

Abstract
We developed a computational framework to robustly identify RNA editing sites using transcriptome and genome deep-sequencing data from the same individual. As compared with previous methods, our approach identified a large number of RNA editing sites with high specificity in both Alu and non-Alu regions. We also found that the editing of non-Alu sites appears to be dependent on nearby edited Alu sites, possibly through the locally formed double-stranded RNA structure.
RNA editing, the post-transcriptional alteration of genome-encoded information by chemical modification of individual RNA bases, provides a powerful way to diversify the transcriptome. In humans, there are two known types of editing, both catalyzed by deaminases. Cytosine-to-uracil editing, which is catalyzed by APOBEC1, appears to be rare and specific to small intestine enterocytes1. The other, much more common type of editing is the adenosine-to-inosine (A-to-I) editing catalyzed by the adenosine deaminases acting on RNA (ADARs)2. ADARs bind double-stranded RNA (dsRNA) and deaminate adenosine bases to inosine, which is recognized as guanosine by the cellular machinery. A-to-I RNA editing is pervasive in Alu repeats because of the dsRNA structure formed by widespread Alu inverted pairs in many genes3.
Identifying human RNA editing events outside of the widely edited Alu repeats has been challenging4. The advent of next-generation sequencing led to our success in identifying hundreds of human A-to-I RNA editing sites in non-Alu regions5. With sequencing data becoming more readily available, a number of groups have recently developed computational approaches and used them to identify numerous RNA editing sites of all 12 possible mismatch types by comparing genomic DNA and RNA sequencing (RNA-seq) data from the same individuals6-9. However, further analyses suggest that many of the identified sites are likely false positives derived mainly from improper analysis of the sequencing data, particularly in non-Alu regions10-13. A major challenge of using short reads from next-generation sequencing is the discrimination of sequencing and mapping errors from true RNA editing events, which we sought to overcome with a robust computational pipeline. In contrast to the previous approaches, our method demonstrates no evidence to support the existence of noncanonical RNA editing.
We developed a framework to robustly identify RNA editing sites by meticulous analyses of genomic DNA and RNA sequences obtained from a single individual (Fig. 1a, Online Methods). The characteristic features that distinguish our approach are (i) the choice of the short read mapper BWA14, which gave high specificity and speed, to map RNA-seq reads to the reference genome and across splicing junctions (Supplementary Note 1, Supplementary Fig. 1); (ii) the careful tuning of parameters to call RNA editing candidate positions (Fig. 1b, Supplementary Note 2) and (iii) the incorporation of several filters to remove false positives, particularly in non-Alu regions where RNA editing appears to be much less frequent and thus far more challenging to accurately identify (Fig. 1a, Supplementary Fig. 2). These filters were designed to remove false discoveries caused by errors introduced during construction and sequencing of RNA-seq libraries, incorrect mapping of short reads, and single-nucleotide polymorphisms (SNPs) in the genome (Supplementary Note 3).
Figure 1. A computational framework to identify RNA editing sites in Alu and non-Alu regions.



(a) Pipeline for the identification of RNA editing sites. RNA-seq reads (short lines) were mapped to the human reference genome (where blue reads map) and regions spanning all known splicing junctions (yellow lines separated by dashes). Boxes denote exons, and striped parts of two adjacent exons are joined together as the splicing junction sequence. (b) Relationship between the percentage of A-to-G mismatches and the minimum number of reads with altered nucleotides in Alu, repetitive non-Alu and nonrepetitive regions in GM12878. For all non-Alu sites, a minimum frequency of 10% for the RNA variant was required, whereas no minimum variant frequency was used for Alu positions. In non-Alu regions at least three variant nucleotides are required to achieve high specificity in RNA editing detection. (c) Percentage of all 12 mismatch types in GM12878.
We applied our method to the lymphoblastoid cell line GM12878, whose genome and RNA have been deeply sequenced (Online Methods and Supplementary Table 1). We identified a total of 147,029 editing sites in Alu repeat regions, 140,825 (95.8%) of which were of the A-to-G type, indicative of A-to-I editing (Fig. 1c, Table 1, Supplementary Tables 2, Supplementary Data 1). In non-Alu regions, we distinguished sites that are located in other repetitive regions (mostly long and short interspersed elements and long terminal repeats) and in nonrepetitive regions. For these two categories, we identified a total of 2,385 and 1,451 mismatches between RNA and DNA sequences, including 2,324 (97.4%) and 1,257 (86.6%) A-to-G sites in repetitive and nonrepetitive regions, respectively (Fig. 1c, Table 1, Supplementary Fig. 3, Supplementary Data 1). To our knowledge, this is the first systematic examination of repetitive non-Alu editing sites in humans, although hundreds of such sites were previously found in mice15. The A-to-G sites that we found in non-Alu regions were associated with two known features of A-to-I RNA editing: double-stranded RNA (dsRNA) structure and the ADAR-binding sequence motif5 (Supplementary Note 4, Supplementary Fig. 4). Furthermore, we successfully validated 11 out of 12 selected A-to-G sites (with >10% editing frequency) in nonrepetitive regions using PCR and Sanger sequencing (Online Methods, Supplementary Tables 3 and 4).
Table 1.
Comparison of our findings with recent efforts by other groups
| No. Alu sites | No. repetitive non-Alu sites | No. nonrepetitive sites | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Total | A>G | A>G (%) |
Total | A>G | A>G (%) |
Total | A>G | A>G (%) |
|
| Li et al. (2011)6 | Not investigated | Not investigated | 10,210 | 2,328 | 22.8 | ||||
| Ju et al. (2011)7 | 1,012 | 806 | 79.6 | 163 | 78 | 47.9 | 646 | 102 | 15.8 |
| Bahn et al. (2012)8 | 3,979 | 3,589 | 90.2 | 477 | 284 | 59.5 | 1,049 | 268 | 25.5 |
| Peng et al. (2012) (YH)9 | 19,408 | 18,919 | 97.5 | 1,544 | 1,390 | 90.0 | 1,734 | 802 | 46.3 |
| This study (GM12878) | 147,029 | 140,825 | 95.8 | 2,385 | 2,324 | 97.4 | 1,451 | 1,257 | 86.6 |
| This study (YH)a | 446,670 | 414,533 | 92.8b | 5,975 | 5,406 | 90.5b | 4,433 | 3,438 | 77.6b |
Union of sites detected from the separate and combined poly(A)+ and poly(A)− sequencing libraries.
A-to-G fraction (A>G) is based on gene annotation. For all three categories, the number of T-to-C changes is higher than that of the remaining mismatch types. If all T-to-C changes are considered to be wrongly annotated A-to-G changes, the A-to-G percentages for the three categories are 95.8%, 93.8% and 83.0%, respectively.
Although unbiased toward identifying A-to-G sites, our pipeline detected a high A-to-G fraction in Alu and non-Alu regions (Table 1). Because A-to-I editing is prevalent in Alu repeats, our method and others7-9 tend to yield results highly enriched for A-to-G mismatches in the Alu regions, although we identified significantly more sites. The advantage of our method over others6-9 is even more striking in nonrepetitive regions, in which identification of editing sites is more challenging; we detected 86.6% of sites as A-to-G mismatches, whereas all other methods returned fractions below 47% (Table 1). We suspect that the 13.4% non-A-to-G sites in our analysis are unlikely to be genuine. We were unable to validate any of a random selection of these sites (with >15% editing frequency) using PCR and Sanger sequencing (n = 7; Supplementary Fig. 5). These are likely to be false positives derived from sequencing and mapping errors as well as undetected SNPs in the genome.
To evaluate the performance of our method on other data sets and to carry out a fair comparison with other methods, we applied our framework to the same data recently used by Peng and colleagues9 to detect RNA editing sites (Supplementary Table 1, Online Methods). Peng et al.9 identified over 22,688 RNA editing sites, of which ~93% are A-to-G changes, from the lymphoblastoid cell line of a Han Chinese individual (YH). This high A-to-G fraction is dominated by repetitive sites, whereas the fraction was only 46.3% in nonrepetitive regions, in sharp contrast to the 86.6% we achieved in GM12878 (Table 1). In repetitive regions (both Alu and non-Alu), our method identified 20 times more A-to-G sites with a comparably high A-to-G fraction. In nonrepetitive regions, our method identified four times more A-to-G sites with significantly higher A-to-G fraction (from 46.3% to 77.6%) (Table 1, Supplementary Data 2). Of note, the A-to-G fraction is slightly lower in the YH data than the counterpart in GM12878, probably for two reasons. First, GM12878 RNA-seq data is strand specific, whereas a subset of YH RNA-seq data is not strand specific. For non–strand-specific RNA-seq data, we used existing gene annotations to infer the editing type. This can erroneously call A-to-G as T-to-C as a result of incorrect or missing annotations of RNA. Second, the removal of genomic variants that are present in the dbSNP database is less effective for an individual of Asian descent due to the heavily biased composition of dbSNP16. Our analysis strongly suggests that effective removal of known SNPs is an important step in reducing the number of false positives even when the sequenced genome from the same individual is available (Supplementary Note 3). In addition, it is evident that the higher RNA-seq coverage in YH (Supplementary Table 1) allows us to detect many more editing sites (see below).
The deeply sequenced transcriptome of GM12878 allowed us to investigate the power of RNA editing detection in relation to RNA-seq depth. We called variants on randomly sampled subsets of reads from the two biological replicates of GM12878, and we observed that the number of identified editing sites, in both Alu and non-Alu regions, depends heavily on the sequencing depth and increases with additional reads (Supplementary Fig. 6). This analysis implies that more sites in both Alu and non-Alu regions could be identified if more RNA-seq reads were obtained, as exemplified by the YH transcriptome with its deeper sequencing coverage.
Most A-to-I RNA editing sites are located in introns (Supplementary Table 2). We speculated that the non-Alu A-to-I editing sites were related to nearby edited Alu sites, and discovered that the two classes of sites indeed tend to significantly co-occur in the same genes (Fig. 2a). The 140,825 Alu, 2,324 repetitive non-Alu and 1,257 nonrepetitive A-to-G sites that we identified in GM12878 (Table 1) fall in 12,764, 891 and 796 genes, respectively. An example of locally clustered sites within the same gene is shown in Figure 2b. These observations prompted us to hypothesize that the dsRNA structure formed by inverted Alu repeats facilitates the editing of the flanking adenosines. Several lines of evidence seem to support this hypothesis. First, edited Alu sites were significantly closer to non-Alu sites than to random adenosines in the same gene (Fig. 2c, Supplementary Fig. 7a). Second, in comparison with genes containing Alu editing sites only, genes containing both Alu and non-Alu sites tended to harbor greater numbers of Alu repeats, edited Alu repeats, invert-repeated Alu pairs, invert-repeated and edited Alu pairs, and total edited Alu sites (Fig. 2d,e; Supplementary Fig. 7b–e). Taken together, these observations suggest that the editing of non-Alu sites depends on the presence of nearby edited Alu sites.
Figure 2. Editing of many non-Alu sites appears to be dependent on nearby edited Alu sites.

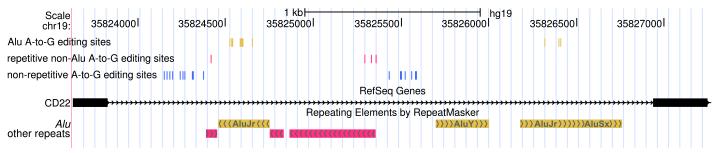



(a) Venn diagram showing the overlap between genes that contain A-to-G editing sites in Alu (yellow), repetitive non-Alu (purple) and nonrepetitive regions (blue). A significant number of genes contain both Alu and repetitive non-Alu A-to-G editing sites (P = 1.9 × 10−83) and Alu and nonrepetitive sites (P = 3.7 × 10−71). (b) Example of a gene that contains all three types of editing. Editing in Alu, repetitive non-Alu and nonrepetitive regions occurs in close proximity to each other. (c) The identified non-Alu A-to-G sites are significantly closer to the nearest Alu A-to-G site than to random adenosines in genes with Alu editing only (nonrepetitive sites versus random adenosines: P = 1.1 × 10−96; repetitive non-Alu sites versus random adenosines: P = 7.9 × 10−160). (d,e) The number of edited Alu repeats is significantly higher in genes with Alu and nonrepetitive editing (P = 2.0 × 10−40) (d) and Alu and repetitive non-Alu editing (P = 2.8 × 10−20) (e), compared to genes with Alu editing only.
An unprecedented large number of RNA editing sites were called in this study. As expected3, the vast majority of sites are promiscuously edited in Alu regions. We identified a total of 493,111 Alu A-to-G sites (140,825 from GM12878, and 414,533 from YH). This is a significant expansion of the previously annotated 36,802 Alu sites17 (Supplementary Fig. 8). RNA editing sites in coding regions seem to be rare in the lymphoblastoid cell line used in our work and others. Nevertheless, our framework can be readily applied to other cell or tissue types in which RNA editing is biologically relevant.
As next-generation sequencing technologies become widely accessible, it will become routine to generate sequencing data for RNA editing discovery. Tools developed for detecting genetic variants in genomes are useful but insufficient to accurately identify RNA editing sites because of the complexity of RNA. Our approach achieved high sensitivity and specificity by implementing meticulous mapping and filtering steps tailored for Alu and non-Alu regions. The identification of RNA editing sites with our approach bypasses several requirements of previous methods, such as clustering of editing sites3 and synthesis of target-capturing probes5, while achieving very high accuracy. In addition, the insights gained in our work will not only allow future endeavors for RNA editing identification, but also benefit other studies that rely on accurate mapping of RNA-seq data.
ONLINE METHODS
Mapping of RNA-seq reads
We obtained poly(A)+ RNA-seq data for whole-cell GM12878 from the ENCODE project (http://genome.ucsc.edu/ENCODE/dataSummary.html). The strand-specific RNA-seq libraries were made as described previously18. The transcriptome was deeply sequenced with Illumina HiSeq in two biological replicates, resulting in 235.8 and 263.7 million paired-end 76-base sequencing reads, respectively (Supplementary Table 1). We chose BWA14 as the mapper for RNA-seq reads due to its demonstrated high accuracy of alignment19. We mapped each of the paired-end reads separately using the commands “bwa aln fastqfile” and “bwa samse -n4”. In contrast to previous approaches, we mapped RNA-seq reads not only to the reference genome8,9 or to the transcriptome6,7 but to a combination of the hg19 reference genome plus exonic sequences surrounding all currently known splicing junctions from gene models available in annotation from Gencode, RefSeq, Ensembl and UCSC Genes. We chose the length of these splicing junction regions to be slightly shorter than the RNA-seq reads to avoid simultaneous hits to the reference genome and the splicing junctions (for 76-bp reads, a region of 75 bp up- and downstream was chosen). When the adjacent exons up- and/or downstream of a splicing junction were shorter than the required length (for 76-bp reads, with exons shorter than 75 bp), the regions were extended across multiple exons. We only considered uniquely mapped reads and used samtools rmdup20 to remove identical reads (PCR duplicates) that mapped to the same location. Of these identical reads, only the read with the highest mapping quality was retained for further analysis.
Identification of RNA editing candidates
After the removal of PCR duplicates, the remaining reads were used to detect mismatches between RNA and DNA that may be putative RNA editing sites. We inspected all positions that showed variation in the RNA and were homozygous in the genomic DNA of the same individual. To determine homozygous positions in the genomic DNA of GM12878, we used read mapping data provided by the 1000 Genomes Project (http://www.1000genomes.org). The genome was sequenced at 44× coverage21, allowing accurate genotype calls. A site was called homozygous if 10 or more reads contained the same base that represented more than 95% of the complete coverage and if only 2 or fewer alternative bases were present at the same position. We only took variant positions in the RNA into consideration if they conformed to our requirements for number, frequency, and quality of bases that vary from the reference genome. We specifically required that each variant be supported by two or more variant bases having a base quality score of ≥25 and a mapping quality score ≥20. We ensured that no variation in the human genome confounded our results by removing all known SNPs present in dbSNP (except SNPs of molecular type “cDNA” database version 135; http://www.ncbi.nlm.nih.gov/SNP/), the 1000 Genomes Project or the University of Washington Exome Sequencing Project (http://evs.gs.washington.edu/EVS/). To avoid false positives at the 5′ read ends due to random-hexamer priming, we truncated the first 6 bases of each read. Subsequently, all variants were separated into Alu and non-Alu regions. Mismatches in Alu regions showed a convincingly high fraction of A-to-G mismatches and did not receive more stringent filtering. Variants in non-Alu regions were subjected to further refinement (see below). The DNA-RNA mismatch type was determined according to the strandedness of RNA-seq reads; we removed sites with conflicting annotation of editing types.
Refinement of non-Alu RNA editing candidates
RNA editing candidates in non-Alu regions were subjected to more stringent variant call criteria than their counterparts in Alu regions (we required at least three variant reads and mismatch frequency ≥0.1). We removed sites in simple repeats according to RepeatMasker annotation, discarded intronic candidates if they were located within 4 bp of all known splicing junctions according to RefGene, UCSC Genes and Gencode (version 7) gene annotations, and removed sites in homopolymer runs of ≥5 bp. Finally, we removed RNA editing candidates if they were located in regions of high similarity to other parts of the genome. For that purpose we applied BLAT to all reads that overlap an RNA candidate site and at the same time show a mismatch from the reference. We required for each read that (i) the best hit overlap the candidate site and (ii) the second-best hit have a score <95% of the best blat hit. We only kept sites for which the number of reads passing the above BLAT criteria was larger than the number of reads that failed the criteria.
Application of our pipeline to YH data
To directly evaluate the performance of our method, we applied our pipeline to the Han Chinese (YH) genome and RNA-seq data obtained from Peng et al.9. The RNA-seq data consists of two different libraries: an unstranded poly(A)+ library and a strand-specific poly(A)- library. Candidate editing sites were called and run through our filtering pipeline using three different subsets of the data: poly(A)+ reads only, poly(A)- reads only, and poly(A)+ reads combined with poly(A)- reads. For the sites obtained from poly(A)- reads only, the DNA-RNA mismatch type was determined according to the strandedness of the edited reads, as we did for GM12878. For the sites obtained from poly(A)+ reads only and from the combination of poly(A)+ and poly(A)- reads, the DNA-RNA mismatch type was determined based on RefSeq, UCSC genes and Gencode v7 gene annotations. Sites with conflicting annotation of editing types were removed.
Validation of sites with PCR and Sanger sequencing
We used PCR to validate whether a subset of candidate sites are edited in vivo. Primer sequences are listed in Supplementary Table 4. Typically, a 25-μl PCR reaction was assembled with 1x iQ SYBR Green Supermix (Bio-Rad), ~50 ng of gDNA (or ~10 ng of cDNA) template, and 200 nM each of the forward and reverse primers. We used the following touch-down PCR program: 95 °C for 5 min, 24 cycles of 95 °C for 30 s, 72 °C for 30 s with a decrement of 0.7 °C every cycle, and 72 °C for 45 s, then 40 cycles of 95 °C for 30 s, 55 °C for 30 s, and 72 °C for 45 s. PCR amplicons were sequenced by Eurofins MWG Operon.
Statistical analysis
To evaluate the significance of the overlap between Alu and non-Alu A-to-G site containing genes, we calculated the cumulative probability of the hypergeometric distribution with the following equation:
where N is the total number of loci, n is the number of genes with Alu A-to-G sites, m is the number of genes with non-Alu A-to-G sites, and k is the number of genes with both Alu and non-Alu A-to-G sites.
The significant difference in (i) the distance between Alu and non-Alu editing sites, (ii) the number of Alu repeats, (iii) the number of edited Alu repeats, (iv) the number of invert-repeated (present on both strands with different orientations) Alu pairs, (v) the number of invert-repeated and edited Alu pairs and (vi) number of edited Alu sites in genes with Alu-only editing versus genes containing Alu and non-Alu editing was determined using a one-tailed Mann-Whitney test.
Supplementary Material
ACKNOWLEDGMENTS
We thank E. Levanon and N. Sanjana for critical reading of the manuscript, C. Pan and J. Sun for technical assistance, and T. Gingeras for support. We appreciate the constructive suggestions made by anonymous reviewers. This work is supported by the Stanford University Department of Genetics and the US National Institutes of Health.
Footnotes
AUTHOR CONTRIBUTIONS
G.R., W.L. and R.P. performed the computational analyses with help from M.H.T. and J.B.L. M.H.T. and G.R. carried out the validation experiments. C.D. generated the GM12878 RNA-seq data. R.P. and J.B.L. wrote the paper with input from the other authors.
COMPETING FINANCIAL INTERESTS
The authors declare no competing financial interests.
References
- 1.Rosenberg BR, Hamilton CE, Mwangi MM, Dewell S, Papavasiliou FN. Nat. Struct. Mol. Biol. 2011;18:230–236. doi: 10.1038/nsmb.1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nishikura K. Annu. Rev. Biochem. 2010;79:321–349. doi: 10.1146/annurev-biochem-060208-105251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Levanon EY, et al. Nat. Biotechnol. 2004;22:1001–1005. doi: 10.1038/nbt996. [DOI] [PubMed] [Google Scholar]
- 4.Silberberg G, Ohman M. Curr. Opin. Genet. Dev. 2011;21:401–406. 1. doi: 10.1016/j.gde.2011.04.009. [DOI] [PubMed] [Google Scholar]
- 5.Li JB, et al. Science. 2009;324:1210–1213. doi: 10.1126/science.1170995. [DOI] [PubMed] [Google Scholar]
- 6.Li M, et al. Science. 2011;333:53–58. doi: 10.1126/science.1207018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ju YS, et al. Nat. Genet. 2011;43:745–752. doi: 10.1038/ng.872. [DOI] [PubMed] [Google Scholar]
- 8.Bahn JH, et al. Genome Res. 2012;22:142–150. doi: 10.1101/gr.124107.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Peng Z, et al. Nat. Biotechnol. 2012 [Google Scholar]
- 10.Schrider DR, Gout JF, Hahn MW. PLoS ONE. 2011;6:e25842. doi: 10.1371/journal.pone.0025842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lin W, Piskol R, Tan MH, Li JB. Science. 2012;335:1302. doi: 10.1126/science.1210419. [DOI] [PubMed] [Google Scholar]
- 12.Kleinman CL, Majewski J. Science. 2012;335:1302. doi: 10.1126/science.1209658. [DOI] [PubMed] [Google Scholar]
- 13.Pickrell JK, Gilad Y, Pritchard JK. Science. 2012;335:1302. doi: 10.1126/science.1210484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li H, Durbin R. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Neeman Y, Levanon EY, Jantsch MF, Eisenberg E. RNA. 2006;12:1802–1809. doi: 10.1261/rna.165106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bustamante CD, Burchard EG, De la Vega FM. Nature. 2011;475:163–165. doi: 10.1038/475163a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kiran A, Baranov PV. Bioinformatics. 2010;26:1772–1776. doi: 10.1093/bioinformatics/btq285. [DOI] [PubMed] [Google Scholar]
- 18.Parkhomchuk D, et al. Nucleic Acids Res. 2009;37:e123. doi: 10.1093/nar/gkp596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li H, Homer N. Brief. Bioinform. 2010;11:473–483. doi: 10.1093/bib/bbq015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li H, et al. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Durbin RM, et al. Nature. 2010;467:1061–1073. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.