

Abstract
Advanced age is one of the most important risk factors for osteoporosis. Accumulation of oxidative DNA damage has been proposed to contribute to age-related deregulation of osteoblastic and osteoclastic cells. ERCC1 (Excision Repair Cross Complementary group 1)-XPF (Xeroderma Pigmentosum Group F) is an evolutionarily conserved structure-specific endonuclease that is required for multiple DNA repair pathways. Inherited mutations affecting expression of ERCC1-XPF cause a severe progeroid syndrome in humans, including early onset of osteopenia and osteoporosis, or anomalies in skeletal development. Herein, we used progeroid ERCC1-XPF deficient mice, including Ercc1-null (Ercc1−/−) and hypomorphic (Ercc1−/Δ) mice, to investigate the mechanism by which DNA damage leads to accelerated bone aging. Compared to their wild-type littermates, both Ercc1−/− and Ercc1−/Δ mice display severe, progressive osteoporosis caused by reduced bone formation and enhanced osteoclastogenesis. ERCC1 deficiency leads to atrophy of osteoblastic progenitors in the bone marrow stromal cell (BMSC) population. There is increased cellular senescence of BMSCs and osteoblastic cells, as characterized by reduced proliferation, accumulation of DNA damage and a senescence-associated secretory phenotype (SASP). This leads to enhanced secretion of inflammatory cytokines known to drive osteoclastogenesis, such as IL-6, TNFα, and RANKL and thereby induces an inflammatory bone microenvironment favoring osteoclastogenesis. Furthermore, we found that the transcription factor NF-κB is activated in osteoblastic and osteoclastic cells of the Ercc1 mutant mice. Importantly, we demonstrated that haploinsufficiency of the p65 NF-κB subunit partially rescued the osteoporosis phenotype of Ercc1−/Δ mice. Finally, pharmacological inhibition of the NF-κB signaling via an IKK inhibitor reversed cellular senescence and SASP in Ercc1−/Δ BMSCs. These results demonstrate that DNA damage drives osteoporosis through an NF-κB-dependent mechanism. Therefore, the NF-κB pathway represents a novel therapeutic target to treat aging-related bone disease.
Keywords: osteoporosis, osteoblasts, osteoclasts, bone, nucleotide excision repair, progeria, aging, NF-κB transcription factor, ERCC1-XPF endonuclease
Introduction
Osteoporosis is the most common metabolic bone disease and is characterized by reduced bone mass and bone mineral density (1). Advanced age is one of the most important risk factors for osteoporosis (2). Age-related cellular changes that impede bone formation and/or promote bone resorption include skeletal stem cell atrophy (3), reduced osteoblast (bone-forming cells) proliferation, impaired osteoblast differentiation and activity, and enhanced osteoclastic (bone-resorbing cells) differentiation and function. The detailed molecular mechanisms underlying age-related osteoblastic and osteoclastic changes remain elusive. However, telomerase deficiency and telomere shortening (4) and accumulation of oxidative DNA damage (5) have been proposed to contribute to age-related deregulation of osteoblastic and osteoclastic cells.
DNA damage is caused by exogenous sources, such as ultraviolet and ionizing radiation, as well as endogenous sources such as reactive oxygen species (ROS), which are byproducts of normal respiration (5). Evidence that DNA damage plays a causal role in skeletal defects comes from the observation that mutations in genes that encode proteins required for DNA repair and/or the DNA damage response lead to compromised bone development and/or deregulation of bone homeostasis. ERCC1 (Excision Repair Cross complementary group 1) -XPF (Xeroderma Pigmentosum Group F) is an evolutionarily conserved structure-specific endonuclease that is required for nucleotide excision repair of helix-distorting DNA lesions (6), the repair of DNA interstrand crosslinks (7), and the repair of some double-strand breaks (DSBs) (8). Genetic deletion of either Ercc1 or Xpf in the mouse leads to what appears to be identical phenotypes (9–11). Although these mice have normal embryonic development, postnatally Ercc1-null mice develop numerous progeroid symptoms, including neurodegeneration, anemia and bone marrow degeneration, osteopenia, and decreased lifespan (10,1213). A human progeroid syndrome caused by ERCC1-XPF deficiency has symptoms strikingly similar to what were observed in ERCC1-deficient mice, including osteopenia (12). Mutations in ERCC1 have been linked to COFS syndrome (cerebro-oculo-facio-skeletal) with severe developmental failure and death in early infancy (14). Skeletal abnormalities include microcephaly, bilateral microphthalmia, micrognathia, short philtrum, and rocker-bottom feet. Given that all of these phenotypes occur in the absence of exposure to exogenous genotoxic stress, the skeletal defects associated with both human and murine ERCC1-XPF deficiency support a critical, yet unexpected role for DNA repair in skeletal development and maintenance of bone homeostasis. What is not known is the mechanism by which failure to repair DNA damage drives deregulation of bone homeostasis.
The NF-κB (Nuclear Factor KappaB) transcription factor is a key regulator of cell death and survival in response to various types of cell stress including genotoxic and inflammatory stimuli (15–17). This leads to the activation of an upstream protein kinase: IκB kinase (IKK). Activated IKK subsequently phosphorylates IκB (18–20) resulting in release of NF-κB from IκB. NF-κB then translocates to the nucleus and induces transcription of a variety of target genes that regulate the cellular response to genotoxic and inflammatory stimuli including cell senescence and apoptosis (21). NF-κB signaling is known to play an essential role in regulating bone homeostasis by inhibiting bone formation (22) and enhancing bone resorption (23). Overexpression of a dominant negative IKKβ subunit or genetic deletion of IKKβ result in increased bone mass (22). In addition, mice deficient for the p65 subunit of NF-κB in the hematopoietic compartment have defective osteoclast formation and thus are resistant to arthritis-induced osteolysis (24). However, it remains unclear whether NF-κB plays a role in aging- related osteoporosis.
In the present study, we systematically analyzed the bones of ERCC1-deficient mice, including both Ercc1-null (Ercc1−/−) and hypomorphic (Ercc1−/Δ) mice at multiple ages. These DNA repair-deficient mice displayed severe and progressive osteoporosisdue to both the loss of bone formation and enhanced bone resorption. Our studies reveal a novel role of SASP in uncoupling bone formation and resorption and NF-κB signaling as a driving force for osteoporosis in response to accumulation of endogenous DNA damage.
Results
ERCC1 deficiency leads to severe, progressive osteoporosis in mice
Humans and mice with reduced expression of ERCC1-XPF develop numerous symptoms associated with old age, including osteopenia (9,10,12). To investigate the cellular and molecular mechanisms underlying these phenotypes, we compared the bones of ERCC1-deficient mice to normal littermates at multiple ages. Three dimensional reconstruction of microcomputed-tomography (μQCT) images of vertebrates of 3-week-old gender-matched Ercc1−/− and WT (Ercc1+/+) mice revealed a dramatic reduction in trabecular structures in bones from DNA repair-deficient mice compared to WT littermates (Fig. 1A, left). Histomorphometric analysis based on the μCT studies indicated that Ercc1−/− mice have a significant reduction in bone volume relative to tissue volume (BV/TV), trabecular thickness (Tb.Th) and trabecular number (Tb.N) and an increase in trabecular space (Tb.Sp) compared to WT littermates (Fig. 1A, right), demonstrating that these mice have osteoporosis. This was confirmed by haematoxylin eosin (H&E) staining of tibias of 2-week-old Ercc1−/− and WT mice (Fig. 1C). Male and female Ercc1−/−mice exhibited similar osteoporotic changes, indicating that ERCC1 deficiency and consequently unrepaired DNA damage drive osteoporosis in a sex-independent manner. The number of osteoblasts per bone perimeter (Ob.N/B.pm) (Fig. 1D, right) was significantly reduced in Ercc1−/− mice, while osteoclast surface (Oc.S/BS) and the number of osteoclasts per bone perimeter (Oc.N/B.pm) was increased (Fig. 2B, right) compared to WT littermates,
Fig. 1. ERCC1 deficiency leads to severe, progressive osteoporosis in mice.

(A) μQCT images (Left) and histomorphometric properties (Right) of lumbar vertebrae of 3-week-old WT (+/+) and Ercc1−/− mice. Upper panel, male WT (n=4) and Ercc1−/− (n=6). Lower panel, female WT (n=6) and Ercc1−/− (n=4). Scale bar, 200μm. (B) μQCT images (Left) and histomorphometric properties (Right) of lumbar vertebrae of 8-week-old (upper panels, n=4) and 22-week-old (lower panels, n=4) WT (+/+) and Ercc1−/Δmice. Scale bar, 200μm. (C) H&E analysis of tibia of 2-week-old WT (+/+) and Ercc1−/− mice. Scale bar, 100μm. (D) H&E of tibia of 8-week-old (left panels) and 22-week-old (right panels) WT (+/+) and Ercc1−/Δmice. Scale bar, 100μm. The number of osteoblasts per bone perimeter (Ob.N/B.pm) of 8-week-old WT (+/+) and Ercc1−/Δmice (n=4) is shown in the right panel. (E) Radiographic images of lumbar vertebrae of 22-week-old WT (+/+) and Ercc1−/Δmice. Scale bar, 1mm. All values are shown as mean ± SEM. * p < 0.05, ** p < 0.01, and *** p < 0.001.
Fig. 2. ERCC1 deficiency leads to reduced bone formation and enhanced osteoclastogenesis.

(A) Calcein double-labeling showing bone formation in 8-week-old (n=4) WT (+/+) (Upper panel) and Ercc1−/Δmice (Lower panel). Scale bar, 20μm. Bone formation rate (BFR) was calculated and presented in right panel. (B) TRAP staining of tibia sections of 8-week-old (n=4) WT (+/+) (Upper panels) and Ercc1−/Δmice (Lower panels). Scale bar, 50μm. The former animals displayed a significant increase in osteoclast surface/bone surface (%) and number of osteoclasts per bone perimeter (Oc.N/B.pm) (right panel, n=4). (C) TRAP staining of tibia sections of 2-week-old WT (+/+) and Ercc1−/− mice. Scale bar, 100μm. Osteoclast surface/bone surface (%) and Oc.N/B.pm were calculated for these animals (right panel, n=4) (D) TRAP staining of pBMMs of WT (+/+) and Ercc1−/Δmice cultured in osteoclastogenic medium in vitro (n=5). Scale bar, 50μm. (E) Quantitative RT-PCR analyses for mRNA levels of osteoclastic differentiation markers in pBMMs from WT and Ercc1−/Δmice (n=3). (F) pBMMs from WT and Ercc1−/Δmice were cultured on bovine cortical bone slices in osteoclastogenic medium for 15 days and stained for toluidine blue to visualize the resorption pits, the number of which in both animals were calculated and presented in the right panel (n=3). Scale bar, 50μm. The experiments in A–C and G were performed three times independently, and representative data are shown. All values are shown as mean ± SEM. **p < 0.01.
Ercc1−/− mice have a short life span and die before 4 weeks of age (9). To determine whether ERCC1 deficiency affects bone homeostasis in adult mice, we studied Ercc1 hypomorphic (Ercc1−/δ) mice. This strain harbors one knock-out and one mutant allele of Ercc1 and express approximately 5% of the normal level of ERCC1 and XPF proteins, leading to a lifespan of 7 months (10,25). μCT analysis of the lumbar vertebra (Fig. 1B) and femurs (Suppl Fig. 1) of age- and gender-matched WT and Ercc1−/Δmice demonstrated that Ercc1−/Δmice also develop osteoporosis. Histomorphometric analysis of the vertebrae of adult 8-week-old Ercc1−/Δmice, which do not have overt symptoms of progeria, revealed a 30% reduction in BV/TV compared to WT littermates. This was due to reduced trabecular thickness, but not number (Fig. 1B, Top). At 22 weeks of age, when Ercc1−/Δmice show severe progeroid symptoms, they had a >60% reduction of BV/TV due to a significantly reduced trabecular thickness and number and an increased trabecular space (Fig. 1B, Bottom). These results demonstrate that osteoporosis is a progressive process in Ercc1−/Δmice. Osteoporosis in Ercc1−/Δmice was confirmed by H&E staining of tibia and vertebrae (Fig. 1D) as well as radiographic examination of vertebrae (Fig. 1E). The similar osteoporotic phenotypes observed in both Ercc1−/− and Ercc1−/δdemonstrate that ERCC1-XPF-dependent DNA repair is required for maintaining normal bone homeostasis.
ERCC1 deficiency leads to reduced bone formation and enhanced osteoclastogenesis
The steady state of bone homeostasis is determined by two tightly coupled, but opposing biological processes: bone formation and bone resorption (26). Immunoblot analysis revealed that ERCC1 is expressed in both osteoblastic and osteoclastic cells of WT mice. (Suppl. Fig. 2A to C). To first determine if ERCC1 deficiency affects bone formation, dynamic histomorphometric analysis was performed by calcein double-labeling of 8-week-old Ercc1−/Δmice and WT littermates, to measure new bone matrix deposition. Ercc1−/Δmice had a significantly reduced rate of bone formation compared to WT littermates (Fig. 2A). This is consistent with their reduced Ob.N/B.pm compared to WT littermates (Fig. 1D, right). Next, we asked if ERCC1 deficiency affects bone resorption (osteoclastogenesis). The tibiae of 8-week-old WT and Ercc1−/Δmice were stained for the osteoclastic marker tartrate-resistant acid phosphatase (TRAP). Ercc1−/Δmice exhibited significantly increased TRAP staining throughout the tibiae, compared to WT mice (Fig. 2B, left). This is consistent with the increased Oc.S/BSand Oc.N/BPm in the spongiosa of Ercc1−/Δtibiae compared to WT (Fig. 2B, right). Increased osteoclastogenesis, Oc.S/BS and Oc.N/BPm were also observed in Ercc1−/− mice. Taken together, these results demonstrate that there is uncoupling of bone formation and resorption, with the latter being enhanced in ERCC1-deficient mice.
Osteoclast progenitor primary bone marrow monocytes (pBMMs) were isolated from the bone marrow of Ercc1−/Δand WT littermates. The pBMMs were induced to undergo osteoclastogenesis ex vivo by exposing them to osteoclastogenic media (27). TRAP positive (TRAP+) multinucleated cells (defined as those having 3 or more nuclei per cell) were counted as mature osteoclasts. Ercc1−/Δcultures contained a significantly greater number of osteoclasts than WT cultures (Fig. 2D). Further, Ercc1−/ΔpBMMs exhibited enhanced mRNA expression of osteoclast differentiation markers, such as cathepsin K (CTSK), nuclear factor of activated T-cells, cytoplasmic C 1 (NFATC1), receptor activator of nuclear factor κB (RANK) and TRAP compared to WT pBMMs (Fig. 2E). Finally, Ercc1−/ΔpBMMs displayed a significantly greater capacity to resorb bovine bone in vitro compared to WT pBMMs (Fig. 2F). This demonstrates that ERCC1-deficient pBMMs are more prone to osteoclastogenesis via a cell autonomous mechanism.
ERCC1 deficiency compromises osteoblastic differentiation
Next we asked if defects in osteoblast lineages required for bone deposition also contribute to osteoporosis in DNA repair-deficient ERCC1 mice. We measured mRNA expression of osteoblastic markers in vertebrae from 5-month-old Ercc1−/Δand WT mice. Expression of Osterix (Osx), a transcription factor required for osteoblastic differentiation and bone sialoprotein (Bsp), a bone extracellular matrix glycoprotein (26), were significantly reduced in Ercc1−/Δmice compared to age-matched WT mice (Fig. 3A). This suggests that DNA repair deficiency affects osteoblast differentiation.
Fig. 3. ERCC1 deficiency leads to atrophy of mesenchymal stem cells and compromises osteoblastic differentiation.

(A) qRT-PCR analyses for expression of osteoblast markers Osx and Bsp in vertebrae extraction of 5-month-old (n=4) WT (+/+) and Ercc1−/Δmice. (B) Bone marrow CFU-F assays (hematoxylin staining) on bone marrow cells isolated from WT (+/+) mice and Ercc1−/Δlittermates. The CFU-F colonies were quantified (n=3). (C) Bone marrow CFU-ALP staining on bone marrow cells isolated from 8-week-old WT (+/+) and Ercc1−/Δlittermates. The CFU-ALP colonies were quantified (right panel, n=3). (D) Bone marrow CFU-OB assays (von-Kossa staining) on bone marrow cells isolated from 8-week-old WT (+/+) and Ercc1−/Δlittermates. Scale bar, 100μm. (E) qRT-PCR analysis of expression of osteoblast markers in adherent BMSCs of isolated from WT (+/+) mice and Ercc1−/− littermates (n=3). (F) ALP staining of WT (+/+) and Ercc1−/− BMSCs under osteogenic induction conditions for the indicated time periods (n=3). All experiments were performed three times independently, and representative data are shown. All values are shown as mean ± SEM. * p < 0.05, ** p < 0.01, and *** p < 0.001.
ERCC1-deficiency has been reported to reduce hematopoietic reserves (13). Thus, we hypothesized that there would be a reduced number of osteoblastic progenitor cells in ERCC1-deficient mice. Osteoblastic progenitors are a clonogenic subset of adherent BMSCs, known as colony-forming unit fibroblasts (CFU-Fs). Indeed, BM of Ercc1−/Δmice contained significantly fewer CFU-Fs than that of WT mice (Fig. 3B). Further, BM of Ercc1−/Δmice contained significantly reduced number of osteogenic alkaline phosphatase positive colonies (CFU-ALP+) than WT littermates (Fig. 3C). Cultures of BM cells from WT mice spontaneously formed mineralized nodules, mimicking bone formation in vitro, whereas the BM cells isolated from Ercc1−/Δmice were defective (Fig. 3D). These results demonstrate that there is a reduced number of osteoblastic progenitor cells in the bone marrow of DNA repair-deficient Ercc1 mutant mice.
We next asked if this was due to failure of BMSCs to differentiate towards osteoblastic lineages. BMSCs were isolated from Ercc1−/Δand WT mice, plated at the same density, and cultured in osteoblastic differentiation media for up to 3 weeks. At weekly time points, cells were harvested and expression of several osteoblastic markers was measured by qRT-PCR. Expression of Osx, Alp and Atf4 and Col1 were significantly reduced in BMSCs of Ercc1−/Δmice compared to WT mice, at least at one time point (Fig. 3E). Furthermore, ALP staining was dramatically reduced in differentiated Ercc1−/ΔBMSCs cultures after 2 and 3 weeks of osteogenic induction (Fig. 3F). These results demonstrate that osteoblastic differentiation of Ercc1−/Δ BMSCs is severely compromised, which likely contributes to the reduced number of osteoblastic progenitor cells in Ercc1−/Δ BMSC population (Figs. 3B to 3D).
ERCC1 deficiency leads to persistent DNA damage and cellular senescence, of primary osteoblasts and BMSCs
ERCC1 plays an essential role in DNA repair. Thus, we predicted that ERCC1 deficiency leads to the accumulation of DNA damage in bone tissues. ATM is a proximal effector of DNA damage, in particular DSBs (28). Upon its activation, ATM phosphorylates several downstream substrates, including H2AX, a nucleosomal histone variant, to facilitate checkpoint activation and DNA repair. Phosphorylated H2AX (γ-H2AX) promptly localizes to DSBs and forms distinct foci, a characteristic feature of persistent DNA damage and cellular senescence (29). Ercc1−/− primary osteoblasts exhibited a greater number and more distinctγ-H2AX foci than WT cells (Fig. 4A). There was also increased γ-H2AX immunostaining in cells lining bone surfaces in Ercc1−/Δmice than WT controls (Fig. 4B). Furthermore, there was increased immunohistochemical staining for phosphorylated ATM substrate in bone surface lining cells, indicative of activated DNA damage response (30) in Ercc1−/Δbone tissues compared to WT animals (Fig. 4C). These data support the conclusion that ERCC1 deficiency results in persistent DNA damage in skeletal tissues.
Fig. 4. ERCC1 deficiency leads to increased DNA damage and cellular senescence in osteoblastic cells.

(A) Immunofluorescent staining for γ-H2AX in cultures of WT (+/+) and Ercc1−/− primary calvarial osteoblasts. Scale bar, 20μm. (B) Staining of γ-H2AX in tibias from 8-week-old WT (+/+) mice and Ercc1−/Δlittermates. Scale bar, 50μm. (C) Staining for p-ATM in tibias of 8-week-old WT (+/+) mice and Ercc1−/Δlittermates. Scale bar, 100μm. (D) Staining for p16INK4A in tibias of 2-week-old WT (+/+) and Ercc1−/− mice. Scale bar, 50μm. (E) Ki67 staining of tibias from 8-week-old WT (+/+) and Ercc1−/Δlittermates. Scale bar, 50μm. (F) Western blot analysis demonstrating expression of cyclin D1 in vertebrae extracts of 5-month-old WT (+/+) mice and Ercc1−/Δlittermates. (G) Population doubling of WT (+/+) and Ercc1−/− primary calvarial osteoblasts. (H) Ki67 staining of primary calvarial osteoblasts isolated from 2-week-old WT (+/+) mice and Ercc1−/− littermates at passage 3 (Upper panel) and passage 6 (Lower panel). The percentage of Ki67 positive cells was quantified (Right panels). Scale bar, 100μm. (I) SA-β-gal staining of BMSCs from 4-week-old WT (+/+) mice and Ercc1−/Δlittermates at passage 2. Scale bar, 50μm. The percentage of positive cells was quantified (Right panel, n=4). All experiments were performed three times independently, and representative data are shown. The values are shown as mean ± SEM. *** p < 0.001.
Persistent DNA damage can drive premature replicative senescence of cells (29,31). To test whether osteoblastic cells in Ercc1−/Δmice undergo premature senescence, we examined expression of several senescence markers in bone tissues. As shown by immunohistochemical staining of 8-week-old tibias of Ercc1−/Δand WT mice, the bone tissues of Ercc1−/Δmice displayed increased expression of p16INK4A (Fig. 4D), a cyclin-dependent kinase inhibitor associated with cellular senescence (32). In addition, expression of Ki67, a marker of cell proliferation (33), was reduced in Ercc1−/Δmice compared to WT animals (Fig. 4E). Finally, Western blot analysis revealed that bone tissues of Ercc1−/Δmice had dramatically decreased cyclin D1 expression compared to WT animals (Fig. 4F). Together these data demonstrate increased cellular senescence and reduced cell proliferation in bone tissues and the related cells of Ercc1−/Δmice.
To confirm these in vivo observations, the proliferation of primary osteoblasts and BMSCs isolated from WT and Ercc1−/− mice was measured in vitro. Despite being plated at the same density, Ercc1−/−osteoblasts had significantly reduced cell number than their WT counterparts at each passage (Fig. 4G). At passage 4, Ercc1−/− osteoblasts stopped proliferating (Fig. 4G) and acquired morphological features of senescence including enlarged cell bodies and nuclei (data not shown), while WT osteoblasts continued to proliferate. At passage 3, 7.4±0.4% Ercc1−/− primary osteoblasts stained positively for the proliferation marker Ki67 compared to 28.4±1.5% of WT cells. At passage 6, there were no Ki67 positive cells in the Ercc1−/− cultures, whereas 12.9±1.8 of WT primary osteoblasts stained positively (Fig. 4H). In accordance, there was an 8-fold increase in the number of Ercc1−/− pBMSCs that stained positively for senescence-associated β-galactosidase (SA β-Gal) compared to WT pBMSCs (Fig. 4I). A similar extent of increased SA β-Gal staining was observed in Ercc1−/− primary osteoblasts compared to WT primary osteoblasts (data not shown). These data demonstrate that DNA repair deficient osteoblasts and BMSCs senesce prematurely. In total, the data support the conclusion that premature senescence of osteoblastic progenitors, in addition to differentiation defects, contribute to osteoporosis in ERCC1-deficient mice.
ERCC1 deficiency triggers a senescence-associated secretory phenotype (SASP) in BMSCs and osteoblasts, creating an inflammatory microenvironment favoring osteoclastogenesis
Persistent DNA damage signaling promotes secretion of senescence-associated inflammatory cytokines, termed senescence-associated secretory phenotype (SASP) characterized by substantial IL-6 secretion (29). Since the osteoblastic lineages of ERCC1-deficient mice exhibited persistent DNA damage, we hypothesized that this induces SASP, thereby creating an inflammatory microenvironment in the bone. In support of this, Ercc1−/ΔBMSCs have significantly increased IL-6 mRNA expression as measured by qRT-PCR (Suppl. Fig. 3A). Further, these cells also secreted a greater level of IL-6 compared to WT counterparts (Fig. 5A). Consistently, the level of IL-6 was also elevated by approximately 1000-fold in the serum of Ercc1−/Δmice compared to WT animals. Since IL-6 is osteoclastogenic (34), we also examined other inflammatory cytokines that regulate osteoclast formation, including TNFα (Tumor necrosis factor α), RANKL and OPG (Osteoprotegerin), a RANKL antagonist (35). RT-PCR analysis on vertebra of 8-week-old Ercc1−/− mice revealed a more than 2-fold up-regulation of TNFα mRNA expression (Suppl. Fig. 3B). Consistently, elevated TNFα secretion was detected in both serum of Ercc1−/Δmice and conditioned medium of Ercc1−/ΔBMSCs compared to WT counterparts (Fig. 5B and C). In addition, expression of RANKL in Ercc1−/Δvertebrae was increased >4-fold, whereas OPG expression was reduced by 70% compared to WT animals (Suppl. Fig. 3B), resulting in an 11-fold increase in the ratio of RANKL to OPG, an indicator for osteoclastogenic potential of BMSCs. The elevated mRNA expression of RANKL was also observed in Ercc1−/Δprimary osteoblasts compared to WT cells as measured by qRT-PCR (Suppl. Fig. 3C). Consistent with that, Ercc1−/− mice exhibited a 2.8-fold elevation of serum RANKL and a 30% reduction of serum OPG compared to WT animals (Fig. 5B). Finally, lentiviral transduction of Ercc1−/Δ BMSCs with murine Ercc1 dramatically attenuated IL-6 and TNFα secretion to levels that are comparable to WT BMSCs (Fig. 5C), supporting the conclusion that failure to repair DNA damage drives cell senescence and SASP in osteoblastic cell lineages.
Fig. 5. ERCC1 deficiency triggers SASP and induces an inflammatory microenvironment favoring bone resorption.

(A) ELISA analysis for IL-6 secretion in the conditioned medium of BMSCs of WT (+/+) and Ercc1−/Δmice. BMSCs were cultured with osteogenic media for 0 or 7 days, respectively. ** p < 0.01 by Student’s t-test. (B) ELISA analyses for the serum levels of IL-6, TNFα, RANKL and OPG of WT (+/+) and Ercc1-deficient mice (n=9). * p < 0.05, ** p < 0.01, and ** p < 0.001 by Student’s t-test. (C) ELISA analysis for IL-6 and TNF-α secretion from WT (+/+) and Ercc1−/ΔBMSCs that were transduced with lentiviruses expressing either empty vector (EV) or Ercc1. * p < 0.05 and ** p < 0.01 compared with +/+ GFP, and # p < 0.05 compared with Ercc1−/Δcells transduced with EV. (D) pBMMs-BMSCs co-culture assays. pBMSCs from WT (+/+) and Ercc1−/Δmice were transduced with lentiviruses expressing either empty vector (EV) or Flag-mErcc1. Then the infected BMSCs were co-cultured with WT (+/+) BMMs in osteoclastogenic differentiation media for 7–8 days prior to TRAP staining for TRAP+ mononuclear cells (MNCs). Scale bar, 50μm. ** p < 0.01 compared to WT (+/+) BMSCs with EV expression and # p < 0.05 compared to Ercc1−/ΔBMSCs with EV expression. All experiments were performed three times independently, and representative data are shown. All values are shown as mean ± SEM.
To determine if cellular senescence and SASP contribute to osteoclastogenesis, primary murine WT BMMs were co-cultured with either primary WT, or Ercc1−/δ, BMSCs for 7 days. TRAP staining revealed that Ercc1−/ΔBMSCs induced formation of a significantly greater number of osteoclasts (Fig. 5D) and more nuclei per osteoclast (data not shown) than WT BMSCs did, despite the fact that there were fewer Ercc1−/ΔBMSCs (data not show). Re-expression of murine Ercc1 in the Ercc1−/ΔBMSCs reduced their enhanced ability to induce osteoclastogenesis (Fig. 5D). These data provide direct experimental evidence that BMSCs from ERCC1-deficient mice also promote osteoclastogenesis through a non-cell autonomous mechanism.
NF-κB is activated in osteoblasts and osteoclasts from DNA repair-deficient mice
Having demonstrated the cellular mechanisms by which unrepaired DNA damage promotes premature osteoporosis, we next examined the underlying molecular events. Induction of NF-κB signaling represents a common molecular change in various tissues and cells of aged animals compared with young animals, such as liver, brain, kidney, bone, and so on (36–38). pBMSCs from aged (28-month-old) WT mice had enhanced NF-κB activity, demonstrated by increased levels of phospho-p65, phospho-IκBα and phospho-IKKα/β in cell lysates after TNFα treatment (Suppl Fig. 4A). In addition there was enhanced immunostaining of nuclear p65 in these cells either in the presence of absence of TNFα treatment (Suppl Fig. 4B) compared to pBMSCs from 2-week-old mice. Similarly, primary osteoblasts from progeroid Ercc1−/− mice displayed enhanced phosphorylation of IκBα (Fig. 6A) as well as increased nuclear localization and levels of the p65 subunit of NF-κB (Fig. 6B) compared to cells from WT littermates. Furthermore, an increase in the phosphorylated-p65 protein level was detected in pBMSCs from Ercc1−/− mice compared to WT BMSCs (Fig. 6C).
Fig. 6. NF-κB is activated in primary osteoblasts, BMSCs and primary bone marrow macrophages from osteoporotic ERCC1-deficient mice.

(A) Western blot analysis demonstrating protein levels of IκBα and phospho-IκBα in primary calvarial osteoblasts isolated from 1-week-old WT (+/+) and Ercc1−/− mice with osteogenic induction for either 0 or 7 days, respectively. β-actin served as a loading control. (B) Immunostaining of p65 in primary calvarial osteoblasts of 1-week-old WT (+/+) and Ercc1−/− mice. Cells were treated with TNFα for either 0 (Upper panel) or 60 minutes (Lower panel). Scale bar, 50μm. (C) Western blot analysis demonstrating protein levels of phospho- and total p65 in WT (+/+) and Ercc1−/Δprimary BMSCs with 7-day osteogenic induction. β-actin served as a loading control. (D) Western blot analysis demonstrating protein levels of phospho- (S85), total IKKγ as well as ATM in WT (+/+) and Ercc1−/ΔpBMSCs. (E) Western blot analysis demonstrating protein levels of phospho- (S85) and total IKKγ in WT (+/+) and Ercc1−/Δprimary BMMs. (F) Western blot analysis demonstrating protein levels of phospho- and total p65 in WT (+/+) and Ercc1−/Δprimary BMMs. The cells were cultured in proliferation medium for 3 days, and then treated with RANKL for the indicated time periods prior to being harvested. β-actin served as a loading control. All experiments were performed three times independently, and representative data are shown.
Next, we determined the molecular changes responsible for the increased NF-κB signaling in ERCC1 deficient mice. Given IKKs are the upstream kinases responsible for phosphorylation of IκBα and consequent activation of NF-κB signaling, we measured the levels of phosphorylated and total IKKα, β, and γ isoforms. Interestingly, both phosphorylated and total IKKα and β were similar in Ercc1-deficient and WT cells (data not shown). However, pBMSCs from Ercc1−/Δ mice displayed increased levels of phosphorylated IKKγ at serine 85 compared to WT counterparts (Fig. 6D). Consistent with these findings, pBMSCs (Fig. 6D) and bone tissues (Fig. 4C) from Ercc1-deficient mice displayed increased levels and activity of ATM, the upstream kinase that phosphorylates IKKγ at serine 85 in response to genotoxic stress. Finally, enhanced phosphorylation of IKKγ at serine 85 (Fig. 6E) leading to elevated baseline and RANKL-induced levels of NF-κB signaling activity (Fig. 6F) were observed in Ercc1−/ΔBMMs. Taken together, these data demonstrate that ERCC1 deficiency leads to increased NF-κB activity in both osteoblastic and osteoclastic cells, potentially through an ATM-dependent increase in IKKγ activity in response to unrepaired endogenous DNA damage.
Heterozygous deletion of the p65 subunit rescues osteoporosis
Having demonstrated increased NF-κB activity in both osteoblasts and osteoclasts of ERCC1-deficient mice, we next asked if this activity contributes to their skeletal defects. First, we observed that p65 overexpression in stable murine osteoblastic cell line MC4 impaired their differentiation in response to ascorbic acid, as reflected by a significant reduction in expression of osteoblast markers Osterix, alkaline phosphatase (Alp) and Osteocalcin (OCN) (Suppl Fig. 5A), as well as reduced alkaline phosphatase staining (Suppl Fig. 5B). Next, we bred ERCC1-deficient mice that were haploinsufficient for the p65 subunit of NF-κB, a strategy previously used to characterize the role of NF-κB in Duchenne muscular dystrophy (39). μQCT on tibia and lumbar vertebrae of age- and gender-matched WT, Ercc1−/Δand Ercc1−/Δ;p65+/− mice revealed that p65 haploinsufficiency partially but significantly rescued osteoporosis in Ercc1−/Δmice (Figs. 7A, B & C). Specifically, Ercc1−/Δ;p65+/− mice showed significantly greater BV/TV compared to Ercc1−/Δmice, which represented 41.7% (vertebrae) or 59.8% (Tibia) rescue of BV/TV to normal level of the WT mice (Fig. 7B). The Ercc1−/Δ;p65+/− mice also showed significantly increased trabecular number and thickness and reduced trabecular space compared to Ercc1−/Δmice (Fig. 7B). Further, histomorphometric analysis demonstrated that p65 haploinsufficiency largely corrected the decrease in Ob.N/B.Pm and enhanced osteoclastogenesis (increased Oc.N/B.Pm and Oc surface) seen in Ercc1−/Δmice (Fig. 7C). These results demonstrate that genetic reduction of NF-κB signaling attenuates osteoporosis in a murine model of accelerated aging.
Fig. 7. Heterozygous deletion of the p65 subunit rescues osteoporosis.


(A–C) μQCT images (A) and histomorphometric analyses (B & C) on tibia (Upper panels) and lumbar vertebrae (Lower panels) from 15-week-old WT (+/+), Ercc1−/Δ, and Ercc1−/Δ;p65+/− age-matched mice (n=3). (D) Visual (left) and quantitative (right) presentations of senescence-associated β-galactosidase staining of BMSCs isolated from 15-week-old WT (+/+), Ercc1−/Δand Ercc1−/Δp65+/− mice at passage 2 (n=4). Scale bar, 100μm. (E) The effects of p65 haploinsufficiency on serum levels of IL-6 (right) and TNFα (left) of 10-week- old Ercc1−/Δmice (n=3), as determined by ELISA assays. (F) Bone marrow CFU-F assays for WT (+/+) and Ercc1−/Δ, Ercc1−/Δp65+/− mice. The number of nodules was quantified (Right panel, n=3). (G) Bone marrow CFU-ALP assays for WT (+/+), Ercc1−/Δ, and Ercc1−/Δp65+/− mice. The number of nodules was quantified (Right panel, n=4). (H) ALP staining of BMSCs isolated from 3-week-old WT (+/+), Ercc1−/−, Ercc1−/− p65+/− mice with osteogenic induction for either 7 or 14 days (n=3). (I) Visual and quantitative presentations of TRAP staining of pBMMs isolated from 15-week-old WT (+/+), Ercc1−/Δ, and Ercc1−/Δp65+/− mice. The cells were cultured in osteoclastogenic medium for 6 days (n=5). Scale bar, 50μm. The experiments were performed at least three times independently, and representative data are shown. All values are shown as mean ± SEM. * p < 0.05, ** p < 0.01, *** p < 0.001 compared with WT and # p < 0.05, ## p < 0.01 compared with Ercc1−/Δ.
To elucidate how p65 haploinsufficiency rescues osteoporosis of ERCC1-deficient mice, we measured senescence, SASP and bone-specific endpoints in cells isolated from age-matched WT, Ercc1−/Δand Ercc1−/Δ;p65+/− mice. p65 haploinsufficiency abolished cellular senescence of BMSCs, as demonstrated by SAβ-Gal staining (Fig. 7D). However, there was nodifference in Ki67 staining between Ercc1−/− and Ercc1−/−;p65+/− BMSCs (Suppl Fig. 6A). p65 haploinsufficiency completely reverted the elevated serum levels of SASP factors IL-6 and TNFα to the levels that were either comparable to, or even lower than, those of WT animals (Fig. 7E). Further, Ercc1−/Δ;p65+/− BMSCs formed significantly more CFU-Fs and CFU-ALP+ colonies than those of Ercc1−/Δmice, although still significantly less colonies than WT BMSCs (Figs. 7F and G). Consistently, alkaline phosphatase staining showed that p65 hapoinsufficiency significantly rescued impaired osteoblastic differentiation of the Ercc1−/− BMSCs (Fig. 7H). These data indicate that p65 haploinsufficiency partially rescued the inhibitory effects of ERCC1 deficiency on osteoblastic cell lineage. Finally, we observed that NF-κB activation also contributes to the enhanced osteoclastogenesis in Ercc1−/Δmice since TRAP staining of BMMs isolated from Ercc1−/Δ;p65+/− mice revealed reduced osteoclast formation compared to Ercc1−/ΔBMMs (Fig. 7I). Taken together, these results support a model where NF-κB mediates osteoporosis in the ERCC1-deficient mice by driving cell autonomous changes that promote increased bone resorption and decreased bone formation.
Pharmacologic inhibition of NF-κB activation rescues osteoporosis
We next asked if pharmacologic inhibition of NF-κB attenuates osteoporosis in ERCC1-deficient mice. IKKiVII is a small molecule inhibitor of the upstream kinase that activates NF-κB (IKK) (40). Addition of IKKiVII to cultures of Ercc1−/ΔBMSCs dramatically reduced the levels of phospho-p65, which represents activated NF-κB (Suppl Fig. 6B). IKKiVII treatment partially, but significantly, reduced cellular senescence in Ercc1−/ΔBMSCs in a dose-dependent manner (Fig. 8A). In addition, IKKiVII significantly restored expression of osteoblastic markers including Osx, Runx2, and Ocn, in Ercc1−/ΔBMSCs in a dose-dependent manner (Fig. 8B). Expression was fully corrected to, and even beyond, the level of WT cells treated with vehicle only (DMSO). Furthermore, IKKiVII abolished IL-6 secretion from Ercc1−/ΔBMSCs (Fig. 8C). IKKiVII treatment also blunted the enhanced capacity of Ercc1−/ΔBMSCs to drive osteoclatogenesis of WT pBMMs (Fig. 8D). Finally, the inhibitor also reduced enhanced osteoclastic differentiation of Ercc1−/ΔBMMs (Fig. 8E). Collectively, these data provide strong experimental evidence that NF-κB is, in part, responsible for the cellular senescence, compromised osteoblastic differentiation, as well as increased inflammatory cytokine secretion in BMSCs that drive osteoclastogenesis in DNA repair-deficient ERCC1 mice, via both cell-autonomous and cell-non-autonomous mechanisms. Importantly, the data also support the conclusion that inhibition of NF-κB with small molecules will be efficacious for preventing and/or attenuating osteoporosis that results from progeria, old age in the general population, and secondary to radiation therapy.
Fig. 8. Pharmacological inhibition of the NF-κB activation rescues the osteoblast and osteoclast defects of ERCC1-deficient mice.

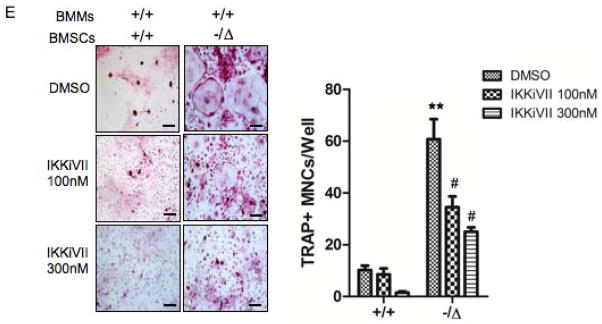
(A) Senescence-associated β-galactosidase staining of WT (+/+) and Ercc1−/ΔBMSCs that were treated with DMSO (vehicle), or the inhibitor of NF-κB activation IKKiVII (100nM, or 300nM) for 3 days. The percent of positive cells was counted (Right panel, n=4). Scale bar, 100μm. (B) qRT-PCR analysis of expression of osteoblastic markers in WT (+/+) and Ercc1−/ΔBMSCs. The cells were cultured in osteogenic media with DMSO, or 100nM, or 300nM IKKiVII for 7 days before harvesting for RNA isolation (n=4). (C) ELISA analysis showing IL-6 secretion from BMSCs isolated from 8-week-old WT (+/+) and Ercc1−/Δmice. The cells were treated with DMSO or 100nM or 300nM IKKiVII for 3 days. Conditioned media was then harvested for ELISA analysis (n=4). (D) TRAP staining of WT (+/+) and Ercc1−/ΔBMMs. The cells were cultured in osteoclastogenic media with DMSO or 100nM or 300nM IKKiVII treatment for 6 days prior to TRAP staining (n=5). Scale bar, 50μm. (E) pBMMs-BMSCs co-culture assays. Primary BMSCs from 4-week-old WT (+/+) and Ercc1−/Δmice were co-cultured with WT (+/+) BMMs for 7–8 days with either DMSO or 100nM or 300nM IKKiVII. The number of TRAP+ MNCs per well was counted (n=5). Scale bar, 100μm. All experiments were performed three times independently, and representative data are shown. All values are shown as mean ± SEM. * p < 0.05 compared to +/+ with DMSO, # p < 0.05 and ## p < 0.01 compared to Ercc1−/Δwith DMSO.
Discussion
Stochastic damage to cellular macromolecules and organelles, including DNA damage, is thought to be a driving force behind ageing and associated degenerative changes. However, how cellular damage drives degenerative diseases is still poorly understood. To address this, we investigated the mechanism(s) underlying the onset and progression of osteoporosis in mice where the primary defect is failure to repair DNA damage, leading to accelerated aging. Here we demonstrate that the mice spontaneously develop osteoporosis as a consequence of damage to DNA. This occurs as a consequence of both cell-autonomous and non-autonomous mechanisms affecting multiple cell types. ERCC1-deficiency leads to persistent DNA damage that causes premature cellular senescence and reduced proliferation of cells of the osteoblastic lineage. This in turn results in a decline in the number of bone marrow mesenchymal progenitors and/or osteoblastic progenitor cells in the bone marrow. Consistent with this, we previously observed premature exhaustion of hematopoietic stem cells (13) and loss of muscle-derived stem/progenitor cells in progeroid ERCC1-deficient and aged WT mice (41). This establishes exhaustion of multiple stem/progenitor cell populations with natural and accelerated ageing.
Osteoclast lineages were also affected in the ERCC1-deficient mice. Our studies suggest that both cell-autonomous and non-autonomous mechanisms drive increased osteoclatogenesis in progeroid ERCC1-deficient mice. In vitro, Ercc1−/ΔBMM cultures formed a significantly greater number of osteoblasts with a greater bone resorbing capacity than WT pBMMs when induced to differentiate (Figs. 2D & F), consistent with a cell autonomous mechanism. In addition, ERCC1-deficienct BMSCs and osteoblasts display a senescence-associated secretory phenotype (SASP), which induces an inflammatory bone microenvironment favoring osteoclastogenesis, suggesting a cell non-autonomous mechanism.
In our study, ERCC1-deficient BMSCs and primary osteoblasts exhibited persistent DNA damage (Figs. 4A to 4C) and significantly increased gene and/or protein expression of not only IL-6 (Suppl. Fig. 3A, Figs. 5A & 5B) and TNFα (Suppl. Fig. 3B, Figs. 5B & 5C), but also RANKL, a potent osteoclastogenic inflammatory cytokine (Suppl. Figs. 3B, 3C, and Figs. 5B). In contrast, both gene expression and protein secretion of OPG (Suppl. Fig. 3B and Fig. 5B), an inhibitor for osteoclastogenesis, were decreased in ERCC1-deficient mice, leading to an increase in the ratio of RANKL to OPG, an indicator for osteoclastogenic potential of BMSCs, in these mice compared to normal mice. Consequently, Ercc1−/ΔBMSCs exhibited enhanced osteoclastic induction capacity than WT BMSCs (Fig. 5D & 8E). These results reveal that RANKL may be a novel SASP factor. Further, it was observed that either lentiviral transduction of ERCC1 or inhibition of NF-κB signaling, via both genetic and pharmacologic approaches, in ERCC1-deficient BMSCs reversed their heightened secretion of IL-6 and TNFα as well as their enhanced capacity to induce osteoclast formation from BMMs (Figs. 5C, 5D, 7C, 7E, 8C & 8E). Taken together, these results provide critical evidence for a novel role of SASP in osteoblastic regulation of osteoclastogenesis and in driving ageing-related osteoporosis. Further, since an increase in senescent cells is a hallmark of many tissues, including bone, with either natural or accelerated aging (42,43), our data suggest that SASP is an important molecular mechanism responsible for uncoupling of decreased bone formation and enhanced bone resorption in ageing-related osteoporosis. Supporting this, induction of mRNA expression of several pro-inflammatory response genes was also detected in bone tissues from telomerase-deficient mice, another model of premature ageing and osteoporosis (44). Furthermore, a recent report revealed that circulating osteoblastic cells found in the peripheral blood from aged postmenopausal women express several osteoclastogenic inflammatory factors that may contribute to the increased bone resorption in these patients (45).
NF-κB activity was induced in both osteoblast and osteoclast lineages isolated from Ercc1-deficient mice. The up-regulation of NF-κB signaling is likely a direct consequence of DNA damage via ATM-dependent activation of the upstream kinase IKK, most likely IKKγ. We also observed increased protein levels (Fig. 6D) and activity of ATM (Fig. 4C) in both Ercc1-deficient osteoblastic and osteoclastic cells compared with WT cells. In addition, we detected an increase in phosphorylation of IKKγ at serine 85 (Figs. 6D & E), a direct target of ATM kinase in responsive to genotoxic stress (17), but not phosphorylation of IKKα/β (data not shown), in both Ercc1-deficient osteoblastic and osteoclastic cells compared with WT cells in vitro. NF-κB activation in turn promotes secretion of inflammatory cytokines, such as IL-6, RANKL, and TNFα into the bone microenvironment. These inflammatory factors also can feed-forward to activate NF-κB through a non-cell autonomous mechanism (27).
To determine the pathophysiological significance of elevated NF-κB signaling in osteoporosis, we generated Ercc1−/Δ;p65+/− mice and found that p65 haploinsufficiency significantly rescued the osteoporotic phenotype of Ercc1−/Δmice (Figs. 7A–C). Specifically, p65 haploinsufficiency rescued premature cellular senescence, reduced SASP and the osteoblastic progenitor cell number and impaired osteoblastic differentiation of ERCC1 deficient BMSCs (Figs. 7D–7H). Furthermore, IKKiVII, a small molecule inhibitor of IKK, rescued senescence (Fig. 8A) and osteoblastic differentiation (Fig. 8B), while attenuating SASP factor IL-6 secretion (Fig. 8C) and osteoclastogenesis (Fig. 8D, E) of Ercc1−/Δcells. Taken together, these data demonstrate that NF-κB signaling plays a pivotal role in mediating the effects of DNA damage on bone homeostasis via affecting both osteoblastic and osteoclastic activity.
The standard treatment for osteoporosis includes bisphosphonate and its derivatives that target osteoclasts to inhibit bone resorption without directly affecting bone formation. In addition, there are limited anabolic agents including PTHrP that restore bone loss (46). NF-κB signaling affects both osteoblastic and osteoclastic lineages. This indicates that inhibitors of NF-κB represent a novel class of drugs that offer the potential to both inhibit bone resorption and promote bone formation using a single agent for treating osteoporosis in the aged and in patients who experience genotoxic stress due to radiation treatment or genetic disorders caused by defective DNA repair.
Materials and methods
Mice
Ercc1−/−, Ercc1−/Δand p65+/− mice were described preciously (12,47,48). Ercc1−/−;p65+/− mice and Ercc1−/Δ;p65+/− mice were generated by crossing p65+/− mice with Ercc1+/− mice and further crossing them with Ercc1+/− or Ercc1+/mice. All mice were in an f1 C57Bl/6:FVB/n genetic background. All procedures involving animals were approved by the University of Pittsburgh Institutional Animal Care and Use Committee.
Cell cultures, in vitro analyses, and reagents
IKKiVII was purchased from Calbiochem. Cells (pObs, BMSCs and BMMs) were cultured with α-MEM containing 10% fetal bovine serum (FBS), 100units/ml penicillin and 100μg/ml streptomycin (P/S) (Invitrogen). Primary calvarial osteoblasts were isolated as previously described (49). Murine bone marrow cells were harvested from long bones and seeded into 100-mm culture dishes.
To induce osteoblastic differentiation in vitro, the cells were cultured in differentiation-inducing media containing 5mM β-glycerophosphate (Sigma-Aldrich), 50μM ascorbic acid (Sigma-Aldrich) for the indicated time periods. The media were changed every 3 days. After induction, the cells were fixed with 10% neutral buffered formalin and stained with an alkaline phosphatase staining kit according to the manufactuer’s protocol (Sigma-Aldrich). To detect biomineralization, the cells were induced in osteoblastic differentiation media for 3 weeks, subsequently fixed with neutral formalin and processed for von Kossa staining (50). For CFU-F assays, single cell suspensions of 1×106 nucleated bone marrow cells were seeded on 6-well plates in α-MEM containing 15% FBS for 14 days. Colonies containing 50 or more cells were counted. For CFU-ALP assays, we induced differentiation of BMSCs by osteogenic media as described above for 14 days followed by ALP staining.
To determine cell proliferation, primary osteoblasts were plated at 1×105 cells/well and cultured for 3 days. The cell number was calculated using a hemacytometer. The cells were then re-plated at 1×105 cells/well and counted 3 days later for each passage.
To induce osteoclast differentiation in vitro, non-adherent BMMs isolated from long bones were incubated with M-CSF (10ng/ml) for 3 days, then were induced to differentiate in media containing M-CSF (10ng/ml) and sRANKL (50ng/ml) for 4–8 days. Osteoclasts were identified as TRAP positive, multinucleated (>3 nuclei) cells. Co-culture experiments were performed as previously described (51). Briefly, primary BMSCs were seed into 96 well plates and cultured in α-MEM containing 10% FBS. BMMs were seeded on top of BMSCs. The media were supplemented with 10−8M 1,25 dihydroxy-vitamin D3. OCLs were identified by TRAP staining and counted.
Supplementary Material
Acknowledgments
We thank Dr. G. David Roodman (Indiana University) and Mr. Kenneth Patrene (University of Pittsburgh) for the bone μCT analyses, Dr. Tao Chen (University of Pittsburgh) for providing the lentiviral construct, as well as Matthew H. Pham and Andrew Stypula (University of Pittsburgh) for preparing histological sections. We thank the VA Pittsburgh Healthcare System for providing common facilities and the support of the Department of Veteran’s Affairs. This work is supported by NIH/NIDCR (RO1DE017439), NIH/NCI (R21CA161150), and the multiple myeloma research foundation (MMRF) tumor microenvironment award to H.O., R01AR055208 to H.C.B., AG033907, AR051456, NS058451, and AG024827 to P.D.R., ES016114, P30AG024827, P30CA047904 and Ellison Medical Foundation AG-NS-0303-05 to L.J.N, as well as the New Investigator Grant of Scoliosis Research Society (SRS) to Q.C. The opinions expressed are not those of the Department of Veteran’s Affairs or of the US Government.
Abbreviations list
- ATM
Ataxia telangiectasia mutated
- CFU-Fs
colony-forming unit fibroblasts
- ERCC1
Excision repair cross complementation group 1
- NF-κB
Nuclear Factor-KappaB
- OCL
osteoclast(s)
- OPG
Osteoprotegerin
- pBMM
primary bone marrow monocytes
- pBMSCs
primary bone marrow stromal cells
- pObs
primary osteoblasts (primary calvarial osteoblastic cells
- RANKL
Receptor activator of nuclear factor kappa-B ligand
- SA β-gal
senescence-associated β-galactosidase. SASP, senescence associated secretory phenotype
- TNFα
Tumor necrosis factor α
- XPF
Xeroderma pigmentosum group F
Footnotes
Disclosure Page
All authors state that they have no conflicts of interest.
Note: Supplementary information is available.
Authorship Contributions: Q.C. designed and performed the experiments, analyzed the data, prepared the figures, as well as wrote the manuscript. K.L. and H.C.B performed experiments. H.C.B provided critical suggestions for the experiments. A.R.R. and C.L.C. maintained the animal colonies of ERCC1 deficient mice, Ercc1−/−;p65+/− and Ercc1−/;p65+/− mice, as well as prepared the bone tissues. L.J.N. and P.D.R. provided critical suggestions to experimental design and data analysis as well as edited the manuscript. H.O. designed and supervised the study, analyzed the data, prepared the figures, wrote and edited the manuscript.
References
- 1.Rachner TD, Khosla S, Hofbauer LC. Osteoporosis: now and the future. Lancet. 2011;377(9773):1276–87. doi: 10.1016/S0140-6736(10)62349-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cheung CL, Xiao SM, Kung AW. Genetic epidemiology of age-related osteoporosis and its clinical applications. Nat Rev Rheumatol. 2010;6(9):507–17. doi: 10.1038/nrrheum.2010.106. [DOI] [PubMed] [Google Scholar]
- 3.Zhang W, Ou G, Hamrick M, Hill W, Borke J, Wenger K, Chutkan N, Yu J, Mi QS, Isales CM, Shi XM. Age-related changes in the osteogenic differentiation potential of mouse bone marrow stromal cells. J Bone Miner Res. 2008;23(7):1118–28. doi: 10.1359/JBMR.080304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stewart SA, Weinberg RA. Telomeres: cancer to human aging. Annu Rev Cell Dev Biol. 2006;22:531–57. doi: 10.1146/annurev.cellbio.22.010305.104518. [DOI] [PubMed] [Google Scholar]
- 5.Hoeijmakers JH. DNA damage, aging, and cancer. N Engl J Med. 2009;361(15):1475–85. doi: 10.1056/NEJMra0804615. [DOI] [PubMed] [Google Scholar]
- 6.Sijbers AM, van der Spek PJ, Odijk H, van den Berg J, van Duin M, Westerveld A, Jaspers NG, Bootsma D, Hoeijmakers JH. Mutational analysis of the human nucleotide excision repair gene ERCC1. Nucleic Acids Res. 1996;24(17):3370–80. doi: 10.1093/nar/24.17.3370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Niedernhofer LJ, Odijk H, Budzowska M, van Drunen E, Maas A, Theil AF, de Wit J, Jaspers NG, Beverloo HB, Hoeijmakers JH, Kanaar R. The structure-specific endonuclease Ercc1-Xpf is required to resolve DNA interstrand cross-link-induced double-strand breaks. Mol Cell Biol. 2004;24(13):5776–87. doi: 10.1128/MCB.24.13.5776-5787.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ahmad A, Robinson AR, Duensing A, van Drunen E, Beverloo HB, Weisberg DB, Hasty P, Hoeijmakers JH, Niedernhofer LJ. ERCC1-XPF endonuclease facilitates DNA double-strand break repair. Mol Cell Biol. 2008;28(16):5082–92. doi: 10.1128/MCB.00293-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.McWhir J, Selfridge J, Harrison DJ, Squires S, Melton DW. Mice with DNA repair gene (ERCC-1) deficiency have elevated levels of p53, liver nuclear abnormalities and die before weaning. Nat Genet. 1993;5(3):217–24. doi: 10.1038/ng1193-217. [DOI] [PubMed] [Google Scholar]
- 10.Weeda G, Donker I, de Wit J, Morreau H, Janssens R, Vissers CJ, Nigg A, van Steeg H, Bootsma D, Hoeijmakers JH. Disruption of mouse ERCC1 results in a novel repair syndrome with growth failure, nuclear abnormalities and senescence. Curr Biol. 1997;7(6):427–39. doi: 10.1016/s0960-9822(06)00190-4. [DOI] [PubMed] [Google Scholar]
- 11.Tian M, Jones DA, Smith M, Shinkura R, Alt FW. Deficiency in the nuclease activity of xeroderma pigmentosum G in mice leads to hypersensitivity to UV irradiation. Mol Cell Biol. 2004;24 (6):2237–42. doi: 10.1128/MCB.24.6.2237-2242.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Niedernhofer LJ, Garinis GA, Raams A, Lalai AS, Robinson AR, Appeldoorn E, Odijk H, Oostendorp R, Ahmad A, van Leeuwen W, Theil AF, Vermeulen W, van der Horst GT, Meinecke P, Kleijer WJ, Vijg J, Jaspers NG, Hoeijmakers JH. A new progeroid syndrome reveals that genotoxic stress suppresses the somatotroph axis. Nature. 2006;444(7122):1038–43. doi: 10.1038/nature05456. [DOI] [PubMed] [Google Scholar]
- 13.Prasher JM, Lalai AS, Heijmans-Antonissen C, Ploemacher RE, Hoeijmakers JH, Touw IP, Niedernhofer LJ. Reduced hematopoietic reserves in DNA interstrand crosslink repair-deficient Ercc1−/− mice. EMBO J. 2005;24(4):861–71. doi: 10.1038/sj.emboj.7600542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jaspers NG, Raams A, Silengo MC, Wijgers N, Niedernhofer LJ, Robinson AR, Giglia-Mari G, Hoogstraten D, Kleijer WJ, Hoeijmakers JH, Vermeulen W. First reported patient with human ERCC1 deficiency has cerebro-oculo-facio-skeletal syndrome with a mild defect in nucleotide excision repair and severe developmental failure. Am J Hum Genet. 2007;80(3):457–66. doi: 10.1086/512486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Miyamoto S. Nuclear initiated NF-kappaB signaling: NEMO and ATM take center stage. Cell Res. 21(1):116–30. doi: 10.1038/cr.2010.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wu ZH, Miyamoto S. Many faces of NF-kappaB signaling induced by genotoxic stress. J Mol Med (Berl) 2007;85(11):1187–202. doi: 10.1007/s00109-007-0227-9. [DOI] [PubMed] [Google Scholar]
- 17.Miyamoto S. Nuclear initiated NF-[kappa]B signaling: NEMO and ATM take center stage. Cell Res. 2011;21(1):116–130. doi: 10.1038/cr.2010.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hsu H, Xiong J, Goeddel DV. The TNF receptor 1-associated protein TRADD signals cell death and NF-kappa B activation. Cell. 1995;81(4):495–504. doi: 10.1016/0092-8674(95)90070-5. [DOI] [PubMed] [Google Scholar]
- 19.Siebenlist U, Franzoso G, Brown K. Structure, regulation and function of NF-kappa B. Annu Rev Cell Biol. 1994;10:405–55. doi: 10.1146/annurev.cb.10.110194.002201. [DOI] [PubMed] [Google Scholar]
- 20.Stancovski I, Baltimore D. NF-kappaB activation: the I kappaB kinase revealed? Cell. 1997;91 (3):299–302. doi: 10.1016/s0092-8674(00)80413-4. [DOI] [PubMed] [Google Scholar]
- 21.Baldwin AS., Jr The NF-kappa B and I kappa B proteins: new discoveries and insights. Annu Rev Immunol. 1996;14:649–83. doi: 10.1146/annurev.immunol.14.1.649. [DOI] [PubMed] [Google Scholar]
- 22.Chang J, Wang Z, Tang E, Fan Z, McCauley L, Franceschi R, Guan K, Krebsbach PH, Wang CY. Inhibition of osteoblastic bone formation by nuclear factor-kappaB. Nat Med. 2009;15 (6):682–9. doi: 10.1038/nm.1954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ruocco MG, Maeda S, Park JM, Lawrence T, Hsu LC, Cao Y, Schett G, Wagner EF, Karin M. I{kappa}B kinase (IKK){beta}, but not IKK{alpha}, is a critical mediator of osteoclast survival and is required for inflammation-induced bone loss. J Exp Med. 2005;201(10):1677–87. doi: 10.1084/jem.20042081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vaira S, Alhawagri M, Anwisye I, Kitaura H, Faccio R, Novack DV. RelA/p65 promotes osteoclast differentiation by blocking a RANKL-induced apoptotic JNK pathway in mice. J Clin Invest. 2008;118(6):2088–97. doi: 10.1172/JCI33392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dolle MET, Kuiper RV, Roodbergen M, Robinson J, Vlugt Sd, Wijnhoven SWP, Beems RB, Fonteyne Ldl, With Pd, Pluijm Ivd, Niedernhofer LJ, Hasty P, Vijg J, Hoeijmakers JHJ, Steeg Hv. Broad segmental progeroid changes in short-lived Ercc1 -/D7 mice. Pathobiology of Aging & Age-related Diseases. 2011;1:7219. doi: 10.3402/pba.v1i0.7219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Karsenty G. Transcriptional control of skeletogenesis. Annu Rev Genomics Hum Genet. 2008;9:183–96. doi: 10.1146/annurev.genom.9.081307.164437. [DOI] [PubMed] [Google Scholar]
- 27.Novack DV, Teitelbaum SL. The osteoclast: friend or foe? Annu Rev Pathol. 2008;3:457–84. doi: 10.1146/annurev.pathmechdis.3.121806.151431. [DOI] [PubMed] [Google Scholar]
- 28.Derheimer FA, Kastan MB. Multiple roles of ATM in monitoring and maintaining DNA integrity. FEBS Lett. 2010;584(17):3675–81. doi: 10.1016/j.febslet.2010.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rodier F, Coppe JP, Patil CK, Hoeijmakers WA, Munoz DP, Raza SR, Freund A, Campeau E, Davalos AR, Campisi J. Persistent DNA damage signalling triggers senescence-associated inflammatory cytokine secretion. Nat Cell Biol. 2009;11(8):973–9. doi: 10.1038/ncb1909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shiloh Y. ATM and related protein kinases: safeguarding genome integrity. Nat Rev Cancer. 2003;3 (3):155–68. doi: 10.1038/nrc1011. [DOI] [PubMed] [Google Scholar]
- 31.Coppe JP, Desprez PY, Krtolica A, Campisi J. The senescence-associated secretory phenotype: the dark side of tumor suppression. Annu Rev Pathol. 2010;5:99–118. doi: 10.1146/annurev-pathol-121808-102144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sharpless NE. Ink4a/Arf links senescence and aging. Exp Gerontol. 2004;39(11–12):1751–9. doi: 10.1016/j.exger.2004.06.025. [DOI] [PubMed] [Google Scholar]
- 33.Scholzen T, Gerdes J. The Ki-67 protein: from the known and the unknown. J Cell Physiol. 2000;182(3):311–22. doi: 10.1002/(SICI)1097-4652(200003)182:3<311::AID-JCP1>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- 34.Dai J, Lin D, Zhang J, Habib P, Smith P, Murtha J, Fu Z, Yao Z, Qi Y, Keller ET. Chronic alcohol ingestion induces osteoclastogenesis and bone loss through IL-6 in mice. J Clin Invest. 2000;106(7):887–95. doi: 10.1172/JCI10483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Simonet WS, Lacey DL, Dunstan CR, Kelley M, Chang MS, Luthy R, Nguyen HQ, Wooden S, Bennett L, Boone T, Shimamoto G, DeRose M, Elliott R, Colombero A, Tan HL, Trail G, Sullivan J, Davy E, Bucay N, Renshaw-Gegg L, Hughes TM, Hill D, Pattison W, Campbell P, Sander S, Van G, Tarpley J, Derby P, Lee R, Boyle WJ. Osteoprotegerin: a novel secreted protein involved in the regulation of bone density. Cell. 1997;89(2):309–19. doi: 10.1016/s0092-8674(00)80209-3. [DOI] [PubMed] [Google Scholar]
- 36.Pasparakis M. Regulation of tissue homeostasis by NF-kappaB signalling: implications for inflammatory diseases. Nat Rev Immunol. 2009;9(11):778–88. doi: 10.1038/nri2655. [DOI] [PubMed] [Google Scholar]
- 37.Zineldeen DH, Uranishi H, Okamoto T. NF-kappa B signature on the aging wall. Curr Drug Metab. 2010;11(3):266–75. doi: 10.2174/138920010791196283. [DOI] [PubMed] [Google Scholar]
- 38.Srinivasan V, Kriete A, Sacan A, Jazwinski SM. Comparing the yeast retrograde response and NF-kappaB stress responses: implications for aging. Aging Cell. 2010;9(6):933–41. doi: 10.1111/j.1474-9726.2010.00622.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Acharyya S, Villalta SA, Bakkar N, Bupha-Intr T, Janssen PM, Carathers M, Li ZW, Beg AA, Ghosh S, Sahenk Z, Weinstein M, Gardner KL, Rafael-Fortney JA, Karin M, Tidball JG, Baldwin AS, Guttridge DC. Interplay of IKK/NF-kappaB signaling in macrophages and myofibers promotes muscle degeneration in Duchenne muscular dystrophy. J Clin Invest. 2007;117(4):889–901. doi: 10.1172/JCI30556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mabb AM, Wuerzberger-Davis SM, Miyamoto S. PIASy mediates NEMO sumoylation and NF-kappaB activation in response to genotoxic stress. Nat Cell Biol. 2006;8(9):986–93. doi: 10.1038/ncb1458. [DOI] [PubMed] [Google Scholar]
- 41.Lavasani M, Robinson AR, Lu A, Song M, Feduska JM, Ahani B, Tilstra JS, Feldman CH, Robbins PD, Niedernhofer LJ, Huard J. Muscle-derived stem/progenitor cell dysfunction limits healthspan and lifespan in a murine progeria model. Nat Commun. 2012;3:608. doi: 10.1038/ncomms1611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kassem M, Marie PJ. Senescence-associated intrinsic mechanisms of osteoblast dysfunctions. Aging Cell. 2011;10(2):191–7. doi: 10.1111/j.1474-9726.2011.00669.x. [DOI] [PubMed] [Google Scholar]
- 43.Wang C, Jurk D, Maddick M, Nelson G, Martin-Ruiz C, von Zglinicki T. DNA damage response and cellular senescence in tissues of aging mice. Aging Cell. 2009;8(3):311–23. doi: 10.1111/j.1474-9726.2009.00481.x. [DOI] [PubMed] [Google Scholar]
- 44.Saeed H, Abdallah BM, Ditzel N, Catala-Lehnen P, Qiu W, Amling M, Kassem M. Telomerase-deficient mice exhibit bone loss owing to defects in osteoblasts and increased osteoclastogenesis by inflammatory microenvironment. J Bone Miner Res. 26(7):1494–505. doi: 10.1002/jbmr.349. [DOI] [PubMed] [Google Scholar]
- 45.Undale A, Srinivasan B, Drake M, McCready L, Atkinson E, Peterson J, Riggs BL, Amin S, Modder UI, Khosla S. Circulating osteogenic cells: characterization and relationship to rates of bone loss in postmenopausal women. Bone. 2010;47(1):83–92. doi: 10.1016/j.bone.2010.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Neer RM, Arnaud CD, Zanchetta JR, Prince R, Gaich GA, Reginster JY, Hodsman AB, Eriksen EF, Ish-Shalom S, Genant HK, Wang O, Mitlak BH. Effect of parathyroid hormone (1–34) on fractures and bone mineral density in postmenopausal women with osteoporosis. N Engl J Med. 2001;344(19):1434–41. doi: 10.1056/NEJM200105103441904. [DOI] [PubMed] [Google Scholar]
- 47.Gregg SQ, Robinson AR, Niedernhofer LJ. Physiological consequences of defects in ERCC1-XPF DNA repair endonuclease. DNA Repair. 2011;10(7):781–791. doi: 10.1016/j.dnarep.2011.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Beg AA, Sha WC, Bronson RT, Ghosh S, Baltimore D. Embryonic lethality and liver degeneration in mice lacking the RelA component of NF-[kappa]B. Nature. 1995;376(6536):167–170. doi: 10.1038/376167a0. [DOI] [PubMed] [Google Scholar]
- 49.Chang J, Wang Z, Tang E, Fan Z, McCauley L, Franceschi R, Guan K, Krebsbach PH, Wang C-Y. Inhibition of osteoblastic bone formation by nuclear factor-[kappa]B. Nat Med. 2009;15 (6):682–689. doi: 10.1038/nm.1954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hilton MJ, Tu X, Wu X, Bai S, Zhao H, Kobayashi T, Kronenberg HM, Teitelbaum SL, Ross FP, Kopan R, Long F. Notch signaling maintains bone marrow mesenchymal progenitors by suppressing osteoblast differentiation. Nat Med. 2008;14(3):306–14. doi: 10.1038/nm1716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mak KK, Bi Y, Wan C, Chuang P-T, Clemens T, Young M, Yang Y. Hedgehog Signaling in Mature Osteoblasts Regulates Bone Formation and Resorption by Controlling PTHrP and RANKL Expression. Developmental Cell. 2008;14(5):674–688. doi: 10.1016/j.devcel.2008.02.003. [DOI] [PubMed] [Google Scholar]
- 52.Xu G, Liu K, Anderson J, Patrene K, Lentzsch S, Roodman GD, Ouyang H. Expression of XBP1s in bone marrow stromal cells is critical for myeloma cell growth and osteoclast formation. Blood. 2012 doi: 10.1182/blood-2011-05-353300. Submitted, in revision. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Blair HC, Yaroslavskiy BB, Robinson LJ, Mapara MY, Pangrazio A, Guo L, Chen K, Vezzoni P, Tolar J, Orchard PJ. Osteopetrosis with micro-lacunar resorption because of defective integrin organization. Lab Invest. 2009;89(9):1007–17. doi: 10.1038/labinvest.2009.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.