

Abstract
Epigenetic genome marking and chromatin regulation are central to establishing tissue-specific gene expression programs, and hence to several biological processes. Until recently, the only known epigenetic mark on DNA in mammals was 5-methylcytosine, established and propagated by DNA methyltransferases and generally associated with gene repression. All of a sudden, a host of new actors—novel cytosine modifications and the ten eleven translocation (TET) enzymes—has appeared on the scene, sparking great interest. The challenge is now to uncover the roles they play and how they relate to DNA demethylation. Knowledge is accumulating at a frantic pace, linking these new players to essential biological processes (e.g. cell pluripotency and development) and also to cancerogenesis. Here, we review the recent progress in this exciting field, highlighting the TET enzymes as epigenetic DNA modifiers, their physiological roles, and their functions in health and disease. We also discuss the need to find relevant TET interactants and the newly discovered TET–O-linked N-acetylglucosamine transferase (OGT) pathway.
Keywords: epigenetics, DNA methylation, hydroxymethylation, TET proteins, OGT
INTRODUCTION
DNA methylation in mammals involves covalent adding of a methyl group, most commonly to the 5′-position of cytosine in a CpG dinucleotide. CpG methylation is essential to normal development, probably because of its importance in transposable element silencing, X chromosome inactivation, genomic imprinting and the regulation of tissue-specific gene expression [1]. The genomic distribution of DNA methylation is gene and tissue specifics, and while the number of methylated CpG sequences is significant, there exist regions of high CpG density, termed CpG islands (CGI), that can be refractory to methylation when associated with active gene promoters [2, 3]. Waves of change affect the global DNA methylation pattern at specific times during early development, while more subtle, localized changes (demethylation or methylation) may occur in somatic cells in response to specific signals or stimuli [4]. DNA methylation patterns are established early in the zygote by the de novo DNA methyltransferases 3A and 3B (DNMT3A/3B) and they are conserved during cell divisions by the maintenance DNA methyltransferase DNMT1. These enzymes transfer the methyl group from S-adenosylmethionine to cytosine (usually in a CpG context), producing 5-methylcytosine (5-mC) [5].
A longstanding mystery in the epigenetic field surrounds the mechanisms allowing transition from the methylated to the unmethylated state. DNA demethylation can occur both passively and actively. ‘Passive’ DNA demethylation means progressive dilution of methylcytosine through mitosis. In this case, DNMT1 is excluded from the replication fork, so that the neosynthesized strand is kept unmethylated. ‘Active’ DNA demethylation states for the rapid, replication independent, enzymatic removal of methylcytosine. Such 5-mC ‘erasers’ have been intensively sought but have long remained elusive [6, 7]. As the methyl group of 5-mC is thermodynamically very stable and thought not to be directly removed from cytosine, one of the many possible mechanisms for active DNA demethylation would be ‘cutting’ of the glycosyl bond between the ribose and the pyrimidine base. However, this would require either selective targeting of glycosylases to the genomic regions that have to be demethylated or the presence of further modifications on the methylcytosine to be removed.
In 1952, Wyatt and Cohen [8] made an important discovery: they found 5-hydroxymethylcytosine (5-hmC) in the DNA of the T-even bacteriophages where C is almost completely replaced by 5-hmC and where 70% of 5-hmC is further glycosylated. These modifications have probably arisen from the ‘battle’ between bacterial restriction enzymes and bacteriophages [9]. For nearly 60 years, researchers studying the role of 5-hmC focused solely on bacteria and bacteriophages, until 2009, when two groups described this so-called ‘sixth base’ in mammalian genomic DNA. A first report described the presence of 5-hmC in Purkinje neurons and granule cells of the mouse brain. The other study showed that the Ten Eleven Translocation 1 enzyme (TET1) (discussed later) can convert 5-mC to 5-hmC, both in cultured cells and in vitro, and that mouse embryonic stem cells (mESCs) contain a significant fraction of 5-hmC (0.032% of all bases) [10, 11]. These two seminal studies opened new avenues in epigenetic research and raised many essential questions regarding the roles of this new epigenetic modification, not only in brain development and pluripotency but also in transcriptional regulation, DNA demethylation and cancer.
In this review, we first introduce the TET proteins, their enzymatic activities and their roles in DNA demethylation. We then present what is known about TETs and 5-hmC genomic localization, notably from epigenomic studies on ESCs and on brain. We further examine the role of TET proteins in transcriptional regulation, as regards both their catalytic activity and their potential ‘non-catalytic’ functions. Finally, we provide an update on the involvement of deregulated hmC and TET levels in cancerogenesis.
THE TET FAMILY OF ENZYMES CATALYZES CONVERSION OF 5-METHYLCYTOSINE TO MULTIPLE MODIFIED CYTOSINES
TET proteins were initially discovered through their involvement in myeloid leukemia where the TET1 gene, located on chromosome 10, can translocate with the H3K4 histone methyltransferase MLL gene on chromosome 11 [12]. TET enzymes are members of the TET/J-binding protein (JBP) family of α-ketoglutarate- and iron (II)-dependent dioxygenases, closely related to the JBP1 and JBP2 proteins found in kinetoplastids such as trypanosomes and leishmanias. In mammals, the TET/JBP family is composed of the founding member TET1 along with TET2 and TET3. These three genes encode proteins sharing a double-stranded β-helix-fold and a cysteine-rich region within the catalytic domain. Although TET1 and TET3 harbor an N-terminal CXXC DNA-binding domain, TET2 seems to have lost it during evolution. Interestingly, TET2 CXXC exists as a separate gene, also called IDAX or CXXC4, which encodes an inhibitor of Wnt signaling. This suggests a connection between the Wnt pathway and the TET proteins [11, 13, 14]. As the thymine hydroxylase activities of JBP1 and JBP2 are involved in producing base J (β-d-glucosyl-hydroxymethyluracil) in kinetoplastids, several labs have investigated the possibility that TET1 might catalyze the conversion of 5-mC to 5-hmC. Furthermore, as overexpression of wt TET1 but not the mutant counterpart was found to decrease methylation in transfected cells, Anjana Rao’s group suggested that TETs might potentiate DNA demethylation and that 5-hmC might be an intermediate in the pathway to unmodified cytosine [11]. This ‘methylcytosine hydroxylase’ activity was extended to Tet2 and Tet3 by Yi Zhang’s lab, which also found that Tet1 knock-down impairs mESC self-renewal and maintenance [15]. In fungi such as Neurospora crassa and Aspergillus nidulans, thymine-7-hydroxylase catalyzes sequential conversion of thymine to 5-hydroxymethyluracil (5-hmU), 5-formyluracil (5-fU) and 5-carboxyuracil (5-caU) [16, 17]. This encouraged researchers to look for further oxidized forms of methylcytosine. In a seminal paper, Ito et al. [18] demonstrated that all three Tets can oxidize 5-hydroxymethylcytosine iteratively to 5-formylcytosine (5-fC) and 5-carboxycytosine (5-caC), and that these ‘adducts’ are physiologically present in several tissues including mESCs. TETs-mediated 5-caC synthesis was further confirmed by He et al. [19], who additionally found thymine DNA glycosylase (TDG) to remove 5-caC from DNA. This suggests that TETs and the base excision repair (BER) machinery might work together to actively remove DNA methylation.
TET PROTEINS AND CYTOSINE MODIFICATIONS: DIFFERENT LEVELS IN DIFFERENT CELLS
Although the proportion of 5-methylcytosine in genomic DNA seems constant (3–4% of total cytosines), 5-hydroxymethylcytosine varies much more between tissues. The brain and spinal cord appear particularly rich in 5-hmC (respectively ≈0.80% and ≈0.45%). Other organs such as the testes, spleen and thymus show very little 5-hmC (<0.10%). ESCs and organs as kidneys, heart and lungs display an intermediate level of this epigenetic modification [15, 20, 21]. Cell cultures show a very small amount of 5-hmC, and unlike 5-mC, this mark gradually decreases upon culturing of normal breast tissue for example [22]. It is noteworthy that 5-fC and 5-caC, although much less abundant than 5-hmC, are both detectable in mESCs, and that 5-fC is also present in tissues such as brain, spleen, liver and pancreatic tissues [18]. TET expression also varies between cells/organs: while TET2 and TET3 are expressed in various tissues, only ESCs appear rich in TET1 [18, 23]. Surprisingly, the TET2 and TET3 expression profiles are often similar, suggesting that these enzymes act in concert [18, 22]. Another point worth stressing is the absence of correlation between TET expression and 5-hmC abundance [22], a fact worthy of future study. In line with observations on cultured cells, the level of 5-hmC is significantly reduced in several sarcomas, notably of the lung, breast, colon, liver, brain and prostate. These reductions might mirror, at least partially, the global hypomethylation phenotype usually found in various cancers [24–26]. Mitochondrial DNA is also rich in both hydroxymethylcytosine and methylcytosine, and although no mitochondrial targeting sequence has been found in TET proteins, western blots of mitochondrial extracts show the presence of Tet1 and Tet2 [27–29]. It thus seems that in mammals, oxidized 5-methylcytosine derivatives are present at various levels according to the cell type, and that in some cells at least, they are likely to have important functions.
DNA DEMETHYLATION AND THE TET PROTEINS
The occurrence of 5-hmC, 5-fC and 5-caC in mammalian tissues led the scientists to investigate what role(s) they might play in 5-mC demethylation. Although focal/local DNA demethylation is a mechanism enabling genes to respond to various stimuli, global DNA demethylation is crucial to erase epigenetic memory and epimutations during development. At this time, the phenomenon appears to give a ‘facelift’ to DNA, enabling it to evolve along with its new environment. This section deals with what is known or suspected about the roles of the TET proteins and the intermediates of 5-mC oxidation in global and focal DNA demethylation.
Global DNA demethylation
In mammals, there are two waves of global DNA demethylation: one occurring when the unipotent primordial germ cells (PGCs) migrate to the future gonads and the other during fertilization. At the time of PGCs migration, the paternal and maternal genomes undergo an active genome-wide demethylation by little-known mechanisms, although a recent publication showed that this phenomenon occurs by 5-hmC conversion of methylcytosine, probably by Tet1 and Tet2, and a subsequent replication-dependent dilution of the 5-hmC mark [30]. De novo methylation patterns and genomic imprints are established by DNMT3 during spermatogenesis at embryonic day 16 (ED16) and in oocytes after birth. The second wave of global demethylation occurs in the zygote soon after fertilization. In this case, two mechanisms must be considered: active demethylation of the paternal pronucleus just after fertilization and delayed passive demethylation of the maternal genome. Finally, methylation profiles are reestablished around the time of fetal implantation. It is worth mentioning that the genomic imprints created during PGC formation are not affected by the demethylation/remethylation events occurring in the zygote [4, 31]. Although the exclusion of Dnmt1o (the oocyte version of somatic Dnmt1) explains how maternal genomic DNA becomes demethylated through multiple rounds of replication, the paternal pronucleus demethylation system has been explained only very recently [32–37]. In 2011, Iqbal et al. [32] showed that the paternal genome is hydroxymethylated upon entry of the sperm into the oocyte, while the maternal counterpart shows no hydroxymethylation and remains methylated. Tet3 is expressed in the mouse oocyte and upregulated upon fertilization. Importantly, the 5-hmC modification remains during the first cell divisions, suggesting that it is not removed. Also, in 2011 were published two other reports showing that Tet3 silencing can impede paternal pronucleus hydroxymethylation causing a hypermethylation phenotype. Interestingly, one group discovered that the Pgc7 protein (also called Dppa3/Stella) maintains methylation in the maternal genome, since Pgc7 knockout renders maternal 5-mC accessible to oxidation in 5-hmC [33, 34]. In 2012, Nakamura et al. [37] revealed that both the maternal pronucleus and the paternal imprinted genes are protected from Tet3-mediated hydroxymethylation/demethylation by binding of Pgc7 to the repressive H3K9me2 histone mark. Finally, it has recently been observed that in the paternal pronucleus, 5-hmC is further oxidized to 5-fC and 5-caC and that these modifications are diluted through multiple cell divisions [35]. A simplified model would thus be as follows: upon fertilization, the paternal pronucleus is actively demethylated through iterative oxidations (5-mC → 5-hmC → 5-fC → 5-caC) mediated by maternal Tet3, and both the imprinted genes and the maternal genome are kept methylated by Pgc7-H3K9me2 binding. We wish to point out that despite the active loss of the methyl group, the successive intermediates of 5-mC oxidation are progressively diluted with mitosis. This remark is further extended to PGCs global demethylation where 5-hmC decreases with replication. Thus, the term ‘active’ DNA demethylation of the paternal genome/PGCs should only refer to the ‘active’ loss of the methyl group per se, keeping in mind that the carbon–carbon bond is passively lost during replication.
Focal DNA demethylation
Focal DNA demethylation events occur primarily upon external stimulation of the cell. Until the discovery of the TET enzymes and of the oxidized forms of 5-mC, the only known ways a gene could be demethylated in mammals were through replication (passive demethylation) or via DNA repair (active demethylation) [9]. There was one report nearly 15 years ago suggesting that MBD2b, a variant of MBD2, can remove the methyl group of 5-mC by breaking the covalent carbon–carbon bond [38], but the results described could not be confirmed by others. DNA repair includes BER and nucleotide excision repair (NER), i.e. the removal, respectively, of a few nucleotides or of a long stretch of DNA from a damaged site. BER and NER are both essential to maintain the integrity of the genome, and their alteration may result in diseases such as neurological syndromes, growth defects, or UV sensitivity and skin cancer [39]. The choice of the pathway used to repair DNA depends on the type of lesion: the BER machinery is recruited to single strand breaks or damaged bases and the NER components act to remove pyrimidine dimers and other bulky adducts. Such damages can have various causes including UV radiation, oxidative stress and chemical agents [39]. The growth arrest and DNA damage protein 45 (GADD45) seems to play a pivotal role in active DNA demethylation, stabilizing the NER and BER components and/or targeting them to methylated cytosines. GADD45 can act in concert with the XPA, XPC, XPG and XPF proteins to remove methylated nucleotides in a NER-dependent fashion [40–42]. GADD45 has also been identified in complexes with AID and TDG or AID and MBD4. In these latter cases, 5-mC is first de-aminated by AID into thymine, introducing a T:G mismatch that is later resolved by the glycosylase activity of TDG or MBD4 and by the BER components (Figure 1A) [43, 44].
Figure 1:

DNA demethylation and the TET proteins. (A) Passive (upper green arrow) and active (lower red arrows) DNA demethylation networks independent of the TET enzymes. 5-methylcytosine can be passively lost upon exclusion of DNMT1 at the time of DNA replication. Active demethylation occurs via the GADD45-mediated BER and NER pathways. The base excision repair (BER) mechanism involves deamination of 5-mC to thymidine by AID; then the T:G mismatch is recognized by the MBD4 or TDG glycosylase, which removes the base, exposing an AP-(apurinic/apyrimidinic) site resolved by the BER machinery. The nucleotide excision repair (NER) pathway involves direct removal of the 5-mC nucleotide by XP enzymes (XPA/XPC/XPG or XPF) and other NER components. (B) Passive (right green arrows) and active (left red arrows) DNA demethylation networks mediated by the TET enzymes. 5-Methylcytosine can be oxidized by TETs to 5-hmC, 5-fC and 5-caC. DNMT1 shows a decreased affinity for 5-hmC-containing and, possibly, 5-fC- and 5-caC-containing DNA, leading to progressive dilution of these modified nucleotides. Cytosine may also be directly decarboxylated by a yet unknown enzyme (referred to here as X). AID/APOBEC-mediated deamination of 5-hmC produces 5-hmU, which can be recognized and removed by the SMUG1 glycosylase and the BER machinery. Finally, the TDG enzyme can react with 5-fC and 5-caC and mediate BER-dependent DNA demethylation.
TETs-mediated oxidation of 5-mC can also lead to local active DNA demethylation (Figure 1B). Some results suggest that Tet1 promotes demethylation of the pluripotency gene Nanog in mESCs: upon Tet1 knockdown, a significant decrease in Nanog expression was observed, accompanied by pluripotency impairment [15]. Although further studies support this view, especially by demonstrating that Nanog is a direct target of Tet1 [45, 46], other reports contradict it [47–52]. Ficz et al. [49] and Williams et al. [51] did not observe a deregulation of Nanog upon Tet1 depletion, and Koh et al. [50] found Tet1 and Tet2 knockdown neither to affect Nanog expression nor to induce mESCs differentiation. Most importantly, Dawlaty et al. [47] showed with a mouse knockout that Tet1 was not required for maintaining pluripotency, and its loss did not effect in embryonic or postnatal development. These discrepancies might be attributable to differences in mESCs background or culture conditions, to RNAi off-targets, or to gene compensation, and therefore it will be important to further study the role of Tet1 in embryonic development [14]. Another example of TETs-mediated DNA demethylation was shown in 2012 by Thomson et al. [53] in mice treated with phenobarbital (PB), a known non-genotoxic carcinogen inducing liver tumors. Upon treatment, a set of 30 genes become upregulated in hepatocytes, and this correlates with an increase in 5-hmC and a decrease in 5-mC. Among the upregulated genes, Cyp2b10 (which is sometimes deregulated in liver tumors) displays a dynamic change in methylation/hydroxymethylation pattern: on day 1 after PB exposure, 5-hmC is already detected throughout the gene body and upstream from the transcription start site, whereas there is no significant change in DNA methylation. On day 7, the gene begins to show a decrease in 5-mC, but there is no substantial change in 5-hmC. On day 91, the gene becomes fully unmethylated, exhibiting no residual 5-hmC. This experiment proves that Cyp2b10 transits progressively from a methylated to a fully unmethylated state through 5-mC oxidation. A last example of TET-mediated demethylation was demonstrated by Wang et al., who found that in mouse brain, TET1 overexpression demethylates the Bdnf and Fgf1 promoters in a BER-dependent manner, causing increased expression of the corresponding genes. Interestingly, the same results are found with AID overexpression. This suggests a possible link between a deaminase activity and 5-hydroxymethylcytosine: the deamination product of 5-hmC would yield 5-hmU, a DNA modification found upon overexpression of TET1 or AID/APOBEC deaminase family members and which can be removed by the SMUG1 glycosylase [54]. It is worth mentioning that AID was initially discovered in B-lymphocytes undergoing antigen-mediated class switch recombination (CSR). In these cells, AID is recruited to the immunoglobulin heavy-chain (IgH) locus and induces double strand breaks by deamination of cytosines to uracils. These lesions are then resolved by the non-homologous end-joining pathway, allowing IgH recombination and antibody isotype switching [55]. Although this has not been fully investigated, some studies have revealed methylation of the IgH locus, showing that demethylation parallels CSR [56, 57]. It is tempting to speculate that methylation of the IgH locus might protect it from AID-mediated deamination, and that upon cell activation, the TET enzymes might hydroxylate 5-mC, producing 5-hmC to be recognized by AID and deaminated to 5-hmU. Subsequent removal of 5-hmU by a DNA glycosylase would lead to double strand breaks and CSR. Further investigations should be conducted to ascertain whether the TET enzymes act on the antibody-mediated immune response.
All in all, it thus seems that there is no consensus ‘path’ to DNA demethylation: it can be passive through repelling of DNMT1, or it can be active and this, dependently or independently of the TET enzymes (Figure 1A and B).
GENOME-WIDE MAPPING OF Tet1 AND OXIDIZED METHYLCYTOSINES: NOT THAT SIMPLE
To understand accurately the functions of 5-hmC, 5-hmU, 5-fC and 5-caC, it is important to develop systems to map these modifications. Until 2009, the gold standard method for analyzing 5-mC was bisulfite DNA sequencing, but after Anjana Rao discovered that TET1 mediates 5-hmC formation, she further found out that 5-hmC is also protected from chemical deamination by bisulfite treatment, rendering this technique unable to distinguish between 5-mC and 5-hmC [58]. The discovery of 5-fC and 5-caC has further complicated the situation, as they are both interpreted as unmodified cytosines after bisulfite treatment [59]. Biologists and chemists have thus united their efforts to develop a plethora of techniques to map modified cytosines, all of which rely on DNA deep sequencing (‘next-generation’ sequencing). Although these methods are reviewed elsewhere [59], we present here some information on 5-hmC and 5-fC patterns in mESCs and the mouse brain and how they relate to Tet1 genomic-binding profiles.
Several groups have reported 5-hmC mapping by deep sequencing in mESCs and showed (Figure 2A and B) that gene-rich regions are particularly abundant in hydroxymethylcytosine, mostly at unmethylated CGI promoters and in the gene bodies of (highly) transcribed genes, especially in exons where methylation is also found. Both 5-hmC and 5-mC are also present within enhancers and repetitive elements such as LINE1 repeats (Figure 2A and B) [49, 51, 52, 60–62]. It seems, however difficult to reach a consensus as recent base-resolution mapping (TAB-seq) in human and mouse ESCs identified 5-hmC mostly at distal regulatory elements and in low CpG content sequences, but not in promoters or gene bodies CGI [62]. These discrepancies could rise from the techniques used to map 5-hmC or the cell types used in the experiments and further high resolution maps will help us to uncover precise and quantitative hydroxymethylcytosine location that is central to understanding its function. It is worth mentioning that 5-hmC can also decorate non-CpG (CpH) dinucleotides. CpA methylation is thought to play a regulatory role in pluripotency, as it disappears when ESCs differentiate and is restored upon reprogramming to obtain induced pluripotent stem cells (iPSCs) [63]. Chromatin immunoprecipitation sequencing (ChIP-seq) of mouse Tet1 has revealed preferential binding of this protein to unmethylated CGI promoters and, to a lesser extent, to exons of poised bivalent (H3K4me3 and H3K27me3-marked) and active (H3K4me3-marked) genes [46, 51, 52]. Interestingly, some hydroxymethylated regions do not appear to be bound by Tet1, suggesting that they are either technically not identified by ChIP-seq or that another Tet (Tet2 for example) regulates these specific territories [51]. In 2012, Raiber et al. [64] described for the first time formylcytosine mapping in mESCs. Along with 5-hmC reports, 5-fC was found in DNA repeats, CGI promoters and gene bodies (mostly in exons) of transcribed H3K4me3-marked genes, paralleling Tet1 binding. These authors also found 5-fC to be regulated by TDG glycosylase, as silencing of the latter leads to 5-fC accumulation (Figure 2A). Finally, 5-fC-containing genes seem to be more expressed than 5-hmC-containing genes [64]. Interestingly, it would seem that some promoters rich in 5-hmC and 5-fC display no 5-mC, and that some exons show localized 5-mC, 5-hmC and 5-fC enrichment [49, 51, 64]. As Tet1 binds to both CGI promoters and exons [46, 51, 52], this suggests that a certain ‘event’ stabilizes 5-mC in gene bodies, especially in exons. It is conceivable that the rate of TET1-catalyzed 5-mC → 5hmC reaction depends on its location, while 5-hmC → 5fC rate does not. If so, the first reaction might be faster in CGI promoters than in gene bodies. This would explain why CGI promoters appear unmethylated, whereas 5-mC plays a regulatory role in exons. There is emerging evidence suggesting that 5-mC act in mRNA splicing: several studies have shown that exons more frequently contain 5-mC than do introns, and that nucleosomes exhibit preferential positioning at exon–intron boundaries [65–68]. Shukla et al. [65] discovered that binding of the CTCF factor to exons flanked by weak splice sites can promote their retention by slowing down the RNA polymerase II (RNAPII). They further revealed that inhibition of CTCF binding by DNA methylation can lead to reciprocal effects on exon inclusion. One might expect 5-mC oxidation intermediates also to be involved in RNA processing. Accordingly, Khare et al. [68] have shown with tiling arrays that in mouse and the human brain, exon–intron boundaries are enriched with 5-hmC and that constitutive exons show a distinct 5-hmC pattern different from that of alternatively spliced exons. Moreover, in vitro studies indicate that the rate of RNAPII elongation is reduced in DNA containing the further oxidized forms 5-fC and 5-caC. These marks might thus, like CTCF, affect the choice of the retained exon [69]. It will be most interesting to learn more about the role of TETs and the 5-mC oxidation status in mRNA splicing as well as the factors regulating TET kinetics [70, 71].
Figure 2:

(A) mESCs epigenetic landscape of an active gene. Tet1 can bind to chromatin at high-CpG-content CGI proximal promoters, where it can maintain an unmethylated state through iterative 5-mC oxidation in concert with TDG glycosylase (note that TDG can also be localised in exons containing CGI). Tet1 and 5-hmC are also found in active regulatory regions such as enhancers marked with the H3K4me1 and H3K27ac histone modifications. Exons are richer than introns in various forms of modified cytosines (5-mC, 5-hmC, 5-fC, and potentially 5-caC), and these might bind specific readers cooperating with the RNAPII and/or mRNA processing machinery (see light blue arrow with a question mark). (B) mESCs epigenetic landscape of an inactive gene (with bivalent promoter). Tet1 binding to the CGI proximal promoter might be associated with indirect recruitment (potentially via DNA hydroxymethylation) of the PRC2-containing Ezh2 complex and also with direct interaction with the Sin3a repressive complex. Sin3a and Ezh2 would subsequently maintain histone H3 in a deacetylated and K27 trimethylated form, thus preventing transcriptional activation.
In 2011, a group produced deep-sequencing maps of hydroxymethylcytosine in the mouse brain (cerebellum and hippocampus). These results are comparable to those obtained for mESCs, except that 5-hmC seems absent from the promoters of neuronal tissue. To date, there are no maps of Tets binding that might link these 5-hmC patterns to the hydroxymethylation machinery [72, 73].
TETs IN TRANSCRIPTIONAL REGULATION: ARE THEY MORE VERSATILE THAN ANTICIPATED?
In addition to their role in DNA demethylation, the TET proteins may regulate expression independently of their enzymatic activity. This view stems from studies by Yi Zhang's and Kristan Helin’s labs [46, 51]. The first group showed that in mESCs, Tet1 can bind to H3K4me3- and H3K27me3-enriched promoters and also recruit the Ezh2 Polycomb group protein (H3K27me3 ‘writer’) (Figure 2B). Although no direct interaction between Ezh2 and Tet1 was detected, Ezh2 binding was found to decrease upon Tet1 knockdown [46]. The second group described the first interactant of TET proteins, showing that the Sin3a histone deacetylase repressive complex associates with Tet1. More importantly, genome-wide Sin3a profiles revealed that this protein occupies a significant fraction of Tet1-bound regions and that, upon silencing of Tet1 or Sin3a, a subset of targets to which they co-bind are upregulated [51]. It thus seems that on repressed promoters (mostly bivalent in mESCs), Tet1 might recruit the Sin3a complex (directly) and the Ezh2 complex (indirectly), enabling these complexes in turn to maintain histones H3 in a deacetylated and K27 trimethylated form, therefore acting on gene repression. Interestingly, the presence of Sin3a is reported as an epigenetic feature linked to alternative mRNA splicing. This strengthens the view that TETs may play a role in RNA processing [74]. Future studies will be needed to identify additional TET partners, notably partners of TET2 and TET3. These are still early days, but we can expect novel interactors to be discovered in the near future, and this should shed light on the mode(s) of action of the TET proteins. In this context, it is worth mentioning some proteomics studies [75] that have identified strong binding between TET2, TET3 and the O-linked N-acetylglucosamine transferase (OGT) glycosyltransferase, which adds regulatory O-GlcNAc moieties to numerous proteins [76]. ChIP-seq studies have shown that TET2, TET3 and OGT co-localize on chromatin, at least in part at active promoters rich in H3K4me3. Furthermore, TET2 and TET3 promote OGT activity, which in turn stabilizes the Set1/COMPASS complex, responsible for bulk deposition of H3K4me3 (Figure 3) [75]. Consistent with these observations, ChIP-seq experiments in bone marrow from Tet2 knockout mice show a strong decrease of O-GlcNAc and H3K4me3 deposition at Tet2 bound genes, among those some key hematopoietic and epigenetic regulators [75 and Deplus, Delatte & Fuks, unpublished data]. Recent findings by Chen et al. [77] support the TET–OGT link in mESCs where they found that TET2 interacts with and recruits OGT to chromatin. OGT in turn glycosylates histone H2B on serine 112 (H2BS112GlcNAc), a modification known to mediate H2B K120 ubiquitinylation (H2BK120ub), a prerequisite for trimethylation of H3K4 by Set1/COMPASS [78, 79]. These results thus suggest a novel means by which TETs might induce transcriptional activation through H3K4me3 by (1) OGT-mediated glycosylation of H2BS112 and further ubiquitinylation of H2BK120 (2) OGT-mediated Set1/COMPASS stabilization, binding of Set1/COMPASS to H2BK120ub and trimethylation of H3K4 (Figure 3). The TET proteins affect gene expression with a dual mode of action, dependently or independently of DNA methylation status. Such dual mode of transcriptional regulation has been proposed by previous studies [14]. One mode might thus act on establishing an H3K4me3 active chromatin landscape through OGT and Set1/COMPASS, and the other on generating an H3K27me3 inactive state of chromatin via, for example Sin3a and Ezh2.
Figure 3:
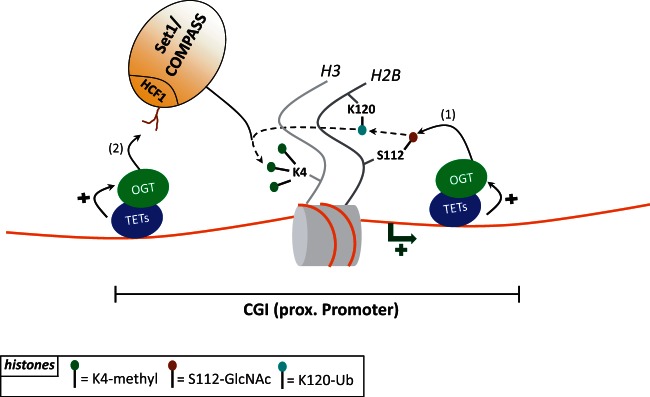
Proposed TETs-OGT-Set1/COMPASS functional link. (1) TET2 and TET3 (but also potentially TET1) interact with the OGT enzyme at GCI promoters and enhance its GlcNAc catalytic activity. TETs-mediated recruitment of OGT potentiates H2B glycosylation on serine 112 (H2BS112GlcNAc) which in turn favors H2B ubiquitinylation on lysine 120 (H2BK120ub). (2) Then, OGT glycosylates the Set1/COMPASS complex on its HCF1 subunit, enhancing the complex stability. This would result in Set1/COMPASS binding to H2BK120ub, trimethylation on H3K4 and in transcriptional activation.
As mentioned in the earlier section, TETs might influence DNA methylation in more than one way. Although 5-mC is supposed to be rapidly erased by oxidation at CGI promoters in mESCs, the 5-hmC and 5-fC intermediates seem quite stable. This is strengthened by the findings of Doege et al. [48], showing that in iPSCs generated from mouse embryonic fibroblasts by overexpression of Oct4, Sox2, Klf4 and c-Myc, Tet2 is recruited to pluripotency genes such as Nanog and Esrrb. Strikingly, although most of the Nanog methylation is lost upon iPSC formation, the 5-hmC modification persists and correlates with changes in histone marks: a decrease in H3K27me3 and an increase in H3K4me3. The authors suggest that 5-hmC is an epigenetic modification per se, distinct from 5-mC. In a manner similar to MBD protein binding to 5-mC, there might exist 5-hmC-specific readers that translate this mark by recruiting other transcription/epigenetic factors to the chromatin. There is evidence in favor of this model: in vitro, it has been shown that UHRF1/NP95, an essential factor in maintenance DNA methylation, binds hemi- or fully hydroxymethylated DNA through its SRA binding-pocket [80]. Studies on mESCs have revealed that Mbd3/NurD can bind 5-hmC DNA via the MBD domain, that the genome-wide profile of Mbd3 is similar to that of Tet1, and that upon Tet1 knockdown, Mbd3 enrichment is decreased on Tet1/Mbd3 targets. This suggests a possible link between 5-hmC and the Mbd3/NurD gene repression machinery [81]. Finally, a recent paper from Mellén et al. [82], showed that MeCP2 is able to bind 5-mC and 5-hmC with high affinity in the nervous system and that the Rett syndrome associated mutation R133C preferentially inhibits 5-hmC binding. These exciting results highlight the potential important roles of the TET proteins in neurological disorders.
The TET enzymes thus appear to have functions beyond DNA demethylation, involving the recruitment of proteins acting on histone modifications and thereby on gene expression. Given the importance of the chromatin landscape in health and disease [83], it is important to conduct further large-scale studies to explore the unappreciated roles of TETs by identifying novel interactants and specific readers of 5-hmC, 5-hmU, 5-fC and 5-caC.
TETs AND HYDROXYMETHYLATION: ARE THESE HALLMARKS OF CANCERS?
Various cancers and transformed cells harbor disturbed epigenomes, as illustrated by DNA bulk hypomethylation and focal promoter hypermethylation [84]. Cultured cell lines and tumors also display a drastic decrease in 5-hmC, possibly related to the global loss of 5-mC [24–26]. In melanoma cells, Lian et al. [85] showed by genome-wide analysis a considerable reduction in 5-hmC marking, correlating with disease progression. They further found, in addition to a lesser-than-expected decrease in 5-mC in intergenic regions, that more than 2000 genes having lost hydroxymethylcytosine were more methylated in melanoma cells than in normal melanocytes. Gene ontology analysis revealed an involvement of these genes in melanoma progression and in various cancer pathways. It is noteworthy that the TET enzymes were not mutated in the melanoma cells; rather, their expression, especially TET2, was significantly reduced. Isocitrate dehydrogenase 2 (IDH2), which converts isocitrate to α-ketoglutarate in the Krebs cycle, also showed markedly decreased expression. As this enzyme produces a cofactor important for TET- mediated 5-mC hydroxylation, IDH2 downregulation might result in a loss of the remaining TET activity, causing global hypohydroxymethylation and hypermethylation of genes involved in melanoma progression. Alongside the generally accepted association between sarcomas and mutations in genes encoding key differentiation/proliferation regulators or involved in signaling (e.g. Ras-MAPK, STAT and PI3K), there is increasing evidence of an association with mutations affecting epigenetic factors and of a link between cell transformation and an altered chromatin landscape [86]. For instance, myeloid malignancies such as myeloproliferative neoplasms (MPNs), myelodysplastic syndromes (MDS) and acute myeloid leukemia (AML) appear associated with mutations in the following genes: MLL and EZH2 (encoding ‘writers’ of H3K4me3 and H3K27me3 mark, respectively), ASXL1 (a protein thought to direct the EZH2 repressive complex to chromatin) and DNMT3A (likely explaining the global hypomethylation phenotype of myeloid malignancies). Interestingly, TET2 and IDH1/IDH2, both involved in DNA demethylation [86], have also been found to be mutated in these pathologies [86]. More precisely, somatic deletions and TET2-inactivating mutations are found in 4–13% of MPNs and 20–25% of MDSs with no clear prognosis [87, 88] and TET2 mutations are found in 7–23% of AMLs, where they correlate with a poor prognosis [89]. To gain insights into the role of TET2 in leukemogenesis, Tet2-knockout mice have been generated by different laboratories. Although with some differences between the mouse strains (possibly due to the use of different knockout methods), the loss of Tet2 appears to increase the hematopoietic stem cell compartment and to skews cell differentiation towards the myeloid compartment, causing symptoms resembling those associated with TET2 mutations [90–92]. One might expect TET2 loss of function to link tumor suppressor gene hypermethylation with an aberrant hydroxymethylation pattern, but results are quite conflicting: while Figueroa et al. [93] found that AML patients with TET2 mutations display decreased hydroxymethylation and increased DNA methylation, Ko et al. [94] found that TET2 loss of function is predominantly associated with reduced methylation at differentially methylated CpG sites. These differences in methylation pattern might be due to technical issues or to the type of disease (AML in the first case, MPNs/MDS in the second). TET2 ChIP-seq and genome-wide profiling of 5-mC and 5-hmC on human MPN, MDS and AML samples will help to shed light on the role of TET2 in myeloid malignancies. IDH1 and IDH2 loss-of-function mutations not only decrease α-ketoglutarate synthesis but also lead to accumulation of 2-hydroxyglutarate (2-HG), an oncometabolite found in the plasma of cancer patients carrying IDH mutations [95, 96]. 2-HG, by competing with α-ketoglutarate, can inhibit various cellular dioxygenases, notably the TET enzymes [93, 97]. IDH1/IDH2 mutations are found in 2.5–5% of MPNs, 3.5% of MDSs and 15–33% of AMLs, and these mutations and TET2 mutations appear to be mutually exclusive [88, 96, 98–101]. In keeping with the view that TET and IDH proteins act in concert, AMLs with IDH mutations display a global and specific hypermethylation signature, potentially due to decreased TET activity [93].
Although decreased 5-hmC seems to be a common feature of various cancers, the link with TET expression/mutation levels and aberrant methylation is not as straightforward as one might expect. Systematic establishment of cancer-associated genome-wide maps of methylation, hydroxymethylation and TETs binding will help scientists to assess the roles these events and enzymes play in cancer progression. The clinical value of using specific TET inhibitors to restore proper hydroxymethylation patterns in cancers where these are altered remains to be explored.
CONCLUSIONS
The discovery of the TET proteins in 2009 sparked feverish interest: for many scientists, here at last was the DNA demethylase activity they had been seeking for so long [10, 11]. Many examples of TET-mediated demethylation have been observed, from paternal pronucleus reprogramming upon fertilization [32–37] to demethylation of cancer-related genes in PB-exposed mice [53] and of the Bdnf/Fgf1 genes upon neuronal stimulation [54]. Yet, it remains a challenge to elucidate the exact pathway followed in each specific context (iterative oxidation, oxidation + deamination or even passive DNA demethylation through repelling of DNMT1 [102]). To further complicate the picture, Schiesser et al. [103] reported an intriguing 5-caC-decarboxylating activity in mESCs (Figure 1B). DNA demethylation through decarboxylation might be less prone to mutations caused by DNA lesions and also less energy-consuming than demethylation mediated by DNA repair, which requires the action of many factors. Finding a 5-caC decarboxylase will thus be one of the next tasks for many epigenetic laboratories. Importantly, the TET proteins seem to be more than just DNA demethylases and transcription regulators. They appear as multitask proteins involved not only in pluripotency and embryonic development [45–52] but also, potentially, in RNA processing (e.g. splicing) [67–69]. With proteins so essential and so versatile, it comes as no surprise that their loss can lead to diseases such as cancer [86]. To fully understand the link with disease, it should be very informative to get genome-wide TETs binding and 5-mC oxidation intermediate profiles for disorders where aberrant methylation patterns are observed. Finally, it seems that oxidized DNA modifications might be ‘epigenetic modifications’ per se [48]. Perhaps other modifications are hiding on nucleotides, waiting for their discovery and whatever the case may be, an epic scientific hunt has been launched. The epigenetic ‘block’ has room for more ‘new kids’ and more exciting discoveries. May future studies help to sketch a more complete picture.
Key Points.
The TET enzymes can convert 5-mC to 5-hmC, 5-fC and 5-caC.
5-mC oxidation products not only are intermediates in active DNA demethylation but may also be epigenetic modifications per se.
The TETs can regulate transcription independently of their catalytic activity.
The DNA methylome and hydroxymethylome are affected in cancers.
FUNDING
B.D. and F.F. are supported by the Belgian F.N.R.S. B. D. is an ‘Aspirant F.N.R.S’. and F.F. is an F.N.R.S. ‘Senior Research Associate’.
Biographies
Benjamin Delatte is a FNRS research fellow working at Dr. François Fuks’ lab of Cancer Epigenetics in Brussels. His work mainly focuses on chromatin modifications with an emphasis on the TET methyldioxygenases.
François Fuks is the head of the Laboratory of Cancer Epigenetics at the Free University of Brussels. His lab is interested in investigating epigenetic mechanisms in cancers, with a focus on DNA modifications and use novel epigenomic technologies in health and disease.
References
- 1.Portela A, Esteller M. Epigenetic modifications and human disease. Nat Biotechnol. 2010;28:1057–68. doi: 10.1038/nbt.1685. [DOI] [PubMed] [Google Scholar]
- 2.Deaton AM, Bird A. CpG islands and the regulation of transcription. Genes Dev. 2011;25:1010–22. doi: 10.1101/gad.2037511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ndlovu MN, Denis H, Fuks F. Exposing the DNA methylome iceberg. Trends Biochem Sci. 2011;36:381–7. doi: 10.1016/j.tibs.2011.03.002. [DOI] [PubMed] [Google Scholar]
- 4.Wu SC, Zhang Y. Active DNA demethylation: many roads lead to Rome. Nat Rev Mol Cell Biol. 2010;11:607–20. doi: 10.1038/nrm2950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Denis H, Ndlovu MN, Fuks F. Regulation of mammalian DNA methyltransferases: a route to new mechanisms. EMBO Rep. 2011;12:647–56. doi: 10.1038/embor.2011.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Patra SK, Patra A, Rizzi F, et al. Demethylation of (Cytosine-5-C-methyl) DNA and regulation of transcription in the epigenetic pathways of cancer development. Cancer Metastasis Rev. 2008;27:315–34. doi: 10.1007/s10555-008-9118-y. [DOI] [PubMed] [Google Scholar]
- 7.Uribesalgo I, Di Croce L. Dynamics of epigenetic modifications in leukemia. Brief Funct Genomics. 2011;10:18–29. doi: 10.1093/bfgp/elr002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wyatt GR, Cohen SS. A new pyrimidine base from bacteriophage nucleic acids. Nature. 1952;170:1072–3. doi: 10.1038/1701072a0. [DOI] [PubMed] [Google Scholar]
- 9.Tsukada Y. Hydroxylation mediates chromatin demethylation. J Biochem. 2012;151:229–46. doi: 10.1093/jb/mvs003. [DOI] [PubMed] [Google Scholar]
- 10.Kriaucionis S, Heintz N. The nuclear DNA base 5-hydroxymethylcytosine is present in Purkinje neurons and the brain. Science. 2009;324:929–30. doi: 10.1126/science.1169786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tahiliani M, Koh KP, Shen Y, et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science. 2009;324:930–5. doi: 10.1126/science.1170116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ono R, Taki T, Taketani T, et al. LCX, leukemia-associated protein with a CXXC domain, is fused to MLL in acute myeloid leukemia with trilineage dysplasia having t(10;11)(q22;q23) Cancer Res. 2002;62:4075–80. [PubMed] [Google Scholar]
- 13.Iyer LM, Tahiliani M, Rao A, et al. Prediction of novel families of enzymes involved in oxidative and other complex modifications of bases in nucleic acids. Cell Cycle. 2009;8:1698–1710. doi: 10.4161/cc.8.11.8580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wu H, Zhang Y. Mechanisms and functions of Tet protein-mediated 5-methylcytosine oxidation. Genes Dev. 2011;25:2436–52. doi: 10.1101/gad.179184.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ito S, D’Alessio AC, Taranova OV, et al. Role of Tet proteins in 5mC to 5hmC conversion, ES-cell self-renewal and inner cell mass specification. Nature. 2010;466:1129–33. doi: 10.1038/nature09303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Smiley JA, Kundracik M, Landfried DA, et al. Genes of the thymidine salvage pathway: thymine-7-hydroxylase from a Rhodotorula glutinis cDNA library and iso-orotate decarboxylase from Neurospora crassa. Biochim Biophys Acta. 2005;1723:256–64. doi: 10.1016/j.bbagen.2005.02.001. [DOI] [PubMed] [Google Scholar]
- 17.Neidigh JW, Darwanto A, Williams AA, et al. Cloning and characterization of Rhodotorula glutinis thymine hydroxylase. Chem Res Toxicol. 2009;22:885–93. doi: 10.1021/tx8004482. [DOI] [PubMed] [Google Scholar]
- 18.Ito S, Shen L, Dai Q, et al. Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science. 2011;333:1300–3. doi: 10.1126/science.1210597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.He YF, Li BZ, Li Z, et al. Tet-mediated formation of 5-carboxylcytosine and its excision by TDG in mammalian DNA. Science. 2011;333:1303–7. doi: 10.1126/science.1210944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Globisch D, Munzel M, Muller M, et al. Tissue distribution of 5-hydroxymethylcytosine and search for active demethylation intermediates. PLoS One. 2010;5:e15367. doi: 10.1371/journal.pone.0015367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Szwagierczak A, Bultmann S, Schmidt CS, et al. Sensitive enzymatic quantification of 5-hydroxymethylcytosine in genomic DNA. Nucleic Acids Res. 2010;38:e181. doi: 10.1093/nar/gkq684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nestor CE, Ottaviano R, Reddington J, et al. Tissue type is a major modifier of the 5-hydroxymethylcytosine content of human genes. Genome Res. 2012;22:467–77. doi: 10.1101/gr.126417.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tollervey JR, Lunyak VV. Epigenetics: judge, jury and executioner of stem cell fate. Epigenetics. 2012;7:823–40. doi: 10.4161/epi.21141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Haffner MC, Chaux A, Meeker AK, et al. Global 5-hydroxymethylcytosine content is significantly reduced in tissue stem/progenitor cell compartments and in human cancers. Oncotarget. 2011;2:627–37. doi: 10.18632/oncotarget.316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jin SG, Jiang Y, Qiu R, et al. 5-Hydroxymethylcytosine is strongly depleted in human cancers but its levels do not correlate with IDH1 mutations. Cancer Res. 2011;71:7360–5. doi: 10.1158/0008-5472.CAN-11-2023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Terragni J, Bitinaite J, Zheng Y, et al. Biochemical characterization of recombinant beta-glucosyltransferase and analysis of global 5-hydroxymethylcytosine in unique genomes. Biochemistry. 2012;51:1009–19. doi: 10.1021/bi2014739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shock LS, Thakkar PV, Peterson EJ, et al. DNA methyltransferase 1, cytosine methylation, and cytosine hydroxymethylation in mammalian mitochondria. Proc Natl Acad Sci U S A. 2011;108:3630–5. doi: 10.1073/pnas.1012311108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen H, Dzitoyeva S, Manev H. Effect of valproic acid on mitochondrial epigenetics. Eur J Pharmacol. 2012;690:51–9. doi: 10.1016/j.ejphar.2012.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dzitoyeva S, Chen H, Manev H. Effect of aging on 5-hydroxymethylcytosine in brain mitochondria. Neurobiol Aging. 2012;33:2881–91. doi: 10.1016/j.neurobiolaging.2012.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hackett JA, Sengupta R, Zylicz JJ, et al. Germline DNA demethylation dynamics and imprint erasure through 5-hydroxymethylcytosine. Science. 2012;339:448–52. doi: 10.1126/science.1229277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Reik W. Stability and flexibility of epigenetic gene regulation in mammalian development. Nature. 2007;447:425–32. doi: 10.1038/nature05918. [DOI] [PubMed] [Google Scholar]
- 32.Iqbal K, Jin SG, Pfeifer GP, et al. Reprogramming of the paternal genome upon fertilization involves genome-wide oxidation of 5-methylcytosine. Proc Natl Acad Sci U S A. 2011;108:3642–7. doi: 10.1073/pnas.1014033108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wossidlo M, Nakamura T, Lepikhov K, et al. 5-Hydroxymethylcytosine in the mammalian zygote is linked with epigenetic reprogramming. Nat Commun. 2011;2:241. doi: 10.1038/ncomms1240. [DOI] [PubMed] [Google Scholar]
- 34.Gu TP, Guo F, Yang H, et al. The role of Tet3 DNA dioxygenase in epigenetic reprogramming by oocytes. Nature. 2011;477:606–10. doi: 10.1038/nature10443. [DOI] [PubMed] [Google Scholar]
- 35.Inoue A, Shen L, Dai Q, et al. Generation and replication-dependent dilution of 5fC and 5caC during mouse preimplantation development. Cell Res. 2011;21:1670–6. doi: 10.1038/cr.2011.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Inoue A, Zhang Y. Replication-dependent loss of 5-hydroxymethylcytosine in mouse preimplantation embryos. Science. 2011;334:194. doi: 10.1126/science.1212483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nakamura T, Liu YJ, Nakashima H, et al. PGC7 binds histone H3K9me2 to protect against conversion of 5mC to 5hmC in early embryos. Nature. 2012;486:415–9. doi: 10.1038/nature11093. [DOI] [PubMed] [Google Scholar]
- 38.Bhattacharya SK, Ramchandani S, Cervoni N, et al. A mammalian protein with specific demethylase activity for mCpG DNA. Nature. 1999;397:579–83. doi: 10.1038/17533. [DOI] [PubMed] [Google Scholar]
- 39.McKinnon PJ. DNA repair deficiency and neurological disease. Nat Rev Neurosci. 2009;10:100–12. doi: 10.1038/nrn2559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Barreto G, Schafer A, Marhold J, et al. Gadd45a promotes epigenetic gene activation by repair-mediated DNA demethylation. Nature. 2007;445:671–5. doi: 10.1038/nature05515. [DOI] [PubMed] [Google Scholar]
- 41.Le May N, Mota-Fernandes D, Velez-Cruz R, et al. NER factors are recruited to active promoters and facilitate chromatin modification for transcription in the absence of exogenous genotoxic attack. Mol Cell. 2010;38:54–66. doi: 10.1016/j.molcel.2010.03.004. [DOI] [PubMed] [Google Scholar]
- 42.Schmitz KM, Schmitt N, Hoffmann-Rohrer U, et al. TAF12 recruits Gadd45a and the nucleotide excision repair complex to the promoter of rRNA genes leading to active DNA demethylation. Mol Cell. 2009;33:344–53. doi: 10.1016/j.molcel.2009.01.015. [DOI] [PubMed] [Google Scholar]
- 43.Cortellino S, Xu J, Sannai M, et al. Thymine DNA glycosylase is essential for active DNA demethylation by linked deamination-base excision repair. Cell. 2011;146:67–79. doi: 10.1016/j.cell.2011.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rai K, Huggins IJ, James SR, et al. DNA demethylation in zebrafish involves the coupling of a deaminase, a glycosylase, and gadd45. Cell. 2008;135:1201–12. doi: 10.1016/j.cell.2008.11.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Freudenberg JM, Ghosh S, Lackford BL, et al. Acute depletion of Tet1-dependent 5-hydroxymethylcytosine levels impairs LIF/Stat3 signaling and results in loss of embryonic stem cell identity. Nucleic Acids Res. 2012;40:3364–77. doi: 10.1093/nar/gkr1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wu H, D’Alessio AC, Ito S, et al. Dual functions of Tet1 in transcriptional regulation in mouse embryonic stem cells. Nature. 2011;473:389–93. doi: 10.1038/nature09934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dawlaty MM, Ganz K, Powell BE, et al. Tet1 is dispensable for maintaining pluripotency and its loss is compatible with embryonic and postnatal development. Cell Stem Cell. 2011;9:166–75. doi: 10.1016/j.stem.2011.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Doege CA, Inoue K, Yamashita T, et al. Early-stage epigenetic modification during somatic cell reprogramming by Parp1 and Tet2. Nature. 2012;488:652–5. doi: 10.1038/nature11333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ficz G, Branco MR, Seisenberger S, et al. Dynamic regulation of 5-hydroxymethylcytosine in mouse ES cells and during differentiation. Nature. 2011;473:398–402. doi: 10.1038/nature10008. [DOI] [PubMed] [Google Scholar]
- 50.Koh KP, Yabuuchi A, Rao S, et al. Tet1 and Tet2 regulate 5-hydroxymethylcytosine production and cell lineage specification in mouse embryonic stem cells. Cell Stem Cell. 2011;8:200–13. doi: 10.1016/j.stem.2011.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Williams K, Christensen J, Pedersen MT, et al. TET1 and hydroxymethylcytosine in transcription and DNA methylation fidelity. Nature. 2011;473:343–8. doi: 10.1038/nature10066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Xu Y, Wu F, Tan L, et al. Genome-wide regulation of 5hmC, 5mC, and gene expression by Tet1 hydroxylase in mouse embryonic stem cells. Mol Cell. 2011;42:451–64. doi: 10.1016/j.molcel.2011.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Thomson JP, Lempiainen H, Hackett JA, et al. Non-genotoxic carcinogen exposure induces defined changes in the 5-hydroxymethylome. Genome Biol. 2012;13:R93. doi: 10.1186/gb-2012-13-10-r93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Guo JU, Su Y, Zhong C, et al. Hydroxylation of 5-methylcytosine by TET1 promotes active DNA demethylation in the adult brain. Cell. 2011;145:423–34. doi: 10.1016/j.cell.2011.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wang JH. The role of activation-induced deaminase in antibody diversification and genomic instability. Immunol Res. 2013;55:287–97. doi: 10.1007/s12026-012-8369-4. [DOI] [PubMed] [Google Scholar]
- 56.Storb U, Arp B. Methylation patterns of immunoglobulin genes in lymphoid cells: correlation of expression and differentiation with undermethylation. Proc Natl Acad Sci U S A. 1983;80:6642–6. doi: 10.1073/pnas.80.21.6642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Storb U, Peters A, Kim N, et al. Molecular aspects of somatic hypermutation of immunoglobulin genes. Cold Spring Harb Symp Quant Biol. 1999;64:227–34. doi: 10.1101/sqb.1999.64.227. [DOI] [PubMed] [Google Scholar]
- 58.Huang Y, Pastor WA, Shen Y, et al. The behaviour of 5-hydroxymethylcytosine in bisulfite sequencing. PLoS One. 2010;5:e8888. doi: 10.1371/journal.pone.0008888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Song CX, Yi C, He C. Mapping recently identified nucleotide variants in the genome and transcriptome. Nat Biotechnol. 2012;30:1107–16. doi: 10.1038/nbt.2398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Stroud H, Feng S, Morey Kinney S, et al. 5-Hydroxymethylcytosine is associated with enhancers and gene bodies in human embryonic stem cells. Genome Biol. 2011;12:R54. doi: 10.1186/gb-2011-12-6-r54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Pastor WA, Pape UJ, Huang Y, et al. Genome-wide mapping of 5-hydroxymethylcytosine in embryonic stem cells. Nature. 2011;473:394–7. doi: 10.1038/nature10102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yu M, Hon GC, Szulwach KE, et al. Base-resolution analysis of 5-hydroxymethylcytosine in the mammalian genome. Cell. 2012;149:1368–80. doi: 10.1016/j.cell.2012.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lister R, Pelizzola M, Dowen RH, et al. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature. 2009;462:315–22. doi: 10.1038/nature08514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Raiber EA, Beraldi D, Ficz G, et al. Genome-wide distribution of 5-formylcytosine in embryonic stem cells is associated with transcription and depends on thymine DNA glycosylase. Genome Biol. 2012;13:R69. doi: 10.1186/gb-2012-13-8-r69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Shukla S, Kavak E, Gregory M, et al. CTCF-promoted RNA polymerase II pausing links DNA methylation to splicing. Nature. 2011;479:74–9. doi: 10.1038/nature10442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Khan DH, Jahan S, Davie JR. Pre-mRNA splicing: role of epigenetics and implications in disease. Adv Biol Regul. 2012;52:377–88. doi: 10.1016/j.jbior.2012.04.003. [DOI] [PubMed] [Google Scholar]
- 67.Huang Y, Rao A. New functions for DNA modifications by TET-JBP. Nat Struct Mol Biol. 2012;19:1061–4. doi: 10.1038/nsmb.2437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Khare T, Pai S, Koncevicius K, et al. 5-hmC in the brain is abundant in synaptic genes and shows differences at the exon-intron boundary. Nat Struct Mol Biol. 2012;19:1037–43. doi: 10.1038/nsmb.2372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kellinger MW, Song CX, Chong J, et al. 5-Formylcytosine and 5-carboxylcytosine reduce the rate and substrate specificity of RNA polymerase II transcription. Nat Struct Mol Biol. 2012;19:831–3. doi: 10.1038/nsmb.2346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Pan Q, Shai O, Lee LJ, et al. Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat Genet. 2008;40:1413–5. doi: 10.1038/ng.259. [DOI] [PubMed] [Google Scholar]
- 71.Maciejewski JP, Padgett RA. Defects in spliceosomal machinery: a new pathway of leukaemogenesis. Br J Haematol. 2012;158:165–73. doi: 10.1111/j.1365-2141.2012.09158.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Song CX, Szulwach KE, Fu Y, et al. Selective chemical labeling reveals the genome-wide distribution of 5-hydroxymethylcytosine. Nat Biotechnol. 2011;29:68–72. doi: 10.1038/nbt.1732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Szulwach KE, Li X, Li Y, et al. 5-hmC-mediated epigenetic dynamics during postnatal neurodevelopment and aging. Nat Neurosci. 2011;14:1607–16. doi: 10.1038/nn.2959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Zhou Y, Lu Y, Tian W. Epigenetic features are significantly associated with alternative splicing. BMC Genomics. 2012;13:123. doi: 10.1186/1471-2164-13-123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Deplus R, Delatte B, Schwinn MK, et al. TET2 and TET3 regulate GlcNAcylation and H3K4 methylation through OGT and SET1/COMPASS. EMBO J. 2013 doi: 10.1038/emboj.2012.357. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Hanover JA, Krause MW, Love DC. Bittersweet memories: linking metabolism to epigenetics through O-GlcNAcylation. Nat Rev Mol Cell Biol. 2012;13:312–21. doi: 10.1038/nrm3334. [DOI] [PubMed] [Google Scholar]
- 77.Chen Q, Chen Y, Bian C, et al. TET2 promotes histone O-GlcNAcylation during gene transcription. Nature. 2013;493:561–4. doi: 10.1038/nature11742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Fujiki R, Hashiba W, Sekine H, et al. GlcNAcylation of histone H2B facilitates its monoubiquitination. Nature. 2011;480:557–60. doi: 10.1038/nature10656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Wu M, Wang PF, Lee JS, et al. Molecular regulation of H3K4 trimethylation by Wdr82, a component of human Set1/COMPASS. Mol Cell Biol. 2008;28:7337–44. doi: 10.1128/MCB.00976-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Frauer C, Hoffmann T, Bultmann S, et al. Recognition of 5-hydroxymethylcytosine by the Uhrf1 SRA domain. PLoS One. 2011;6:e21306. doi: 10.1371/journal.pone.0021306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Yildirim O, Li R, Hung JH, et al. Mbd3/NURD complex regulates expression of 5-hydroxymethylcytosine marked genes in embryonic stem cells. Cell. 2011;147:1498–510. doi: 10.1016/j.cell.2011.11.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Mellén M, Ayata P, Dewell S, et al. MeCP2 binds to 5hmC enriched within active genes and accessible chromatin in the nervous system. Cell. 2012;151:1417–1430. doi: 10.1016/j.cell.2012.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Choo KB. Epigenetics in disease and cancer. Malays J Pathol. 2011;33:61–70. [PubMed] [Google Scholar]
- 84.Sandoval J, Esteller M. Cancer epigenomics: beyond genomics. Curr Opin Genet Dev. 2012;22:50–5. doi: 10.1016/j.gde.2012.02.008. [DOI] [PubMed] [Google Scholar]
- 85.Lian CG, Xu Y, Ceol C, et al. Loss of 5-hydroxymethylcytosine is an epigenetic hallmark of melanoma. Cell. 2012;150:1135–46. doi: 10.1016/j.cell.2012.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Shih AH, Abdel-Wahab O, Patel JP, et al. The role of mutations in epigenetic regulators in myeloid malignancies. Nat Rev Cancer. 2012;12:599–612. doi: 10.1038/nrc3343. [DOI] [PubMed] [Google Scholar]
- 87.Tefferi A, Pardanani A, Lim KH, et al. TET2 mutations and their clinical correlates in polycythemia vera, essential thrombocythemia and myelofibrosis. Leukemia. 2009;23:905–11. doi: 10.1038/leu.2009.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Bejar R, Stevenson K, Abdel-Wahab O, et al. Clinical effect of point mutations in myelodysplastic syndromes. N Engl J Med. 2011;364:2496–506. doi: 10.1056/NEJMoa1013343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Patel JP, Gonen M, Figueroa ME, et al. Prognostic relevance of integrated genetic profiling in acute myeloid leukemia. N Engl J Med. 2012;366:1079–89. doi: 10.1056/NEJMoa1112304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Li Z, Cai X, Cai CL, et al. Deletion of Tet2 in mice leads to dysregulated hematopoietic stem cells and subsequent development of myeloid malignancies. Blood. 2011;118:4509–18. doi: 10.1182/blood-2010-12-325241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Moran-Crusio K, Reavie L, Shih A, et al. Tet2 loss leads to increased hematopoietic stem cell self-renewal and myeloid transformation. Cancer Cell. 2011;20:11–24. doi: 10.1016/j.ccr.2011.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Quivoron C, Couronne L, Della Valle V, et al. TET2 inactivation results in pleiotropic hematopoietic abnormalities in mouse and is a recurrent event during human lymphomagenesis. Cancer Cell. 2011;20:25–38. doi: 10.1016/j.ccr.2011.06.003. [DOI] [PubMed] [Google Scholar]
- 93.Figueroa ME, Abdel-Wahab O, Lu C, et al. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell. 2010;18:553–67. doi: 10.1016/j.ccr.2010.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Ko M, Huang Y, Jankowska AM, et al. Impaired hydroxylation of 5-methylcytosine in myeloid cancers with mutant TET2. Nature. 2010;468:839–43. doi: 10.1038/nature09586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Dang L, White DW, Gross S, et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature. 2009;462:739–44. doi: 10.1038/nature08617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Ward PS, Patel J, Wise DR, et al. The common feature of leukemia-associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting alpha-ketoglutarate to 2-hydroxyglutarate. Cancer Cell. 2010;17:225–34. doi: 10.1016/j.ccr.2010.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Xu W, Yang H, Liu Y, et al. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of alpha-ketoglutarate-dependent dioxygenases. Cancer Cell. 2011;19:17–30. doi: 10.1016/j.ccr.2010.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Mardis ER, Ding L, Dooling DJ, et al. Recurring mutations found by sequencing an acute myeloid leukemia genome. N Engl J Med. 2009;361:1058–66. doi: 10.1056/NEJMoa0903840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Marcucci G, Maharry K, Wu YZ, et al. IDH1 and IDH2 gene mutations identify novel molecular subsets within de novo cytogenetically normal acute myeloid leukemia: a cancer and leukemia group B study. J Clin Oncol. 2010;28:2348–55. doi: 10.1200/JCO.2009.27.3730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Tefferi A, Lasho TL, Abdel-Wahab O, et al. IDH1 and IDH2 mutation studies in 1473 patients with chronic-, fibrotic- or blast-phase essential thrombocythemia, polycythemia vera or myelofibrosis. Leukemia. 2010;24:1302–1309. doi: 10.1038/leu.2010.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Tefferi A, Jimma T, Sulai NH, et al. IDH mutations in primary myelofibrosis predict leukemic transformation and shortened survival: clinical evidence for leukemogenic collaboration with JAK2V617F. Leukemia. 2012;26:475–80. doi: 10.1038/leu.2011.253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Valinluck V, Sowers LC. Endogenous cytosine damage products alter the site selectivity of human DNA maintenance methyltransferase DNMT1. Cancer Res. 2007;67:946–50. doi: 10.1158/0008-5472.CAN-06-3123. [DOI] [PubMed] [Google Scholar]
- 103.Schiesser S, Hackner B, Pfaffeneder T, et al. Mechanism and stem-cell activity of 5-carboxycytosine decarboxylation determined by isotope tracing. Angew Chem Int Ed Engl. 2012;51:6516–20. doi: 10.1002/anie.201202583. [DOI] [PubMed] [Google Scholar]