to HSP70i prevented complete dendritic cell activation as measured by expression of CD80, CD83, CD86 and MHC class II molecules. It is thus entirely possible that preventing HSP70i from binding to dendritic cells will provide an effective prophylactic measure in vitiligo. In conclusion, the current data provide evidence of a unique and pivotal role for HSP70i in progressive vitiligo, suggesting that targeting this heat shock protein can contribute to new treatment of the autoimmune depigmentation process.
MATERIALS AND METHODS
Mice
C57BL/6 mice were purchased from Jackson Labs (Bar Harbor, ME, USA) and inducible hsp70 (Hsp70-1) knockout mice (B6;129S7-Hspa1a/Hspa1btm1Dix/Mmcd) were purchased from Mutant Mouse Resource Center (UC Davis, CA, USA). Constitutive hsp70 (Hsp70-2) knockout mice were contributed by Dr. E.M. Eddy and maintained as heterozygotes; male Hsp70-2 knockout mice are infertile, whereas females are not (Eddy, 1998). A neomycin insert precedes the Hsp70-2 gene; DNA was isolated from mice tail snips, followed by PCR with the appropriate primers to determine homozygosity of the offspring with Hsp70-2 knockout mice displaying the presence of the neomycin gene insert that obliterates Hsp70-2 expression, and complete absence of Hsp70-2-encoding DNA. Wild-type mice were also selected and used in the Hsp70-2 knockout experiments. Vitiligo-prone, 78B6.Cg-Thy1a/Cy Tg (TcraTcrb) 8Rest/J (Pmel-1) gp100 reactive T cell receptor transgenic mice were purchased from Jackson Labs. All mice were 6-8 weeks old when included in experiments, excluding the Hsp70-2 knockouts and associated control animals which were 18-24 months of age. All mice were gender matched to wild-type animals. Experiments included 10 mice per group unless stated otherwise in the figure legends. Mice were maintained in facilities approved by institutional IACUC regulations.
Gene gun vaccination
Vaccinations for all experiments were prepared as described previously (Denman et al, 2008) unless stated otherwise. Briefly, bullets were prepared by precipitating endotoxin-free plasmid DNA onto spermidine-coated gold beads (Fluka Biochemika and Sigma-Aldrich, St. Louis, MO, USA), and used to coat silicone tubing (Bio-Rad, Hercules, CA, USA). All bullets were used within 14 days of preparation and maintained under vacuum before use. Mice were prepared for vaccination by biweekly abdominal hair removal with Nair (Church and Dwight Co., Princeton, NJ, USA) and vaccinated using a Helios Gene Gun (Bio-Rad) under isofluorane (E-Z Euthanex gas chamber, E-Z Systems Corp, Palmer, PA, USA). Unless otherwise stated, mice were ventrally vaccinated weekly for 4 consecutive weeks with a 4.8 μg dose of total DNA. Where combinations of plasmids were used, such DNA was mixed 1:1 to the same total dose of DNA and combined to prepare bullets.
Evaluating depigmentation
Depigmentation was quantified by flatbed scanning of the ventral and dorsal sides of the mice under anesthesia, immediately prior to the initial vaccination, and 4 weeks after final vaccination (6 months after the final vaccination in Pmel-1 mice) and subsequent image analysis using Adobe Photoshop software (Adobe Systems, Inc, San Jose, CA, USA). The percent depigmentation was calculated from the largest evaluable area as the percentage of pixels among >150,000 evaluated with a luminosity above the cutoff level set to include 95% of pixels for untreated mice (Denman et al, 2008). Mice were also photographed using a DSC-S950 SteadyShot digital camera (Sony, Tokyo, Japan) as indicated.
Assessment of CTL activation
In vivo cytotoxicity assays were performed as described (Denman et al, 2008). Briefly, 4 weeks after evaluating depigmentation, mice received 2 booster gene gun vaccinations 3 days apart. Separate batches of splenocytes were pulsed from the respective knockout mice with TRP-1-derived, K(b)-restricted peptide 455-463 TAPDNLGYM, TRP-2-derived, K(b)-restricted peptide 180-188 SVYDFFVWL, or control HPV16E7-derived, D(b)-restricted peptide 49-57 RAHYNIVTF (Pi Proteomics, Huntsville, AL, USA). Then cells were loaded with 1, 3, and 9 μM carboxyfluorescein succimidyl ester (CFSE) (Invitrogen, Carlsbad, CA, USA), respectively. 1 and 9 μm concentrations were used where only two peptides were analyzed in the assay. Labeled cells were periorbitally injected 4 days after the final booster gene gun vaccination, and splenocytes were harvested after 18 hours. Samples representing 1,000 irrelevantly pulsed splenocytes were acquired using FACS Canto equipment (BD Biosciences, Sparks, MD, USA) comparing the ratio of TRP-1 or TRP-2 peptide-pulsed splenocytes between vaccinated and naïve mice from shifts in the area under the curves.
Immunohistology
At euthanasia, mouse skin biopsies from the vaccinated site were snap-frozen in OCT compound (Sakura Finetek, Torrance, CA, USA). Eight-μm cryostat sections were fixed in cold acetone and indirect immunoperoxidase staining procedures were performed as described previously (Le Poole et al, 1993). The tissue sections were treated with Super Block (ScyTek, Logan, UT, USA) to prevent non-specific antibody binding. Primary antibodies Ta99 to mouse monoclonal TRP-1 (mouse monoclonal; COVANCE, Princeton, NJ, USA), biotinylated 145-2C11 to CD3ε (Armenian Hamster monoclonal; BD Pharmigen, Sparks, MD, USA), or biotinylated 53-6.7 to CD8α (mouse monoclonal; Biolegend, San Diego, CA, USA), followed by horseradish peroxidase conjugated secondary antibodies (Streptavidin, goat anti-mouse IGg2a, or goat anti-rat; Santa Cruz Biotechnology Inc., Santa Cruz, CA, USA) were utilized. Enzymatic detection was finalized using amino ethyl carbazole as a substrate (Sigma). For CD4 detection, primary antibody RM4-5 to CD4 (BD Pharmigen) was followed by an alkaline phosphatase anti-rat secondary antibody (Santa Cruz Biotechnology). Staining was quantified as the number of cells per mm2 of skin observed by light microscopy (Nikon, Melville, NY, USA) and Adobe Photoshop software (Adobe Systems Inc.). Fluorescent staining was performed with APC/Cy7 conjugated 145-2C11 to mouse CD3ε (Biolegend), FITC conjugated 53-6.7 to mouse CD8α (BD Pharmigen), and V450 conjugated RM4-5 to mouse CD4 (BD Pharmigen). Confocal images were captured with a Zeiss LSM 510 scanning electron microscope (Zeiss, Maple Grove, MN, USA). Image analysis was performed using Adobe Photoshop (Adobe Systems Inc.) and ImageJ (U.S. National Institutes of Health, Bethesda, Maryland, USA) software. For visualizing colocalization, the following channels were used: APC/Cy7 visualized as red, FITC as green, and V450 as blue.
Activating immature dendritic cells
Bone marrow-derived monocytes were prepared by culturing murine bone marrow cells using a modified protocol (Boudreau et al, 2008). Briefly, bone marrow cells were flushed aseptically from the femurs and the tibia of mice. Monocytes were enriched using the EasySep Mouse Monocyte Enrichment Kit (Vancouver, British Columbia, Canada) according to the manufacturer’s instructions. To generate dendritic cells, non-adherent cells were maintained in Teflon containers (Savillex, Minnetonka, Minnesota, USA), in Roswell Park Memorial Institute medium (RPMI) (Mediatech) supplemented with 10% fetal bovine serum, 100U/ml penicillin, 100 mg/ml streptomycin, 50 ng/ml murine granulocyte macrophage colony stimulating factor (GM-CSF) (Peprotech, Rocky Hill, New Jersey, USA), and 12.5 ng/ml murine interleukin-4 (IL-4) (Peprotech). On day 7, 1 μg/ml of either lipopolysaccharide (Sigma), or HSP70i (Catalogue number 4859-25, Biovision, San Francisco, CA, USA) was added. To block HSP70i, the cells were first incubated with 2μl/ml of Fc receptor block (rat anti-mouse CD16/CD32, BD Pharmigen) followed by addition of HSP70i (Biovision) and SPA-810 to HSP70i (mouse monoclonal, Enzo Life Sciences Inc., Farmingdale, NY, USA). After 24 h, expression of activation markers was assessed as described under flow cytometry.
Flow Cytometry
For experiments including the Pmel-1 mice, splenocytes were recovered twenty-five weeks after the final vaccination, and stained for the following surface markers CD3, CD8, CD11b, CD11c, F4/80, and IgM-Thy1.2 (BD Biosciences). Initial gating was performed on live non-debris singlets, with subsequent gating towards CD11c versus CD11b cells using FACS LSR-II equipment (BD Biosciences). IgM-Thy1.2, and CD3-positive cells were excluded from the final gating. For experiments using in vitro generated immature dendritic cells from C57BL/6 mice, the cells were gently spun and stained with Hamster antimouse CD11c, CD80, CD83, CD86, and MHCII labeled with V450, allophycocyanin, phycoerythrin, phycoerythrin-cyan and fluorescein isothiocyanate fluorochromes, respectively (BD Biosciences, Sparks, Maryland, USA). Initial gating was performed on live non-debris singlets, with subsequent gating toward CD11c+ cells using FACS LSR-II equipment (BD Biosciences).
Statistical analyses
All data were presented as the means ± SEM unless otherwise indicated. Numeric data were analyzed for statistical significance using Student’s unpaired 1-tailed t-test with Prism software (Graphpad, La Jolla, CA, USA). P values of less than 0.05 were considered statistically significant.
Supplementary Material
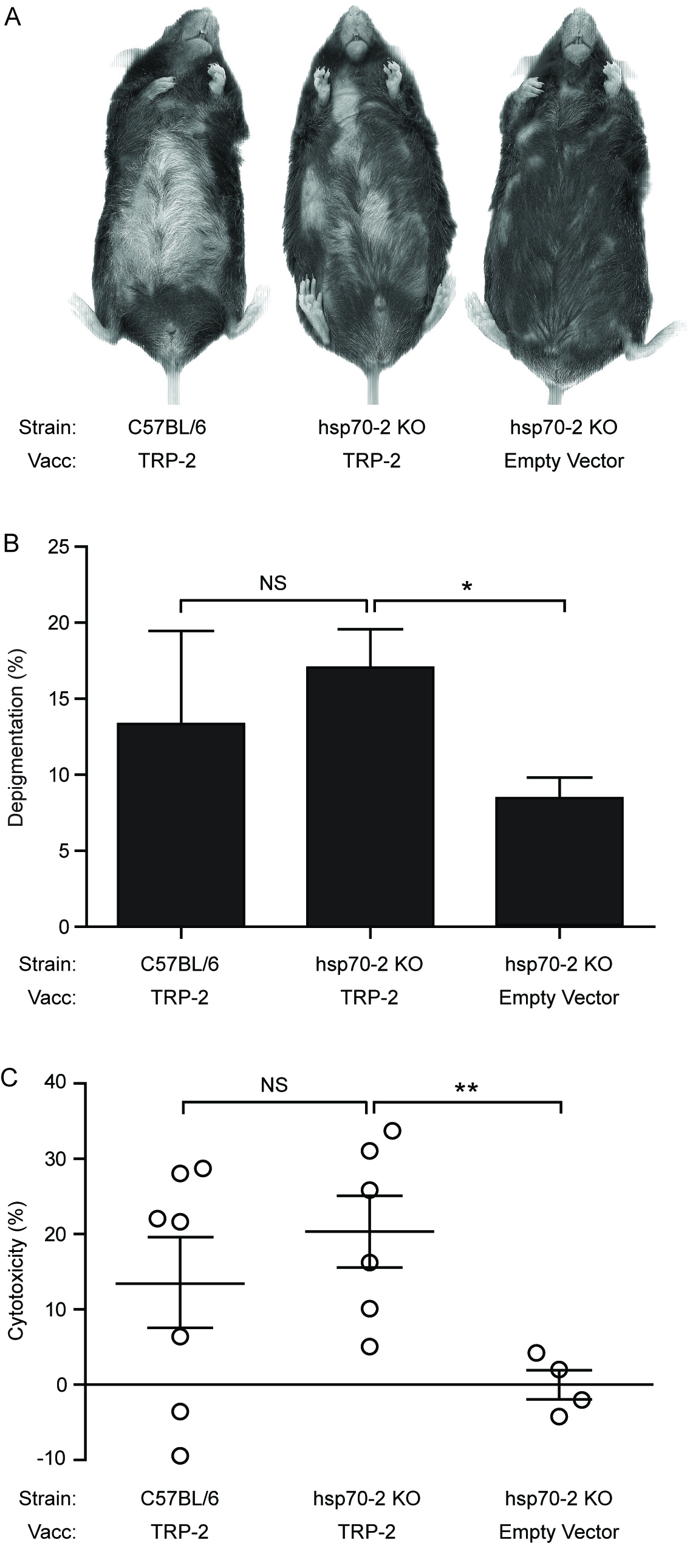
Alternative HSP70 isoform Hsp70-2 is irrelevant for inducing autoimmune responses. (A) Wild-type (WT) and constitutive Hsp70 knockout (Hsp70-2 KO) mice were gene gun vaccinated five times every six days with 6 μg of either TRP-2 or empty vector (EV; plasmid containing no gene insert) control DNA. (B) No difference in depigmentation was observed between vaccinated (vacc) wild-type and Hsp70-2 KO mice. (C) No differences in cytotoxicity were observed in wild-type or Hsp70-2 KO mice towards TRP-2-pulsed splenocytes. Data are presented as means ± SEM. (NS [no significance]; *P < 0.05; **P < 0.01; n = 10 per group.)
{kind=link}
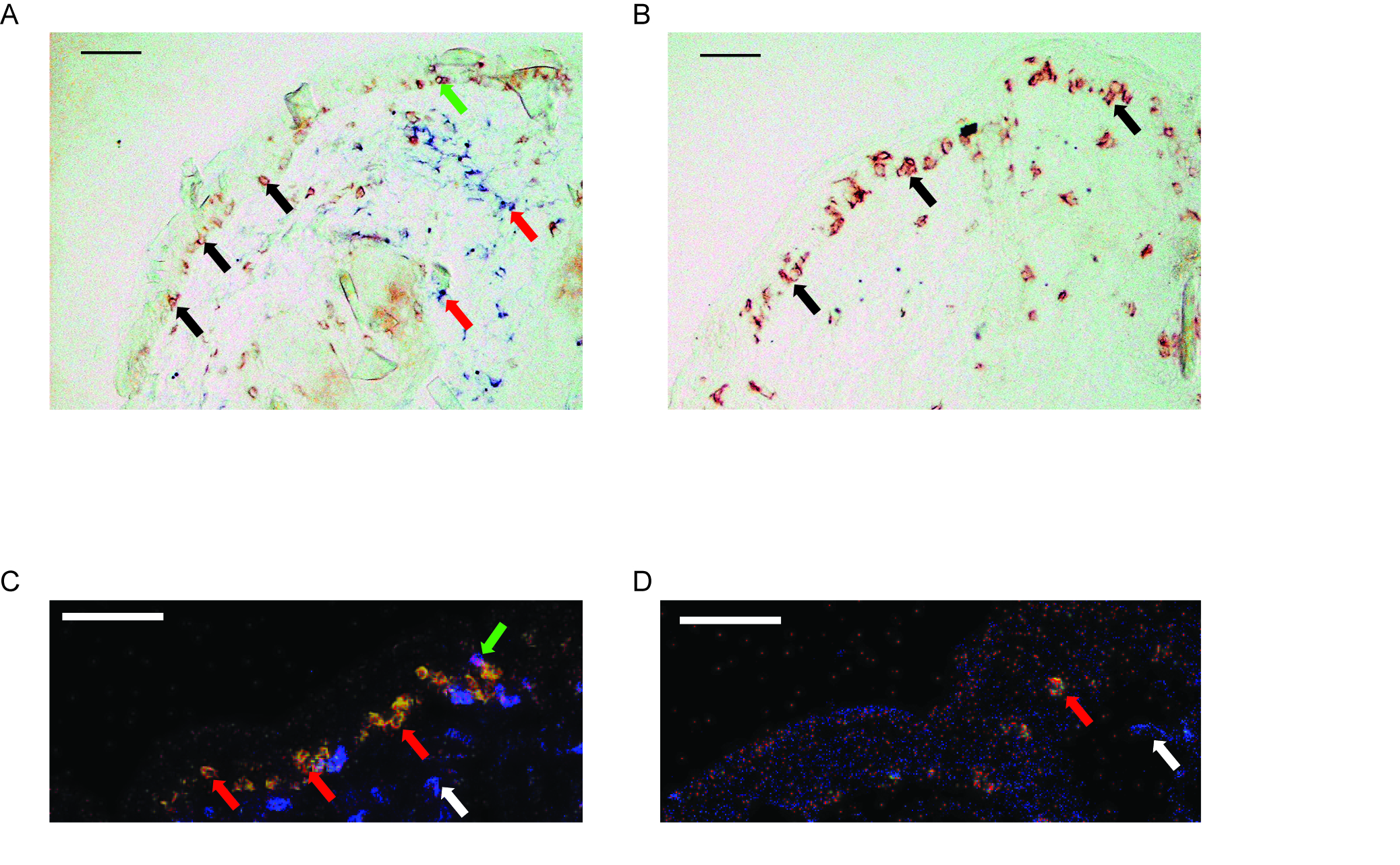
Immunohistology reveal a greater influx of CD8+ T cells in wild-type versus Hsp70-1 knockout mice. (A) Image of skin from a C57BL6 mouse vaccinated with HSP70i + TRP-1 reveal an abundant number of CD3+ T cells (black arrows) in the dermoepidermal junction. CD3-CD4+ non-T cells (red arrows) were mainly localized to the dermis. A single CD3+CD4+ T cell is shown in the epidermis (green arrow). (B) A serial section from the same tissue as (A) indicates the vast majority of the T cells are CD8+ (black arrows). (C) Fluorescent staining of skin from a C57BL6 mouse vaccinated with HSP70i + TRP-1 further demonstrate the colocalization of CD3 (red) and CD8 (green) as indicated by yellow (red arrows). CD3-CD4+ non-T cells were mainly confined to the dermis (white arrow). A single CD3+CD4+ T cell is shown (green arrow). (D) Skin from an Hsp70-1 knockout mouse vaccinated with HSP70i + TRP-1 reveal less CD3+CD8+ T cell infiltration (red arrow). CD3-CD4+ non-T cells were also present in the dermis (white arrow). Scale equal 50 μm.
{kind=link}
Gene-gun vaccination alone does not accelerate depigmentation in vitiligo-prone mice. This supplementary image of the Pmel-1 mice from Figure 4 demonstrates similar levels of depigmentation in empty vector vaccinated and unvaccinated Pmel-1 mice. Ventral and dorsal views are shown.
{kind=link}
Antibodies can block HSP70i induced dendritic cell activation in vitro. This supplementary image displays the fluorocytometry histograms representing activation marker expression by CD11c+, cultured mouse dendritic cells as summarized in Figure 6. Whereas exposure to HSP70i increases expression of CD80, CD83, CD86, and MHCII, in presence of antibodies to HSP70i, dendritic cell activation is largely prevented.
Significance.
We have demonstrated that HSP70i plays a unique and indispensable role in progressive autoimmune depigmentation. Since overexpression of HSP70i funnels the stress response that ultimately leads to vitiligo development, this work identifies HSP70i as a targetable treatment entity in the disease.
Acknowledgments
We thank Borean Pharma and Anaphore Inc. for supporting the initial maintenance of the Hsp70 knockout animals. These studies were supported by NIH/ NIAMS grant RO1 AR054749 to CLP.
Abbreviations
- CTL
cytotoxic T lymphocyte
- HSP
heat shock protein
- KO
knockout
- TRAIL
tumor necrosis factor-related apoptosis-inducing ligand
- Tregs
regulatory T cells
- TRP
tyrosinase related protein
- WT
wild-type
Footnotes
The authors have no additional financial interests.
References
- Asea A. Mechanisms of HSP72 release. J Biosci. 2007;32:579–584. doi: 10.1007/s12038-007-0057-5. [DOI] [PubMed] [Google Scholar]
- Asea A, Kraeft SK, Kurt-Jones EA, Stevenson MA, Chen LB, Finberg RW, Koo GC, Calderwood SK. HSP70 stimulates cytokine production through a CD14-dependant pathway, demonstrating its dual role as a chaperone and cytokine. Nat Med. 2000;6:435–442. doi: 10.1038/74697. [DOI] [PubMed] [Google Scholar]
- Asea A, Rehli M, Kabingu E, Boch JA, Bare O, Auron PE, Stevenson MA, Calderwood SK. Novel signal transduction pathway utilized by extracellular HSP70: Role of toll-like receptor (TLR) 2 and TLR4. J Biol Chem. 2002;277:15028–15034. doi: 10.1074/jbc.M200497200. [DOI] [PubMed] [Google Scholar]
- Avogadri F, Merghoub T, Maughan MF, Hirschhorn-Cymerman D, Morris J, Ritter E, Olmsted R, Houghton AN, Wolchok JD. Alphavirus replicon particles expressing TRP-2 provide potent therapeutic effect on melanoma through activation of humoral and cellular immunity. PLoS One. 2010;5:e12670. doi: 10.1371/journal.pone.0012670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badri AM, Todd PM, Garioch JJ, Gudgeon JE, Stewart DG, Goudie RB. An immunohistological study of cutaneous lymphocytes in vitiligo. J Pathol. 1993;170:149–155. doi: 10.1002/path.1711700209. [DOI] [PubMed] [Google Scholar]
- Basu S, Binder RJ, Suto R, Anderson KM, Srivastava PK. Necrotic but not apoptotic cell death releases heat shock proteins, which deliver a partial maturation signal to dendritic cells and activate the NF-kappa B pathway. Int Immunol. 2000;12:1539–1546. doi: 10.1093/intimm/12.11.1539. [DOI] [PubMed] [Google Scholar]
- Boissy RE, Manga P. On the etiology of contact/occupational vitiligo. Pigment Cell Res. 2004;17:208–214. doi: 10.1111/j.1600-0749.2004.00130.x. [DOI] [PubMed] [Google Scholar]
- Boudreau J, Koshy S, Cummings D, Wan Y. Culture of myeloid dendritic cells from bone marrow precursors. J Vis Exp. 2008;17 doi: 10.3791/769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman PB, Wolchok JD. Melanoma antigens: vaccines and monoclonal antibodies. In: Borden EC, editor. Melanoma: biologically targeted therapeutics. Totowa, NJ: Humana Press Inc; 2002. pp. 157–183. [Google Scholar]
- Cedercreutz K, Denman CJ, Klarquist J, Vaitla R, Boissy RE, Westerhof W, Hernandez C, Le Poole IC. Vitiligo Etiology and Treatment: Parameters Derived From a Patient Survey. J Dermatol Nurses’ Assoc. 2010;2:265–272. [Google Scholar]
- Chen T, Guo J, Han C, Yang M, Cao X. Heat shock protein 70, released from heat-stressed tumor cells, initiates antitumor immunity by inducing tumor cell chemokine production and activating dendritic cells via TLR4 pathway. J Immunol. 2009;182:1449–1459. doi: 10.4049/jimmunol.182.3.1449. [DOI] [PubMed] [Google Scholar]
- Daniels G, Sanchez-Perez L, Kottke T, Diaz RM, Thompson J, Lai M, Gough M, Karim M, Bushell A, Chong H, Melcher A, Harrington K, Vile RG. Intentional killing of normal melanocytes by gene transfer generates curative anti melanoma without obligate autoimmunity. Nature Biotech. 2004;22:1125–1132. doi: 10.1038/nbt1007. [DOI] [PubMed] [Google Scholar]
- Das PK, van den Wijngaard RM, Wankowicz-Kalinska A, Le Poole IC. A symbiotic concept of autoimmunity and tumour immunity: Lessons from vitiligo. Trends Immunol. 2001;22:130–136. doi: 10.1016/s1471-4906(00)01844-5. [DOI] [PubMed] [Google Scholar]
- Daugaard M, Rohde M, Jaattela M. The heat shock protein 70 family: Highly homologous proteins with overlapping and distinct functions. FEBS Lett. 2007;581:3702–3710. doi: 10.1016/j.febslet.2007.05.039. [DOI] [PubMed] [Google Scholar]
- Denman CJ, McCracken J, Hariharan V, Klarquist J, Oyarbide-Valencia K, Guevara-Patino JA, Le Poole IC. HSP70i accelerates depigmentation in a mouse model of autoimmune vitiligo. J Invest Dermatol. 2008;128:2041–2048. doi: 10.1038/jid.2008.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denning TL, Wang YC, Patel SR, Williams IR, Pulendran B. Lamina propria macrophages and dendritic cells differentially induce regulatory and interleukin 17-producing T cell responses. Nat Immunol. 2007;8:1086–1094. doi: 10.1038/ni1511. [DOI] [PubMed] [Google Scholar]
- Eddy EM. HSP70-2 heat-shock protein of mouse spermatogenic cells. J Exp Zool. 1998;282:261–271. [PubMed] [Google Scholar]
- Gregg RK, Nichols L, Chen Y, Lu B, Engelhard VH. Mechanisms of spatial and temporal development of autoimmune vitiligo in tyrosinase-specific TCR transgenic mice. J Immunol. 2010;184:1909–1917. doi: 10.4049/jimmunol.0902778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimes PE. White patches and bruised souls: Advances in the pathogenesis and treatment of vitiligo. J Am Acad Dermatol. 2004;51:S5–7. doi: 10.1016/j.jaad.2004.01.007. [DOI] [PubMed] [Google Scholar]
- Guevara-Patino JA, Engelhorn ME, Turk MJ, Liu C, Duan F, Rizzuto G, Cohen AD, Merghoub T, Wolchok JD, Houghton AN. Optimization of a self antigen for presentation of multiple epitopes in cancer immunity. J Clin Invest. 2006;116:1382–1390. doi: 10.1172/JCI25591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han JM, Kwon NH, Lee JY, Jeong SJ, Jung HJ, Kim HR, Li Z, Kim S. Identification of gp96 as a novel target for treatment of autoimmune disease in mice. PLoS One. 2010;5:e9792. doi: 10.1371/journal.pone.0009792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho RC, Neo LF, Chua AN, Cheak AA, Mak A. Research on psychoneuroimmunology: Does stress influence immunity and cause coronary artery disease? Ann Acad Med Singapore. 2010;39:191–196. [PubMed] [Google Scholar]
- Hu Y, Shen F, Crellin NK, Ouyang W. The IL-17 pathway as a major therapeutic target in autoimmune diseases. Ann N Y Acad Sci. 2011;1217:60–76. doi: 10.1111/j.1749-6632.2010.05825.x. [DOI] [PubMed] [Google Scholar]
- Hultqvist M, Backlund J, Bauer K, Gelderman KA, Holmdahl R. Lack of reactive oxygen species breaks T cell tolerance to collagen type II and allows development of arthritis in mice. J Immunol. 2007;179:1431–1437. doi: 10.4049/jimmunol.179.3.1431. [DOI] [PubMed] [Google Scholar]
- Hunt CR, Dix DJ, Sharma GG, Pandita RK, Gupta A, Funk M, Pandita TK. Genomic instability and enhanced radiosensitivity in Hsp70.1- and Hsp70.3-deficient mice. Mol Cell Biol. 2004;24:899–911. doi: 10.1128/MCB.24.2.899-911.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin Y, et al. Variant of TYR and autoimmunity susceptibility loci in generalized vitiligo. N Engl J Med. 2010;362:1686–1697. doi: 10.1056/NEJMoa0908547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabani M, Martineau CN. Multiple Hsp70 isoforms in the eukaryotic cytosol: Mere redundancy or functional specificity? Curr Genomics. 2008;9:338–248. doi: 10.2174/138920208785133280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroll TM, Bommiasamy H, Boissy RE, Hernandez C, Nickoloff BJ, Mestril R, Caroline Le Poole I. 4-tertiary butyl phenol exposure sensitizes human melanocytes to dendritic cell-mediated killing: Relevance to vitiligo. J Invest Dermatol. 2005;124:798–806. doi: 10.1111/j.0022-202X.2005.23653.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuppner MC, Gastpar R, Gelwer S, Nossner E, Ochmann O, Scharner A, Issels RD. The role of heat shock protein (hsp70) in dendritic cell maturation: Hsp70 induces the maturation of immature dendritic cells but reduces DC differentiation from monocyte precursors. Eur J Immunol. 2001;31:1602–1609. doi: 10.1002/1521-4141(200105)31:5<1602::AID-IMMU1602>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- Lane C, Leitch J, Tan X, Hadjati J, Bramson JL, Wan Y. Vaccination-induced autoimmune vitiligo is a consequence of secondary trauma to the skin. Cancer Res. 2004;64:1509–1514. doi: 10.1158/0008-5472.can-03-3227. [DOI] [PubMed] [Google Scholar]
- Le Poole IC, Das PK, van den Wijngaard RM, Bos JD, Westerhof W. Review of the etiopathomechanism of vitiligo: A convergence theory. Exp Dermatol. 1993;2:145–153. doi: 10.1111/j.1600-0625.1993.tb00023.x. [DOI] [PubMed] [Google Scholar]
- Le Poole IC, van den Wijngaard RM, Westerhof W, Dutrieux RP, Das PK. Presence or absence of melanocytes in vitiligo lesions: An immunohistochemical investigation. J Invest Dermatol. 1993b;100:816–822. doi: 10.1111/1523-1747.ep12476645. [DOI] [PubMed] [Google Scholar]
- Le Poole IC, Luiten RM. Autoimmune etiology of generalized vitiligo. Curr Dir Autoimmun. 2008;10:227–243. doi: 10.1159/000131485. [DOI] [PubMed] [Google Scholar]
- Li Z, Dai J, Zheng H, Liu B, Caudill M. An integrated view of the roles and mechanisms of heat shock protein gp96-peptide complex in eliciting immune response. Front Biosci. 2002;7:731–51. doi: 10.2741/a808. [DOI] [PubMed] [Google Scholar]
- Mambula SS, Stevenson MA, Ogawa K, Calderwood SK. Mechanisms for Hsp70 secretion: Crossing membranes without a leader. Methods. 2007;43:168–175. doi: 10.1016/j.ymeth.2007.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millar DG, Garza KM, Odermatt B, Elford AR, Ono N, Li Z, Ohashi PS. Hsp70 promotes antigen-presenting cell function and converts T-cell tolerance to autoimmunity in vivo. Nat Med. 2003;9:1469–1476. doi: 10.1038/nm962. [DOI] [PubMed] [Google Scholar]
- Millar Douglas G, Ohashi Pamela S. Hsp70 family members, danger signals and autoimmunity. In: Asea Alexzander A, De Maio Antonio., editors. Heat Shock Proteins: Potent Mediators of Inflammation and Immunity. Netherlands: Springer; 2007. pp. 189–211. [Google Scholar]
- Millington GW, Levell NJ. Vitiligo: The historical curse of depigmentation. Int J Dermatol. 2007;46:990–995. doi: 10.1111/j.1365-4632.2007.03195.x. [DOI] [PubMed] [Google Scholar]
- Multhoff G. Heat shock proteins in immunity. Handb Exp Pharmacol. 2006;172:279–304. doi: 10.1007/3-540-29717-0_12. [DOI] [PubMed] [Google Scholar]
- Muranski P, et al. Tumor-specific Th17-polarized cells eradicate large established melanoma. Blood. 2008;112:362–373. doi: 10.1182/blood-2007-11-120998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murshid A, Gong J, Calderwood SK. Heat-shock proteins in cancer vaccines: Agents of antigen cross-presentation. Expert Rev Vaccines. 2008;7:1019–1030. doi: 10.1586/14760584.7.7.1019. [DOI] [PubMed] [Google Scholar]
- Namazi MR. Neurogenic dysregulation, oxidative stress, autoimmunity, and melanocytorrhagy in vitiligo: Can they be interconnected? Pigment Cell Res. 2007;20:360–363. doi: 10.1111/j.1600-0749.2007.00408.x. [DOI] [PubMed] [Google Scholar]
- Nicchitta CV. Re-evaluating the role of heat-shock protein-peptide interactions in tumour immunity. Nat Rev Immunol. 2003;3:427–432. doi: 10.1038/nri1089. [DOI] [PubMed] [Google Scholar]
- Ongenae K, Van Geel N, Naeyaert JM. Evidence for an autoimmune pathogenesis of vitiligo. Pigment Cell Res. 2003;16:90–100. doi: 10.1034/j.1600-0749.2003.00023.x. [DOI] [PubMed] [Google Scholar]
- Overwijk WW, et al. Tumor regression and autoimmunity after reversal of a functionally tolerant state of self-reactive CD8+ T cells. J Exp Med. 2003;198:569–580. doi: 10.1084/jem.20030590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez-Perez L, Kottke T, Daniels GA, Diaz RM, Thompson J, Pulido J, Melcher A, Vile RG. Killing of normal melanocytes, combined with heat shock protein 70 and CD40L expression, cures large established melanomas. J Immunol. 2006;177:4168–4177. doi: 10.4049/jimmunol.177.6.4168. [DOI] [PubMed] [Google Scholar]
- Snoek M, Jansen M, Olavesen MG, Campbell RD, Teuscher C, van Vugt H. Three Hsp70 genes are located in the C4-H-2D region: Possible candidates for the orch-1 locus. Genomics. 1993;15:350–356. doi: 10.1006/geno.1993.1067. [DOI] [PubMed] [Google Scholar]
- Srivastava P. Interaction of heat shock proteins with peptides and antigen presenting cells: Chaperoning of the innate and adaptive immune responses. Ann Rev Immunol. 2002;20:395–425. doi: 10.1146/annurev.immunol.20.100301.064801. [DOI] [PubMed] [Google Scholar]
- Strbo N, Podack ER. Secreted heat shock protein gp96-ig: An innovative vaccine approach. Am J Reprod Immunol. 2008;59:407–416. doi: 10.1111/j.1600-0897.2008.00594.x. [DOI] [PubMed] [Google Scholar]
- Tosti G, di Pietro A, Ferrucci PF, Testori A. HSPPC-96 vaccine in metastatic melanoma patients: From the state of the art to a possible future. Exp Rev Vaccines. 2009;8:1513–1526. doi: 10.1586/erv.09.108. [DOI] [PubMed] [Google Scholar]
- Townsend A, Bodmer H. Antigen recognition by class I-restricted T lymphocytes. Annu Rev Immunol. 1989;7:601–624. doi: 10.1146/annurev.iy.07.040189.003125. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Alternative HSP70 isoform Hsp70-2 is irrelevant for inducing autoimmune responses. (A) Wild-type (WT) and constitutive Hsp70 knockout (Hsp70-2 KO) mice were gene gun vaccinated five times every six days with 6 μg of either TRP-2 or empty vector (EV; plasmid containing no gene insert) control DNA. (B) No difference in depigmentation was observed between vaccinated (vacc) wild-type and Hsp70-2 KO mice. (C) No differences in cytotoxicity were observed in wild-type or Hsp70-2 KO mice towards TRP-2-pulsed splenocytes. Data are presented as means ± SEM. (NS [no significance]; *P < 0.05; **P < 0.01; n = 10 per group.)
Immunohistology reveal a greater influx of CD8+ T cells in wild-type versus Hsp70-1 knockout mice. (A) Image of skin from a C57BL6 mouse vaccinated with HSP70i + TRP-1 reveal an abundant number of CD3+ T cells (black arrows) in the dermoepidermal junction. CD3-CD4+ non-T cells (red arrows) were mainly localized to the dermis. A single CD3+CD4+ T cell is shown in the epidermis (green arrow). (B) A serial section from the same tissue as (A) indicates the vast majority of the T cells are CD8+ (black arrows). (C) Fluorescent staining of skin from a C57BL6 mouse vaccinated with HSP70i + TRP-1 further demonstrate the colocalization of CD3 (red) and CD8 (green) as indicated by yellow (red arrows). CD3-CD4+ non-T cells were mainly confined to the dermis (white arrow). A single CD3+CD4+ T cell is shown (green arrow). (D) Skin from an Hsp70-1 knockout mouse vaccinated with HSP70i + TRP-1 reveal less CD3+CD8+ T cell infiltration (red arrow). CD3-CD4+ non-T cells were also present in the dermis (white arrow). Scale equal 50 μm.
Gene-gun vaccination alone does not accelerate depigmentation in vitiligo-prone mice. This supplementary image of the Pmel-1 mice from Figure 4 demonstrates similar levels of depigmentation in empty vector vaccinated and unvaccinated Pmel-1 mice. Ventral and dorsal views are shown.
Antibodies can block HSP70i induced dendritic cell activation in vitro. This supplementary image displays the fluorocytometry histograms representing activation marker expression by CD11c+, cultured mouse dendritic cells as summarized in Figure 6. Whereas exposure to HSP70i increases expression of CD80, CD83, CD86, and MHCII, in presence of antibodies to HSP70i, dendritic cell activation is largely prevented.