

Abstract
Tumors have long been viewed as a population in which all cells have the equal propensity to form new tumors, the so called conventional stochastic model. The cutting-edge theory on tumor origin and progression, tends to consider cancer as a stem cell disease. Stem cells are actively involved in the onset and maintenance of colon cancer. This review is intended to examine the state of the art on colon cancer stem cells (CSCs), with regard to the recent achievements of basic research and to the corresponding translational consequences. Specific prominence is given to the hypothesized origin of CSCs and to the methods for their identification. The growing understanding of CSC biology is driving the optimization of novel anti-cancer targeted drugs.
Keywords: Colon cancer stem cells, Colorectal cancer, CD133, Therapy, Chemoresistance
Core tip: According to the “cancer stem cell” (CSC) theory, tumor growth and spread are driven by a minority of cancer cells which exhibit characteristics similar to normal stem cells. Although CSCs have been implicated in colon carcinogenesis, due to the complexity of their biology and unsolved technical issues, an unequivocally approved identification and isolation strategy is still a matter of debate. Several markers have been used to identify colon CSCs but the function of these proteins in CSC biology has not yet been clarified. Moreover, the possibility that CSCs might contribute to the failure of existing chemotherapies to eradicate malignant tumors, indicate that targeting of CSCs may represent a promising strategy to eradicate chemoresistant cancers. Aim of this review was to acquire more information on the biology of human colon CSCs and shed light on the role of this cells in the onset and the maintenance of colon cancer.
INTRODUCTION
The intestine epithelium is subjected to a rapid and continuous regeneration, supported by crypt intestinal stem cells (ISCs). This feature severely increases the risk for malignant conversion[1]. Indeed, colorectal cancer (CRC) is one of the most common type of neoplasm worldwide, representing the second leading cause of morbidity and mortality from cancer in both Europe and the United States. This means that CRC can also be considered one of the main national emergencies, both in terms of morbidity and in terms of social and economic costs[2].
Studies on CRC pathogenesis have been originally focusing on the clonal selection process, a model of carcinogenesis postulated in 1975[3]. The characterization of the genetic mechanisms underlying this process has been performed, for the first time, in the early 90s by Bert Vogelstein, who developed the molecular model of CRC progression known as “Vogelstein model”. According to it, the CRC develops from epithelial cells lining the gastrointestinal tract, which undergo sequential mutations in specific DNA sequences that disrupt normal mechanisms of proliferation and self-renewal[3,4]. This pathogeneic model still represents a paradigm of tumor growth and provides a standard rationale to dissect the molecular mechanisms responsible for CRC. Though, current anticancer treatments are often unable, even those based on molecular targeted strategies, to eradicate the disease. Otherwise, the cellular origin of human cancers is still controversial and the mechanisms responsible for the complexity and heterogeneity of tumors remain to be defined[3,4]. In recent years, converging evidence has suggested that human cancer can be considered as a stem cell disease. Therefore, the “stochastic” theory for the cellular origin of cancer, based upon the assumption that all cancer cells are equally malignant and able to give rise to tumors, has been abandoned in favor of the “hierarchical” theory. The latter assumes that: (1) tumors are hierarchically organized; and (2) only a rare subpopulation of undifferentiated cells at the apex of this hierarchy have the unique biological properties necessary for tumor initiation, maintenance and spreading. Given the similarities between tumor-initiating cells and normal stem cells (SC), the tumor-initiating cells have been termed “cancer stem cells” (CSCs)[5]. In its simplicity, this hypothesis suggests that tumorigenesis is an “aberrant organogenesis”, supported by a minority of cancer cells, that by consecutive genetic changes, can differentiate to give rise to different phenotypes in the neoplastic population[6].
The hierarchical model implies that within tumors there are cells with different tumorigenic potential: cells that have lost the ability to propagate the tumor and cells that retain their clonogenic ability. Indeed, biologically distinct populations of CSCs have been identified within hematological malignancies[7] and in most solid tumors, including colon cancer[8,9]. The origin of CSCs is still unclear but the discovery of stem cells in the majority of normal tissues, including colon crypts, support the hypothesis that normal SC might represent a possible target for tumorigenic mutations, due to both their long life and their capacity of self-renewal[10].
Cancer stem cells theory has profound translational implications. Current treatments are hardly able to completely eradicate cancer cells, being often complicated by the occurrence of tumor recurrence and/or metastasis and are burdened by toxicity issues. The failure of chemotherapy may at least in part lie in its capacity of targeting the bulk of cancer without affecting stem cells. These can on turn replicate after treatment and, eventually, develop selective features responsible for the occurrence of drug-resistance, which usually characterize and complicate the course of the disease[11-13].
Several studies have suggested that the CSC fraction may be identified within a variety of human cancers, including CRC, by the expression of the CD133 surface marker[8,9,14,15]. CD133 (also known as prominin-1 in rodents or AC133 in humans), a 120 kDa transmembrane and cell surface protein, has been shown to characterize normal and cancer stem cells in several human tissues, including the colonic mucosa. Hence, CD133 has been used to identify and isolate tumor initiating cells from human colon cancers. CD133+ cells are able to maintain themselves as well as to differentiate and re-establish tumor heterogeneity upon serial transplantation in vivo[8,9]. However, despite constant research efforts, the molecular mechanisms and signaling pathways that regulate the behavior of CD133-expressing cells remain unknown. Aim of this review was to acquire more information on the biology of human colon CSCs and shed light on the role of CD133+ tumor-initiating cells in the onset and the maintenance of colon cancer.
CANCER STEM CELLS: PROPERTIES
It is widely accepted the concept that tumor is an heterogeneous entity derived from a small subpopulation of undifferentiated cancer cells, the CSCs. CSCs are defined as cells having three unique properties: the capacity of self-renew indefinitely, the ability to recreate the full repertoire of cancer cells of the parent tumor and the expression of a distinctive set of surface biomarkers[16]. It must be emphasized that self-renewal is not synonymous with proliferation. Self-renewal involves the ability of SC populations to precisely maintain their numbers through a combination of symmetric and asymmetric SC division, giving rise to one or both daughter cells identical to the mother and retaining SC properties[17]. In the case of CSCs, mechanisms involved in self-renewal are deregulated and seem to lead to CSC overpopulation. The underlying mechanisms for generating excess of CSC numbers are believed to relate to increases in symmetric CSC division (which produces two CSC daughters) relative to asymmetric CSC division (which produces one CSC daughter and one non-CSC daughter). Many authors have documented this concept quantitatively, using mathematical modeling[17] or fluorescent dye assay[18]. These authors have showed that only increased symmetric division of CSCs could account both for the biologic observation that there is an exponential increase in CSC populations in tumoral tissues and for the known long lag phase, which is typical in the development of many cancers, including colorectal cancer[17]. Although CSCs have the capacity for self-renewal, they are relatively quiescent; that is, they have proliferative capacity but are often not cycling. Indeed, they have been shown to have significantly longer cell cycle times than proliferating non-SCs. This is presumably due to the arrest of SCs at a G0-like cell cycle phase or checkpoint[19].
Another property of CSCs is their potential for multilineage differentiation. This is consistent with the concept that CSCs, like normal SCs, give rise to a hierarchical organization of cell populations that underlie organogenesis[20].
CSCs also display alterations of DNA repair, due to the presence of cytoprotective properties (including telomerase activation and high expression of anti-apoptotic factors), and a relatively low proliferative potential. In addition, they express high levels of proteins belonging to the ABC membrane transporters family, involved in chemotherapeutic resistance (i.e., to paclitaxel, cisplatin, 5-fluoruracilee, mitoxantrone, methotrexate, anthracyclines, etoposide, vinca alkaloids, camptothecins, topotecan, imatinib)[20]. These unique properties of CSCs would explain the failure of many antitumor therapies, which affect rapidly dividing cells, determining only a reduction of tumor cells number, while CSCs divide slowly and are not sensitive to the cytotoxic drugs. New insight in the molecular mechanisms that underlie these processes were obtained by studying the intracellular regulator of gene expression[21].
On this regard, Bitarte and colleagues showed that the micro RNA miR-451 was downregulated in colonspheres, obtained from different colon carcinoma cells, versus parental cells. The expression of miR-451 caused a decrease in self-renewal, tumorigenicity, and chemoresistance to irinotecan of colonspheres. Authors demonstrated that miR-451 downregulation allows the expression of the target gene macrophage migration inhibitory factor, which induces cyclooxygenase-2 (COX-2) expression. In turn, COX-2 allows Wnt activation, which is essential for CSC growth. Furthermore, miR-451 restoration decreased expression of the ATP-binding cassette drug transporter ABCB1 and resulted in irinotecan sensitization. These findings correlated well with the lower expression of miR-451 observed in patients who did not respond to irinotecan-based first-line therapy compared with patients who did[22]. Moreover, various signaling pathways have already been identified and described in CSCs. It is known that standard pathways for self-renewal of normal stem cells, such as Wnt, Notch and Hedgehog signaling, are also present in CSCs and have an important role in their function. Targeting critical steps in those pathways, however, will be complicated by intense crosstalks as well as main safety issues related to the pleiotropic effects of these signaling molecules[23]. Nevertheless, there are already several reports indicating that CSCs can be selectively targeted without damaging normal stem cells[24]. These and other findings could reasonably pave the way to the development of novel, more efficient and less toxic anti-cancer medications.
CANCER STEM CELLS: DEFINITION
In operational terms, CSCs can only be defined experimentally on the basis of their ability to regenerate continuously the tumor. The implementation of this approach, explains the use of alternative terms in the literature. For example, the term “tumor-initiating cells” is frequently used to describe putative CSCs. However, both these terms (cancer stem cell and tumor-initiating cells) can cause confusion about the cell type to which they relate[16]. In fact, the term CSCs might suggest that these cells arise from normal stem cells, which have acquired a number of genetic mutations sufficient to induce malignant transformation. This assumption is probably true in many cancers, but may not be the case of all tumors. It is plausible, indeed, that in some tumors, a number of differentiated cells can acquire the capacity for self-renewal, through multiple mutagenic events, and thus ‘‘reacquire’’ stem-like properties[25].
On the other hand, the term “tumor-initiating cells”, according to experimental evidence, refers to the ability of these cells to initiate tumors when transplanted in a xenograft model. In this case, it could be incorrectly inferred that the cell that gives rise to the xenograft tumor is the same cell in which the first oncogenic mutation occurred. This is unlikely, since it is clear that the cells that drive aberrant growth at one precise moment may differ from those acting during different stages in tumor evolution or during metastasis. Furthermore, both genetic and epigenetic instability can induce cellular heterogeneity within the stem and non-stem cell populations of the tumor[26]. It has been argued that species differences alone might account for the selective growth of subpopulations of cells in xenotransplantations. Indeed, the great majority of cells in a mouse lymphoma were shown recently to possess tumor initiating capacity when allografted into syngenic mice[27].
WHICH IS THE ORIGIN OF COLON CANCER STEM CELLS?
Although, as mentioned earlier, the sequence of events in CRC has been intensively studied, the cell of origin for cancer formation is still poorly known. Two possible hypotheses have been suggested: the so called “bottom-up” and the “top-down” theories. The first proposes that an ISC, either a progenitor or a differentiated cell, is the first transformed cell, as a consequence of anomalous differentiation, giving rise directly to cancer cells or reprogramming itself, acquiring SC behavior before inducing cancer[28-30] (Figure 1).
Figure 1.
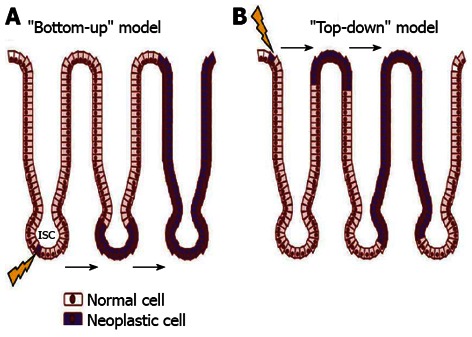
Schematic presentation of two possible ways of colon adeocarcinoma formation. A: “Bottom-up” theory: intestinal stem cells (ISCs, arrow) at the base of the crypt, within the stem cell zone, are the cell of origin of neoplasia as a consequence of anomalous differentiation; B: “Top-down” theory: a progenitor or differentiated cell is the first transformed cell that can acquire stem cells (arrow) behavior before inducing cancer.
ISCs represents the ideal target for neoplastic transformation, due to their extended life span that alter their behavior as a result of genetic and epigenetic changes. Also, similar signalling pathways may regulate self-renewal in stem cells and cancer cell, so that the transformation of an SC would require fewer mutations compared to a progenitor cell[31,32]. Conversely, histological evidence suggested that colon cancer might arise from late progenitors or even an early differentiated cell[33], sustaining the “top-down model” of CRC development. By contrast, genetic observation recently support strongly the bottom-up theory. Indeed, the identification of specific genes expressed in the stem cells of the intestine has recently allowed to show that the cell of origin of adenomas, induced by a constitutively active Wnt signaling, is the stem cell of the small intestine[30,34,35]. This studies have demonstrated that ISC-specific deletion of both functional adenomatous polyposis coli (APC) alleles using Bmi1-, CD133- and (leucine-rich-repeat containing G-protein-coupled receptor 5) Lgr5- CRE recombinase mice leads to efficient tumor formation. Interestingly, Barker and colleagues also showed that tumor formation does not occur when APC is deleted in progenitors or differentiated cells[30]. These results show that the cell giving rise to adenomas in the small intestine is the stem cell. It is still pending, though, whether BMI-, LGR5-expressing cells or CD133-expressing cells of the tumor, are able alone to induce tumor progression and therefore are markers of the intestinal CSC.
Recently, Schwitalla et al[36] have suggested that these models do not exclude each other and that tumor-initiating mutations can occur in both Lgr5+ crypt stem cells or in more differentiated Lgr5-cells, as long as these initially negative cells dedifferentiate and re-express Lgr5. Indeed, the authors have demonstrated, in a genetic model of intestinal tumor initiation, that epithelial non-stem cells can re-express stem cell markers and be converted into tumor-initiating cells. This phenomenon is strictly dependent on the degree of Wnt activation and can only be observed when Wnt signaling is markedly elevated[37].
It has also been discussed that even cells from outside the tumor, for example, bone marrow-derived cells, might also serve as CSC’s ancestors. This phenomenon has been firstly demonstrated in a murine model of gastric cancer induced by Helicobacter pylori, in which SC derived from bone marrow were able to generate the tumor[37,38].
Emerging evidence suggests that bone-marrow-derived mesenchymal stem cells (BM-MSCs) contribute to tissue regeneration in the colon partly by promoting neovascularization or arteriogenesis[39,40]. Although tumor stromal fibroblasts are mainly recruited from local tissue fibroblasts, it has been proposed that BM-MSCs are recruited into the stroma of developing tumors[41-43]. Several studies have demonstrated that BM-MSCs can selectively migrate to sites of mucosal damage and wound healing including colorectal cancers, where a number of tumor-related inflammatory reactions and abnormal tissue regeneration phenomena take place actively. It also has been shown that cancer cells release specific factors that induce BM-MSC mobilization and recruitment to the tumor stroma where they eventually contribute to the formation of a tumor-supportive microenvironment[44] (Table 1).
Table 1.
Summary of characteristics and controversies about colon cancer stem cells
| Origin | “Bottom-up” theory[30-32,34,35] |
| “Top-down” theory[33] | |
| CSCs can derive from epithelial non-stem cells that re-express stem cell markers[36] | |
| CSCs can derive from bone marrow cells[37-43] | |
| Identification assays | Serial transplantation in immune-compromised mice[8,9,27] |
| Expression of cell surface marker CD133[5,8,9,14,31,44-46,48,49] | |
| Side Population[52-61] | |
| Therapeutic strategies | Induction of CSC differentiation by salinomycin or BMP4[72,73] |
| Monoclonal antibodies directed against cell surface molecules, such as CD133, CD44, EGFR (cetuximab) and VEGF (bevacizumab)[74,76-79] | |
| Blockage of self-renewal pathways, including Wnt, Notch, PTEN, Hedgehog , EMT and IL-4 pathway by microRNA or selected small-molecule antagonists[65-70,80-86] | |
| Target the Warburg effect[87,88] |
CSC: Colon cancer stem cell; EGFR: Epidermal growth factor receptor; VEGF: Vascular endothelial growth factor; EMT: Epithelial-to-mesenchymal transition.
IDENTIFICATION OF COLON CANCER STEM CELLS: LIMITS AND CONTROVERSIES
The correct identification and isolation of the cells responsible for tumor formation is always challenging in cancer research. Although CSCs have been implicated in colon carcinogenesis, due to the complexity of their biology and unsolved technical issues, an unequivocally approved identification and isolation strategy is still a matter of debate[32]. The gold standard for identifying a CSC is the capacity to propagate tumors as xenografts in immuno-compromised mice. However, it has been argued that species differences alone might account for the selective growth of subpopulations of cells in these assays. Indeed, the great majority of cells in a mouse lymphoma were shown recently to possess tumor initiating capacity when allografted into syngenic mice[27]. Moreover, serial transplantation experiments with animal models are laborious and time-consuming, hence the need to develop reliable surrogate assays.
Several in vitro assays have been used to identify CSCs can derive, including sphere assays, surface cell markers and the Hoechst dye efflux properties, which identify the so-called Side-Population (SP). Studies have also been performed to define putative CSC genetic signatures. However, each of these methods has potential pitfalls that complicate the interpretation of results[25].
It is clearly not sufficient to define a stem cell based only on surface markers. Moreover, none of the markers used to isolate stem cells in various normal and cancerous tissues is expressed exclusively by the stem cell fraction. Indeed most markers used for colon CSC isolation are chosen either because they are expressed in normal stem cells or as they were found to identify CSCs in other malignancies, either hematological or solid. The disadvantage of choosing markers in this fashion is that the functional effect of expression of the marker in CSCs is usually unknown.
For instance, focusing on CRC, several studies have suggested that the CSC fraction within colon cancer might be identified by the expression of the cell surface marker CD133[8,9]. CD133 is a trans-membrane glycoprotein, expressed by normal progenitors belonging to neuronal, hematopoietic, epithelial and endothelial lineages. In the last years, CD133 has become the “molecule of the moment”, being recognized as a putative CSC marker for many human solid tumors, including liver, pancreas and colon neoplasms[14,45]. However, despite constant research efforts, the molecular mechanisms and signaling pathways that regulate the behavior of CD133-expressing CSC, remain unknown.
In particular, we demonstrated the existence of a population of self renewing cells expressing CD133 within primary and metastatic human CRC[5]. This antigen was expressed in significantly higher percentage in CRC samples, compared to the respective normal tissues. CD133-positive cells were also found in liver metastases (up to 10%), while they were hardly detectable in the healthy liver tissue[5]. In addition, CD133+ cells, isolated from different human colonic adenocarcinoma lines (CaCo-2, HT29, LoVo), were highly clonogenic in vitro and gave rise to tumors following transplantation in mice. Conversely, the CD133-negative fraction of all cell lines had a lower clonogenic potential in soft agar assays and did not generate tumors in secondary recipients[45], confirming the tumor initiating properties of CD133+ CSC. Interestingly, we also provided the original demonstration that modulation of CD133 expression in the CaCo-2 colon cancer cell line was associated with corresponding variations in the expression levels of both Endothelin-1 and nuclear receptor subfamily 4, group A, member 2[46], both known to play an important role in the proliferation and metastasis processes. This modulation was associated with a significant inhibition of the cells’ clonogenic and migration ability, thus further confirming a role of the CD133 molecule in the definition of the CSC phenotype[46].
There are though still some controversies on the role of CD133 as a CSC marker in CRC; the opposing theories emerge from the evidence that most CD133 antibodies target glycosylation-dependent epitopes[35], whose presence is related to the differentiation stage of the cell. Experimental data from colon and glioblastoma cells suggested that the differential glycosylation of specific epitopes may mask the presence of CD133 on cells previously characterized as negative[47,48]. Moreover, CD133 has been found to be expressed by the full spectrum of undifferentiated and differentiated colonic epithelial cells, both in humans and in mice[49]. Shmelkov et al[49] have demonstrated that primary and metastatic colon cancers contain CD133+ and CD133- parenchymal tumor cells, and both types of cells are capable of tumor initiation, as observed in a xenotransplantation model. A similar lack of specificity has been also observed for other potential CSC markers of CRC, such as CD44, CD166, CD29, CD24, Lgr5, and nuclear beta-catenin[50]. In fact, the vast majority of cells that express these markers are not stem cells[51].
Another approach to identify CSCs is their presence within the so-called “Side Population”. SP cells have been first described within the hematopoietic system. In particular, bone marrow stem cells contain a subpopulation that extrude the DNA-binding dye, Hoechst 33342, out of the cell membrane. Comparing the fluorescence intensity on the wavelength of blue against the red, the SP appears as a tail of cells with low fluorescence. This phenotype is attributed to the activity of the ABC membrane transporter proteins that can confer drug resistance to stem cells. These proteins can be blocked by inhibitors of efflux pumps, such as verapamil[52-54]. The SP fractions have been identified in various human tissues[55,56], cancer specimens[57,58] and tumor cell lines[59,60] and it has been suggested that they may represent the true stem cell population. However, as with cell surface markers, the SP phenotype is not synonymous with stemness. Indeed, a recent article claimed that both SP and non-SP cells, isolated from gastrointestinal cancer cell lines, displayed similar clonogenic and tumourigenic potential in vitro and in vivo, and showed identical expression of putative stem cell markers. Therefore, the Authors concluded that the SP does not enrich for stem cells in gastrointestinal cell lines[61]. Also, possible toxicity of the dye, in cells not capable of extrusion, should also be considered as a caveat to interpreting functional assays of SP cells.
Without a better understanding of normal tissue stem cells and their susceptibility to neoplastic transformation, it will be difficult to conduct conclusive studies of the existence and origins of CSCs (Table 1).
WHICH STRATEGY TO TARGET COLON CSCS?
According to the CSC model, the few self-renewing CSCs that mediate tumor growth are difficult to kill and their persistence might explain tumor recurrence after therapy[62]. Indeed, chemotherapeutics interfere with the ability of rapidly growing cells to divide, so CSCs may be spared, leading to tumor recurrence and metastasis[11-13]. Therefore, to assess the efficacy of therapeutics, it is necessary to accurately distinguish tumorigenic from non-tumorigenic cancer cells and to understand which progression model occurs in the tumor. Unfortunately, the complex network of mechanisms that regulate SC renewal and carcinogenesis is not clear.
Chemotherapeutic resistance is exerted either through a shift from active state to quiescence or through a wide spectrum of protective mechanisms that characterize CSC biology; these include altered DNA damage repair, altered cell-cycle checkpoint control, malfunction of apoptosis, expression of drug transporters and detoxifying enzymes, and a high expression of proteins belonging to the ABC membrane transporters famil[63,64].
Moreover, the plasticity of CSCs and the epithelial-to-mesenchymal transition (EMT) complicate therapeutic approaches because participate in the acquisition of both de novo and acquired drug resistance[65]. Indeed, EMT can trigger reversion to a CSC-like phenotype, providing an association between EMT, CSCs and drug resistance[66]. Several key signaling pathways contribute to this process, such as transforming growth factor β and Wnt, that are well known to induce EMT and promote stem cell maintenance[67,68]. Recent studies have implicated microRNA functionality in these processes; indeed the dysregulation of microRNA expression is likely to be a major contributing factor in the etiology of some cancers[69,70].
It is therefore essential to renovate the therapeutic repertoire by designing new treatments that specifically target CSCs and, at the same time, also eliminate the non-CSC population, intervening in the process of EMT[65,71]. Novel therapeutic strategy based on targeting EMT pathways and CSC maintenance might be a promising tool for CRC defeat.
Moreover, CSCs can be functionally antagonized by inducing their differentiation. Differentiation therapy forces cells to shift into a mature phenotype, lose their self-renewal abilities, and therefore become vulnerable to conventional treatment. For instance, salinomycin, a highly selective potassium ionophore, was recently described as the first compound that can selectively eradicate the tumor through induction of terminal epithelial differentiation of CSCs[72]. Gupta et al[72] revealed that salinomycin decreases the proportion of CD44high/CD24low breast cancer cells, whereas paclitaxel has opposing effects. Importantly, cells exposed to salinomycin were less capable of inducing tumors following injection into mice; salinomycin also slowed the growth of the animals’ tumors through unknown mechanisms[71]. Salinomycin is thought to inhibit potassium-positive channel-regulated migration and interfere with EMT and metastasis[72]. Also induction of differentiation in colon CSCs by exposing these cells to Bone Morphogenetic Protein 4 (BMP4), which can initiate a differentiation program as well as mediate apoptosis, sensitizes CRC cells to the effects of 5-Fluorouracil (5-FU) or oxaliplatin in vivo, resulting in complete and long term regression of colon xenografts[73].
The potential toxic effect, that might occur from the impact on normal SCs, can be minimized by targeting molecules or pathways that are preferentially active in CSCs[73]. Monoclonal antibodies could be directed against cell surface molecules, such as CD133, CD44, or even drug transporters, resulting in reduction of tumor size, metastatic potential, and resistance to chemoradiotherap[74,75].
Advances in high-throughput technologies and bioinformatics will allow developing additional compounds targeting CSC signaling pathways. Currently there are two established targets for such therapies: epidermal growth factor receptor (EGFR), which belongs to the ErbB family of tyrosine kinase receptors and is abnormally activated in many tumors[76], and vascular endothelial growth factor (VEGF), which is known to promote formation of new vessels by inducing growth and differentiation of endothelial cells[77,78]. Several clinical trials have demonstrated that introduction of targeted therapies with monoclonal antibodies against EGFR (cetuximab) and VEGF (bevacizumab) in addition to 5-FU, resulted in a significant survival increase in patients with advanced disease[79].
Another rational target includes blockage of various self-renewal pathways, including Wnt, Notch, PTEN, and Hedgehog[80]. Small-molecules that inhibit the Wnt pathway and γ-secretases that inhibit the Notch pathway have been recently identified as novel approaches to CRC[73]. The Wnt/β-catenin pathway has been implicated in the maintenance of the intestinal crypt stem cell phenotype and Wnt signaling dysregulation through either loss of APC function or oncogenic β-catenin mutations has been shown to cause the majority of sporadic cancer cases[81]. Disruption of Tcf/β-catenin complexes by selected small-molecule antagonists has been shown to antagonize cellular effects of β-catenin and to result in inhibition of cellular proliferation in colon cancer cells[82]. Similarly, the Notch signaling pathway has been reported to be overexpressed in colon CSCs, where it was found to play a role in colon CSC viability, tumorigenicity, and self-renewal[83-87]. Van Es et al[87] have demonstrated that blocking the Notch cascade with a gamma-secretase inhibitor induced goblet cell differentiation in adenomas in mice carrying a mutation of the APC tumor suppressor gene and subsequent tumor growth arrest. Moreover, Hoey et al[88] have demonstrated that by inhibiting delta-like 4 ligand (DLL4), an important component of the Notch pathway, with human monoclonal antibody 21M18 in colon carcinoma xenografts, the tumor growth as well as the CSC frequency, was decreased compared to control. Interestingly, even though treatment of the xenografts with irinotecan, a chemotherapeutic often used in colon cancer, slowed down tumor growth, and the clonogenicity was increased. Combination treatment of irinotecan with anti-hDLL4 reduced again the tumor growth and stem cell frequency, at even higher levels than the anti-DLL4 treatment alone. This indicates that inhibiting Notch signaling reduces CSC frequencies and sensitizes tumor cells for irinotecan treatment.
It has recently been observed that the inhibition of the interleukin (IL)-4 pathway with an anti-IL-4 antibody or an IL-4 receptor antagonist in CD133+ colorectal CSCs augmented the antitumor effects of conventional chemotherapeutics[89-90]. Indeed, colon carcinomas produce IL-4 that functions in an autocrine manner, promoting antiapoptotic pathways in these tumors. Inhibiting IL-4 by blocking antibodies sensitizes the cells for killing by 5-FU and oxaliplatin IL-4[89-90].
Reversing chemoresistance and radioresistance represents a promising proposal. This can be achieved through interference with a plethora of cellular components, including inactivation of drug transporters and DNA checkpoint kinases, depletion of reactive oxygen species scavengers, and inhibition of signal transduction pathways.
Interestingly, little is as of yet known with regard to the metabolism of CSC population, leaving an exciting avenue unstudied in the dawn of the emerging field of metabolomics. The Warburg effect, the premise of which is that cancer cells restrict use of fatty-acid oxidation in favor of glycolysis as an ATP energy source, can also be harnessed to create novel broad-spectrum anticancer agents[91-94]. A recent study by Akao et al[95] provided initial evidence of metabolic changes in therapy-resistant cell populations by demonstrating significant overexpression of a metabolic “master-regulator” Sirt1 in DLD-1 5-FU-resistant cells.
Altogether, these data illustrate the therapeutic utility of the cancer stem cell concept, which, by enabling specific examination of more aggressive cancer-initiating and cancer propagating subpopulations, provides the tools for discovery of novel mechanisms of cancer therapeutic resistance (Table 1).
CONCLUSION
Understanding the details of CSCs’ biology is a primary goal in basic oncology research but would also pave the way for a better clarification of CRC progression. The translation implication of such information is clearly deducible. In particular, the combination of previously known and new markers defining CSC specificity, could lead to the development of a better oriented anticancer therapy, possibly targeting CSCs.
Footnotes
P- Reviewers Bommireddy R, Wu W, Zhu YL S- Editor Zhai HH L- Editor A E- Editor Zhang DN
References
- 1.Heath JP. Epithelial cell migration in the intestine. Cell Biol Int. 1996;20:139–146. doi: 10.1006/cbir.1996.0018. [DOI] [PubMed] [Google Scholar]
- 2.Jemal A, Siegel R, Ward E, Murray T, Xu J, Smigal C, Thun MJ. Cancer statistics, 2006. CA Cancer J Clin. 2006;56:106–130. doi: 10.3322/canjclin.56.2.106. [DOI] [PubMed] [Google Scholar]
- 3.Fearon ER, Vogelstein B. A genetic model for colorectal tumorigenesis. Cell. 1990;61:759–767. doi: 10.1016/0092-8674(90)90186-i. [DOI] [PubMed] [Google Scholar]
- 4.McDonald SA, Preston SL, Lovell MJ, Wright NA, Jankowski JA. Mechanisms of disease: from stem cells to colorectal cancer. Nat Clin Pract Gastroenterol Hepatol. 2006;3:267–274. doi: 10.1038/ncpgasthep0473. [DOI] [PubMed] [Google Scholar]
- 5.Puglisi MA, Sgambato A, Saulnier N, Rafanelli F, Barba M, Boninsegna A, Piscaglia AC, Lauritano C, Novi ML, Barbaro F, et al. Isolation and characterization of CD133+ cell population within human primary and metastatic colon cancer. Eur Rev Med Pharmacol Sci. 2009;13 Suppl 1:55–62. [PubMed] [Google Scholar]
- 6.Vermeulen L, Sprick MR, Kemper K, Stassi G, Medema JP. Cancer stem cells--old concepts, new insights. Cell Death Differ. 2008;15:947–958. doi: 10.1038/cdd.2008.20. [DOI] [PubMed] [Google Scholar]
- 7.Bonnet D, Dick JE. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat Med. 1997;3:730–737. doi: 10.1038/nm0797-730. [DOI] [PubMed] [Google Scholar]
- 8.Ricci-Vitiani L, Lombardi DG, Pilozzi E, Biffoni M, Todaro M, Peschle C, De Maria R. Identification and expansion of human colon-cancer-initiating cells. Nature. 2007;445:111–115. doi: 10.1038/nature05384. [DOI] [PubMed] [Google Scholar]
- 9.O’Brien CA, Pollett A, Gallinger S, Dick JE. A human colon cancer cell capable of initiating tumour growth in immunodeficient mice. Nature. 2007;445:106–110. doi: 10.1038/nature05372. [DOI] [PubMed] [Google Scholar]
- 10.Li F, Tiede B, Massagué J, Kang Y. Beyond tumorigenesis: cancer stem cells in metastasis. Cell Res. 2007;17:3–14. doi: 10.1038/sj.cr.7310118. [DOI] [PubMed] [Google Scholar]
- 11.NIH consensus conference. Adjuvant therapy for patients with colon and rectal cancer. JAMA. 1990;264:1444–1450. [PubMed] [Google Scholar]
- 12.Efficacy of adjuvant fluorouracil and folinic acid in colon cancer. International Multicentre Pooled Analysis of Colon Cancer Trials (IMPACT) investigators. Lancet. 1995;345:939–944. [PubMed] [Google Scholar]
- 13.Comparison of flourouracil with additional levamisole, higher-dose folinic acid, or both, as adjuvant chemotherapy for colorectal cancer: a randomised trial. QUASAR Collaborative Group. Lancet. 2000;355:1588–1596. [PubMed] [Google Scholar]
- 14.Yin S, Li J, Hu C, Chen X, Yao M, Yan M, Jiang G, Ge C, Xie H, Wan D, et al. CD133 positive hepatocellular carcinoma cells possess high capacity for tumorigenicity. Int J Cancer. 2007;120:1444–1450. doi: 10.1002/ijc.22476. [DOI] [PubMed] [Google Scholar]
- 15.Suetsugu A, Nagaki M, Aoki H, Motohashi T, Kunisada T, Moriwaki H. Characterization of CD133+ hepatocellular carcinoma cells as cancer stem/progenitor cells. Biochem Biophys Res Commun. 2006;351:820–824. doi: 10.1016/j.bbrc.2006.10.128. [DOI] [PubMed] [Google Scholar]
- 16.Maenhaut C, Dumont JE, Roger PP, van Staveren WC. Cancer stem cells: a reality, a myth, a fuzzy concept or a misnomer? An analysis. Carcinogenesis. 2010;31:149–158. doi: 10.1093/carcin/bgp259. [DOI] [PubMed] [Google Scholar]
- 17.Boman BM, Wicha MS, Fields JZ, Runquist OA. Symmetric division of cancer stem cells--a key mechanism in tumor growth that should be targeted in future therapeutic approaches. Clin Pharmacol Ther. 2007;81:893–898. doi: 10.1038/sj.clpt.6100202. [DOI] [PubMed] [Google Scholar]
- 18.Cicalese A, Bonizzi G, Pasi CE, Faretta M, Ronzoni S, Giulini B, Brisken C, Minucci S, Di Fiore PP, Pelicci PG. The tumor suppressor p53 regulates polarity of self-renewing divisions in mammary stem cells. Cell. 2009;138:1083–1095. doi: 10.1016/j.cell.2009.06.048. [DOI] [PubMed] [Google Scholar]
- 19.Boman BM, Fields JZ, Cavanaugh KL, Guetter A, Runquist OA. How dysregulated colonic crypt dynamics cause stem cell overpopulation and initiate colon cancer. Cancer Res. 2008;68:3304–3313. doi: 10.1158/0008-5472.CAN-07-2061. [DOI] [PubMed] [Google Scholar]
- 20.Botchkina G. Colon cancer stem cells - From basic to clinical application. Cancer Lett. 2012:Epub ahead of print. doi: 10.1016/j.canlet.2012.04.006. [DOI] [PubMed] [Google Scholar]
- 21.Pantic I. Cancer stem cell hypotheses: impact on modern molecular physiology and pharmacology research. J Biosci. 2011;36:957–961. doi: 10.1007/s12038-011-9155-5. [DOI] [PubMed] [Google Scholar]
- 22.Bitarte N, Bandres E, Boni V, Zarate R, Rodriguez J, Gonzalez-Huarriz M, Lopez I, Javier Sola J, Alonso MM, Fortes P, et al. MicroRNA-451 is involved in the self-renewal, tumorigenicity, and chemoresistance of colorectal cancer stem cells. Stem Cells. 2011;29:1661–1671. doi: 10.1002/stem.741. [DOI] [PubMed] [Google Scholar]
- 23.Takebe N, Harris PJ, Warren RQ, Ivy SP. Targeting cancer stem cells by inhibiting Wnt, Notch, and Hedgehog pathways. Nat Rev Clin Oncol. 2011;8:97–106. doi: 10.1038/nrclinonc.2010.196. [DOI] [PubMed] [Google Scholar]
- 24.Visvader JE, Lindeman GJ. Cancer stem cells in solid tumours: accumulating evidence and unresolved questions. Nat Rev Cancer. 2008;8:755–768. doi: 10.1038/nrc2499. [DOI] [PubMed] [Google Scholar]
- 25.Clarke MF, Dick JE, Dirks PB, Eaves CJ, Jamieson CH, Jones DL, Visvader J, Weissman IL, Wahl GM. Cancer stem cells--perspectives on current status and future directions: AACR Workshop on cancer stem cells. Cancer Res. 2006;66:9339–9344. doi: 10.1158/0008-5472.CAN-06-3126. [DOI] [PubMed] [Google Scholar]
- 26.Tan BT, Park CY, Ailles LE, Weissman IL. The cancer stem cell hypothesis: a work in progress. Lab Invest. 2006;86:1203–1207. doi: 10.1038/labinvest.3700488. [DOI] [PubMed] [Google Scholar]
- 27.Kelly PN, Dakic A, Adams JM, Nutt SL, Strasser A. Tumor growth need not be driven by rare cancer stem cells. Science. 2007;317:337. doi: 10.1126/science.1142596. [DOI] [PubMed] [Google Scholar]
- 28.Vicente-Dueñas C, Gutiérrez de Diego J, Rodríguez FD, Jiménez R, Cobaleda C. The role of cellular plasticity in cancer development. Curr Med Chem. 2009;16:3676–3685. doi: 10.2174/092986709789105019. [DOI] [PubMed] [Google Scholar]
- 29.Krivtsov AV, Twomey D, Feng Z, Stubbs MC, Wang Y, Faber J, Levine JE, Wang J, Hahn WC, Gilliland DG, et al. Transformation from committed progenitor to leukaemia stem cell initiated by MLL-AF9. Nature. 2006;442:818–822. doi: 10.1038/nature04980. [DOI] [PubMed] [Google Scholar]
- 30.Barker N, Ridgway RA, van Es JH, van de Wetering M, Begthel H, van den Born M, Danenberg E, Clarke AR, Sansom OJ, Clevers H. Crypt stem cells as the cells-of-origin of intestinal cancer. Nature. 2009;457:608–611. doi: 10.1038/nature07602. [DOI] [PubMed] [Google Scholar]
- 31.Vries RG, Huch M, Clevers H. Stem cells and cancer of the stomach and intestine. Mol Oncol. 2010;4:373–384. doi: 10.1016/j.molonc.2010.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Papailiou J, Bramis KJ, Gazouli M, Theodoropoulos G. Stem cells in colon cancer. A new era in cancer theory begins. Int J Colorectal Dis. 2011;26:1–11. doi: 10.1007/s00384-010-1022-6. [DOI] [PubMed] [Google Scholar]
- 33.Shih IM, Wang TL, Traverso G, Romans K, Hamilton SR, Ben-Sasson S, Kinzler KW, Vogelstein B. Top-down morphogenesis of colorectal tumors. Proc Natl Acad Sci USA. 2001;98:2640–2645. doi: 10.1073/pnas.051629398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sangiorgi E, Capecchi MR. Bmi1 is expressed in vivo in intestinal stem cells. Nat Genet. 2008;40:915–920. doi: 10.1038/ng.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhu L, Gibson P, Currle DS, Tong Y, Richardson RJ, Bayazitov IT, Poppleton H, Zakharenko S, Ellison DW, Gilbertson RJ. Prominin 1 marks intestinal stem cells that are susceptible to neoplastic transformation. Nature. 2009;457:603–607. doi: 10.1038/nature07589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schwitalla S, Fingerle AA, Cammareri P, Nebelsiek T, Göktuna SI, Ziegler PK, Canli O, Heijmans J, Huels DJ, Moreaux G, et al. Intestinal tumorigenesis initiated by dedifferentiation and acquisition of stem-cell-like properties. Cell. 2013;152:25–38. doi: 10.1016/j.cell.2012.12.012. [DOI] [PubMed] [Google Scholar]
- 37.Houghton J, Stoicov C, Nomura S, Rogers AB, Carlson J, Li H, Cai X, Fox JG, Goldenring JR, Wang TC. Gastric cancer originating from bone marrow-derived cells. Science. 2004;306:1568–1571. doi: 10.1126/science.1099513. [DOI] [PubMed] [Google Scholar]
- 38.Varon C, Dubus P, Mazurier F, Asencio C, Chambonnier L, Ferrand J, Giese A, Senant-Dugot N, Carlotti M, Mégraud F. Helicobacter pylori infection recruits bone marrow-derived cells that participate in gastric preneoplasia in mice. Gastroenterology. 2012;142:281–291. doi: 10.1053/j.gastro.2011.10.036. [DOI] [PubMed] [Google Scholar]
- 39.Okamoto R, Yajima T, Yamazaki M, Kanai T, Mukai M, Okamoto S, Ikeda Y, Hibi T, Inazawa J, Watanabe M. Damaged epithelia regenerated by bone marrow-derived cells in the human gastrointestinal tract. Nat Med. 2002;8:1011–1017. doi: 10.1038/nm755. [DOI] [PubMed] [Google Scholar]
- 40.de Jong JH, Rodermond HM, Zimberlin CD, Lascano V, De Sousa E Melo F, Richel DJ, Medema JP, Vermeulen L. Fusion of intestinal epithelial cells with bone marrow derived cells is dispensable for tissue homeostasis. Sci Rep. 2012;2:271. doi: 10.1038/srep00271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mishra PJ, Mishra PJ, Humeniuk R, Medina DJ, Alexe G, Mesirov JP, Ganesan S, Glod JW, Banerjee D. Carcinoma-associated fibroblast-like differentiation of human mesenchymal stem cells. Cancer Res. 2008;68:4331–4339. doi: 10.1158/0008-5472.CAN-08-0943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Quante M, Tu SP, Tomita H, Gonda T, Wang SS, Takashi S, Baik GH, Shibata W, Diprete B, Betz KS, et al. Bone marrow-derived myofibroblasts contribute to the mesenchymal stem cell niche and promote tumor growth. Cancer Cell. 2011;19:257–272. doi: 10.1016/j.ccr.2011.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Saulnier N, Puglisi MA, Lattanzi W, Castellini L, Pani G, Leone G, Alfieri S, Michetti F, Piscaglia AC, Gasbarrini A. Gene profiling of bone marrow- and adipose tissue-derived stromal cells: a key role of Kruppel-like factor 4 in cell fate regulation. Cytotherapy. 2011;13:329–340. doi: 10.3109/14653249.2010.515576. [DOI] [PubMed] [Google Scholar]
- 44.Hall B, Andreeff M, Marini F. The participation of mesenchymal stem cells in tumor stroma formation and their application as targeted-gene delivery vehicles. Handb Exp Pharmacol. 2007;(180):263–283. doi: 10.1007/978-3-540-68976-8_12. [DOI] [PubMed] [Google Scholar]
- 45.Mizrak D, Brittan M, Alison M. CD133: molecule of the moment. J Pathol. 2008;214:3–9. doi: 10.1002/path.2283. [DOI] [PubMed] [Google Scholar]
- 46.Puglisi MA, Barba M, Corbi M, Errico MF, Giorda E, Saulnier N, Boninsegna A, Piscaglia AC, Carsetti R, Cittadini A, et al. Identification of Endothelin-1 and NR4A2 as CD133-regulated genes in colon cancer cells. J Pathol. 2011;225:305–314. doi: 10.1002/path.2954. [DOI] [PubMed] [Google Scholar]
- 47.Kemper K, Sprick MR, de Bree M, Scopelliti A, Vermeulen L, Hoek M, Zeilstra J, Pals ST, Mehmet H, Stassi G, et al. The AC133 epitope, but not the CD133 protein, is lost upon cancer stem cell differentiation. Cancer Res. 2010;70:719–729. doi: 10.1158/0008-5472.CAN-09-1820. [DOI] [PubMed] [Google Scholar]
- 48.Osmond TL, Broadley KW, McConnell MJ. Glioblastoma cells negative for the anti-CD133 antibody AC133 express a truncated variant of the CD133 protein. Int J Mol Med. 2010;25:883–888. doi: 10.3892/ijmm_00000418. [DOI] [PubMed] [Google Scholar]
- 49.Shmelkov SV, Butler JM, Hooper AT, Hormigo A, Kushner J, Milde T, St Clair R, Baljevic M, White I, Jin DK, et al. CD133 expression is not restricted to stem cells, and both CD133+ and CD133- metastatic colon cancer cells initiate tumors. J Clin Invest. 2008;118:2111–2120. doi: 10.1172/JCI34401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chu P, Clanton DJ, Snipas TS, Lee J, Mitchell E, Nguyen ML, Hare E, Peach RJ. Characterization of a subpopulation of colon cancer cells with stem cell-like properties. Int J Cancer. 2009;124:1312–1321. doi: 10.1002/ijc.24061. [DOI] [PubMed] [Google Scholar]
- 51.Vermeulen L, Todaro M, de Sousa Mello F, Sprick MR, Kemper K, Perez Alea M, Richel DJ, Stassi G, Medema JP. Single-cell cloning of colon cancer stem cells reveals a multi-lineage differentiation capacity. Proc Natl Acad Sci USA. 2008;105:13427–13432. doi: 10.1073/pnas.0805706105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Goodell MA, Rosenzweig M, Kim H, Marks DF, DeMaria M, Paradis G, Grupp SA, Sieff CA, Mulligan RC, Johnson RP. Dye efflux studies suggest that hematopoietic stem cells expressing low or undetectable levels of CD34 antigen exist in multiple species. Nat Med. 1997;3:1337–1345. doi: 10.1038/nm1297-1337. [DOI] [PubMed] [Google Scholar]
- 53.Zhou S, Schuetz JD, Bunting KD, Colapietro AM, Sampath J, Morris JJ, Lagutina I, Grosveld GC, Osawa M, Nakauchi H, et al. The ABC transporter Bcrp1/ABCG2 is expressed in a wide variety of stem cells and is a molecular determinant of the side-population phenotype. Nat Med. 2001;7:1028–1034. doi: 10.1038/nm0901-1028. [DOI] [PubMed] [Google Scholar]
- 54.Budak MT, Alpdogan OS, Zhou M, Lavker RM, Akinci MA, Wolosin JM. Ocular surface epithelia contain ABCG2-dependent side population cells exhibiting features associated with stem cells. J Cell Sci. 2005;118:1715–1724. doi: 10.1242/jcs.02279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Alvi AJ, Clayton H, Joshi C, Enver T, Ashworth A, Vivanco Md, Dale TC, Smalley MJ. Functional and molecular characterisation of mammary side population cells. Breast Cancer Res. 2003;5:R1–R8. doi: 10.1186/bcr547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Asakura A, Rudnicki MA. Side population cells from diverse adult tissues are capable of in vitro hematopoietic differentiation. Exp Hematol. 2002;30:1339–1345. doi: 10.1016/s0301-472x(02)00954-2. [DOI] [PubMed] [Google Scholar]
- 57.Hirschmann-Jax C, Foster AE, Wulf GG, Nuchtern JG, Jax TW, Gobel U, Goodell MA, Brenner MK. A distinct “side population” of cells with high drug efflux capacity in human tumor cells. Proc Natl Acad Sci USA. 2004;101:14228–14233. doi: 10.1073/pnas.0400067101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Szotek PP, Pieretti-Vanmarcke R, Masiakos PT, Dinulescu DM, Connolly D, Foster R, Dombkowski D, Preffer F, Maclaughlin DT, Donahoe PK. Ovarian cancer side population defines cells with stem cell-like characteristics and Mullerian Inhibiting Substance responsiveness. Proc Natl Acad Sci USA. 2006;103:11154–11159. doi: 10.1073/pnas.0603672103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Berlin JB. Richard Beer-Hofmann and Theodor Reik: a letter in exile. Psychoanal Rev 1981- 1982;68:479–486. [PubMed] [Google Scholar]
- 60.Dekaney CM, Rodriguez JM, Graul MC, Henning SJ. Isolation and characterization of a putative intestinal stem cell fraction from mouse jejunum. Gastroenterology. 2005;129:1567–1580. doi: 10.1053/j.gastro.2005.08.011. [DOI] [PubMed] [Google Scholar]
- 61.Burkert J, Otto WR, Wright NA. Side populations of gastrointestinal cancers are not enriched in stem cells. J Pathol. 2008;214:564–573. doi: 10.1002/path.2307. [DOI] [PubMed] [Google Scholar]
- 62.Saigusa S, Tanaka K, Toiyama Y, Yokoe T, Okugawa Y, Ioue Y, Miki C, Kusunoki M. Correlation of CD133, OCT4, and SOX2 in rectal cancer and their association with distant recurrence after chemoradiotherapy. Ann Surg Oncol. 2009;16:3488–3498. doi: 10.1245/s10434-009-0617-z. [DOI] [PubMed] [Google Scholar]
- 63.Schatton T, Frank NY, Frank MH. Identification and targeting of cancer stem cells. Bioessays. 2009;31:1038–1049. doi: 10.1002/bies.200900058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Chaffer CL, Weinberg RA. A perspective on cancer cell metastasis. Science. 2011;331:1559–1564. doi: 10.1126/science.1203543. [DOI] [PubMed] [Google Scholar]
- 65.Singh A, Settleman J. EMT, cancer stem cells and drug resistance: an emerging axis of evil in the war on cancer. Oncogene. 2010;29:4741–4751. doi: 10.1038/onc.2010.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Polyak K, Weinberg RA. Transitions between epithelial and mesenchymal states: acquisition of malignant and stem cell traits. Nat Rev Cancer. 2009;9:265–273. doi: 10.1038/nrc2620. [DOI] [PubMed] [Google Scholar]
- 67.Thiery JP, Acloque H, Huang RY, Nieto MA. Epithelial-mesenchymal transitions in development and disease. Cell. 2009;139:871–890. doi: 10.1016/j.cell.2009.11.007. [DOI] [PubMed] [Google Scholar]
- 68.Shipitsin M, Campbell LL, Argani P, Weremowicz S, Bloushtain-Qimron N, Yao J, Nikolskaya T, Serebryiskaya T, Beroukhim R, Hu M, et al. Molecular definition of breast tumor heterogeneity. Cancer Cell. 2007;11:259–273. doi: 10.1016/j.ccr.2007.01.013. [DOI] [PubMed] [Google Scholar]
- 69.Kong W, Yang H, He L, Zhao JJ, Coppola D, Dalton WS, Cheng JQ. MicroRNA-155 is regulated by the transforming growth factor beta/Smad pathway and contributes to epithelial cell plasticity by targeting RhoA. Mol Cell Biol. 2008;28:6773–6784. doi: 10.1128/MCB.00941-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Gibbons DL, Lin W, Creighton CJ, Rizvi ZH, Gregory PA, Goodall GJ, Thilaganathan N, Du L, Zhang Y, Pertsemlidis A, et al. Contextual extracellular cues promote tumor cell EMT and metastasis by regulating miR-200 family expression. Genes Dev. 2009;23:2140–2151. doi: 10.1101/gad.1820209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Gupta PB, Chaffer CL, Weinberg RA. Cancer stem cells: mirage or reality? Nat Med. 2009;15:1010–1012. doi: 10.1038/nm0909-1010. [DOI] [PubMed] [Google Scholar]
- 72.Gupta PB, Onder TT, Jiang G, Tao K, Kuperwasser C, Weinberg RA, Lander ES. Identification of selective inhibitors of cancer stem cells by high-throughput screening. Cell. 2009;138:645–659. doi: 10.1016/j.cell.2009.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Todaro M, Francipane MG, Medema JP, Stassi G. Colon cancer stem cells: promise of targeted therapy. Gastroenterology. 2010;138:2151–2162. doi: 10.1053/j.gastro.2009.12.063. [DOI] [PubMed] [Google Scholar]
- 74.Dou J, Gu N. Emerging strategies for the identification and targeting of cancer stem cells. Tumour Biol. 2010;31:243–253. doi: 10.1007/s13277-010-0023-y. [DOI] [PubMed] [Google Scholar]
- 75.Frank NY, Schatton T, Frank MH. The therapeutic promise of the cancer stem cell concept. J Clin Invest. 2010;120:41–50. doi: 10.1172/JCI41004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Cunningham D, Humblet Y, Siena S, Khayat D, Bleiberg H, Santoro A, Bets D, Mueser M, Harstrick A, Verslype C, et al. Cetuximab monotherapy and cetuximab plus irinotecan in irinotecan-refractory metastatic colorectal cancer. N Engl J Med. 2004;351:337–345. doi: 10.1056/NEJMoa033025. [DOI] [PubMed] [Google Scholar]
- 77.Takahashi Y, Kitadai Y, Bucana CD, Cleary KR, Ellis LM. Expression of vascular endothelial growth factor and its receptor, KDR, correlates with vascularity, metastasis, and proliferation of human colon cancer. Cancer Res. 1995;55:3964–3968. [PubMed] [Google Scholar]
- 78.Fan F, Wey JS, McCarty MF, Belcheva A, Liu W, Bauer TW, Somcio RJ, Wu Y, Hooper A, Hicklin DJ, et al. Expression and function of vascular endothelial growth factor receptor-1 on human colorectal cancer cells. Oncogene. 2005;24:2647–2653. doi: 10.1038/sj.onc.1208246. [DOI] [PubMed] [Google Scholar]
- 79.Meyerhardt JA, Mayer RJ. Systemic therapy for colorectal cancer. N Engl J Med. 2005;352:476–487. doi: 10.1056/NEJMra040958. [DOI] [PubMed] [Google Scholar]
- 80.Huang EH, Wicha MS. Colon cancer stem cells: implications for prevention and therapy. Trends Mol Med. 2008;14:503–509. doi: 10.1016/j.molmed.2008.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Fodde R, Smits R, Clevers H. APC, signal transduction and genetic instability in colorectal cancer. Nat Rev Cancer. 2001;1:55–67. doi: 10.1038/35094067. [DOI] [PubMed] [Google Scholar]
- 82.Lepourcelet M, Chen YN, France DS, Wang H, Crews P, Petersen F, Bruseo C, Wood AW, Shivdasani RA. Small-molecule antagonists of the oncogenic Tcf/beta-catenin protein complex. Cancer Cell. 2004;5:91–102. doi: 10.1016/s1535-6108(03)00334-9. [DOI] [PubMed] [Google Scholar]
- 83.Sikandar SS, Pate KT, Anderson S, Dizon D, Edwards RA, Waterman ML, Lipkin SM. NOTCH signaling is required for formation and self-renewal of tumor-initiating cells and for repression of secretory cell differentiation in colon cancer. Cancer Res. 2010;70:1469–1478. doi: 10.1158/0008-5472.CAN-09-2557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Miyamoto S, Rosenberg DW. Role of Notch signaling in colon homeostasis and carcinogenesis. Cancer Sci. 2011;102:1938–1942. doi: 10.1111/j.1349-7006.2011.02049.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Shao H, Huang Q, Liu ZJ. Targeting Notch signaling for cancer therapeutic intervention. Adv Pharmacol. 2012;65:191–234. doi: 10.1016/B978-0-12-397927-8.00007-5. [DOI] [PubMed] [Google Scholar]
- 86.Kim HA, Koo BK, Cho JH, Kim YY, Seong J, Chang HJ, Oh YM, Stange DE, Park JG, Hwang D, et al. Notch1 counteracts WNT/β-catenin signaling through chromatin modification in colorectal cancer. J Clin Invest. 2012;122:3248–3259. doi: 10.1172/JCI61216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.van Es JH, van Gijn ME, Riccio O, van den Born M, Vooijs M, Begthel H, Cozijnsen M, Robine S, Winton DJ, Radtke F, et al. Notch/gamma-secretase inhibition turns proliferative cells in intestinal crypts and adenomas into goblet cells. Nature. 2005;435:959–963. doi: 10.1038/nature03659. [DOI] [PubMed] [Google Scholar]
- 88.Hoey T, Yen WC, Axelrod F, Basi J, Donigian L, Dylla S, Fitch-Bruhns M, Lazetic S, Park IK, Sato A, et al. DLL4 blockade inhibits tumor growth and reduces tumor-initiating cell frequency. Cell Stem Cell. 2009;5:168–177. doi: 10.1016/j.stem.2009.05.019. [DOI] [PubMed] [Google Scholar]
- 89.Todaro M, Alea MP, Di Stefano AB, Cammareri P, Vermeulen L, Iovino F, Tripodo C, Russo A, Gulotta G, Medema JP, et al. Colon cancer stem cells dictate tumor growth and resist cell death by production of interleukin-4. Cell Stem Cell. 2007;1:389–402. doi: 10.1016/j.stem.2007.08.001. [DOI] [PubMed] [Google Scholar]
- 90.Todaro M, Perez Alea M, Scopelliti A, Medema JP, Stassi G. IL-4-mediated drug resistance in colon cancer stem cells. Cell Cycle. 2008;7:309–313. doi: 10.4161/cc.7.3.5389. [DOI] [PubMed] [Google Scholar]
- 91.Kim JW, Dang CV. Cancer’s molecular sweet tooth and the Warburg effect. Cancer Res. 2006;66:8927–8930. doi: 10.1158/0008-5472.CAN-06-1501. [DOI] [PubMed] [Google Scholar]
- 92.Bartrons R, Caro J. Hypoxia, glucose metabolism and the Warburg’s effect. J Bioenerg Biomembr. 2007;39:223–229. doi: 10.1007/s10863-007-9080-3. [DOI] [PubMed] [Google Scholar]
- 93.Stubbs M, Griffiths JR. The altered metabolism of tumors: HIF-1 and its role in the Warburg effect. Adv Enzyme Regul. 2010;50:44–55. doi: 10.1016/j.advenzreg.2009.10.027. [DOI] [PubMed] [Google Scholar]
- 94.Robey IF, Lien AD, Welsh SJ, Baggett BK, Gillies RJ. Hypoxia-inducible factor-1alpha and the glycolytic phenotype in tumors. Neoplasia. 2005;7:324–330. doi: 10.1593/neo.04430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Akao Y, Noguchi S, Iio A, Kojima K, Takagi T, Naoe T. Dysregulation of microRNA-34a expression causes drug-resistance to 5-FU in human colon cancer DLD-1 cells. Cancer Lett. 2011;300:197–204. doi: 10.1016/j.canlet.2010.10.006. [DOI] [PubMed] [Google Scholar]