

Abstract
A structure-based approach was used to design irreversible, cysteine-targeted inhibitors of the human centrosomal kinase, Nek2. Potent inhibition of Nek2 kinase activity in biochemical and cell-based assays required a noncatalytic cysteine residue (Cys22), located near the glycine-rich loop in a subset of human kinases. Elaboration of an oxindole scaffold led to our most selective compound, oxindole propynamide 16 (JH295). Propynamide 16 irreversibly inhibited cellular Nek2 without affecting the mitotic kinases, Cdk1, Aurora B, or Plk1. Moreover, 16 did not perturb bipolar spindle assembly or the spindle assembly checkpoint. To our knowledge, 16 is the first small molecule shown to inactivate Nek2 kinase activity in cells.
Introduction
Nek2a is a homodimeric serine/threonine kinase that localizes to centrosomes, the microtubule organizing centers of the cell.1,2 At the onset of mitosis, Nek2 phosphorylates the inter-centrosome linker proteins, C-Nap1 and rootletin.3,4 Nek2 is thus thought to play a role in bipolar spindle assembly driven by the microtubule motor protein, Eg5 (also known as “kinesin-5” or “kinesin spindle protein”).5 Moreover, Nek2 knockdown by RNA interference (RNAi) was found to partially compromise the spindle assembly checkpoint (SAC).6 The SAC pathway functions early in mitosis (metaphase) to monitor the strength and orientation of microtubule/chromosome connections and mediates mitotic arrest in response to inhibitors of Eg57 and microtubule dynamics.8 It is subject to regulation by multiple protein kinases (e.g. Plk1, AurB, and Mps1)8-12 and is of great interest as a potential point of intervention for anti-cancer drugs. The cellular roles of Nek2, including its putative role in the SAC pathway, have been defined primarily by RNAi-mediated knockdown approaches. The lack of cell-active Nek2 inhibitors has hindered attempts to elucidate its kinase activity-dependent functions.
Like many protein kinases with roles in mitosis, Nek2 has been implicated in cancer. Knockdown of Nek2 inhibited the proliferation of cholangiocarcinoma and breast cancer cell lines in tissue culture and in mouse tumor xenografts, while having no effect on normal fibroblasts.13,14 Nek2 knockdown also abrogated the ability of oncogenic H-Ras(G12V) to induce centrosome amplification.15 Forced overexpression of Nek2 in non-transformed breast epithelial cells induced the formation of multinucleated cells with increased numbers of centrosomes, a phenotype associated with mitotic errors, aneuploidy, and oncogenesis.16 Finally, Nek2 overexpression at the mRNA and/or protein level has been detected in primary breast tumors,16 cholangiocarcinoma,13 testicular seminoma,17 and diffuse large B-cell lymphoma.18 These studies have motivated the development of Nek2 inhibitors as potential therapeutic leads.
Previously reported Nek2 inhibitors include a series of aminopyrazines,19 a thiophene-based Plk1 inhibitor,20 a wortmannin-like series,21 and the sunitinib-like oxindole inhibitor 1 (SU11652, Figure 1A).22 The aminopyrazines were extensively characterized in biochemical assays and were found to bind to an inactive conformation of the isolated Nek2 kinase domain by x-ray crystallography. However, none of the aminopyrazines were active in cells, possibly due to insufficient membrane permeability conferred by a critical carboxylic acid moiety.19 The wortmannin-like compounds were reported to antagonize the effects of Nek2 overexpression on centrosome separation in cells;21 however, it is not clear whether these effects were caused by inhibition of Nek2, or of other cellular targets.
Figure 1.

(A) Oxindole pyrrole 1 guides the design of irreversible Nek2 inhibitors. E = Electrophile. (B) Crystal structure of 1 bound to Nek2 (PDB: 2JAV),22 showing the key cysteine (Cys22), the gatekeeper (Met86), and hydrogen bonds to the hinge region.
The selective alkylation of poorly conserved, noncatalytic cysteines has emerged as a powerful strategy for enhancing the potency and especially the selectivity of kinase inhibitors.23-26 At least six cysteine-targeted kinase inhibitors have entered clinical trials for various cancer indications.24.27,28 Moreover, several useful tool compounds have resulted from this strategy.29-31 A kinome-wide structural bioinformatics analysis carried out by our group revealed a previously untargeted cysteine located near the glycine-rich loop in 11 out of the ~500 human kinases, including Rsk1-4, Msk1/2, Plk1-3, Mekk1, and Nek2. Based on the presence of this cysteine, along with a threonine in the gatekeeper position, we designed an irreversible fluoromethylketone inhibitor that is highly selective for Rsk1/2/4.29,30,32 Herein, we report the structure-based design of propynamide oxindole 16 (JH295), which to our knowledge is the first reported inhibitor that irreversibly inactivates Nek2 kinase activity in cells.
Results and Discussion
Structure-based design of electrophilic oxindoles
A crystal structure of the Nek2 kinase domain bound to oxindole 1 provided a starting point for the design of irreversible inhibitors (Figure 1).22 Because this structure represents an unusual inactive conformation of the isolated monomeric kinase domain, its relevance to full-length Nek2 is unclear. We therefore used this structure as a rough guide to predict the orientation of key residues relative to the oxindole scaffold. Our basic design started with the oxindole-pyrrole core found in 1, which forms three hydrogen bonds to the Nek2 hinge region (Figure 1). This structural feature, found in many kinase inhibitors, is predicted to be critical for binding. Alkylation of the oxindole NH group should thus prevent binding to Nek2 and most other kinases, a property we exploited to control for nonspecific effects of the reactive electrophiles (see below).
Oxindole positions 6 and 7 form close contacts with Met86, the “gatekeeper” residue, and were therefore left unsubstituted. By contrast, the 5-chloro substituent of oxindole 1 is ~6 Å from the thiol of Cys22 (Fig. 1B). We hypothesized that Cys22 would be within striking distance of electrophilic substituents placed at the oxindole 5-position. Finally, the pyrrole 3-, 4-, and 5-positions (Figure 1) represent opportunities for increasing the selectivity for Nek2. The pyrrole 5-methyl group of 1 forms close contacts with Nek2 (~3.8 Å from Tyr88, Figure 1D) and points toward a small cleft near the hinge region. Alkyl groups at this position may be tolerated by Nek2 to a greater extent than off-target kinases.
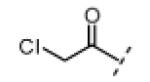
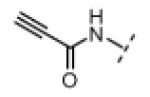
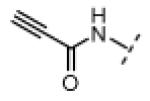
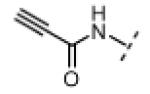
Based on the design in Figure 1A, we first synthesized and tested a series of seven oxindole pyrroles, each substituted with a different electrophile at the oxindole 5-position (Figure 2A). The electrophiles included a chloromethylketone (2), a chloroacetamide (3), and five Michael acceptors (4-8). In vitro kinase assays using a single compound concentration of 5 μM and a fixed 30-min pre-incubation period revealed that chloromethylketone 2 and propynamide 8 were the most potent inhibitors in this initial series, inhibiting Nek2 by more than 80% (Figure 2B). Intrinsic chemical reactivity, coupled with the location of the electrophilic carbon relative to the oxindole, likely account for the observed potency differences. Notably, the acrylamide electrophile (compound 7), used extensively in the context of irreversible EGFR and BTK inhibitors, was ineffective against Nek2. By contrast, the more reactive propynamide (compound 8) provided nearly complete inhibition.
Figure 2.

(A) First-generation electrophilic oxindoles. (B) Percent inhibition of Nek2 kinase activity by compounds 2-8 (5 μM, 30 min). (C) Dose-response curves from in vitro kinase assays with 2 and 8 vs. WT and C22V Nek2. Specific kinase activities of WT and C22V Nek2 were indistinguishable.
To test the requirement of Cys22 for potent inhibition by 2 and 8, we prepared a C22V mutant of Nek2 in which the target cysteine was replaced by valine (found at the equivalent position in most kinases). Comparison of dose-response curves for 2 and 8 against WT and C22V Nek2 revealed a strong dependence on Cys22 for potent inhibition. Whereas chloromethylketone 2 and propynamide 8 inhibited WT Nek2 with submicromolar potency, they were significantly less potent against the C22V mutant (Figure 2C). Under our standard in vitro kinase assay conditions, chloromethylketone 2 was much more potent than propynamide 8, but this advantage disappeared in cell-based assays (see below). The increased potency of both compounds toward WT Nek2 suggests that they function through alkylation of Cys22. However, the data do not rule out other possible explanations, such as steric occlusion by the bulkier valine side chain or enhanced affinity of the C22V mutant for ATP.
To test for Cys22-dependent alkylation of Nek2, we employed an electrospray mass spectrometry assay that reports the molecular mass of the Nek2 kinase domain (amino acids 1-271), plus that of any covalent adducts, with high accuracy. This assay does not detect reversible Nek2/inhibitor complexes, as the protein is denatured during the reversed-phase liquid chromatography step prior to mass spectrometric detection. At physiological pH, a ten-fold molar excess of 2 or 8 alkylated WT Nek2 at a single site, giving the predicted increase in mass (Figure 3). No labeling of the C22V mutant was detected under identical conditions, consistent with the unique site of modification being Cys22. These results are remarkable given that the Nek2 kinase domain has five cysteines, at least three of which are exposed to solvent. Thus, alkylation of Nek2 by 2 and 8 likely occurs via formation of an initial complex in which the electrophilic carbon is proximal to Cys22, as predicted by the orientation of oxindole 1 in the Nek2 crystal structure (Figure 1).
Figure 3.

Electrospray mass spectrometry reveals stoichiometric alkylation of WT, but not C22V, Nek2 kinase domain (aa 1-271) by compounds 2 and 8 (10 equiv). Note that the observed molecular weight of C22V Nek2 kinase domain differs from that predicted by +79 Da, presumably due to efficient autophosphorylation.
2′-imidazole substituents confer improved selectivity for Nek2
Several oxindole kinase inhibitors have been shown to potently inhibit Cdk1.33 Because Cdk1 inhibitors block mitotic entry34 and rapidly trigger mitotic exit,35 oxindoles that inhibit Cdk1 would not be useful for studying the mitotic roles of Nek2. Based on the Nek2/compound 1 crystal structure (Figure 1), as well as the structure of an imidazole oxindole bound to Cdk2 (PDB: 1PF8),33 it seemed plausible that alkyl substituents on the 5′- or 2′-position of the pyrrole or imidazole ring, respectively, could destabilize interactions with a subset of off-target kinases, including the cyclin-dependent kinases. Consistent with this idea, sunitinib, which contains a 5′-methyl group (similar to 1) did not bind detectably to the Cdk1-related kinase, Cdk2 (Kd > 10 μM).36
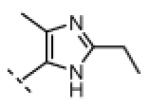
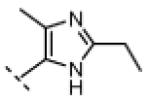
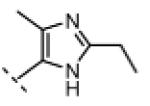
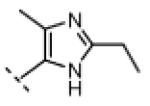
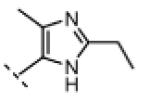
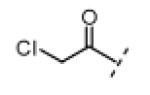
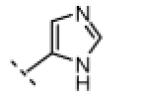
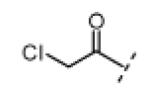
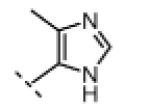
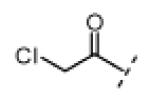
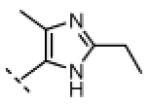
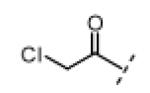
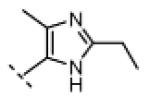
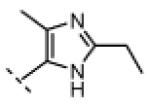
Seeking alternatives to the pyrrole found in compounds 2 and 8, we prepared two series of imidazole-substituted oxindoles (Table 1). Each series retained either the chloromethylketone (9-11) or propynamide (14-16) at the oxindole 5-position. We noted that the IC50 values of these inhibitors were time dependent, as expected for irreversible inhibitors. To make meaningful comparisons among inhibitors, we therefore strictly standardized the duration of the assays. In vitro kinase assays with the imidazole-based inhibitors revealed different potency trends depending on the electrophile (Table 1). In the chloromethylketone series, replacement of the pyrrole of 2 with the imidazoles of 9-11 led to a 10–20-fold decrease in potency. By contrast, the same imidazole replacements had little effect on potency in the propynamide series. Thus, propynamide 16, which contains a 2′-ethyl, 4′-methyl imidazole appended to the oxindole core, inhibited Nek2 with an IC50 of ~800 nM, similar to the unsubstituted pyrrole 8. Mass spectrometry analysis revealed that both chloromethylketone 11 and propynamide 16 reacted stoichiometrically with WT but not C22V Nek2 (Supporting Information).
Table 1.
In vitro kinase assays with Nek2 and Cdk1/cyclin B.a
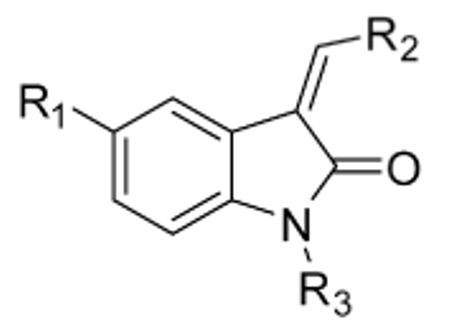
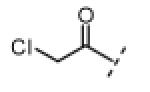
| compound | R1 | R2 | R3 | Nek2 IC50 (nM) |
Cdk1/CycB IC50 (nM) |
|---|---|---|---|---|---|
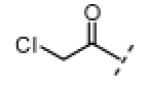
| 2 |
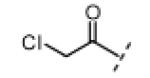
|
|
H | <6 | 10 |
| 9 |
|
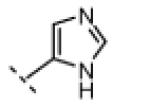
|
H | 60 | <1 |
| 10 |
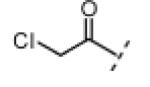
|
|
H | 20 | <1 |
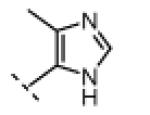
| 11 |
|
|
H | 130 | 2500 |
| 12 |
|
|
Me | 4900 | >20,000 |
| 13 |
|
|
H | >20,000 | 3600 ±1400 |
| 8 |
|
|
H | 920 | 90 ±42 |
| 14 |
|
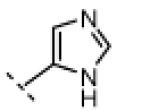
|
H | 620 | <1 |
| 15 |
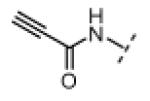
|
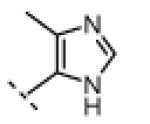
|
H | 390 | <1 |
| 16 |
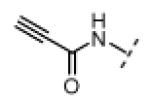
|
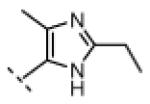
|
H | 770 | >20,000 |
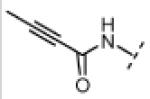
| 17 |
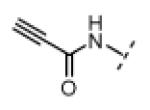
|
|
Me | >20,000 | >20,000 |
| 18 |
|
|
H | 20,000 | >20,000 |
Each IC50 value is the mean of three determinations (S.D. < 20%, unless indicated).
A counterscreen against Cdk1 (bound to its activator, cyclin B) revealed that the 2-ethyl group in compounds 11 and 16 is essential for selective Nek2 inhibition (Table 1). Propynamide 16, which exhibited the highest selectivity toward Nek2, was completely inactive against Cdk1 (IC50 > 20 μM). By contrast, propynamide 15 lacks the 2-ethyl group and demonstrated inverse selectivity, being 400-fold more potent toward Cdk1 compared to Nek2. Comparison of propynamides 15 and 16 reveals that the 2-ethyl group confers a >20,000-fold decrease in potency toward Cdk1 (Table 1). All of the oxindoles lacking a 2′-ethyl group proved to be low or subnanomolar Cdk1 inhibitors.
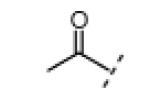
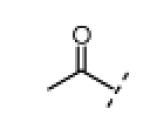
In vitro kinase assays with compounds 12 and 17 (Table 1, R3 = Me) confirmed the importance of the oxindole NH (Table 1), which forms a hydrogen bond with the Nek2 hinge region in the crystal structure with compound 1 (Figure 1). An electrophilic moiety was also essential for potent Nek2 inhibition, as revealed by the sharply reduced activities of methyl ketone 13 (IC50 > 20 μM) and 2-butynamide 18 (IC50 ~ 20 μM). We envisioned that these “negative control” compounds would be useful for ruling out off-target effects in cell-based assays, including assays for cellular Nek2 kinase activity, as described below.
Finally, we tested propynamide 16, which had the highest selectivity for Nek2 vs. Cdk1, against the kinases Rsk2 and Plk1. Both kinases have a cysteine that is structurally equivalent to Cys22 of Nek2. It was especially important to test Plk1, as this mitotic kinase is an essential regulator of bipolar spindle assembly, among other mitotic functions also proposed to be regulated by Nek2.9 In vitro kinase assays (under identical conditions to the Nek2 assays) revealed that 16 was ~5-fold selective for Nek2 vs. Rsk2 and was essentially inactive toward Plk1 (IC50 > 20 μM, see Supporting Information). With the exception of Nek2 and the polo-like kinases (Plk1-3), kinases containing a cysteine equivalent to Cys22 of Nek2 (i.e., Rsk1-4, Msk1/2, and Mekk1) are not thought to be critical mitotic regulators in human cells.
Irreversible inhibition of Nek2 kinase activity in cells
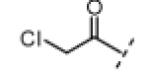
We next tested our inhibitors against endogenous Nek2 in human A549 lung cancer cells. This was accomplished by treating live cells with inhibitors for 45 min at a concentration of 5 μM, followed by cell lysis and immunoprecipitation (IP) of endogenous Nek2. Immunoprecipitated Nek2 was then subjected to in vitro kinase assays (Table 2). It is important to note that the IP process, which includes several wash and dilution steps, allows reversibly bound inhibitors to dissociate and is thus well suited for detecting irreversible kinase inhibition in cells that had previously been treated with inhibitors. Electrophilic oxindoles with potent activity against Nek2 in vitro (IC50 < 1 μM) demonstrated up to 95% inhibition of cellular Nek2 at 5 μM. By contrast, the negative control compounds (12, 13, 17, and 18), including the N-methyl oxindoles bearing reactive electrophiles (12 and 17), were significantly less active (10-25% inhibition at 5 μM). Despite being much more potent against purified Nek2 in vitro, the chloromethylketones were somewhat less efficacious than the propynamides in intact cells (Table 2), most likely due to their reduced stability toward cellular thiols (e.g., glutathione). Compared to the propynamides, the chloromethylketones are ~20 times more reactive toward 10 mM β-mercaptoethanol (pH 7.5; chloromethylketone 11, t1/2 ~3 min; propynamide 16, t1/2 ~60 min).
Table 2.
Cellular effects of electrophilic oxindoles.
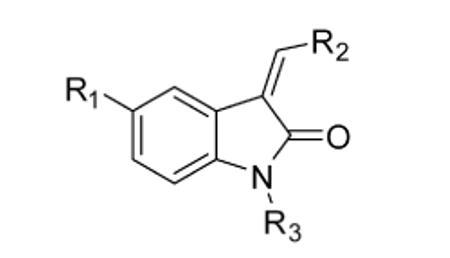
| compound | R1 | R2 | R3 | Nek2a % Inhib. (5 μM) |
% mitoticb exit (5 μM) |
|---|---|---|---|---|---|
| 2 |
|
|
H | 90 | 98 |
| 9 |
|
|
H | 73 | 92 |
| 10 |
|
|
H | 75 | 100 |
| 11 |
|
|
H | 89 | 50 |
| 12 |
|
|
Me | 12 | 0.9 |
| 13 |
|
|
H | 14 | 27 |
| 8 |
|
|
H | 94 | 78 |
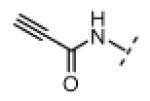
| 14 |
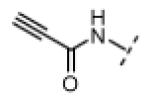
|
|
H | 95 | 80 |
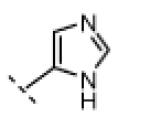
| 15 |
|
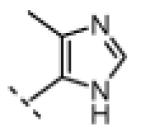
|
H | 95 | 80 |
| 16 |
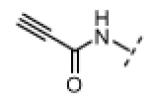
|
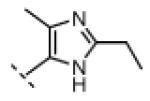
|
H | 92 | 4 |
| 17 |
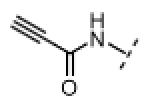
|
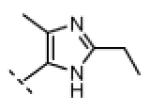
|
Me | 19 | 0 |
| 18 |
|
|
H | 26 | 33 |
IP kinase assays of Nek2 from cells that had been pre-treated with 5 μM of the indicated compounds for 45 min (single measurement).
Percentage of cells that exited mitosis after treatment with 5 μM of the indicated compounds for 45 min, assessed by immunofluorescence microscopy (see text and Figure 5 for details).
To confirm that propynamide 16 inhibits cellular Nek2 in a Cys22-dependent manner, we generated cell lines that stably express hemagglutinin (HA) epitope-tagged WT or C22V Nek2 under the control of a tetracycline-inducible promoter. Using an IP kinase assay similar to that used for endogenous Nek2, we found that 16 inhibited WT Nek2 in cells with an IC50 of ~1.3 μM, whereas it had little effect on the C22V mutant (Figure 4). These results suggest that inhibition of cellular Nek2 by 16 occurs via alkylation of Cys22 and is not an indirect effect of inhibiting kinases that regulate Nek2.
Figure 4.

Hek293 cells containing tetracycline (tet.)-inducible, HA-tagged WT (A) and C22V (B) Nek2, were pre-treated with propynamide 16 (45 min) prior to cell lysis and IP kinase assays. After separation by SDS-PAGE, 32P-labeled β-casein was visualized by phosphor-imaging. Anti-HA Western blot and Coomassie Blue (CB) staining confirmed equivalent amounts of Nek2-HA and β-casein, respectively.
Effect of electrophilic oxindoles on the spindle assembly checkpoint
The spindle assembly checkpoint (SAC) is a complex signaling network that prevents progression into anaphase (the stage of mitosis in which chromosome separation occurs, just prior to cell division) until proper attachment of sister chromatids to spindle microtubules occurs in metaphase.8 Small molecules that prevent bipolar spindle assembly, such as microtubule or Eg5 kinesin inhibitors (e.g., monastrol7), arrest cells in metaphase in a SAC-dependent manner. Inhibitors of kinases required for SAC maintenance, such as AurB, promote rapid exit from mitosis without cell division.10 As Nek2 had been previously implicated in SAC regulation by RNAi-mediated knockdown,6 we were motivated to test the effect of our inhibitors on this process.
To test for a role of Nek2 in SAC maintenance, we treated monastrol-arrested A549 cells with the electrophilic oxindoles shown in Table 2 for 45 min, at which point the cells were fixed, stained for immunofluorescence analysis, and counted to assess the percentage of cells that had exited mitosis (Table 2; >75 cells were counted for each condition). We included the Cdk1 inhibitor 19 (RO3306) as a positive control for inducing mitotic exit.34 Representative images of cells treated with 19 and propynamides 15 and 16 are shown in Figure 5. Whereas cells treated with DMSO (control) and the more selective Nek2 inhibitor 16 stained brightly with antibodies against phospho-Ser10 histone H3, cells treated with 19 and propynamide 15 were devoid of this mitotic marker (Figure 5). Consistent with their potent activity toward Cdk1 in vitro (Table 1), compounds lacking a 2′-ethyl group, including propynamide 15, triggered mitotic exit in 78-99% of the monastrol-arrested cells (Table 2). The strong correlation between Cdk1 inhibition in vitro and rapid induction of mitotic exit in monastrol-arrested cells suggests that Cdk1 is a relevant target of these compounds in cells. Conversely, the 2′-ethyl group of 16 is sufficient to prevent inhibition of Cdk1 in cells, consistent with the results from Cdk1 kinase assays (Table 1).
Figure 5.

A549 cells were arrested in metaphase (monastrol, 18 h) and treated with the indicated compounds (5 μM) for 45 min. Cells were then fixed and processed for immunofluorescence microscopy for detection of phospho-Ser10 histone H3 (red), chromatin (blue), and tubulin (green). Scale bar, 10 μm.
Taken together, our results demonstrate that inhibition of Nek2 kinase activity (Figure 4) is not sufficient to override the SAC or induce mitotic exit (Figure 5). We further conclude that 16 does not significantly inhibit cellular kinases required for SAC maintenance, such as AurB or Mps1, since inhibitors of these kinases rapidly trigger mitotic exit under similar conditions.10-12
Effect of propynamide 16 on bipolar spindle assembly and chromosome congression
RNAi knockdown and dominant negative overexpression experiments have implicated Nek2 in two critical mitotic functions: bipolar spindle assembly and chromosome congression at the “metaphase plate”.37,38 These conclusions were challenged in a recent study (also based on RNAi), which argued that Nek2 plays a supporting rather than an obligatory role in mitosis.5 To test whether propynamide 16 perturbs these processes, we first arrested cells in late G2 phase (after DNA replication and just prior to mitosis) with the Cdk1 inhibitor, 19 (Figure 5). We then treated the arrested cells with 16 (5 μM) for 30 min, conditions that irreversibly inhibit Nek2, as revealed by parallel IP kinase assays (Figure 6A). Finally, the cells were released from G2-arrest by washing into fresh media containing a proteasome inhibitor (MG132), which allows cells to transit mitosis until they stably arrest in late metaphase with bipolar spindles and chromosomes aligned along the equator (termed “congression”). This well-established protocol provides a sensitive test for bipolar spindle assembly and chromosome congression.35
Figure 6.

Propynamide 16 does not perturb bipolar spindle assembly or chromosome congression. (A) Nek2 IP kinase assay from A549 cells arrested with CDK1 inhibitor 19 and treated with the indicated inhibitors for 30 min. (B) Quantification of cells with bipolar spindles 1 h after 19 washout into MG132-containing media. N > 160 cells/condition; mean ± SD from 3 experiments. (C) Immunofluorescence images of bipolar spindles and chromosomes aligned at the metaphase plate. Cells were treated as in (a) fixed and stained 1 h after release from 19 into MG132-containing media. Scale bar, 10 μm.
In the presence of sufficient concentrations of 16 to inhibit cellular Nek2 kinase activity by greater than 95%, we observed no defects in bipolar spindle assembly or chromosome congression, nor any reduction in the total number of mitotic cells (Figure 6). Thus, Nek2 kinase activity may not be required for these processes, although it is possible that the trace amount of active Nek2 remaining after inhibitor treatment is sufficient to carry out important mitotic functions. Previously reported mitotic defects may have resulted from depletion of the entire Nek2 protein or from off-target effects caused by RNAi or Nek2 overexpression. The lack of mitotic defects also supports the conclusion that 16 does not significantly inhibit Plk1 in cells, as Plk1 inhibition causes cells to arrest in early metaphase with monopolar spindles39-41 Selectivity vs. Plk1 is not readily explained by available crystal structures and is surprising given the presence of a cysteine equivalent to Cys22 in Nek2.
Conclusions
Starting with an oxindole scaffold that has been frequently used to generate reversible kinase inhibitors, we have developed cell-active, irreversible inhibitors of the centrosomal kinase, Nek2. A crystal structure of Nek2 bound to oxindole 1 guided the design of compounds with electrophilic substituents at the 5-position, which we hypothesized would be within striking distance of Cys22, located near the C-terminal end of the glycine-rich loop in only 11 human kinases. From a set of seven electrophilic functional groups tested at this position, propynamides proved superior in terms of balancing potency, selectivity, and chemical stability. Both propynamide- and chloromethylketone-based inhibitors irreversibly alkylated Nek2 with 1:1 stoichiometry in a Cys22-dependent manner, consistent with our structural hypothesis.
Our primary goal was to generate a chemical tool that could be used to inhibit Nek2 acutely in living cells without inhibiting other kinases that have critical mitotic functions. Our initial compounds were problematic in that they inhibited Cdk1 with subnanomolar potency and caused cells to rapidly exit mitosis, precluding their use as Nek2 inhibitors. This problem was solved by adding a 2′-ethyl group to the imidazole moiety. The resulting propynamide 16 was inactive against Cdk1 in vitro. Moreover, at concentrations that resulted in complete and irreversible inhibition of Nek2, propynamide 16 did not significantly inhibit mitotic entry, the spindle assembly checkpoint, bipolar spindle assembly, or chromosome congression. These data strongly suggest that 16 does not inhibit cellular Cdk1, Plk1, AurB, or Mps1. Unraveling the cellular roles of Nek2 will likely benefit from acute small-molecule inhibition and high-resolution live imaging experiments, which may reveal effects on centrosome, microtubule, and chromosome dynamics. Propynamide 16, along with Nek2-inactive compounds 17 and 18, provide an initial toolkit for undertaking these studies.
Chemistry
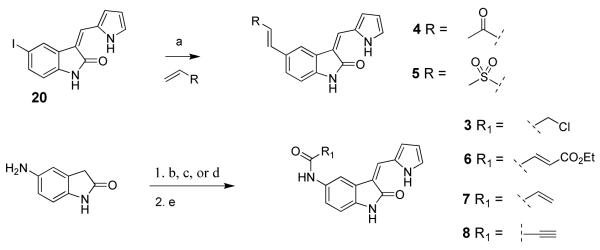
Vinyl sulfone 4 and vinyl ketone 5 were synthesized by coupling the requisite olefins to iodide 20 under Heck conditions (Scheme 1). Amide-linked electrophilic oxindoles 3 and 6-8 were prepared from 5-amino oxindole in two steps: (1) acylation of the 5-amino group, and (2) Knoevenagel condensation with pyrrole-2-carboxaldehyde (Scheme 1). To prepare imidazole-substituted oxindoles 9-11, the 5-chroroacetyl group was first protected to give dixolane 21 prior to Knoevenagel condensation with the requisite imidazole 5′-carboxaldehydes (Scheme 2). Finally, imidazole-substituted propynamides 14-16 were synthesized from Boc-protected 5-amino oxindole 22 in three steps: (1) Knoevenagel condensation, (2) Boc deprotection, and (3) coupling of the resultant amine with propiolic acid (Scheme 2). Methyl ketone 13, 2-butynamide 18, and N-methyl-oxindoles 12 and 17 were synthesized by analogous routes (see Experimental section for details).
Scheme 1.
Synthesis of pyrrole-substituted electrophilic oxindoles 3-5 and 6-8.a
aReagents and conditions: (a) R-CH=CH2 (4 equiv), Ph3P (0.6 equiv), Pd(OAc)2 (0.3 equiv), Et3N (5 equiv), DMF, 85 °C, 2 h (yield: 4, 48%; 5, 34%); (b) ClCH2COCl (1.3 equiv), pyridine (1.5 equiv), THF, 0 °C to rt, 30 min; (c) CH2=CHCOCl (1.2 equiv), THF, 0 °C to rt, 10 min; (d) R1-CO2H (1.2 equiv), EDC (2 equiv), acetonitrile, rt, 0.5-2 h; (e) pyrrole-2-carboxaldehyde (5-10 equiv), piperidine (0.1-0.2 equiv), THF, 75 °C, 4-20 h, (yield, 2 steps: 3, 28%; 6, 28%; 7, 23%; 8, 12%).
Scheme 2.

Synthesis of imidazole-substituted electrophilic oxindoles 9-11 and 14-16.a
aReagents and conditions: (a) R-CHO (1.2 equiv), piperidine (0.3 equiv), THF, 75 °C, 7-20 h; (b) aq. HCl, THF, rt, 5-24 h (yield, 2 steps: 9, 41%; 10, 49%; 11, 49%); (c) R1-CHO (1.1 equiv), piperidine (0.2 equiv), THF, 75 °C, 20 h; (d) anhydrous HCl (excess), MeOH, rt, 23-48 h; (e) propiolic acid (1.5-6 equiv), DIPEA (2 equiv), EDC (2 equiv), DMF, 0 °C, 2-2.5 h (yield, 3 steps: 14, 7%; 15, 27%; 16, 32%).
Experimental Section
In vitro kinase assays with Nek2, Cdk1, Plk1, and Rsk2
Inhibitors were incubated for 30 min at room temperature with WT or C22V Nek2 (15 nM), in 20 μL of kinase reaction buffer (100 μM ATP, 20 mM HEPES pH 7.5, 5 mM MgCl2, 0.1 mM EDTA, 0.08 mg/mL BSA) with 3% DMSO. Next, 5 μL of kinase reaction buffer containing 0.5 mg/mL β-casein (Sigma) and 2 μCi [γ-32P]ATP (PerkinElmer) was added. Kinase reactions were allowed to proceed for 30 min at room temperature. Next, 5 μL of the mixture was blotted onto nitrocellulose membrane (Bio-Rad) that had been pre-washed with wash buffer (1 M NaCl, 0.1 % H3PO4) and dried. The membrane was washed three times with wash buffer and dried under a heat lamp. The dry blots were exposed to a storage-phosphor screen (GE Healthcare), imaged using a Typhoon Scanner (GE Healthcare), quantified using Image Quant software (GE Healthcare), and the data processed and plotted using Excel (Microsoft) and GraphPad (Prism) software.
Cdk1 assays were conducted using the method for Nek2 with the following substitutions: Cdk1/Cyclin-B complex (2 nM, Millipore), 38 μM ATP, 0.2 mg/mL histone-H1 (Millipore) as the substrate, and 4 μCi [γ-32P]ATP.
Plk1 assays were conducted using the method for Nek2 with the following substitutions: Plk1 (8.5 nM, Millipore), 3.8% DMSO, 2.5 mg/mL dephosphorylated α-casein as the substrate (Sigma), and 0.75 μCi [γ-32P]ATP.
For Rsk2 assays, inhibitors were incubated for 30 min at room temperature with the Rsk2 C-terminal kinase domain29 (15 nM) in 15 μL of kinase reaction buffer (100 μM ATP, 20 mM HEPES pH 7.5, 5 mM MgCl2, 0.02 mg/mL BSA) with 5% DMSO. Next, 10 μL of kinase reaction buffer containing 0.2 mg/mL BSA, 417 μM substrate peptide (RRQLFRGFSFVAK), and 3 μCi [γ-32P]ATP was added. Kinase reactions were allowed to proceed for 30 min at room temperature, and 5 μL of the mixture was blotted onto P81 filtermat (Whatman). The filtermat was washed once with 10% acetic acid, and twice with 0.1% phosphoric acid, and dried under a heat lamp, and further processed exactly as for Nek2 kinase assays.
Mass spectrometry-based covalent binding assays
Inhibitors (500 nM) were incubated with 50 nM WT or C22V Nek2 kinase domain in pH 7.4 PBS with 5% DMSO for 5 h at room temperature, then acidified to pH 3 with 0.5 M HCl. The samples were analyzed by LC-MS using linear gradient elution (5% to 95% acetonitrile with 0.2% formic acid over 3 min) on a Microtrap C18 protein column (Michrom Bioresources) with a Waters 1525 μ solvent delivery system and a Waters LCT Premier ESI mass spectrometer for detection. Proteins or protein-adducts were detected as multiply charged ions and computationally deconvoluted to provide a single mass for each species using MassLynx software (Waters).
Immunoprecipitation kinase assays
All cells were grown in media (high glucose DMEM, Gibco 11965) containing 10% fetal bovine serum (Culture Pure, Axenia Biologix) and penicillin/streptomycin (Gibco 15140) at 37 °C in an atmosphere of 10% CO2. For IP kinase assays with endogenous Nek2, A549 cells were plated at 400,000/well in six-well plates (Nunc) and grown for 20 h. Cells (three wells per condition) were treated with 5 μM Nek2 inhibitors for 45 min, washed twice with DPBS (Gibco), and stored at −80 °C. Alternatively, cells (three wells per condition) were arrested in late G2 phase by incubation with 5 μM 19 for 20 h. Cells were then treated with 5 μM Nek2 inhibitors plus 5 μM 19 for 30 min, washed twice with DPBS (Gibco), and stored at −80 °C. The cells were thawed at room temperature for 5 min, treated with 200 μL/well of ice-cold lysis buffer (50 mM HEPES pH 7.6, 150 mM NaCl, 0.1% Triton X100, 1 mM DTT, 1× Roche PhosSTOP phosphatase inhibitors, 1× Complete Roche protease inhibitors) and bath sonicated for 30 sec. Lysates from a single condition were combined and clarified for 10 min (20,000 g) at 4 °C. The clarified lysates were normalized for total protein concentration. Next, 600 μL of normalized lysate was incubated with 0.4 μg anti-Nek2 antibody (H-235, Santa Cruz) with rocking for 1.5 h at 4 °C. Next, 30 μL of protein A Dynabeads (Invitrogen) was added, and rocking was continued for an additional 1.5 h at 4 °C. The beads were washed (200 μL per wash for 5 min with rocking at 4 °C) once with lysis buffer, once with lysis buffer plus 0.5 M NaCl, and twice with kinase buffer (20 mM HEPES pH 7.5, 5 mM MgCl2, 0.1 mM EDTA, 1 mM DTT, 1× PhosSTOP). The beads were suspended in the last wash buffer and divided in half. One half was used for Nek2 western blot analysis (Supporting Information), and the other half was used for a kinase assay. For the kinase assay, beads were incubated in 30 μL kinase buffer containing 0.5 mg/mL β-casein (Sigma), 100 μM ATP, and 4 μCi [γ-32P]ATP for 1 h at room temperature. Reactions were quenched with 8 μL of 5× sample buffer (300 mM TRIS pH 6.8, 10% SDS, 50% glycerol, 0.01% bromophenol blue, 0.5 M DTT). Substrate proteins were resolved by SDS-PAGE and detected by Coomassie staining and phosphor-imaging.
IP kinase assays of HA-tagged Nek2 from tetracycline-inducible Hek293 cell lines were carried out exactly as for endogenous Nek2 with the following changes. Tet-inducible Hek293 cells expressing either WT or C22V Nek2-3HA were plated at 630,000 cells per well in six-well plates (Nunc), grown for 20 h, and then treated with 1 μg/mL tetracycline for 7 h to induce Nek2 expression. Cells (one well per condition) were treated with propynamide 16 in 2 mL of media for 45 min, washed twice with DPBS (Gibco), and stored at −80 °C. Normalized cell lysates were immunoprecipitated with 1.6 μg of 12CA5 anti-HA antibody (Roche). Western blot conditions and methods for generating these cell lines are presented in the Supporting Information.
Mitosis assays in A549 cells
For assessing mitotic exit induced by acute treatment with kinase inhibitors, A549 cells (180,000 in 1 mL media) were plated on 18 mm round poly-lysine-coated coverslips in 12-well plates (Nunc), grown for 24 h, and treated with 100 μM monastrol for 18 h. The monastrol-arrested cells were then treated with 5 μM kinase inhibitors in the continued presence of 100 μM monastrol for an additional 45 min. Cells were then processed for immunofluorescence microscopy as described in the Supporting Information.
For assessing mitotic defects induced by propynamide 16, A549 cells (180,000 in 1 mL media) were plated on 18 mm round poly-lysine-coated coverslips in 12-well plates (Nunc), grown for 24 h, and treated with 5 μM 19 for 20 h. Cells arrested with 19 were treated with 5 μM 16 for 30 min in the presence of 5 μM 19, washed twice compound-free media, then treated with 10 μM MG132 for 1 h. Cells were then processed for immunofluorescence microscopy as described in the Supporting Information.
General chemistry
All reactions were conducted in oven-dried glassware under an atmosphere of air unless otherwise noted. Reagent grade solvents DCM, DMF, and THF were dried over molecular sieves prior to use. Reagent grade solvents carbon disulfide, EtOAc, Et2O, hexanes, methanol, and acetonitrile were used as purchased. The organic bases Et3N and DIPEA were dried over solid NaOH prior to use. All other reagents and starting materials were purchased and used as obtained. Reactions were monitored by TLC using K65 250 μm glass-backed silica gel plates with a fluorescent indicator (Whatman). Flash column chromatography was carried out on 140-400 mesh silica gel as the solid phase (Fisher Scientific). Preparative reverse phase HPLC was preformed on a Peeke Scientific Combi-A 5 μm preparative C18 column (50 × 22 mm, flow rate 10 mL/min) using a Varian Prostar 210 solvent delivery system equipped with a Varian Prostar 335 full spectrum UV/VIS detector. Analytical HPLC was preformed using the same system equipped with a Varian Pursuit XRs 5 C18 column (150 × 4.6 mm, flow rate 1 mL/min), using acetonitrile/water linear gradient elution, 5% to 80% acetonitrile over 20 min. Using this method the purity of all compounds used in bioassays was determined to be >95%. 1H NMR spectra were obtained using a Varian Inova 400 MHz spectrometer. Spectral data are reported as follows: chemical shift (as ppm referenced to the residual DMSO solvent signal at 2.50 ppm), multiplicity (s=singlet, d=doublet, dd=doublet of doublets, t=triplet, q=quartet, m=multiplet), coupling constant, and integration value. Fully assigned 1H NMR spectra are reported for every compound synthesized. High resolution mass spectra (HRMS) were recorded on a Thermo Electron Corporation LTQFT using electrospray ionization with FT resolution set to 30,000. HRMS spectra are reported for every compound synthesized. The following compounds were synthesized according to published procedures: 5-amino-oxindole,42 5-acetyl-oxindole,42 t-butyl hypochlorite,43 monastrol,44 and 19.45
(Z)-3-((1H-Pyrrol-2-yl)methylene)-5-(2-chloroacetyl)indolin-2-one (2)
Chloroacetyl chloride (21.7 g, 192 mmol, 1.7 equiv) was added dropwise to a stirred suspension of anhydrous AlCl3 (90.0 g, 675 mmol, 6 equiv) in carbon disulfide (420 mL). Stirring was allowed to continue at room temperature for 15 min. Indolin-2-one (15.0 g, 113 mmol, 1 equiv) was then added, and the mixture was brought to reflux for 2.5 h, then cooled on ice. After removal of the solvent by decantation the remaining thick brown residue was treated with ice cold water (500 mL). The resulting precipitate was recovered by filtration, washed with water, and dried under vacuum to provide pure 5-(2-chloroacetyl)indolin-2-one (21.6 g, 92%) as a white solid. 1H NMR (DMSO-d6): δ 3.57 (s, 2H), 5.08 (s, 2H), 6.93 (d, J = 8.4 Hz, 1H), 7.83 (s, 1H), 7.88 (d, J = 8.1 Hz, 1H), 10.8 (bs, 1H); HRMS calcd for C10H9ClNO2 210.0316; found (ESI+) (M + H) 210.0319.
5-(2-chloroacetyl)indolin-2-one (750 mg, 3.58 mmol, 1 equiv) was added to a solution of pyrrole-2-carboxaldehyde (3.42 g, 36.3 mmol, 10 equiv) and piperidine (61.0 mg, 0.716 mmol, 0.2 equiv) in THF (125 mL). The resulting mixture was heated to 60 °C in a sealed jar for 45 min. After cooling to room temperature, the solvent was removed under vacuum to give a thick red oil that was dissolved in EtOAc. Treating this solution with hexanes (30 mL) produced a tarry red precipitate that was removed by filtration and discarded. The filtrate was then cooled on ice to produce an orange precipitate that was collected by filtration. This crude product was further purified by flash chromatography on silica gel (hexanes/EtOAc 3:1) followed by recrystallization from EtOAc to give 2 (200 mg, 20%) as a crystalline orange solid. 1H NMR (DMSO-d6): δ 5.16 (s, 2H), 6.40-6.42 (m, 1H), 6.92-6.93 (m, 1H), 7.01 (d, J = 8.1 Hz, 1H), 7.42-7.44 (m, 1H), 7.84 (dd, J = 8.3, 1.7 Hz, 1H), 7.99 (s, 1H), 8.34 (d, J = 1.5 Hz, 1H), 11.3 (s, 1H), 13.2 (bs, 1H); HRMS calcd for C15H12ClN2O2 287.0587; found (ESI+) (M + H) 287.0584.
(Z)-N-(3-((1H-Pyrrol-2-yl)methylene)-2-oxoindolin-5-yl)-2-chloroacetamide (3)
Pyridine (120 mg, 1.52 mmol, 1.5 equiv) was added to a solution of 5-aminoindolin-2-one (150 mg, 1.01 mmol, 1 equiv) in anhydrous THF (50 mL). The mixture was cooled to 0 °C, and chloroacetyl chloride (148 mg, 1.31 mmol, 1.3 equiv) was added dropwise over the course of 5 min. The mixture was allowed to come to room temperature for 30 min. Next water (10 mL) was added and most of the THF was removed under vacuum. The resulting precipitate was recovered by filtration and washed with water to give pure 2-chloro-N-(2-oxoindolin-5-yl)acetamide (102 mg, 45%) as a white solid. 1H NMR (DMSO-d6): δ 3.47 (s, 2H), 4.20 (s, 2H), 6.76 (d, J = 8.4 Hz, 1H), 7.33 (dd, J = 8.4, 1.8 Hz, 1H), 7.49 (bs, 1H), 10.1 (s, 1H), 10.3 (s, 1H); HRMS calcd for C10H10ClN2O2 225.0425; found (ESI+) (M + H) 225.0429.
2-Chloro-N-(2-oxoindolin-5-yl)acetamide (40.0 mg, 0.178 mmol, 1 equiv) was added to a solution of pyrrole-2-carboxaldehyde (84.7 mg, 0.891 mmol, 5 equiv), and piperidine (1.5 mg, 0.018 mmol, 0.1 equiv) in THF (6 mL). The resulting mixture was heated to 75 °C in a sealed jar for 5 h. After cooling to room temperature, the solvent was removed under vacuum. The residue was then triturated in EtOAc, recovered by filtration, and dried under vacuum to give pure 3 (34 mg, 63%) as a yellow/orange solid. 1H NMR (DMSO-d6): δ 4.24 (s, 2H), 6.34-6.37 (m, 1H), 6.85 (d, J = 8.4 Hz, 1H), 6.91-6.94 (m, 1H), 7.20 (d, J = 8.4 Hz, 1H), 7.35-7.38 (m, 1H), 7.65 (s, 1H), 7.92 (s, 1H), 10.2 (s, 1H), 10.9 (s, 1H), 13.3 (bs, 1H); HRMS calcd for C15H13ClN3O2 302.0696; found (ESI+) (M + H) 302.0694.
(Z)-3-((1H-Pyrrol-2-yl)methylene)-5-((E)-3-oxobut-1-enyl)indolin-2-one (4)
Under an atmosphere of argon, indolinone 20 (50.0 mg, 0.149 mmol, 1 equiv), triphenylphosphine (23.0 mg, 0.0876 mmol, 0.6 equiv), triethylamine (75.0 mg, 0.741 mmol, 5 equiv), methyl vinyl ketone (42.0 mg, 0.599 mmol, 4 equiv), and palladium(II) acetate (10.0 mg, 0.0445 mmol, 0.3 equiv) were added to anhydrous DMF (2 mL). The mixture was sparged with dry argon, then heated to 85 °C for 2 h. After cooling to room temperature, the mixture was diluted with water (50 mL) and extracted four times with EtOAc (40 mL). The combined EtOAc extract was washed with water, then saturated NaCl, and concentrated under vacuum to give a crude orange solid. This material was further purified by flash chromatography on silica gel (hexanes/EtOAc 1:1) to give 4 (20 mg, 48%) as a yellow solid. A small amount of this material was further purified by preparative HPLC (acetonitrile/water, linear gradient elution, 5% to 80% acetonitrile over 20 min) for use in bioassays. 1H NMR (DMSO-d6): δ 2.33 (s, 3H), 6.38-6.40 (m, 1H), 6.80 (d, J = 16 Hz, 1H), 6.84-6.86 (m, 1H), 6.93 (d, J = 8.1 Hz, 1H), 7.39-7.41 (m, 1H), 7.47 (dd, J = 8.2, 1.6 Hz, 1H), 7.61 (d, J = 16 Hz, 1H), 7.91 (s, 1H), 8.15 (d, J = 1.5 Hz, 1H), 11.1 (s, 1H), 13.2 (bs, 1H); HRMS calcd for C17H15N2O2 279.1134; found (ESI+) (M + H) 279.1130.
(Z)-3-((1H-Pyrrol-2-yl)methylene)-5-((E)-2-(methylsulfonyl)vinyl)indolin-2-one (5)
Under an atmosphere of argon, indolinone 20 (50.0 mg, 0.149 mmol, 1 equiv), triphenylphosphine (23.0 mg, 0.0876 mmol, 0.6 equiv), triethylamine (75.0 mg, 0.741 mmol, 5 equiv), methyl vinyl sulfone (63.0 mg, 0.594 mmol, 4 equiv), and palladium(II) acetate (10.0 mg, 0.0445 mmol, 0.3 equiv) were added to anhydrous DMF (2 mL). The mixture was sparged with dry argon, then heated to 85 °C for 2 h. After cooling to room temperature, the DMF was removed under vacuum and the remaining black residue was purified by flash chromatography on silica gel (hexanes/EtOAc 2:1 then hexanes/EtOAc 1:1) to give pure 5 (16 mg, 34%) as a yellow solid. 1H NMR (DMSO-d6): δ 3.09 (s, 3H), 6.39-6.41 (m, 1H), 6.84-6.86 (m, 1H), 6.94 (d, J = 8.1 Hz, 1H), 7.34 (d, J = 15 Hz, 1H), 7.40-7.42 (m, 1H), 7.47 (d, J = 15 Hz, 1H), 7.49 (dd, J = 8.1, 1.5 Hz, 1H), 7.84 (s, 1H), 8.14 (d, J = 1.1 Hz, 1H), 11.2 (s, 1H), 13.2 (s, 1H); HRMS calcd for C16H15N2O3S 315.0803; found (ESI+) (M + H) 315.0798.
(E)-Ethyl 4-((Z)-3-((1H-pyrrol-2-yl)methylene)-2-oxoindolin-5-ylamino)-4-oxobut-2-enoate (6)
A solution of monoethyl fumarate (175 mg, 1.21 mmol, 1.2 equiv) in acetonitrile (3 mL) was added dropwise over 10 min to a stirred solution of 5- aminoindolin-2-one (150 mg, 1.01 mmol, 1 equiv) and EDC (387 mg, 2.02 mmol, 2 equiv) in acetonitrile (50 mL) at room temperature. After stirring for 105 min, water (5 mL) was added and most of the acetonitrile removed under vacuum. The resulting precipitate was recovered by filtration, washed with water, and dried under vacuum to give pure (E)-ethyl 4-oxo-4-(2-oxoindolin-5-ylamino)but-2-enoate (153 mg, 55%) as a white solid. 1H NMR (DMSO-d6): δ 1.26 (t, J = 7.1 Hz, 3H), 3.48 (s, 2H), 4.21 (q, J = 7.2 Hz, 2H), 6.67 (d, J = 15 Hz, 1H), 6.78 (d, J = 8.4 Hz, 1H), 7.19 (d, J = 15 Hz, 1H), 7.45 (dd, J = 8.2, 2.0 Hz, 1H), 7.61 (s, 1H), 10.3 (s, 1H), 10.4 (s, 1H); HRMS calcd for C14H15N2O4 275.1032; found (ESI ) (M + H) 275.1029.
(E)-Ethyl 4-oxo-4-(2-oxoindolin-5-ylamino)but-2-enoate (40.0 mg, 0.198 mmol, 1 equiv) was added to a solution of pyrrole-2-carboxaldehyde (188 mg, 1.98 mmol, 10 equiv), and piperidine (3.5 mg, 0.041 mmol, 0.2 equiv) in THF (6 mL). The resulting mixture was heated to 75 °C in a sealed jar for 20 h. After cooling to room temperature the solvent was removed under vacuum. The residue was then triturated in 1:1 hexanes/EtOAc, recovered by filtration, and dried under vacuum. This crude product was further purified by dissolving in THF (2 mL) and treating with hexanes (2.5 mL). The resulting precipitate was recovered by filtration, washed with hexanes, and dried under vacuum to give pure 6 (35 mg, 50%) as an orange solid. 1H NMR (DMSO-d6): δ 1.27 (t, J = 7.1 Hz, 3H), 4.22 (q, J = 7.1 Hz, 2H), 6.35-6.38 (m, 1H), 6.7 (d, J = 15 Hz, 1H), 6.87 (d, J = 8.1 Hz, 1H), 6.94-6.97 (m, 1H), 7.24 (d, J = 15 Hz, 1H), 7.27 (dd, J = 8.2, 1.6 Hz, 1H), 7.35-7.38 (m, 1H), 7.65 (s, 1H), 8.08 (d, J = 1.8, 1H), 10.5 (s, 1H), 10.9 (s, 1H), 13.3 (bs, 1H); HRMS calcd for C19H18N3O4 352.1297; found (ESI+) (M + H) 352.1296.
(Z)-N-(3-((1H-Pyrrol-2-yl)methylene)-2-oxoindolin-5-yl)acrylamide (7)
Acryloyl chloride (191 mg, 2.11 mmol, 1.2 equiv) was added dropwise over the course of 5 min to a solution of 5-aminoindolin-2-one (261mg, 1.76 mmol, 1 equiv) in anhydrous THF (100 mL) at 0 °C. The mixture was allowed to warm to room temperature for 5 min, then filtered to remove some insoluble material. The filtrate was then concentrated under vacuum, and the remaining residue dissolved in EtOAc. This solution was washed with 5% NaHCO3, then water, and concentrated under vacuum to give pure N-(2-oxoindolin-5-yl)acrylamide (147 mg, 41%) as a white solid. 1H NMR (DMSO-d6): δ 3.47 (s, 2H), 5.71 (dd, J = 9.9, 2.2 Hz, 1H), 6.21 (dd, J = 17, 2.2 Hz, 1H), 6.40 (dd, J = 17, 10 Hz, 1H), 6.75 (d, J = 8.4 Hz, 1H), 7.41 (dd, J = 8.4, 2.2 Hz, 1H), 7.59 (d, J = 1.8 Hz, 1H), 9.98 (s, 1H), 10.3 (bs, 1H); HRMS calcd for C11H11N2O2 203.0821; found (ESI+) (M + H) 203.0818.
N-(2-oxoindolin-5-yl)acrylamide (40.0 mg, 0.198 mmol, 1 equiv) was added to a solution of pyrrole-2-carboxaldehyde (94.2 mg, 0.990 mmol, 5 equiv) and piperidine (1.7 mg, 0.020 mmol, 0.1 equiv) in THF (6 mL). The resulting mixture was heated to 75 °C in a sealed jar for 4 h. After cooling to room temperature, the solvent was removed under vacuum. The residue was then triturated in EtOAc, recovered by filtration, and dried under vacuum to give pure 7 (31 mg, 56%) as a yellow/orange solid. 1H NMR (DMSO-d6): δ 5.74 (d, J = 10 Hz, 1H), 6.25 (d, J = 17 Hz, 1H), 6.34-6.37 (m, 1H), 6.44 (dd, J = 17, 10 Hz, 1H), 6.84 (d, J = 8.4 Hz, 1H), 6.91-6.94 (m, 1H), 7.26 (d, J = 8.1 Hz, 1H), 7.34-7.38 (m, 1H), 7.62 (s, 1H), 8.04 (s, 1H), 10.1 (s, 1H), 10.9 (s, 1H), 13.3 (bs, 1H); HRMS calcd for C16H14N3O2 280.1086; found (ESI+) (M + H) 280.1083.
(Z)-N-(3-((1H-Pyrrol-2-yl)methylene)-2-oxoindolin-5-yl)propiolamide (8)
A solution of propiolic acid (114 mg, 1.63 mmol, 1.2 equiv) in acetonitrile (10 mL) was added dropwise over 10 min to a stirred solution of 5-aminoindolin-2-one (200 mg, 1.35 mmol, 1 equiv), and EDC (518 mg, 2.70 mmol, 2 equiv) in acetonitrile (70 mL) at room temperature. Stirring was continued for 30 min; water (20 mL) was added and most of the acetonitrile removed under vacuum. The resulting precipitate was recovered by filtration, washed with water, and dried under vacuum to give pure N-(2-oxoindolin-5-yl)propiolamide (108 mg, 40%) as a white solid. 1H NMR (DMSO-d6): δ 3.46 (s, 2H), 4.35 (s, 1H), 6.75 (d, J = 8.4 Hz, 1H), 7.37 (dd, J = 8.4, 1.8 Hz, 1H), 7.49 (d, J = 1.8 Hz, 1H), 10.3 (s,1H), 10.7 (s, 1H); HRMS calcd for C11H9N2O2 201.0659; found (ESI+) (M + H) 201.0661.
N-(2-Oxoindolin-5-yl)propiolamide (40.0 mg, 0.200 mmol, 1 equiv) was added to a solution of pyrrole-2-carboxaldehyde (190 mg, 2.00 mmol, 10 equiv), and piperidine (3.3 mg, 0.039 mmol, 0.2 equiv) in THF (6 mL). The resulting mixture was heated to 75 °C in a sealed jar for 18 h. After cooling to room temperature, the solvent was removed under vacuum. The residue was purified by flash chromatography on silica gel (hexanes/EtOAc 2:1) to give 8 (16 mg, 29%) as an orange solid. A small amount of this material was further purified by preparative HPLC (acetonitrile/water, linear gradient elution, 5% to 80% acetonitrile over 20 min) for use in bioassays. 1H NMR (DMSO-d6): δ 4.37 (s, 1H), 6.35-6.37 (m, 1H), 6.84 (d, J = 8.4 Hz, 1H), 6.93-6.95 (m, 1H), 7.22 (dd, J = 8.2, 2.0 Hz, 1H), 7.35-7.38 (m, 1H), 7.61 (s, 1H), 7.91 (d, J = 1.8 Hz, 1H), 10.7 (s, 1H), 10.9 (s, 1H), 13.3 (bs, 1H); HRMS calcd for C16H11N3O2 277.0851; found (ESI+) (M +) 277.0848.
(Z)-3-((1H-Imidazol-5-yl)methylene)-5-(2-chloroacetyl)indolin-2-one (9)
Indolinone 21 (50.0 mg, 0.197 mmol, 1 equiv) was added to a suspension of 1H-imidazole-5-carbaldehyde (20.8 mg, 0.216 mmol, 1.1 equiv), and piperidine (5.0 mg, 0.059 mmol, 0.3 equiv) in THF (4 mL). The resulting mixture was heated to 75 °C in a sealed jar for 7 h. After cooling to room temperature, conc. HCl (300 μL) was added and stirring continued for 4.5 h. The mixture was treated with EtOAc (5 mL) and the resulting precipitate collected by filtration, triturated in 2 N NaOH, washed with water, and dried under vacuum to give pure 9 (23 mg, 41%) as a yellow solid. 1H NMR (DMSO-d6): δ 5.16 (s, 2H), 7.03 (d, J = 8.4 Hz, 1H), 7.72 (s, 1H), 7.89 (d, J = 8.4 Hz, 1H), 8.06 (s, 1H), 8.09 (s, 1H), 8.37 (s, 1H), 11.4 (s, 1H), 13.5 (bs, 1H); HRMS calcd for C14H11ClN3O2 288.0540; found (ESI+) (M + H) 288.0532.
(Z)-5-(2-Chloroacetyl)-3-((4-methyl-1H-imidazol-5-yl)methylene)indolin-2-one (10)
Indolinone 21 (50.0 mg, 0.197 mmol, 1 equiv) was added to a suspension of 4-methyl-1H-imidazole-5-carbaldehyde (23.9 mg, 0.216 mmol, 1.1 equiv) and piperidine (5.0 mg, 0.059 mmol, 0.3 equiv) in THF (3 mL). The resulting mixture was heated to 75 °C in a sealed jar for 9 h. After cooling to room temperature, conc. HCl (300 μL) was added and stirring continued for 24 h. The resulting precipitate was collected by filtration, washed with EtOAc, triturated in 2 N NaOH, washed with water, and dried under vacuum to give pure 10 (29 mg, 49%) as a yellow solid. 1H NMR (DMSO-d6): δ 2.51 (s, 3H), 5.18 (s, 2H), 7.02 (d, J = 8.4 Hz, 1H), 7.86 (dd, J = 8.3, 1.7 Hz, 1H), 7.97 (s, 1H), 7.99 (s, 1H), 8.52 (d, J = 1.8 Hz, 1H), 11.4 (s, 1H), 13.7 (bs, 1H); HRMS calcd for C15H13ClN3O2 302.0696; found (ESI+) (M + H) 302.0693.
(Z)-5-(2-Chloroacetyl)-3-((2-ethyl-4-methyl-1H-imidazol-5-yl)methylene)indolin-2-one (11)
Indolinone 21 (50.0 mg, 0.197 mmol, 1 equiv) was added to a suspension of 2-ethyl-4-methyl-1H-imidazole-5-carbaldehyde (33.0 mg, 0.239 mmol, 1.2 equiv) and piperidine (5.0 mg, 0.059 mmol, 0.3 equiv) in THF (5 mL). The resulting mixture was heated to 75 °C in a sealed jar for 20 h. After cooling to room temperature, conc. HCl (300 μL) was added and stirring continued for 20 h. The resulting precipitate was collected by filtration, triturated in 1 N NaOH, washed with water, and dried under vacuum to give pure 11 (32 mg, 49%) as a yellow solid. 1H NMR (DMSO-d6): δ 1.28 (t, J = 7.5 Hz, 3H), 2.47 (s, 3H), 2.78 (q, J = 7.6 Hz, 2H), 5.18 (s, 2H), 7.02 (d, J = 8.4 Hz, 1H), 7.84 (dd, J = 8.2, 1.6 Hz, 1H), 7.94 (s, 1H), 8.49 (d, J = 1.6 Hz, 1H), 11.4 (s, 1H), 13.7 (bs, 1H); HRMS calcd for C17H17ClN3O2 330.1009; found (ESI+) (M + H) 330.1000.
(Z)-5-(2-Chloroacetyl)-3-((2-ethyl-4-methyl-1H-imidazol-5-yl)methylene)-1-methylindolin-2-one hydrochloride (12)
Chloroacetyl chloride (1.95g, 17.3 mmol, 1.7 equiv) was added dropwise to a stirred suspension of anhydrous AlCl3 (8.10 g, 60.9 mmol, 6 equiv) in carbon disulfide (40 mL). Stirring was allowed to continue at room temperature for 15 min. 1-Methylindolin-2-one (1.50 g, 10.2 mmol, 1 equiv) was then added and the mixture was brought to reflux for 2 h, then cooled on ice. After decanting the solvent, the remaining thick brown residue was treated with ice cold water. The resulting precipitate was recovered by filtration, washed with water, and dried under vacuum to provide pure 5-(2-chloroacetyl)-1-methylindolin-2-one (2.0 g, 88%) as a white solid. 1H NMR (DMSO-d6): δ 3.17 (s, 3H), 3.64 (s, 2H), 5.11 (s, 2H), 7.12 (d, J = 8.4 Hz, 1H), 7.86 (d, J = 1.1 Hz, 1H), 7.99 (dd, J = 8.4, 1.8 Hz, 1H); HRMS calcd for C11H11ClNO2 224.0478; found (ESI+) (M + H) 224.0475.
Ethylene glycol (555 mg, 8.94 mmol, 2 equiv) and pTSA·H2O (170 mg, 0.894 mmol, 0.2 equiv) were added to a suspension of indolinone 5-(2-chloroacetyl)-1-methylindolin-2-one (1.00 g, 4.47 mmol, 1 equiv) in benzene (200 mL). The mixture was brought to reflux using a Dean Stark trap and stirred vigorously for 5 h. After cooling to room temperature, the mixture was washed with 5% NaHCO3, then saturated NaCl, and concentrated under vacuum to give 5-(2-(chloromethyl)-1,3-dioxolan-2-yl)-1-methylindolin-2-one (1.2 g, 100%) as a tan solid. 1H NMR (DMSO-d6): δ 3.06 (s, 3H), 3.50 (s, 2H), 3.76-3.80 (m, 2H), 3.82 (s, 2H), 4.02-4.06 (m, 2H), 6.91 (d, J = 8.1 Hz, 1H), 7.31 (s, 1H), 7.33 (d, J = 8.4 Hz, 1H); HRMS calcd for C13H15ClNO3 268.0740; found (ESI+) (M + H) 268.0737.
5-(2-(Chloromethyl)-1,3-dioxolan-2-yl)-1-methylindolin-2-one (50.0 mg, 0.187 mmol, 1 equiv) was added to a suspension of 2-ethyl-4-methyl-1H-imidazole-5-carbaldehyde (25.0 mg, 0.181 mmol, 0.97 equiv), and piperidine (4.8 mg, 0.056 mmol, 0.3 equiv) in THF (6 mL). The resulting mixture was heated to 75 °C in a sealed jar for 18 h. After cooling to room temperature conc. HCl (300 μL) was added, and stirring was allowed to continue for an additional 18 h. The resulting precipitate was collected by filtration, washed with THF, and dried under vacuum to give pure 12 (40 mg, 60%) as an orange solid. 1H NMR (DMSO-d6): δ 1.30 (t, J = 7.7 Hz, 3H), 2.41 (s, 3H), 2.76 (q, J = 7.7 Hz, 2H), 3.18 (s, 3H), 4.29 (s, 1H), 6.89 (d, J = 8.4 Hz, 1H), 7.24 (dd, J = 8.4, 2.2 Hz, 1H), 7.43 (s, 1H), 9.70 (d, J = 1.8 Hz, 1H), 10.5 (s, 1H), 12.3 (bs, 1H); HRMS calcd for C18H19ClN3O2 344.1166; found (ESI+) (M + H) 344.1153.
(Z)-5-Acetyl-3-((2-ethyl-4-methyl-1H-imidazol-5-yl)methylene)indolin-2-one (13)
5-Acetylindolin-2-one (100 mg, 0.571 mmol, 1 equiv) was added to a suspension of 2-ethyl-4-methyl-1H-imidazole-5-carbaldehyde (71.0 mg, 0.514 mmol, 0.9 equiv) and piperidine (14.0 mg, 0.164 mmol, 0.3 equiv) in THF (8 mL). The resulting mixture was heated to 75 °C in a sealed jar for 16 h. After cooling to room temperature, the product was collected by filtration and washed with THF and EtOAc to give 13 (57 mg, 34%) as an orange solid. A small amount of this material was further purified by preparative HPLC (acetonitrile/water, linear gradient elution, 5% to 80% acetonitrile over 20 min) for use in bioassays. 1H NMR (DMSO-d6): δ 1.28 (t, J = 7.7 Hz, 3H), 2.46 (s, 3H), 2.59 (s, 3H), 2.78 (q, J = 7.6 Hz, 2H), 6.98 (d, J = 8.4 Hz, 1H), 7.82 (dd, J = 8.2, 1.6 Hz, 1H), 7.94 (s, 1H), 8.46 (d, J = 1.8 Hz, 1H), 11.3 (bs, 1H), 13.7 (bs, 1H); HRMS calcd for C17H18N3O2 296.1394; found (ESI+) (M + H) 296.1396.
(Z)-N-(3-((1H-Imidazol-5-yl)methylene)-2-oxoindolin-5-yl)propiolamide (14)
Indolinone 22 (750 mg, 3.02 mmol, 1 equiv) was added to a suspension of 1H-imidazole-5-carbaldehyde (320 mg, 3.33 mmol, 1.1 equiv) and piperidine (50.4 mg, 0.592 mmol, 0.2 equiv) in THF (45 mL). The resulting mixture was heated to 75 °C in a sealed jar for 20 h, then concentrated under vacuum. The residue was triturated in EtOAc, collected by filtration, washed with 1:1 hexanes/EtOAc, and then treated with 80 mL of methanolic HCl (prepared fresh by dissolving 4% v/v acetyl chloride in MeOH) for 23 h. The mixture was concentrated under vacuum to give pure (Z)-3-((1H-imidazol-5-yl)methylene)-5-aminoindolin-2-one dihydrochloride (750 mg, 83%) as a yellow solid. 1H NMR (DMSO-d6): δ 7.02 (d, J = 8.1 Hz, 1H), 7.26 (d, J = 8.4 Hz, 1H), 7.59 (s, 1H), 7.94 (s, 1H), 8.36 (s, 1H), 8.99 (s, 1H), 11.4 (s, 1H); HRMS (free base) calcd for C12H11N4O 227.0933; found (ESI+) (M + H) 227.0926.
Under an atmosphere of argon, DIPEA (45.5 mg, 0.352 mmol, 2.1 equiv) was added to a stirred suspension of (Z)-3-((1H-imidazol-5-yl)methylene)-5-aminoindolin-2- one dihydrochloride (50.0 mg, 0.167 mmol, 1 equiv) in DMF (5 mL). The resulting red solution was cooled to 0 °C then treated with propiolic acid (70.2 mg, 1.00 mmol, 6 equiv) followed by EDC (67.5 mg, 0.352 mmol, 2.1 equiv). After stirring for 2 h, the mixture was diluted with EtOAc (150 mL), washed three times with water (50 mL), then with saturated NaCl, and concentrated under vacuum to give a crude orange solid. This material was further purified by preparative HPLC (acetonitrile/water, linear gradient elution, 5% to 80% acetonitrile over 20 min) to give pure 14 (3.8 mg, 8.2%) as a yellow/orange solid. 1H NMR (DMSO-d6): δ 4.38 (s, 1H), 6.86 (d, J = 8.4 Hz, 1H), 7.27 (d, J = 8.4 Hz, 1H), 7.74 (s, 1H), 7.74 (s, 1H), 7.96 (s, 1H), 8.02 (s, 1H), 10.7 (s, 1H), 11.0 (bs, 1H), 13.7 (bs, 1H); HRMS calcd for C15H11N4O2 279.0882; found (ESI+) (M + H) 279.0876.
(Z)-N-(3-((4-Methyl-1H-imidazol-5-yl)methylene)-2-oxoindolin-5-yl)propiolamide (15)
Indolinone 22 (750 mg, 3.02 mmol, 1 equiv) was added to a suspension of 4-methyl-1H-imidazole-5-carbaldehyde (366 mg, 3.32 mmol, 1.1 equiv) and piperidine (50.4 mg, 0.592 mmol, 0.2 equiv) in THF (45 mL). The resulting mixture was heated to 75 °C in a sealed jar for 20 h, then concentrated under vacuum. The residue was triturated in EtOAc, collected by filtration, washed with 1:1 hexanes/EtOAc, and then treated with 80 mL of methanolic HCl (prepared fresh by dissolving 4% v/v acetyl chloride in MeOH) for 48 h. The mixture was concentrated to dryness under vacuum to give pure (Z)-5-amino-3-((4-methyl-1H-imidazol-5-yl)methylene)indolin-2-one dihydrochloride (781 mg, 83%) as a yellow solid. 1H NMR (DMSO-d6): δ 2.63 (s, 3H), 7.02 (d, J = 8.1 Hz, 1H), 7.30 (dd, J = 8.1, 1.6 Hz, 1H), 7.86 (d, J = 1.8 Hz, 1H), 8.00 (s, 1H), 9.01 (s, 1H), 11.5 (bs, 1H); HRMS (free base) calcd for C13H13N4O 241.1089; found (ESI+) (M + H) 241.1081.
DIPEA (41.4 mg, 0.320 mmol, 2 equiv) was added to a suspension of (Z)-5-amino-3-((4-methyl-1H-imidazol-5-yl)methylene)indolin-2-one dihydrochloride (50.0 mg, 0.160 mmol, 1 equiv) in DMF (5 mL) at 0 °C with stirring. The resulting red solution was treated with propiolic acid (16.8 mg, 0.240 mmol, 1.5 equiv), then EDC (61.3 mg, 0.320 mmol, 2 equiv). After stirring for 2.5 h, the mixture was diluted with 5% NaHCO3 (50 mL) and extracted three times with EtOAc (50 mL). The combined EtOAc extract was washed with water, then saturated NaCl, and concentrated under vacuum. The residue was triturated in 1:1 hexanes/EtOAc, recovered by filtration, and washed with EtOAc, to give 15 (15 mg, 32%) as an orange solid. A small amount of this material was further purified by preparative HPLC (acetonitrile/water, linear gradient elution, 5% to 80% acetonitrile over 20 min) for use in bioassays. 1H NMR (DMSO-d6): δ 2.44 (s, 3H), 4.36 (s, 1H), 6.85 (d, J = 8.4 Hz, 1H), 7.27 (dd, J = 8.4, 2.2 Hz, 1H), 7.62 (s, 1H), 7.93 (s, 1H), 7.94 (d, J = 1.8 Hz, 1H), 10.7 (s, 1H), 11.0 (s, 1H), 13.8 (bs, 1H); HRMS calcd for C16H12N4O2 292.0960; found (ESI+) (M +) 292.0965.
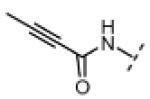
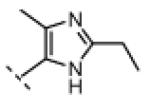
(Z)-N-(3-((2-ethyl-4-methyl-1H-imidazol-5-yl)methylene)-2-oxoindolin-5-yl)propiolamide (16)
Indolinone 22 (750 mg, 3.02 mmol, 1 equiv) was added to a suspension of 2-ethyl-4-methyl-1H-imidazole-5-carbaldehyde (459 mg, 3.32 mmol, 1.1 equiv) and piperidine (50.4 mg, 0.592 mmol, 0.2 equiv) in THF (45 mL). The resulting mixture was heated to 75 °C in a sealed jar for 20 h, then concentrated under vacuum. The residue was triturated in EtOAc, collected by filtration, washed with 1:1 hexanes/EtOAc, and treated with 80 mL of methanolic HCl (prepared fresh by dissolving 4% v/v acetyl chloride in MeOH) for 48. The mixture was concentrated to dryness under vacuum to give pure (Z)-5-amino-3-((2-ethyl-4-methyl-1H-imidazol-5-yl)methylene)indolin-2-one dihydrochloride (740 mg, 72%) as a yellow solid. 1H NMR (DMSO-d6): δ 1.36 (t, J = 7.5 Hz, 3H), 2.60 (s, 3H), 3.04 (q, J = 7.6 Hz, 2H), 7.01 (d, J = 8.4 Hz, 1H), 7.23 (d, J = 8.4 Hz, 1H), 7.78 (bs, 1H), 7.99 (s, 1H), 11.5 (s, 1H); HRMS (free base) calcd for C15H17N4O 269.1402; found (ESI+) (M + H) 269.1394.
DIPEA (45.5 mg, 0.352 mmol, 2 equiv) was added to a stirred suspension of (Z)-5-amino-3-((2-ethyl-4-methyl-1H-imidazol-5-yl)methylene)indolin-2-one dihydrochloride (60.0 mg, 0.176 mmol, 1 equiv) in DMF (5 mL) at 0 °C. The resulting red solution was treated with propiolic acid (18.5 mg, 0.264 mmol, 1.5 equiv), followed by EDC (67.5 mg, 0.352 mmol, 2 equiv). After stirring was continued for 2.5 h, then the mixture was allowed to warm to room temperature over 1.5 h. The mixture was then diluted with 5% NaHCO3 (50 mL) and extracted three times with EtOAc (50 mL). The combined EtOAc extract was washed with water, then saturated NaCl, and concentrated under vacuum. The resulting residue was triturated in EtOAc, recovered by filtration, washed with EtOAc, and dried under vacuum to give pure 16 (25 mg, 44%) as a yellow/orange solid. 1H NMR (DMSO-d6): δ 1.27 (t, J = 7.5 Hz, 3H), 2.40 (s, 3H), 2.76 (q, J = 7.6 Hz, 2H), 4.36 (s, 1H), 6.85 (d, J = 8.4 Hz, 1H), 7.24 (dd, J = 8.4, 1.5 Hz, 1H), 7.56 (s, 1H), 7.92 (d, J = 1.8 Hz, 1H), 10.6 (s, 1H), 10.9 (bs, 1H), 13.8 (bs, 1H); HRMS calcd for C18H17N4O2 321.1352; found (ESI+) (M + H) 321.1368.
(Z)-N-(3-((2-Ethyl-4-methyl-1H-imidazol-5-yl)methylene)-1-methyl-2-oxoindolin-5-yl)propiolamide (17)
HNO3 (70% aqueous, 1.41 g, 22.4 mmol, 1.7 equiv) was slowly added to a solution of 1-methylindolin-2-one (2.00 g, 13.6 mmol, 1 equiv) in conc. H2SO4 (8 mL) at 0 °C with stirring. After stirring for 30 min, the reaction was poured onto a water/ice mixture (200 mL). The resulting precipitate was collected by filtration and washed with water (250 mL) to give pure 1-methyl-5-nitroindolin-2-one (2.3 g, 88%) as a tan solid. 1H NMR (DMSO-d6): δ 3.19 (s, 3H), 3.70 (s, 2H), 7.19 (d, J = 8.4 Hz, 1H), 8.14 (s, 1H), 8.26 (d, J = 8.8 Hz, 1H); HRMS calcd for C9H7N2O3 191.0462; found (ESI−) (M − H) 191.0464.
1-Methyl-5-nitroindolin-2-one (100 mg, 0.520 mmol, 1 equiv) was added to a suspension of 2-ethyl-4-methyl-1H-imidazole-5-carbaldehyde (68.3 mg, 0.494 mmol, 0.95 equiv) and piperidine (13.3 mg, 0.156 mmol, 0.3 equiv) in THF (7 mL). The resulting mixture was heated to 75 °C in a sealed jar for 18 h. After cooling to room temperature, the resulting precipitate was collected by filtration and washed with EtOAc to give pure (Z)-3-((2-Ethyl-4-methyl-1H-imidazol-5-yl)methylene)-1-methyl-5- nitroindolin-2-one (113 mg, 73%) as an orange solid. 1H NMR (DMSO-d6): δ 1.43 (t, J = 7.5 Hz, 3H), 2.45 (s, 3H), 2.75 (q, J = 7.6 Hz, 2H), 3.29 (s, 3H), 7.16 (d, J = 8.4 Hz, 1H), 7.55 (s, 1H), 8.15 (dd, J = 8.8, 2.6 Hz, 1H), 10.6 (d, J = 2.6 Hz, 1H), 12.5 (bs, 1H); HRMS calcd for C16H17N4O3 313.1301; found (ESI+) (M + H) 313.1293.
Zinc dust (1.02 g, 15.6 mmol, 47 equiv) was added to a suspension of (Z)-3-((2-ethyl-4-methyl-1H-imidazol-5-yl)methylene)-1-methyl-5-nitroindolin-2-one (105 mg, 0.336 mmol, 1 equiv) in THF (40 mL) and saturated aqueous NH4Cl (20 mL) at room temperature. The resulting mixture was vigorously stirred for 10 min, then diluted with EtOAc (150 mL), washed with water, then saturated NaCl, and concentrated under vacuum to give pure (Z)-5-amino-3-((2-ethyl-4-methyl-1H-imidazol-5-yl)methylene)-1-methylindolin-2-one (67 mg, 71%) as an red/orange solid. 1H NMR (DMSO-d6): δ 1.32 (t, J = 7.7 Hz, 3H), 2.38 (s, 3H), 2.75 (q, J = 7.6 Hz, 2H), 3.11 (s, 3H), 4.50 (bs, 2H), 6.50 (dd, J = 8.1, 2.6 Hz, 1H), 6.62 (d, J = 8.1 Hz, 1H), 7.32 (s, 1H), 8.81 (d, J = 2.2 Hz, 1H), 12.2 (bs, 1H); HRMS calcd for C16H19N4O 283.1553; found (ESI+) (M + H) 283.1552.
EDC (80.3 mg, 0.419 mmol, 2 equiv) was added to a solution of indolinone (Z)-5-amino-3-((2-ethyl-4-methyl-1H-imidazol-5-yl)methylene)-1-methylindolin-2-one (59.1 mg, 0.209 mmol, 1 equiv) and propiolic acid (22.2 mg, 0.317 mmol, 1.5 equiv) in DMF (4 mL) at 0 °C. After stirring for 2 h, the mixture was diluted with EtOAc (100 mL), washed with water, then saturated NaCl, and concentrated under vacuum to give a crude orange solid. This material was further purified by preparative HPLC (acetonitrile/water, linear gradient elution, 5% to 80% acetonitrile over 20 min) to give pure 17 (16 mg, 23%) as an orange solid. 1H NMR (DMSO-d6): δ 1.30 (t, J = 7.7 Hz, 3H), 2.41 (s, 3H), 2.76 (q, J = 7.7 Hz, 2H), 3.18 (s, 3H), 4.29 (s, 1H), 6.89 (d, J = 8.4 Hz, 1H), 7.24 (dd, J = 8.4, 2.2 Hz, 1H), 7.43 (s, 1H), 9.70 (d, J = 1.8 Hz, 1H), 10.5 (s, 1H), 12.3 (bs, 1H); HRMS calcd for C19H19N4O2 335.1508; found (ESI+) (M + H) 335.1493.
(Z)-N-(3-((2-Ethyl-4-methyl-1H-imidazol-5-yl)methylene)-2-oxoindolin-5-yl)but-2-ynamide (18)
To a solution of (Z)-5-amino-3-((2-ethyl-4-methyl-1H-imidazol-5-yl)methylene)indolin-2-one dihydrochloride (50.0 mg, 0.147 mmol, 1 equiv), EDC (112 mg, 0.584 mmol, 4 equiv), HOBt (20.0 mg, 0.148 mmol, 1 equiv), and 2-butynoic acid (74.0 mg, 0.880 mmol, 6 equiv), in DMF (6 mL) was added DIPEA (57.0 mg, 0.441 mmol, 3 equiv). After stirring for 2 h, the mixture was diluted with 5% NaHCO3 (20 mL) and extracted with EtOAc. The EtOAc extract was washed with water, then saturated NaCl, and concentrated to dryness to give pure 18 (25 mg, 51%) as an orange solid. 1H NMR (DMSO-d6): δ 1.27 (t, J = 7.7 Hz, 3H), 2.04 (s, 3H), 2.40 (s, 3H), 2.75 (q, J = 7.6 Hz, 2H), 6.83 (d, J = 8.4. 1H), 7.22 (dd, J = 8.2, 2.0 Hz, 1H), 7.53 (s, 1H), 7.92 (d, J = 1.8 Hz, 1H), 10.4 (s, 1H), 10.9 (bs, 1H), 13.8 (s, 1H); HRMS calcd for C19H19N4O2 335.1503; found (ESI+) (M + H) 335.1496.
Supplementary Material
Acknowledgment
We gratefully acknowledge the UCSF Mass Spectrometry Facility (A.L. Burlingame, Director, NIH NCRR P41RR001614), Stefan Knapp (University of Oxford) for Nek2 expression plasmids, Nick Hertz (UCSF) for RO3306 and Rand Miller (UCSF) for help with kinase assays. J.H. acknowledges the ARCS Foundation for funding and members of the Taunton lab for support. This work was funded by the NIH (GM071434).
Footnotes
Supporting Information Available. Experimental protocols for purification of Nek2 constructs, generation of stable cell lines, immunofluorescence microscopy, and Western blotting; mass spectrometry data of Nek2 adducts with compounds 12 and 16, and raw data from IP kinase assays. This material is available free of charge via the Internet at http://pubs.acs.org.
Abbreviations: BME, beta-mercaptoethanol; Cdk1, cyclin dependent kinase 1; C-Nap1, centrosomal Nek2 associated protein 1; DCM, dichloromethane; DIPEA, N,N-diisopropylethylamine; DMF, dimethylformamide; EDC, 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide hydrochloride; Eg5, kinesin family member 11; HA, hemagglutinin; IP, immunoprecipitation; Mps1, monopolar spindle kinase 1; Nek2, NIMA related kinase 2; Plk1, polo-like kinase 1; Rsk2, p90 ribosomal S6 protein kinase 2; SAC, spindle assembly checkpoint; SDS-PAGE, sodium dodecylsulfate-polyacrylamide gel electrophoresis; TLC, thin layer chromatography; WT, wild type.
References
- (1).Fry AM, Meraldi P, Nigg EA. A centrosomal function for the human Nek2 protein kinase, a member of the NIMA family of cell cycle regulators. EMBO J. 1998;17:470–481. doi: 10.1093/emboj/17.2.470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).O’Regan L, Blot J, Fry AM. Mitotic regulation by NIMA-related kinases. Cell Div. 2007;2:25. doi: 10.1186/1747-1028-2-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Bahe S, Stierhof YD, Wilkinson CJ, Leiss F, Nigg EA. Rootletin forms centriole-associated filaments and functions in centrosome cohesion. J. Cell Biol. 2005;171:27–33. doi: 10.1083/jcb.200504107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Fry AM, Mayor T, Meraldi P, Stierhof YD, Tanaka K, Nigg EA. C-Nap1, a Novel centrosomal coiled-coil protein and candidate substrate of the cell cycle–regulated protein kinase Nek2. J. Cell Biol. 1998;141:1563–1574. doi: 10.1083/jcb.141.7.1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Mardin BR, Lange C, Baxter JE, Hardy T, Scholz SR, Fry AM, Schiebel E. Components of the Hippo pathway cooperate with Nek2 kinase to regulate centrosome disjunction. Nature Cell Biol. 2010;12:1166–1176. doi: 10.1038/ncb2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Draviam VM, Stegmeier F, Nalepa G, Sowa ME, Chen J, Liang A, Hannon GJ, Sorger PK, Harper JW, Elledge SJ. A functional genomic screen identifies a role for TAO1 kinase in spindle-checkpoint signalling. Nature. Cell Biol. 2007;9:556–564. doi: 10.1038/ncb1569. [DOI] [PubMed] [Google Scholar]
- (7).Kapoor TM, Mayer TU, Coughlin ML, Mitchison TJ. Probing spindle assembly mechanisms with monastrol, a small molecule inhibitor of the mitotic kinesin, Eg5. J. Cell Biol. 2000;150:975–988. doi: 10.1083/jcb.150.5.975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Musacchio A, Salmon ED. The spindle-assembly checkpoint in space and time. Nature Rev. Mol. Cell Biol. 2007;8:379–393. doi: 10.1038/nrm2163. [DOI] [PubMed] [Google Scholar]
- (9).Strebhardt K. Multifaceted polo-like kinases: drug targets and antitargets for cancer therapy. Nature Rev. Drug Discovery. 2010;9:643–660. doi: 10.1038/nrd3184. [DOI] [PubMed] [Google Scholar]
- (10).Hauf S, Cole RW, LaTerra S, Zimmer C, Schnapp G, Walter R, Heckel A, van Meel J, Rieder CL, Peters J-M. The small molecule Hesperadin reveals a role for Aurora B in correcting kinetochore–microtubule attachment and in maintaining the spindle assembly checkpoint. J. Cell Biol. 2003;161:281–294. doi: 10.1083/jcb.200208092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Tighe A, Staples O, Taylor S. Mps1 kinase activity restrains anaphase during an unperturbed mitosis and targets Mad2 to kinetochores. J. Cell Biol. 2008;181:893–901. doi: 10.1083/jcb.200712028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Kwiatkowski N, Jelluma N, Filippakopoulos P, Soundararajan M, Manak MS, Kwon M, Choi HG, Sim T, Deveraux QL, Rottmann S, Pellman D, Shah JV, Kops GJ, Knapp S, Gray NS. Small-molecule kinase inhibitors provide insight into Mps1 cell cycle function. Nature Chem. Biol. 2010;6:359–368. doi: 10.1038/nchembio.345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Kokuryo T, Senga T, Yokoyama Y, Nagino M, Nimura Y, Hamaguchi M. Nek2 as an effective target for inhibition of tumorigenic growth and peritoneal dissemination of cholangiocarcinoma. Cancer Res. 2007;67:9637–9642. doi: 10.1158/0008-5472.CAN-07-1489. [DOI] [PubMed] [Google Scholar]
- (14).Tsunoda N, Kokuryo T, Oda K, Senga T, Yokoyama Y, Nagino M, Nimura Y, Hamaguchi M. Nek2 as a novel molecular target for the treatment of breast carcinoma. Cancer Sci. 2009;100:111–116. doi: 10.1111/j.1349-7006.2008.01007.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Zeng X, Shaikh FY, Harrison MK, Adon AM, Trimboli AJ, Carroll KA, Sharma N, Timmers C, Chodosh LA, Leone G, Saavedra HI. The Ras oncogene signals centrosome amplification in mammary epithelial cells through cyclin D1/Cdk4 and Nek2. Oncogene. 2010;29:5103–5112. doi: 10.1038/onc.2010.253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Hayward DG, Clarke RB, Faragher AJ, Pillai MR, Hagan IM, Fry AM. The centrosomal kinase Nek2 displays elevated levels of protein expression in human breast cancer. Cancer Res. 2004;64:7370–7376. doi: 10.1158/0008-5472.CAN-04-0960. [DOI] [PubMed] [Google Scholar]
- (17).Barbagallo F, Paronetto MP, Franco R, Chieffi P, Dolci S, Fry AM, Geremia R, Sette C. Increased expression and nuclear localization of the centrosomal kinase Nek2 in human testicular seminomas. J. Pathol. 2009;217:431–441. doi: 10.1002/path.2471. [DOI] [PubMed] [Google Scholar]
- (18).Andréasson U, Dictor M, Jerkeman M, Berglund M, Sundström C, Linderoth J, Rosenquist R, Borrebaeck CA, Ek S. Identification of molecular targets associated with transformed diffuse large B cell lymphoma using highly purified tumor cells. Am. J. Hematol. 2009;84:803–808. doi: 10.1002/ajh.21549. [DOI] [PubMed] [Google Scholar]
- (19).Whelligan DK, Solanki S, Taylor D, Thomson DW, Cheung KM, Boxall K, Mas-Droux C, Barillari C, Burns S, Grummitt CG, Collins I, van Montfort RL, Aherne GW, Bayliss R, Hoelder S. Aminopyrazine inhibitors binding to an unusual inactive conformation of the mitotic kinase Nek2: SAR and structural characterization. J. Med. Chem. 2010;53:7682–7698. doi: 10.1021/jm1008727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Emmitte KA, Adjabeng GM, Andrews CW, Alberti JG, Bambal R, Chamberlain SD, Davis-Ward RG, Dickson HD, Hassler DF, Hornberger KR, Jackson JR, Kuntz KW, Lansing TJ, Mook RA, Jr., Nailor KE, Pobanz MA, Smith SC, Sung CM, Cheung M. Design of potent thiophene inhibitors of polo-like kinase 1 with improved solubility and reduced protein binding. Bioorg. Med. Chem. Lett. 2009;19:1694–1697. doi: 10.1016/j.bmcl.2009.01.094. [DOI] [PubMed] [Google Scholar]
- (21).Hayward DG, Newbatt Y, Pickard L, Byrne E, Mao G, Burns S, Sahota NK, Workman P, Collins I, Aherne W, Fry AM. Identification by high-throughput screening of viridin analogs as biochemical and cell-based inhibitors of the cell cycle-regulated Nek2 kinase. J. Biomol. Screening. 2010;15:918–927. doi: 10.1177/1087057110376537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Rellos P, Ivins FJ, Baxter JE, Pike A, Nott TJ, Parkinson D-M, Das S, Howell S, Fedorov O, Shen QY, Fry AM, Knapp S, Smerdon SJ. Structure and regulation of the human Nek2 centrosomal kinase. J. Biol. Chem. 2006;282:6833–6842. doi: 10.1074/jbc.M609721200. [DOI] [PubMed] [Google Scholar]
- (23).Singh J, Dobrusin EM, Fry DW, Haske T, Whitty A, McNamara DJ. Structure-based design of a potent, selective, and irreversible inhibitor of the catalytic domain of the ErbB receptor subfamily of protein tyrosine kinases. J. Med. Chem. 1997;40:1130–1135. doi: 10.1021/jm960380s. [DOI] [PubMed] [Google Scholar]
- (24).Singh J, Petter RC, Kluge AF. Targeted covalent drugs of the kinase family. Curr. Opin. Chem. Biol. 2010;14:475–480. doi: 10.1016/j.cbpa.2010.06.168. [DOI] [PubMed] [Google Scholar]
- (25).Zhou W, Ercan D, Chen L, Yun CH, Li D, Capelletti M, Cortot AB, Chirieac L, Iacob RE, Padera R, Engen JR, Wong KK, Eck MJ, Gray NS, Jänne PA. Novel mutant-selective EGFR kinase inhibitors against EGFR T790M. Nature. 2009;462:1070–1074. doi: 10.1038/nature08622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Leproult E, Barluenga S, Moras D, Wurtz JM, Winssinger N. Cysteine mapping in conformationally distinct kinase nucleotide binding sites: application to the design of selective covalent inhibitors. J. Med. Chem. 2011;54:1347–1355. doi: 10.1021/jm101396q. [DOI] [PubMed] [Google Scholar]
- (27).Zhang J, Yang PL, Gray NS. Targeting cancer with small molecule kinase inhibitors. Nature Rev. Cancer. 2009;9:28–39. doi: 10.1038/nrc2559. [DOI] [PubMed] [Google Scholar]
- (28).Honigberg LA, Smith AM, Sirisawad M, Verner E, Loury D, Chang B, Li S, Pan Z, Thamm DH, Miller RA, Buggy JJ. The Bruton tyrosine kinase inhibitor PCI-32765 blocks B-cell activation and is efficacious in models of autoimmune disease and B-cell malignancy. PNAS. 2010;107:13075–13080. doi: 10.1073/pnas.1004594107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Cohen MS, Zhang C, Shokat KM, Taunton J. Structural bioinformatics-based design of selective, irreversible kinase inhibitors. Science. 2005;308:1318–1321. doi: 10.1126/science1108367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Cohen MS, Hadjivassiliou H, Taunton J. A clickable inhibitor reveals context-dependent autoactivation of p90 RSK. Nature Chem. Biol. 2007;3:156–160. doi: 10.1038/nchembio859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Zhou W, Hur W, McDermott U, Dutt A, Xian W, Ficarro SB, Zhang J, Sharma SV, Brugge J, Meyerson M, Settleman J, Gray NS. A structure-guided approach to creating covalent FGFR inhibitors. Chem. Biol. 2010;17:285–295. doi: 10.1016/j.chembiol.2010.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Doehn U, Hauge C, Frank SR, Jensen CJ, Duda K, Nielsen JV, Cohen MS, Johansen JV, Winther BR, Lund LR, Winther O, Taunton J, Hansen SH, Frödin M. RSK is a principal effector of the RAS-ERK pathway for eliciting a coordinate promotile/invasive gene program and phenotype in epithelial cells. Mol. Cell. 2009;35:511–22. doi: 10.1016/j.molcel.2009.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Moshinsky DJ, Bellamacina CR, Boisvert DC, Huang P, Hui T, Jancarik J, Kim SH, Rice AG. SU9516: biochemical analysis of Cdk inhibition and crystal structure in complex with Cdk2. Biochem. Biophys. Res. Commun. 2003;310:1026–1031. doi: 10.1016/j.bbrc.2003.09.114. [DOI] [PubMed] [Google Scholar]
- (34).Vassilev LT, Tovar C, Chen S, Knezevic D, Zhao X, Sun H, Heimbrook DC, Chen L. Selective small-molecule inhibitor reveals critical mitotic functions of human Cdk1. PNAS. 2006;103:10660–10665. doi: 10.1073/pnas.0600447103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Potapova TA, Daum JR, Pittman BD, Hudson JR, Jones TN, Satinover DL, Stukenberg PT, Gorbsky GJ. The reversibility of mitotic exit in vertebrate cells. Nature. 2006;440:954–958. doi: 10.1038/nature04652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Fabian MA, Biggs WH, 3rd., Treiber DK, Atteridge CE, Azimioara MD, Benedetti MG, Carter TA, Ciceri P, Edeen PT, Floyd M, Ford JM, Galvin M, Gerlach JL, Grotzfeld RM, Herrgard S, Insko DE, Insko MA, Lai AG, Lélias JM, Mehta SA, Milanov ZV, Velasco AM, Wodicka LM, Patel HK, Zarrinkar PP, Lockhart DJ. A small molecule-kinase interaction map for clinical kinase inhibitors. Nat. Biotechnol. 2005;23:329–336. doi: 10.1038/nbt1068. [DOI] [PubMed] [Google Scholar]
- (37).Fu G, Ding X, Yuan K, Aikhionbare F, Yao J, Cai X, Jiang K, Yao X. Phosphorylation of human Sgo1 by NEK2A is essential for chromosome congression in mitosis. Cell Res. 2007;17:608–618. doi: 10.1038/cr.2007.55. [DOI] [PubMed] [Google Scholar]
- (38).Faragher AJ, Fry AM. Nek2A kinase stimulates centrosome disjunction and is required for formation of bipolar mitotic spindles. Mol. Biol. Cell. 2003;14:2876–2889. doi: 10.1091/mbc.E03-02-0108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Peters U, Cherian J, Kim JH, Kwok BH, Kapoor TM. Probing cell-division phenotype space and Polo-like kinase function using small molecules. Nature Chem. Biol. 2006;2:618–626. doi: 10.1038/nchembio826. [DOI] [PubMed] [Google Scholar]
- (40).McInnes C, Mazumdar A, Mezna M, Meades C, Midgley C, Scaerou F, Carpenter L, Mackenzie M, Taylor P, Walkinshaw M, Fischer PM, Glover D. Inhibitors of Polo-like kinase reveal roles in spindle-pole maintenance. Nature Chem. Biol. 2006;2:608–617. doi: 10.1038/nchembio825. [DOI] [PubMed] [Google Scholar]
- (41).Lénárt P, Petronczki M, Steegmaier M, Di Fiore B, Lipp JJ, Hoffmann M, Rettig WJ, Kraut N, Peters JM. The small-molecule inhibitor BI 2536 reveals novel insights into mitotic roles of polo-like kinase 1. Curr. Biol. 2007;17:304–315. doi: 10.1016/j.cub.2006.12.046. [DOI] [PubMed] [Google Scholar]
- (42).Tang PC, Sun Li., Mcmahon G, Blake RA. 3-(Cyclohexano-heteroarylidenyl)-2-indolinone protein tyrosine kinase inhibitors. 6114371 US Patent. 2000
- (43).Mintz MJ, Walling C. t-Butyl hypochlorite. Org. Syn. Coll. 1969;49:9. [Google Scholar]
- (44).Dallinger D, Kappe CO. Rapid preparation of the mitotic kinesin Eg5 inhibitor monastrol using controlled microwave-assisted synthesis. Nature Protoc. 2007;2:317–321. doi: 10.1038/nprot.2006.436. [DOI] [PubMed] [Google Scholar]
- (45).Chen S, Chen L, Le NT, Zhao C, Sidduri A, Lou JP, Michoud C, Portland L, Jackson N, Liu JJ, Konzelmann F, Chi F, Tovar C, Xiang Q, Chen Y, Wen Y, Vassilev LT. Synthesis and activity of quinolinyl-methylene-thiazolinones as potent and selective cyclin-dependent kinase 1 inhibitors. Bioorg. Med. Chem. Lett. 2007;17:2134–2138. doi: 10.1016/j.bmcl.2007.01.081. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.