

The crystal structures of two circularly permuted streptavidins probe the role of a flexible loop in the tight binding of biotin. Molecular-dynamics calculations for one of the mutants suggests that increased fluctuations in a hydrogen bond between the protein and biotin are associated with cleavage of the binding loop.
Keywords: biotin-binding protein, biotin, circular permutation
Abstract
Circular permutation of streptavidin was carried out in order to investigate the role of a main-chain amide in stabilizing the high-affinity complex of the protein and biotin. Mutant proteins CP49/48 and CP50/49 were constructed to place new N-termini at residues 49 and 50 in a flexible loop involved in stabilizing the biotin complex. Crystal structures of the two mutants show that half of each loop closes over the binding site, as observed in wild-type streptavidin, while the other half adopts the open conformation found in the unliganded state. The structures are consistent with kinetic and thermodynamic data and indicate that the loop plays a role in enthalpic stabilization of the bound state via the Asn49 amide–biotin hydrogen bond. In wild-type streptavidin, the entropic penalties of immobilizing a flexible portion of the protein to enhance binding are kept to a manageable level by using a contiguous loop of medium length (six residues) which is already constrained by its anchorage to strands of the β-barrel protein. A molecular-dynamics simulation for CP50/49 shows that cleavage of the binding loop results in increased structural fluctuations for Ser45 and that these fluctuations destabilize the streptavidin–biotin complex.
1. Introduction
The strong binding of biotin by streptavidin (K ≃ 2.5 × 1013 M −1; Green, 1990 ▶) forms the basis for various molecular-labeling, molecular-localization and separation methods in biotechnical applications. Determining the interatomic interactions that give rise to the tight binding has been a focus of structural and biophysical studies since the first structure determinations of streptavidin (Hendrickson et al., 1989 ▶; Weber et al., 1989 ▶). Subsequent examinations of biotin, streptavidin and avidin have concentrated on three contributors to ligand binding: hydrogen bonds between the protein and the ureido and carboxylate groups of biotin, van der Waals interactions between tryptophan residues and biotin, and the closure of a flexible protein loop over the ligand-binding pocket. These interactions have been teased apart using mutagenesis techniques (Klumb et al., 1998 ▶; Freitag et al., 1997 ▶, 1998 ▶; Chu et al., 1998 ▶; Marttila et al., 2003 ▶; Laitinen et al., 2006 ▶; Maata et al., 2008 ▶) and binding studies of biotin and nonbiotin ligands (Weber et al., 1995 ▶; Schmidt et al., 1996 ▶; Korndörfer & Skerra, 2002 ▶; Katz & Cass, 1997 ▶; Schmidt & Skerra, 2007 ▶). Molecular-dynamics simulations have also probed the effects of these interactions on the thermodynamics and kinetics of ligand association and dissociation (Freitag et al., 1999 ▶; Hyre et al., 2002 ▶; Cerutti et al., 2009 ▶; Baugh et al., 2012 ▶).
One protein–biotin interaction that is difficult to address using mutagenesis is the hydrogen bond between the biotin carboxylate and the main-chain amide of Asn49. To investigate this, we built on the circularly permuted streptavidin reagents used to delete the flexible binding loop (Chu et al., 1998 ▶). Using appropriate primers, we expressed two circularly permuted forms of streptavidin with the N-terminus moved to positions 49 and 50 in the sequence of core streptavidin (see Fig. 1 ▶). These mutants are named CP49/48 and CP50/49, respectively, where the residue numbers of core streptavidin (Sano et al., 1995 ▶) have been retained. The first number denotes the new N-terminus of the protein and the second is the new C-terminal residue. Both mutations result in single-chain proteins with what would constitute breaks in the flexible binding loop of core streptavidin. The first mutant, CP49/48, makes Asn49 the new N-terminus of the protein and places it with the C-terminal half of the cleaved loop. The other mutant, CP50/49, makes Asn49 the C-terminus of the protein and places it with the N-terminal portion of the loop. The biophysical and crystallographic information provided here probes the effects of shifting the position of Asn49 on biotin binding and conformations of the flexible binding loop.
Figure 1.

Schematic showing the locations of the N- and C-termini for wild-type streptavidin, CP49/48 and CP50/49. N-termini are shown in blue and C-termini in red. The flexible binding-loop residues are shown in black. Linker residues in the circularly permuted mutants are shown in green.
2. Materials and methods
2.1. Protein expression and biophysical characterization
The design of CP49/48 and CP50/49 was based on previous studies of circularly permuted proteins (Goldenberg & Creighton, 1983 ▶; Yang & Schachman, 1993 ▶) and the method used to generate the engineered proteins was an adaptation of the procedure of Horlick et al. (1992 ▶). The materials and methods used to generate these circularly permuted streptavidins have been described in detail elsewhere (Chu et al., 1998 ▶; Chu, 1998 ▶) and are summarized briefly here.
Starting with a synthetic gene for core streptavidin (residues 13–139; Chilkoti et al., 1995 ▶), a tandem gene was constructed containing two copies of the streptavidin sequence joined by DNA coding for four linker residues (Gly140-Gly-Gly-Ser143). The CP49/48 and CP50/49 mutants were created utilizing primers designed and synthesized to terminate the protein at the new N- and C-termini. The sense primer adds an NdeI site to the beginning of the gene, while the antisense primer adds stop codons and a HindIII site at the other end. PCR mutagenesis was used to produce the circularly permuted genes, which were ligated into pT7Blue plasmids (Novagen, Madison, Wisconsin, USA) and transformed into NovaBlue maintenance hosts. The genes were subcloned into pET-21a plasmids (Novagen) in Escherichia coli BL21(DE3) hosts for expression. The integrity of the mutant genes was confirmed by DNA sequencing.
The BL21(DE3) cells containing the pET-21a plasmids were cultured overnight at 310 K in Luria–Bertani (LB) medium. After washing in fresh LB, the cell pellet was used to inoculate 5 l 2×YT medium containing 100 µg ml−1 ampicillin. The culture was incubated at 310 K with shaking until the A 600 reached 1.0. Protein expression was then induced by the addition of 1 mM isopropyl β-d-1-thiogalactoside. The cells were cultured for 3 h before being harvested by centrifugation.
Inclusion bodies containing the mutant proteins were obtained by sonicating and centrifuging cells after resuspending them in 50 mM Tris–HCl, 200 mM NaCl, 5 mM EDTA, 8% sucrose, 1% Triton X-100, 1 mM phenylmethylsulfonyl fluoride (PMSF) pH 8.0. The cells were lysed by sonication and centrifuged at 17 700g for 20 min three times. Three further cycles of sonication and centrifugation were carried out in the same buffer but without Triton X-100.
The remaining insoluble inclusion bodies were dissolved in 6 M guanidine, 50 mM Tris–HCl pH 7.5 to a concentration of no greater than 10 mg ml−1. After equilibration for several hours at 277 K, solubilized protein was then diluted dropwise with stirring in a 50× volume of 50 mM Tris–HCl pH 7.5, 100 mM NaCl, 5 mM EDTA, 0.1 mM PMSF and equilibrated overnight. Residual insoluble material was removed by centrifugation and the solution was concentrated in a stirred ultrafiltration cell (Amicon, Beverly, Massachusetts, USA). The proteins were purified by affinity chromatography over iminobiotin-agarose (Pierce, Rockford, Illinois, USA; Hofmann et al., 1980 ▶). Protein-containing fractions were pooled and exchanged into a storage buffer consisting of 50 mM phosphate, 100 mM NaCl at pH 7.75.
Equilibrium constants for the mutants were measured in an equilibrium competition assay developed by Klumb et al. (1998 ▶). In summary, six histidine residues were added to the C-terminus of wild-type streptavidin to generate a protein (WT-Tag) that could be immobilized on Ni–NTA resin (Superflow, Qiagen). [3H]-biotin was equilibrated with a mixture of His-tagged and mutant streptavidins. WT-Tag can be removed via the Ni–NTA resin, and the partitioning of the tritiated ligand can be determined by measuring the radioactivity of the separated proteins. A competition curve is generated by varying the concentration of the competing streptavidin while keeping the concentrations of the His-tagged protein and [3H]-biotin constant. In a typical experiment, [3H]-biotin (21 µM; Amersham, Chicago, Illinois, USA) was added to a final concentration of 20 nM to mixtures of WT-Tag (50 nM) and the mutant in phosphate buffer pH 7. Imidazole (20 mM) was added to minimize any nonspecific adsorption of the mutant that might result from the presence of the two native histidines (His87 and His127) in each monomer. The protein and ligand mixtures were equilibrated over several half-lives to determine the wild-type streptavidin–biotin dissociation rate (t 1/2 is 4.5 h at 310 K) with typical incubation times of 24 h at 310 K. After equilibration, WT-Tag streptavidin was removed by batch incubation for 1 h with 50 µl of a 50% slurry of washed Ni–NTA resin. The mixture was then cleared via centrifugation. The supernatants were collected, boiled in 5% SDS to liberate bound [3H]-biotin and assayed for radioactivity. Under these conditions 98% of the WT-Tag streptavidin was removed from solution by the Ni–NTA resin and nonspecific binding of the mutant streptavidin to the resin varied between 2 and 5%. A negligible amount of [3H]-biotin in the absence of streptavidin was found to bind to the Ni–NTA resin, and in the presence of protein the free [3H]-biotin was less than 3% of the total ligand concentration. The ratio of the equilibrium constants for the mutant and WT-Tag proteins was obtained from the expressions for the equilibrium constants and the mass-balance equations.
Isothermal titration calorimetry (ITC) experiments on the mutants were carried out using a Calorimetry Science Corporation 4200 Calorimeter (Provo, Utah, USA). Protein solutions at 30–40 µM concentration were titrated with 20 × 5 µl aliquots of 750 µM biotin dissolved in the same buffer as the protein. All calorimetry experiments used either phosphate (50 mM sodium phosphate, 100 mM NaCl pH 7.75) or Tris (50 mM Tris–HCl, 100 mM NaCl pH 7.75) buffers. Biotin concentrations were determined gravimetrically.
The calorimetry data were analyzed using the proprietary software supplied by Calorimetry Science Corporation. Heats of dilution for each injection were subtracted from the reaction heats before data analysis. Nonlinear fitting of the data allowed the number of binding sites (n), association constant (K a) and binding enthalpy (ΔH°) to be determined assuming noncooperative binding and one site per subunit.
Samples for differential scanning calorimetry (DSC) were prepared in the same manner as those for ITC. Briefly, a 25–30 µM solution of the protein of interest in phosphate buffer was degassed and injected into the DSC sample chamber. Buffer without protein was injected into the reference cell. Both solutions were placed under a pressure of 284 kPa and subjected to two or three heating/cooling cycles over a temperature range of 273–284 K. The heating and cooling rate was 1 K min−1. Data processing and extraction of the melting point was accomplished using software from Calorimetry Sciences Corporation.
The off-rate (k off) of biotin from the mutants was determined using a radiometric competition assay (Piran & Riordan, 1990 ▶; Chilkoti & Stayton, 1995 ▶). In a sodium phosphate solution (50 mM NaH2PO4, 100 mM NaCl at pH 7.0), streptavidin at 50 nM concentration was preloaded with 10 nM tritiated biotin. At the beginning of the experiment, non-tritiated biotin was introduced into the solution to a final concentration of 50 µM. Sampling intervals were chosen so that each experiment would span three to four half-lives. The dissociated [3H]-biotin at each time point was immediately separated from the protein via centrifugation using Microcon-10 filters (Amicon, Beverly, Massachusetts, USA) at 280.3 K in a microcentrifuge (Eppendorf, Beverly, Massachusetts, USA) at maximum speed (14 000 rev min−1). The total spin time including acceleration and deceleration of the rotor was 47 s. The amount of dissociated [3H]-biotin was determined by adding 30 µl of the filtrate to 5 ml EcoLume scintillation cocktail (ICN Pharmaceuticals, Costa Mesa, California, USA) and using an LS-7000 liquid scintillation counter (Beckman, Fullerton, California, USA) to measure the counts (1 min collection times).
The dissociation rate for the release of biotin from streptavidin was calculated from a plot of the logarithm of the fraction of biotin bound versus time according to the integrated expression for a first-order reaction. The transition-state enthalpy, ΔH ‡, and entropy, ΔS ‡, were determined from measurement of the off-rate values at several temperatures.
2.2. Crystallization and diffraction data collection
Crystals of the biotin complex of the CP49/48 mutant were obtained by hanging-drop vapor-diffusion experiments. Drops of a protein solution containing biotin were mixed with equal volumes of 1.0 M ammonium phosphate, 0.1 M Tris–HCl pH 7.5, which also served as the reservoir solution. The resulting crystals were cryoprotected with 30% glycerol. The resulting crystals belonged to space group P212121, with unit-cell parameters a = 54.72, b = 85.89, c = 100.14 Å and with a complete tetramer in the asymmetric unit.
Crystals of CP50/49 complexed with biotin were obtained using the same techniques but with 25% saturated ammonium sulfate (1.0 M), 0.1 M Tris–HCl pH 8.5, 0.2 M lithium sulfate as the reservoir solution. 35% glycerol served as a cryoprotectant for these crystals. The CP50/49 crystals belonged to space group I222, with unit-cell parameters a = 46.81, b = 94.03, c = 104.28 Å. Two subunits are found in the asymmetric unit.
Diffraction data for both complexes were collected at 100 K at Stanford Synchrotron Radiation Lightsource. The data sets were processed using DENZO and SCALEPACK (Otwinowski & Minor, 1997 ▶). A summary of the data-set statistics are presented in Table 1 ▶.
Table 1. Diffraction data statistics.
Values in parentheses are for the last shell.
| CP49/48 | CP50/49 | |
|---|---|---|
| Unit-cell parameters (Å) | a = 54.72, b = 85.89, c = 100.14 | a = 46.81, b = 94.03, c = 104.28 |
| Space group | P212121 | I222 |
| Beamline | SSRL BL11-1 | SSRL BL9-1 |
| No. of unique reflections | 83070 | 135318 |
| Resolution range (Å) | 50–1.50 (1.54–1.50) | 50–0.97 (0.99–0.97) |
| Completeness (%) | 88.1 (73.3) | 99.8 (99.6) |
| 〈I〉/〈σ(I)〉 | 26.5 (3.6) | 20.7 (0.4) |
| Multiplicity | 7.4 (3.5) | 4.8 (4.4) |
| R merge | 0.066 (0.276) | 0.076 (0.997) |
2.3. Structure solution and refinement
The structure of CP49/48 was solved by BALBES (Long et al., 2008 ▶) using PDB entry 2qcb (Creus et al., 2008 ▶) as the test structure. Refinement of the model made use of REFMAC5 (Murshudov et al., 2011 ▶) from the CCP4 program suite (Winn et al., 2011 ▶). Attempts to refine the model against all reflections to 1.3 Å resolution with individual atomic anisotropic displacement parameters (ADPs) resulted in systematically distorted thermal ellipsoids. The reflections were weak and the data-set completeness was low at high resolution. Accordingly, the data set was limited to 1.5 Å resolution (see Table 1 ▶).
The smaller data set required a revision of the temperature-factor model used in refinement. The ADPs were analyzed using the TLSMD server (Painter & Merritt, 2006 ▶; Zucker et al., 2010 ▶) and each molecule in the asymmetric unit was divided into 12 TLS groups. These were refined, along with isotropic temperature factors for each atom, using REFMAC5. 5% of the reflections were reserved for the calculation of R free (Brünger, 1992 ▶).
The unit cell of CP50/49 is isomorphous to that of the S45A mutant of streptavidin (Hyre et al., 2000 ▶). The initial structural model for CP50/49 was that of S45A with residues 48–51 removed. The two subunits in the asymmetric unit were refined with REFMAC5, but with individual ADPs for each atom. Data to 0.97 Å resolution were used for refinement. The statistics for the highest resolution shell indicated that many reflections had intensities near or below the noise level (see Table 1 ▶), but there were still individual reflections in the shell that were significantly above their standard deviations.
XtalView (McRee, 1999 ▶) was used to view σA-weighted |F o| − |F c| and 2|F o| − |F c| electron-density maps (Read, 1986 ▶) and manipulate the molecular model. The structural models were evaluated during and after refinement using PARVATI (Merritt, 1999a ▶,b ▶), MolProbity (Davis et al., 2007 ▶) and ADIT (Berman et al., 2000 ▶). Superposition of the wild-type and mutant molecules was carried out using the method of Ferro & Hermans (1977 ▶). Figs. 2–5 were generated using MolScript (Kraulis, 1991 ▶) and Raster3D (Merritt & Bacon, 1997 ▶).
The final model for CP49/48 contains residues 49–133 and 14–48 for subunit A, 49–134 and 14–48 for subunit B, 49–134 and 14–48 for subunit C and 49–134 and 13–48 for subunit D. The final model of CP50/49 contains residues 50–135 and 14–49 for subunit A and 50–135 and 13–49 for subunit B. Electron density for the linker residues between Ser139 and Ala13 is only observed for chain B of CP49/48. This linker is involved in crystal-packing interactions with a neighboring tetramer. The thermal parameters for the linker are high compared with the rest of the subunit, but presumably the inter-tetramer interactions are sufficient to limit its conformational flexibility. A similar situation occurred in the CP51/46 mutant, in which the linker was ordered in only one of the four subunits (Chu et al., 1998 ▶). The conformation of the linker in this structure differs from that observed in CP49/48, although both are involved in intermolecular interactions.
Sequencing information indicates that the initiator methionine was not removed for CP49/48. No density was observed for the methionine residue.
Refinement statistics for each structure are given in Table 2 ▶. The coordinates and structure factors for the two circularly permuted mutants have been deposited in the Protein Data Bank (PDB entries 4gd9 for CP49/48 and 4gda for CP50/49). (In the released PDB files, the residue numbers have been incremented by 200 for residues 13–48 of CP49/48 and residues 13–49 of CP50/49.)
Table 2. Refinement statistics.
| CP49/48 | CP50/49 | |
|---|---|---|
| Resolution (Å) | 36.9–1.50 | 69.0–1.00 |
| No. of reflections, working set | 60383 | 111150 |
| No. of reflections, test set | 3343 | 6225 |
| R cryst, all data | 0.202 | 0.119 |
| R cryst, working set | 0.201 | 0.118 |
| R free, all data | 0.230 | 0.143 |
| Bond r.m.s.d. (restrained) (Å) | 0.013 | 0.011 |
| No. of protein atoms | 3720 | 2009 |
| No. of heteroatoms | 70 | 74 |
| No. of solvent atoms | 366 | 372 |
| Ramachandran outliers as determined by MolProbity (%) | 0.00 | 0.42 |
| Ramachandran favored (%) | 98.13 | 97.49 |
| PDB code | 4gd9 | 4gda |
2.4. Molecular-dynamics simulation
The molecular-dynamics simulation protocol was identical to that reported in previous studies in order to facilitate direct comparisons with previous results. Starting coordinates for the tetramer complex were generated using the current CP50/49 crystal structure. All histidine residues were singly protonated to model the ionization state expected at the neutral pH used for most of our experimental measurements. H atoms were added to all heavy atoms using the LEaP module of the Amber12 package (Case et al., 2005 ▶) and the complex was solvated in a truncated octahedral box with 11 983 water molecules and eight sodium cations to maintain charge neutrality for the system.
The AMBER 99ffSB force field (Hornak et al., 2006 ▶) was used for the simulations, together with the SPC/E water model (Berendsen et al., 1987 ▶), a sodium-cation model from Åqvist (1990 ▶) and biotin parameters from Izrailev et al. (1997 ▶). Periodic boundary conditions were used for energy and force calculations, with a 9.0 Å cutoff for real-space interactions, a smooth particle-mesh Ewald for long-range electrostatics (Essmann et al., 1995 ▶) and a homogeneity assumption to approximate long-range Lennard–Jones force contributions to the virial tensor. All covalent bonds involving H atoms were constrained using the SHAKE algorithm (Ryckaert et al., 1977 ▶) and the rigid internal geometry for the SPC/E water molecules was constrained using the SETTLE algorithm (Miyamoto & Kollman, 1992 ▶). The system temperature was maintained at 300 K using a Langevin thermostat (Izaguirre et al., 2001 ▶) with a 3 ps−1 collision frequency.
The fully solvated system was first minimized by 2000 steps of conjugate-gradient energy minimization to relax water molecules, sodium cations and added H atoms, using 1000 kcal mol−1 Å−2 (1 cal = 4.186 J) positional restraints for all crystallographically resolved atoms. All protein atoms were then minimized for 2000 steps while solvent and cations were restrained. Finally, the entire system was energy-minimized for an additional 3000 steps with no positional restraints on any atoms. Next, a 150 ps canonical ensemble molecular-dynamics simulation was run with a 0.5 fs integration time step and with positional restraints applied to all crystallographically resolved atoms. The restraints were gradually reduced from 16.0 to 1.0 kcal mol−1 Å−2 during the simulation. The simulation was then extended for an additional 300 ps in the isothermal isobaric (TPN) ensemble with a 1.5 fs integration time step, while further reducing the positional restraints to 0.0625 kcal mol−1 Å−2. The simulation was then continued in the TPN ensemble for an additional 250 ns with no position restraints. All energy-minimization and molecular-dynamics simulations were performed using the PMEMD module in Amber12.
3. Results and discussion
An overview of the structural issues addressed by CP49/48 and CP50/49 is provided in Fig. 2 ▶. Streptavidin is a tetrameric protein with a β-barrel serving as the core of each subunit. Biotin is bound in the barrel, and a flexible-binding loop, L3,4 (residues 45–52), when closed over the ligand, provides a number of protein–biotin interactions including a hydrogen bond from the main-chain amide of Asn49 to the carboxylate of biotin. In the CP49/48 crystal form there is a complete tetramer in the asymmetric unit (chains A, B, C and D). The streptavidin tetramer has 222 (D 2) symmetry. In the CP50/49 crystal one of the molecular twofold axes superposes with a crystallographic dyad; thus, two subunits (chains A and B) comprise the asymmetric unit.
Figure 2.
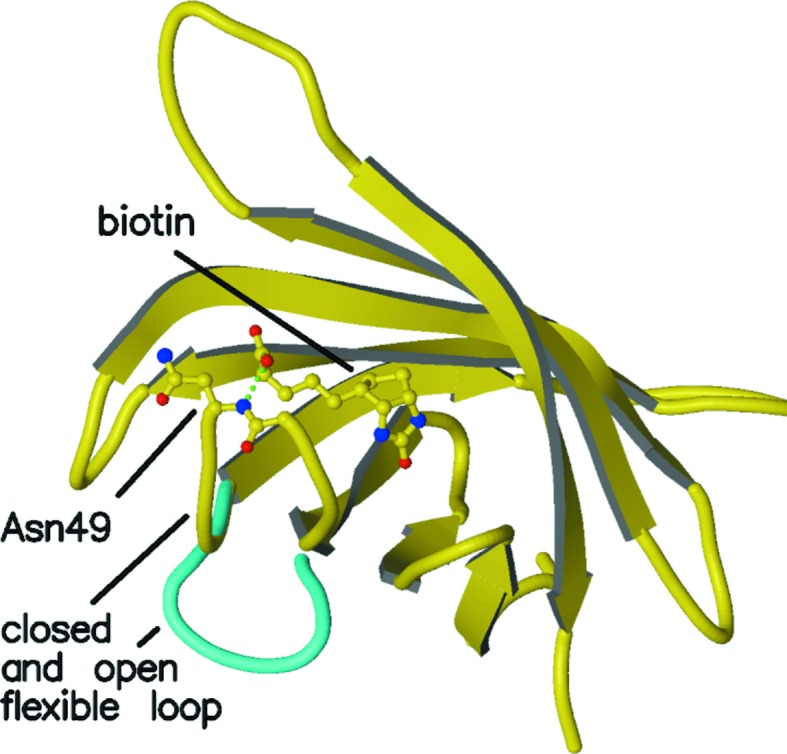
Overview of the polypeptide trace of a streptavidin subunit with a closed flexible binding loop shown in yellow (subunit A of PDB entry 3ry2). A bound biotin and Asn49 are shown in ball-and-stick mode. The hydrogen bond between the biotin carboxylate and the main-chain amide of Asn49 is shown by a green dotted line, as is the hydrogen bond between Ser45 and the ureido N atom of biotin. The open conformation of the flexible binding loop in unliganded streptavidin is shown in cyan (subunit A of PDB entry 3ry1).
CP49/48 and CP50/49 were designed to investigate the contributions of the Asn49 hydrogen bond to ligand binding and to determine how the flexible binding loop responds if cleaved before and after residue 49. Cleavage of a peptide bond causes a number of perturbations in a folded protein. Addition of H and O atoms to form the amine and carboxylate, and concomitant van der Waals repulsions, will effectively wedge the new termini apart. At most pH values of interest the new termini will also be charged, but how their electrostatic interactions with neighboring groups on the protein (or the solvent) will stabilize or destabilize structures similar to the native protein is impossible to predict. The folded protein might accommodate the chain cleavage by small local structural changes or by larger, more global, changes.
In the cases of CP49/48 and CP50/49, the structures are altered in both ways. In both mutants the residues leading up to Gly48 take on the conformation of the closed binding loop and interact with biotin very much like the wild-type protein (see Fig. 3 ▶). The equilibrium crystal structure of these residues is the same as in wild-type streptavidin. After the cleavage point, the residues beyond Ala50 are found in the open loop conformation that is more commonly found in unliganded streptavidin. In CP50/49, Asn49 is located at the C-terminus of the closed loop conformation and the mutant retains the hydrogen bond between the backbone amide and biotin. In CP49/48, in which Asn49 is the designed N-terminus of the protein (the initiator methionine is retained in this mutant), it is found at the end of the protein in the open conformation. After superposition of the A chains from the two CP mutants, the distance between the Cα atoms of Asn49 in the two mutants is 14.4 Å; thus, the location of residue 49 is sensitive to whether it is the C- or N-terminus of the mutant protein.
Figure 3.

Stereo figure showing residues 49–52 and 44–48 of subunit A of CP49/48 (green) and 50–52 and 44–49 of subunit A of CP50/49 (magenta) with the closed and open loops of the liganded (yellow) and unliganded (cyan) wild-type streptavidins. Biotin is included as described in Fig. 2 ▶.
Apart from the interactions between biotin and Asn49, the protein–ligand interactions observed for these two mutants in their crystal structures are the same as in the wild-type complex. In wild-type streptavidin there are two hydrogen bonds between the flexible-loop residues and biotin. In addition to the Asn49 amide hydrogen bond to O11 of biotin, Ser45 OG forms a hydrogen bond to N2 of biotin. The only significant difference in the hydrogen-bonding pattern between the wild-type and mutant proteins is the absence of the N⋯O11 interaction in CP49/48. The van der Waals interactions between the protein and biotin are also the same in the wild-type and mutant proteins.
Figs. 4 ▶ and 5 ▶ show the polypeptide chains for the loop residues in more detail for CP49/48 and CP50/49. The hydrogen bonds between biotin and Tyr43 and Ser45 tether the residues in the closed conformation and there is little variability in residues 45–48 in CP49/48 and residues 45–49 in CP50/49. The additional hydrogen bond involving the main-chain amide of Asn49 further stabilizes the closed conformation.
Figure 4.
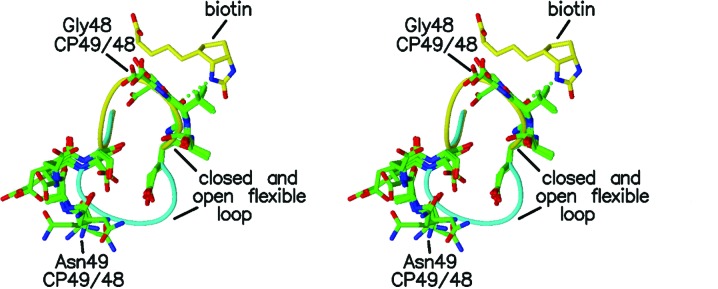
Stereo figure showing residues 49–52 and 44–48 for the four subunits of CP49/48 (green) with the closed and open loops of the liganded (yellow) and unliganded (cyan) wild-type streptavidins. Biotin is included as described in Fig. 2 ▶ except that the Asn49 hydrogen bond is not present in CP49/48.
Figure 5.
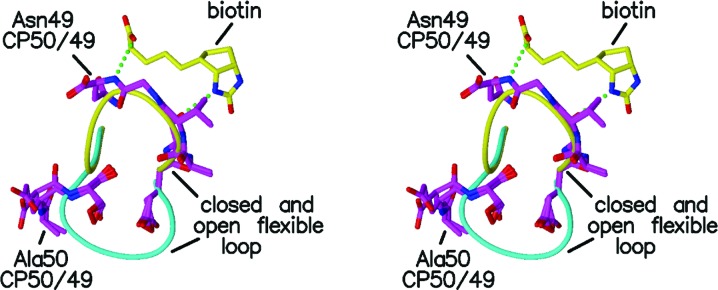
Stereo figure showing residues 50–52 and 44–49 for the two subunits of CP50/49 (magenta) with the closed and open loops of the liganded (yellow) and unliganded (cyan) wild-type streptavidins. Biotin is included as described in Fig. 2 ▶.
The portions of the mutants in the open conformation (residues 49–52 in CP49/48 and residues 50–52 in CP50/49) are more varied in their structures. While similar, their conformations are not as restrained as they would be if the polypeptide chain were uncleaved. The fact that these residues readily take on the open loop conformation suggests that cleavage of the loop releases strain in the structure. Alternatively, it might be that the open conformation is preferred by these residues. A strong interaction that is observed in the open conformation and not in the closed form is a hydrogen bond between Asn81 NE2 and Glu51 O. This is observed in all of the subunits in CP49/48 and CP50/49. In addition, in two of the subunits in CP49/48, Asn49 O forms a hydrogen bond to the amide or side chain of Ser52. Some sequences of amino acids forming the loop can stabilize the open form of the loop even in the presence of ligand, as shown by Korndörfer & Skerra (2002 ▶). In their streptavidin mutants 1 and 2, residues 44, 45 and 47 were mutated to optimize binding of the Strep-Tag II peptide. In the resulting liganded and unliganded structures the binding loops retained the open conformation, showing that the sequence of the loop residues can stabilize the open conformation and counter any preference for the closed conformation associated with the bound ligand.
The hydrogen bonds involving residues 49, 51 and 52 stabilize the open conformation and generate an ordered structure for the new N-termini of the mutants. Although sufficiently ordered to have visible electron density, residues in the open loop conformations are not as well ordered as most of the rest of the protein subunits. The side chains of the loop residues are not involved in many interactions with other protein atoms, so there is more variability in their locations, and this is reflected in their average temperature factors. When the flexible binding loop closes over the biotin ligand, the temperature factors for the loop residues drop to the average value for the core of the subunit. For CP49/48, the loop residues in both the closed and open conformation have high temperature factors consistent with the open loop and the loss of a hydrogen bond to biotin. In the case of CP50/49, the first part of the loop, including residue 49, has temperature factors appropriate for the closed conformation and similar to those of preceding parts of the protein. The biotin–Asn49 hydrogen bond stabilizes the closed loop, which results in lower temperature factors. The temperature factors are once again large for the latter part of the loop, which is the part in the more flexible open conformation.
The location of Asn49 and its effects on the loop structure, dynamics and biotin interactions are consistent with the observed binding thermodynamics. Table 3 ▶ compares the thermodynamics of biotin binding to wild-type streptavidin for the two circularly permuted proteins. In both CP49/48 and CP50/49 the binding enthalpy (ΔH°) is reduced by 7.5–8.0 kcal mol−1 at 310 K relative to the wild type. Further experiments on CP50/49 showed that its binding affinity is decreased by a factor of 417 from the wild-type value (ΔΔG° = 3.7 kcal mol−1, TΔΔS° = 3.8 kcal mol−1). The K a for CP49/48 could not be determined owing to insufficient data in the steepest part of the titration curve. The ΔC p values of both CP50/49 and CP49/48 fall between those of wild-type streptavidin and the CP51/46 mutant (in which the entire loop is excised), implying ordering of at least a portion of the cleaved loop during binding.
Table 3. Thermodynamic comparison of wild type, CP49/48 and CP50/49 at 310 K.
| Wild type | CP49/48 | CP50/49 | |
|---|---|---|---|
| K a ratio (mutant:wt) | — | — | 417 ± 14 |
| ΔΔG° (kcal mol−1) | — | — | +3.7 ± 0.1 |
| ΔΔH° (kcal mol−1) | — | +7.9 ± 0.1 | +7.6 ± 0.1 |
| TΔΔS° (kcal mol−1) | — | — | +3.8 ± 0.1 |
| ΔC p (cal mol−1 K−1) | −345 ± 12 | −166 ± 3 | −145 ± 3 |
| T melting (K) | 352 | — | 360 |
| k off (s−1) | 2.9 × 10−5 | 0.33 | 0.024 |
| ΔG ‡ (kcal mol−1) | 24.6 | 18.8 ± 1.3 | 20.5 ± 0.9 |
| ΔH ‡ (kcal mol−1) | 30.4 | 39.3 ± 0.6 | 29.5 ± 0.4 |
| TΔS ‡ (kcal mol−1) | 5.8 | 20.5 ± 0.7 | 9.0 ± 0.5 |
The mutants have significantly higher dissociation rates than does wild-type streptavidin. For CP49/48, k off is about 10 000 times greater than for the wild type. For CP50/48, the rate is about 1000 times greater. The lowering of the dissociation barriers is entropically driven. The entropic gain for CP49/48 is 14.7 kcal mol−1, while that for CP50/49 is 3.2 kcal mol−1. CP49/48 has a compensating increase in ΔH ‡, while CP50/49 retains the wild-type ΔH ‡. As stated elsewhere, CP49/48 retains the initiator methionine, and although no electron density was observed for it, its presence could complicate the thermodynamic and kinetic behavior of the mutant.
Complete thermodynamic and kinetic characterization of CP50/49 reveals that a nicked binding loop retains partial function when compared with the deleted loop of CP51/46 (Chu et al., 1998 ▶). The nature of this function is manifested as a reduction of enthalpy for the binding reaction with no change in the enthalpic component of the dissociation barrier. This implies that the loop is already open and does not contact biotin in the transition from the bound to the unbound state, but it does contribute to the stability of the bound complex. The entropic penalty for binding biotin to CP50/49 is lower than that of the wild type by 3.8 kcal mol−1. Had both loop segments become ordered on binding, we would expect the entropic penalty to increase as a result of the additional degrees of freedom lost in immobilization of the chain termini versus immobilization of the intact loop. The value of TΔΔS° is still lower than that for deletion of the full loop in CP51/46 (Chu et al., 1998 ▶), which is consistent with partial ordering of one of the loop segments as observed in the crystal structure of CP50/49.
Despite the lowered entropic penalty for binding, the overall binding affinity for CP50/49 decreases by approximately 400-fold compared with the wild type as a result of changes in the enthalpy of binding. It is not apparent from the crystal structure which structural features contribute to the enthalpic destabilization of the bound complex since the Asn49–biotin hydrogen bond is retained in this mutant. Likewise, no obvious changes in the van der Waals interactions with biotin are apparent.
We analyzed the molecular-dynamics simulation results to determine whether these data might suggest some basis for the dramatically reduced binding enthalpy for the CP50/49 mutant. The first segment of the L3,4 loop, residues 42–49, generally retains the closed loop conformation observed for wild-type streptavidin when bound to biotin, while the second segment, residues 50–52, maintains an open conformation observed for unliganded streptavidin. Both of these structural trends are consistent with our crystal structure results. However, both segments display larger structural fluctuations for the CP50/49 mutant compared with our earlier wild-type streptavidin simulations (Cerutti et al., 2009 ▶), as shown in Fig. 6 ▶. This plot displays the fluctuation differences for the wild-type streptavidin complex compared with the CP50/49 mutant. The large negative peak in this difference plot centered near residue 50 indicates that the atomic fluctuations for these residues are much larger in the mutant than in the wild-type complex. The open conformation adopted by residues 50–52 diminishes the side-chain packing interactions with neighboring residues, including Trp79, which is a key biotin binding-site contact. With fewer side-chain packing restraints, Trp79 is not held as tightly fixed in close juxtaposition to biotin in the mutant compared with the wild-type complex during the course of the simulation. This behavior is quite similar to that observed previously for the Y54F mutant (Baugh et al., 2012 ▶). We proposed that the diminished Trp79–biotin interactions we observed for the Y54F mutant could explain the reduced biotin-binding enthalpy that we measured for this mutant, and it appears the same effect is also a factor for the CP50/49 mutant. While diminished Trp79–biotin interactions can partially account for the dramatic binding-enthalpy reduction that we measure for CP50/49, there must be other factors that also contribute to the large enthalpy loss.
Figure 6.

Difference plot for atomic fluctuations averaged over all atoms in each residue for wild-type versus CP50/49 mutant streptavidin complexes. Positive values indicate greater mobility in the wild-type complex versus the mutant. The large negative peak observed for residues 45–53 indicates that these residues exhibit significantly increased motion in the CP50/49 mutant complex versus the wild-type complex. The positive peaks present for residues 64–69 (loop L4,5) and residues 115–122 (loop L7,8) indicate larger ranges of motion for these large flexible loops in the wild-type simulation. Large fluctuations are typically observed for these loop residues in simulations of all of the wild-type and mutant complexes that we have studied previously, and as the simulations are propagated for longer time periods the atom fluctuation differences in these loop regions decrease asymptotically to baseline. The CP50/49 mutant simulation reported here is relatively short (∼250 ns) compared with the reference wild-type simulation, so the positive peaks for loops L4,5 and L7,8 reflect the more extensive conformational sampling achieved in the longer wild-type simulation.
We next analyzed the primary streptavidin–biotin hydrogen-bonding interactions as a function of time over the duration of the MD trajectory and discovered an extremely interesting result. We chose a discrete donor–acceptor cutoff distance of 4.75 Å to define a ‘broken’ hydrogen bond and then compared the results for CP50/49 versus the wild-type and F130L (Baugh et al., 2010 ▶), Y54F (Baugh et al., 2012 ▶) and T106V mutant trajectories studied previously. There are no discernible differences for four of five key hydrogen-bonding residues (Asn23, Ser27, Tyr43 and Asp128). Ser27 and Tyr43 form strong interactions with the ureido carbonyl O atom in biotin and rarely exceed the cutoff distance for CP50/49, the wild type or any other point-mutant complexes analyzed in previous studies (fewer than 0.01% of configurations exhibit a ‘broken’ hydrogen bond for any complex). Asn23 donates a hydrogen bond to the biotin ureido carbonyl O atom and Asp128 accepts a hydrogen bond from the biotin N1 N atom. We observe increased fluctuations for these hydrogen bonds compared with Ser27 or Tyr43 interactions, suggesting that these hydrogen bonds break and reform more frequently in the complex, but the behavior of these two hydrogen bonds is indistinguishable for CP50/49 versus the wild type and all other single-site point-mutant complexes.
However, we observed a dramatically different profile for Ser45, which accepts a hydrogen bond from N2 of biotin. In the wild-type complex, as well as all previous single-site point-mutant trajectories that we have analyzed, this hydrogen bond is broken in fewer than 0.05% of snapshots, while for the CP50/49 trajectory this hydrogen bond is broken in more than 10% of the configurations. The exact numerical results are obviously somewhat dependent on the specific cutoff distance that we choose to define a ‘broken’ hydrogen bond, but any cutoff value in the range 3.8–5.0 Å yields comparable statistics, while shorter cutoff values would increase the percentage of broken Ser45 hydrogen bonds in the CP50/49 mutant versus the wild-type and single-site point-mutant complexes. Electron density for an alternative Ser45 conformation is not observed, but the reduced occupancy of the hydrogen-bonded conformation for Ser45 would not easily be observed, even at high resolution.
Analysis of the CP50/49 simulation and comparison with the wild-type and F130L, Y54F and T106V mutant trajectories provides a clear explanation for these differences in Ser45 hydrogen bonding. Ser45 is near the beginning of the L3,4 loop and this loop adopts the closed conformation in the wild-type and single-site point-mutant biotin complexes. As a result, the residues in this loop, including Ser45, are largely immobilized, as demonstrated by small B values in the crystal structures and small-magnitude structural fluctuations in all of the MD simulations. The relative immobility of this loop ensures that the Ser45–biotin hydrogen bond is well maintained in all complexes studied previously. In the CP50/49 mutant, the L3,4 loop has been cleaved between Asn49 and Ala50, and while the first segment of the loop (residues 42–49) adopts the closed conformation in the biotin complex, the residues nearer the cleavage site exhibit larger amplitude structural fluctuations compared with their counterparts in the wild type and all other mutant complexes (see Fig. 6 ▶). Therefore, Ser45 is not anchored as tightly in perfect position to accept a hydrogen bond from N2 of biotin in the CP50/49 mutant.
The reduction of the Ser45–biotin hydrogen-bonding interaction in CP50/49 compared with the wild type and other mutant complexes is substantial, and we propose that the attenuation of this important hydrogen-bonding interaction is a primary factor contributing to the reduced binding enthalpy. This weakened hydrogen bond and the diminished Trp79–biotin van der Waals interaction provide a logical explanation for the dramatic binding-enthalpy decrease that we measured for CP50/49. These increased-amplitude structural fluctuations that negatively impact binding enthalpy in CP50/49 are also perfectly consistent with the more favorable binding-entropy results, as well as the increased biotin dissociation rate that we measured.
4. Conclusions
Creation of nicked-loop variants of streptavidin using circular permutation highlights the importance of the flexible loop in the high-affinity interaction of streptavidin and biotin. Simple cleavage of the protein in the flexible binding loop, without mutation or removal of any binding-site residues, results in a drop in affinity and a large increase in biotin dissociation.
Crystal structure analyses of CP49/48 and CP50/49 show that in each mutant the portion of the flexible loop that interacts with the bound ligand closes over the binding site. The other part of the loop adopts an open conformation similar to that found in the unbound protein. The partially closed structure is not as effective in slowing the dissociation rate as is the intact loop. Thus, the mutants have significantly reduced binding.
While it is difficult to completely rationalize the thermodynamic data using only the crystal structures, the molecular-dynamics simulations provide a logical explanation for the experimental results that nicely reconciles the thermodynamic and the structural information. Cleavage of the Asn49-Ala50 peptide bond leads to increased mobility for most of these residues in the L3,4 loop, even though a portion of this loop can still adopt the closed conformation when biotin is bound. What is not easy to predict without the aid of the molecular-dynamics simulations is the impact that the increased mobility of the L3,4 loop residues has on important, specific streptavidin interactions with biotin, i.e. residues Ser45 and Trp79 have clearly degraded interactions with biotin in the CP50/49 mutant when compared to the wild-type and single-site point-mutant complexes studied previously.
Supplementary Material
PDB reference: CP49/48, 4gd9
PDB reference: CP50/49, 4gda
Acknowledgments
The research reported in this publication was supported by the National Institute of General Medical Sciences of the National Institutes of Health under award No. R01GM080214 (TPL). Portions of this research were carried out at the Stanford Synchrotron Radiation Lightsource, a Directorate of SLAC National Accelerator Laboratory and an Office of Science User Facility operated for the US Department of Energy Office of Science by Stanford University. The SSRL Structural Molecular Biology Program is supported by the DOE Office of Biological and Environmental Research and by the National Institutes of Health, National Institute of General Medical Sciences (including P41GM103393) and the National Center for Research Resources (P41RR001209). The contents of this publication are solely the responsibility of the authors and do not necessarily represent the official views of NIGMS, NCRR or NIH.
References
- Åqvist, J. (1990). J. Phys. Chem. 94, 8021–8024.
- Baugh, L., Le Trong, I., Cerutti, D. S., Gülich, S., Stayton, P. S., Stenkamp, R. E. & Lybrand, T. P. (2010). Biochemistry, 49, 4568–4570. [DOI] [PMC free article] [PubMed]
- Baugh, L., Le Trong, I., Cerutti, D. S., Mehta, N., Gülich, S., Stayton, P. S., Stenkamp, R. E. & Lybrand, T. P. (2012). Biochemistry, 51, 597–607. [DOI] [PMC free article] [PubMed]
- Berendsen, H. J. C., Grigera, J. R. & Straatsma, T. P. (1987). J. Phys. Chem. 91, 6269–6271.
- Berman, H. M., Westbrook, J., Feng, Z., Gilliland, G., Bhat, T. N., Weissig, H., Shindyalov, I. N. & Bourne, P. E. (2000). Nucleic Acids Res. 28, 235–242. [DOI] [PMC free article] [PubMed]
- Brünger, A. T. (1992). Nature (London), 355, 472–475. [DOI] [PubMed]
- Case, D. A., Cheatham, T. E., Darden, T., Gohlke, H., Luo, R., Merz, K. M., Onufriev, A., Simmerling, C., Wang, B. & Woods, R. J. (2005). J. Comput. Chem. 26, 1668–1688. [DOI] [PMC free article] [PubMed]
- Cerutti, D. S., Le Trong, I., Stenkamp, R. E. & Lybrand, T. P. (2009). J. Phys. Chem. B, 113, 6971–6985. [DOI] [PMC free article] [PubMed]
- Chilkoti, A. & Stayton, P. S. (1995). J. Am. Chem. Soc. 117, 10622–10628.
- Chilkoti, A., Tan, P. H. & Stayton, P. S. (1995). Proc. Natl Acad. Sci. USA, 92, 1754–1758. [DOI] [PMC free article] [PubMed]
- Chu, V. (1998). PhD thesis, University of Washington.
- Chu, V., Freitag, S., Le Trong, I., Stenkamp, R. E. & Stayton, P. S. (1998). Protein Sci. 7, 848–859. [DOI] [PMC free article] [PubMed]
- Creus, M., Pordea, A., Rossel, T., Sardo, A., Letondor, C., Ivanova, A., Le Trong, I., Stenkamp, R. E. & Ward, T. R. (2008). Angew. Chem. Int. Ed. 47, 1400–1404. [DOI] [PubMed]
- Davis, I. W., Leaver-Fay, A., Chen, V. B., Block, J. N., Kapral, G. J., Wang, X., Murray, L. W., Arendall, W. B., Snoeyink, J., Richardson, J. S. & Richardson, D. C. (2007). Nucleic Acids Res. 35, W375–W383. [DOI] [PMC free article] [PubMed]
- Essmann, U., Perera, L., Berkowitz, M. L., Darden, T., Lee, H. & Pedersen, L. G. (1995). J. Chem. Phys. 103, 8577.
- Ferro, D. R. & Hermans, J. (1977). Acta Cryst. A33, 345–347.
- Freitag, S., Chu, V., Penzotti, J. E., Klumb, L. A., To, R., Hyre, D., Le Trong, I., Lybrand, T. P., Stenkamp, R. E. & Stayton, P. S. (1999). Proc. Natl Acad. Sci. USA, 96, 8384–8389. [DOI] [PMC free article] [PubMed]
- Freitag, S., Le Trong, I., Chilkoti, A., Klumb, L. A., Stayton, P. S. & Stenkamp, R. E. (1998). J. Mol. Biol. 279, 211–221. [DOI] [PubMed]
- Freitag, S., Le Trong, I., Klumb, L., Stayton, P. S. & Stenkamp, R. E. (1997). Protein Sci. 6, 1157–1166. [DOI] [PMC free article] [PubMed]
- Goldenberg, D. P. & Creighton, T. E. (1983). J. Mol. Biol. 165, 407–413. [DOI] [PubMed]
- Green, N. M. (1990). Methods Enzymol. 184, 51–67. [DOI] [PubMed]
- Hendrickson, W. A., Pähler, A., Smith, J. L., Satow, Y., Merritt, E. A. & Phizackerley, R. P. (1989). Proc. Natl Acad. Sci. USA, 86, 2190–2194. [DOI] [PMC free article] [PubMed]
- Hofmann, K., Wood, S. W., Brinton, C. C., Montibeller, J. A. & Finn, F. M. (1980). Proc. Natl Acad. Sci. USA, 77, 4666–4668. [DOI] [PMC free article] [PubMed]
- Horlick, R. A., George, H. J., Cooke, G. M., Tritch, R. J., Newton, R. C., Dwivedi, A., Lischwe, M., Salemme, F. R., Weber, P. C. & Horuk, R. (1992). Protein Eng. 5, 427–431. [DOI] [PubMed]
- Hornak, V., Abel, R. A. O., Strockbine, B., Roitberg, A. & Simmerling, C. (2006). Proteins, 65, 712–725. [DOI] [PMC free article] [PubMed]
- Hyre, D. E., Amon, L. M., Penzotti, J. E., Le Trong, I., Stenkamp, R. E., Lybrand, T. P. & Stayton, P. S. (2002). Nature Struct. Biol. 9, 582–585. [DOI] [PubMed]
- Hyre, D. E., Le Trong, I., Freitag, S., Stenkamp, R. E. & Stayton, P. S. (2000). Protein Sci. 9, 878–885. [DOI] [PMC free article] [PubMed]
- Izaguirre, J. A., Catarello, D. P., Wozniak, J. M. & Skeel, R. D. (2001). J. Chem. Phys. 114, 2090.
- Izrailev, S., Stepaniants, S., Balsera, M., Oono, Y. & Schulten, K. (1997). Biophys. J. 72, 1568–1581. [DOI] [PMC free article] [PubMed]
- Katz, B. A. & Cass, R. T. (1997). J. Biol. Chem. 272, 13220–13228. [DOI] [PubMed]
- Klumb, L. A., Chu, V. & Stayton, P. S. (1998). Biochemistry, 37, 7657–7663. [DOI] [PubMed]
- Korndörfer, I. P. & Skerra, A. (2002). Protein Sci. 11, 883–893. [DOI] [PMC free article] [PubMed]
- Kraulis, P. J. (1991). J. Appl. Cryst. 24, 946–950.
- Laitinen, O. H., Hytönen, V. P., Nordlund, H. R. & Kulomaa, M. S. (2006). Cell. Mol. Life Sci. 63, 2992–3017. [DOI] [PMC free article] [PubMed]
- Long, F., Vagin, A. A., Young, P. & Murshudov, G. N. (2008). Acta Cryst. D64, 125–132. [DOI] [PMC free article] [PubMed]
- Maata, J. A. E., Airenne, T. T., Nordlund, H. R., Janis, J., Paldanius, T. A., Vainiotalo, P., Johnson, M. S., Kulomaa, M. S. & Hytonen, V. R. (2008). Chembiochem, 9, 1124–1135. [DOI] [PubMed]
- Marttila, A. T., Hytönen, V. P., Laitinen, O. H., Bayer, E. A., Wilchek, M. & Kulomaa, M. S. (2003). Biochem. J. 369, 249–254. [DOI] [PMC free article] [PubMed]
- McRee, D. E. (1999). J. Struct. Biol. 125, 156–165. [DOI] [PubMed]
- Merritt, E. A. (1999a). Acta Cryst. D55, 1109–1117. [DOI] [PubMed]
- Merritt, E. A. (1999b). Acta Cryst. D55, 1997–2004. [DOI] [PubMed]
- Merritt, E. A. & Bacon, D. J. (1997). Methods Enzymol. 277, 505–524. [DOI] [PubMed]
- Miyamoto, S. & Kollman, P. A. (1992). J. Comput. Chem. 13, 952–962.
- Murshudov, G. N., Skubák, P., Lebedev, A. A., Pannu, N. S., Steiner, R. A., Nicholls, R. A., Winn, M. D., Long, F. & Vagin, A. A. (2011). Acta Cryst. D67, 355–367. [DOI] [PMC free article] [PubMed]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol. 276, 307–326. [DOI] [PubMed]
- Painter, J. & Merritt, E. A. (2006). J. Appl. Cryst. 39, 109–111.
- Piran, U. & Riordan, W. J. (1990). J. Immunol. Methods, 133, 141–143. [DOI] [PubMed]
- Read, R. J. (1986). Acta Cryst. A42, 140–149.
- Ryckaert, J., Ciccotti, G. & Berendsen, H. J. (1977). J. Comput. Phys. 23, 327–341.
- Sano, T., Pandori, M. W., Chen, X., Smith, C. L. & Cantor, C. R. (1995). J. Biol. Chem. 270, 28204–28209. [DOI] [PubMed]
- Schmidt, T. G., Koepke, J., Frank, R. & Skerra, A. (1996). J. Mol. Biol. 255, 753–766. [DOI] [PubMed]
- Schmidt, T. G. & Skerra, A. (2007). Nature Protoc. 2, 1528–1535. [DOI] [PubMed]
- Weber, P. C., Ohlendorf, D. H., Wendoloski, J. J. & Salemme, F. R. (1989). Science, 243, 85–88. [DOI] [PubMed]
- Weber, P. C., Pantoliano, M. W. & Salemme, F. R. (1995). Acta Cryst. D51, 590–596. [DOI] [PubMed]
- Winn, M. D. et al. (2011). Acta Cryst. D67, 235–242.
- Yang, Y. R. & Schachman, H. K. (1993). Proc. Natl Acad. Sci. USA, 90, 11980–11984. [DOI] [PMC free article] [PubMed]
- Zucker, F., Champ, P. C. & Merritt, E. A. (2010). Acta Cryst. D66, 889–900. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: CP49/48, 4gd9
PDB reference: CP50/49, 4gda