

The yellow fluorescent protein phiYFPv with improved folding has been developed from the spectrally identical wild-type phiYFP found in the marine jellyfish Phialidium.
Keywords: yellow fluorescent protein, Phialidium, structure–function relationship, chromophores, oligomeric structure, intersubunit surface
Abstract
The yellow fluorescent protein phiYFPv (λem max ≃ 537 nm) with improved folding has been developed from the spectrally identical wild-type phiYFP found in the marine jellyfish Phialidium. The latter fluorescent protein is one of only two known cases of naturally occurring proteins that exhibit emission spectra in the yellow–orange range (535–555 nm). Here, the crystal structure of phiYFPv has been determined at 2.05 Å resolution. The ‘yellow’ chromophore formed from the sequence triad Thr65-Tyr66-Gly67 adopts the bicyclic structure typical of fluorophores emitting in the green spectral range. It was demonstrated that perfect antiparallel π-stacking of chromophore Tyr66 and the proximal Tyr203, as well as Val205, facing the chromophore phenolic ring are chiefly responsible for the observed yellow emission of phiYFPv at 537 nm. Structure-based site-directed mutagenesis has been used to identify the key functional residues in the chromophore environment. The obtained results have been utilized to improve the properties of phiYFPv and its homologous monomeric biomarker tagYFP.
1. Introduction
In the last decade, green fluorescent proteins (GFPs) and GFP-like proteins (FPs) have become important noninvasive tools for visualization and monitoring of internal biological processes in cell biology, biotechnology and biomedicine (Chudakov et al., 2010 ▶; Lam et al., 2012 ▶; Passamaneck et al., 2006 ▶; Remington, 2006 ▶; Shcherbo et al., 2007 ▶; Stepanenko et al., 2011 ▶; Stewart, 2006 ▶; Wacker et al., 2007 ▶; Wang et al., 2008 ▶; Wiedenmann et al., 2009 ▶; Wu et al., 2011 ▶; Zubova & Savitsky, 2005 ▶). The extensive spectral diversity of GFP-like proteins enabled multicolour labelling and the development of specific biosensors based on fluorescent donor–acceptor FRET (Förster resonance energy transfer) pairs (Carlson & Campbell, 2009 ▶; Chudakov et al., 2010 ▶; Davidson & Campbell, 2009 ▶; Miyawaki, 2011 ▶; Piston & Kremers, 2007 ▶; Shaner et al., 2007 ▶). Several efficient blue–green and cyan–yellow FRET pairs (Chudakov et al., 2010 ▶; Shaner et al., 2007 ▶) are currently available, whereas efficient yellow/orange–red/far-red FRET pairs, with a greater emission transparency in living tissues, are still in great demand.
The optical emission window of the known FPs covers a spectral range of 425–670 nm (Chudakov et al., 2010 ▶; Shcherbo et al., 2010 ▶). Each colour range is populated by a relatively large number of FPs, with the exception of the yellow–orange range (535–555 nm), which is of significant importance for the design of yellow–red FRET pairs. At present, this spectral range is represented by only two wild-type yellow fluorescent proteins: zYFP538 from the marine button polyp Zoanthus sp. (with a Lys66-Tyr67-Gly68 chromophore-forming sequence; Matz et al., 1999 ▶) and phiYFP from the jellyfish Phialidium (Thr65-Tyr66-Gly67; Shagin et al., 2004 ▶). In spite of their significantly different chromophore structures, zYFP538 and phiYFP have almost identical spectral characteristics (λex max ≃ 525 nm and λem max ≃ 537 nm).
The crystal structure of zYFP538 has previously been reported at resolutions of 2.7 Å (Remington et al., 2005 ▶) and 1.8 Å (Pletneva et al., 2007 ▶). Here, we present the results of a crystallographic study of the yellow fluorescent protein phiYFPv, which differs from its progenitor wild-type phiYFP by ten amino-acid residues introduced by random mutagenesis: Met1Gly, Glu88Asp, Val103Asn, Met166Cys, Glu174Gly, Ile201Met, Thr202Ser, Thr206Lys, Val221Lys and Leu234Asp. The variant phiYFPv is spectrally identical to its progenitor but exhibits faster and more complete maturation in bacteria. All replacements are outside the chromophore area and are positioned mostly on the protein surface exposed to solvent, enhancing its hydrophilicity and thus presumably leading to favourable crystallization properties. Particular attention has been paid to stereochemical features in the chromophore area. Extensive structure-based mutagenesis has been applied in order to identify the most important sites affecting the spectral characteristics, and the obtained results have been utilized to improve the properties of phiYFPv and its homologous monomeric biomarker tagYFP. We have carefully analyzed the surface amino-acid residues of phiYFPv responsible for protein oligomerization in phiYFPv and its wild-type progenitor, as they provide important information that is required for the development of the monomeric variants that are needed for FRET-based biosensors.
2. Materials and methods
2.1. Expression, purification and crystallization
For protein expression, the fragment encoding phiYFPv with an N-terminal His tag was cloned into a pQE30 vector (Qiagen, USA) and transformed into Escherichia coli XL1 Blue strain (Invitrogen, USA). Bacterial cultures were grown overnight at 310 K. No IPTG induction was necessary since promoter leakage was sufficient for effective expression. The cells were pelleted by centrifugation, resuspended in phosphate-buffered saline and lysed by sonication. phiYFPv was purified by immobilized metal-affinity chromatography using TALON resin (Clontech Laboratories, USA) followed by size-exclusion chromatography using a Superdex 200 (16/60) column (GE Healthcare, USA). For crystallization, phiYFPv was transferred into a buffer consisting of 20 mM Tris–HCl pH 8.0, 200 mM NaCl, 5 mM EDTA and concentrated to 9 mg ml−1. Crystallization trials were set up by the hanging-drop method at room temperature. phiYFPv crystals suitable for data acquisition were obtained in two weeks from 0.16 M NaH2PO4, 16% PEG 3350.
2.2. Data collection, structure solution and crystallographic refinement
X-ray diffraction data were collected from a single crystal flash-cooled in a 100 K nitrogen stream. Prior to cooling, the crystal of phiYFPv was transferred into a cryoprotectant solution consisting of 20% glycerol and 80% reservoir solution. Data were collected using a MAR 300 CCD detector on SER-CAT beamline 22-ID at the Advanced Photon Source, Argonne National Laboratory, Argonne, Illinois, USA and were processed with HKL-2000 (Otwinowski & Minor, 1997 ▶).
The crystal structure of phiYFPv was solved using the molecular-replacement method with MOLREP (Vagin & Teplyakov, 2010 ▶) from the CCP4 package (Winn et al., 2011 ▶), using the coordinates of the crystal structure of mutant GFP-R96A (Aequorea victoria; PDB entry 1qy3; Barondeau et al., 2003 ▶) as a search model. Structure refinement was performed with REFMAC5 (Murshudov et al., 2011 ▶) and was alternated with manual correction of the model using Coot (Emsley & Cowtan, 2004 ▶). Noncrystallographic symmetry restraints were not applied during refinement. Water molecules were located with Coot. Crystallographic data and refinement statistics are presented in Table 1 ▶.
Table 1. Data-collection and refinement statistics for phiYFPv (PDB entry 4he4).
Values in parentheses are for the highest resolution shell.
| Data collection | |
| Space group | H32 |
| Unit-cell parameters (Å) | a = b = 102.9, c = 242.5 |
| Z/Z′ | 36/2 |
| Estimated solvent content (%) | 46 |
| Temperature (K) | 100 |
| Wavelength (Å) | 1.00 |
| Resolution range (Å) | 28.7–2.05 (2.12–2.05) |
| Total reflections measured | 226374 |
| Unique reflections observed | 29786 |
| Multiplicity | 7.6 (7.1) |
| 〈I/σ(I)〉 | 22.1 (3.3) |
| R merge | 0.078 (0.562) |
| Completeness (%) | 99.7 (98.2) |
| Refinement statistics | |
| Non-H atoms in model | |
| Protein | 3663 [2 × residues 1–234] |
| Water | 190 |
| R work | 0.196 [95.0% of data] |
| R free | 0.256 [5.0% of data] |
| Mean B factor/r.m.s.d. (Å2) | |
| Protein atoms | |
| Main chain | 40.5/0.8 |
| Side chain | 42.1/2.2 |
| Chromophore | 35.2/5.9 |
| Water | 45.2 |
| Geometry r.m.s.d. | |
| Bond lengths (Å) | 0.026 |
| Bond angles (°) | 1.872 |
| General planes (Å) | 0.009 |
| Cα r.m.s.d. (A and B subunits) (Å) | 0.335 |
| Ramachandran statistics (for non-Gly/Pro residues) (%) | |
| Most favourable/additional allowed | 89.5 |
| Generously allowed | 10.5 |
Structure validation was performed with Coot and PROCHECK (Laskowski et al., 1993 ▶). The figures were prepared with LIGPLOT/HBPLUS (McDonald & Thornton, 1994 ▶; Wallace et al., 1995 ▶), PyMOL (DeLano, 2002 ▶), SETOR (Evans, 1993 ▶) and ChemDraw (CambridgeSoft). The coordinates and structure factors were deposited in the Protein Data Bank under accession code 4he4.
2.3. Mutagenesis and photophysical characterization
Preparation of mutant variants by site-directed mutagenesis was carried out by PCR using the overlap extension method with primers containing appropriate target substitutions (Ho et al., 1989 ▶). N-terminally His6-tagged variants were expressed in E. coli XL1 Blue strain (Invitrogen, USA) and purified using TALON metal-affinity resin (Clontech Laboratories, USA). Absorbance and fluorescence spectra of the purified proteins were recorded using a Varian Cary 100 UV–Vis spectrophotometer and a Varian Cary Eclipse fluorescence spectrophotometer, respectively.
3. Results
3.1. Protomer structure
The principal structural fold of the phiYFPv subunit, which is shared with all members of the GFP family, is an 11-stranded β-barrel with loop caps on both sides and a chromophore (matured from the sequence Thr65-Tyr66-Gly67) embedded in the middle of an internal helix which consists of a combination of distorted α-helical and 310-helical turns. The peptide group preceding Pro87 adopts a cis conformation. Interestingly, the structure of the spectrally identical zYFP538 shows the existence of a pore on the cylindrical β-barrel surface with a chain of hydrogen-bonded water molecules passing from the outside to the hydroxyphenyl moiety of the chromophore (Pletneva et al., 2007 ▶). In TurboGFP this water-filled pore was suggested to be essential for the enhanced chromophore maturation rate as it enables additional access for molecular oxygen (Evdokimov et al., 2006 ▶). In phiYFPv, a similar pore formed by the backbone atoms of Phe143, Thr144, Pro145, His204 and Val205 contains only one water molecule bound to the hydroxyl of Tyr66. It appears that the origin of the phenomenon observed in phiYFPv is the insufficient resolution of the diffraction data (2.05 Å). The pore size in the phiYFPv structure was found to be almost identical to its counterparts in TurboGFP (1.6 Å; Evdokimov et al., 2006 ▶), zYFP538 (1.8 Å; Pletneva et al., 2007 ▶) and mKate (1.8, 1.75 and 2.6 Å at different pH values; Pletnev et al., 2008 ▶). The continuous chain of water molecules passing through the pore was identified in structures at a resolution of ∼1.8 Å or better. Only a few water molecules (0–2) were found in the pores of the eight crystallographically independent subunits of mKate at a resolution of 2.6 Å.
3.2. Oligomeric structure
The biological unit of phiYFPv is a dimer both in solution and in the crystalline state. The two identical subunits, A and B (Cα r.m.s.d. of 0.335 Å), are related by a noncrystallographic twofold symmetry axis with side-to-side packing at ∼60°. The buried contact area between the monomers (∼1350 Å2 per monomer) is formed by the side chains of 13 residues, His146, Cys147, Tyr149, Trp151, Lys160, Lys164, Asp175, Phe176, Asp180, Tyr198, Arg225, Arg230 and Asp233, which contribute 12 hydrogen bonds and six salt bridges to stabilize the dimeric assembly (Fig. 1 ▶ and Table 2 ▶). The irregular C-terminal tail 227–234 extends away from the β-barrel towards to the cylindrical surface of the interacting counterpart, contributing to interface stabilization. Removal of the dimer-stabilizing contacts, accompanied by deletion of the C-terminal segment, typically favours the monomeric state of a biomarker, which is highly desirable for protein labelling and FRET techniques.
Figure 1.

Stereoview of the interface between the A and B subunits in the dimeric structure of phiYFPv. This figure was produced using SETOR (Evans, 1993 ▶).
Table 2. The unique stabilizing intersubunit interactions in the phiYFPv dimer.
The number of stabilizing contacts is doubled by the twofold noncrystallographic symmetry axis.
| Bond type | Interacting residues and atoms |
|---|---|
| Hydrogen bonds | Pro145 CO⋯His200 NE2 |
| Pro145 CO⋯Tyr198 OH | |
| Tyr149 OH⋯Cys147 NH | |
| Tyr149 OH⋯Lys164 CO | |
| Arg225 NH2⋯Asp234 CO | |
| Arg225 NH2⋯Thr232 CO | |
| Salt bridges | Lys160 NZ–Asp175 OD1 |
| Lys164 NZ–Asp180 OD1 | |
| Lys164 NZ–Asp180 OD2 |
3.3. Structural features of the chromophore area
The structure of the chromophore and the stereochemistry of its environment are the key factors that determine the photophysical properties of GFP-like proteins. PhiYFPv exhibits fluorescence in a yellow spectral range with excitation and emission maxima at 524 and 537 nm, respectively. No indication of any disorder in the relatively tightly packed chromophore environment was observed. The atomic displacement factors of the chromophore are below the average for the protein (Table 1 ▶).
The post-translational modification of the chromophore-forming sequence Thr65-Tyr66-Gly67 results in a coplanar two-ring chromophore structure consisting of a five-membered imidazolinone heterocycle with a p-hydroxybenzylidene substituent (Fig. 2 ▶ a) typical of the green-emitting FPs. Tyr66 is found in a cis conformation described by torsion angles of ∼0.15° and ∼2.0° around the Cα—Cβ and Cβ—Cγ bonds, respectively. The geometry of the first chromophore residue, Thr65, is consistent with that of a ‘green’ chromophore and is characterized by an sp 3-hybridized Cα centre with the standard trans configuration of the preceding peptide linkage.
Figure 2.

Chemical structures of the chromophores of the spectrally identical phiYFPv (a) and zYFP538 (b) matured from the chromophore-forming sequences Thr-Tyr-Gly and Lys-Tyr-Gly, respectively. This figure was produced using ChemDraw (CambridgeSoft).
The nearest shell of the chromophore (within 3.9 Å) is composed of 19 residues, most of which are involved in an extensive network of hydrogen bonds with active participation of mediating water molecules. This network is functionally important in the maturation and fluorescence as a potential proton wire. It forms five direct and six water-mediated hydrogen bonds to the chromophore (Fig. 3 ▶). Direct and water-mediated hydrogen bonds of His146, Thr144 and Val205 to the hydroxyl of Tyr66 stabilize the fluorescent cis form of the chromophore. These interactions, together with the other three specific hydrogen bonds between the chromophore Thr65 and the catalytic Glu222, as well as between the imidazolinone carbonyl and Gln92 and the catalytic Arg94, greatly affect the distribution of the electron density of the chromophore.
Figure 3.

A schematic diagram illustrating the nearest amino-acid environment of the chromophore in the phiYFPv structure. Hydrogen bonds (≤3.3 Å) are shown as blue dashed lines, water molecules (W) as red spheres and van der Waals contacts (≤3.9 Å) as black ‘eyelashes’. This figure was prepared using LIGPLOT/HBPLUS (McDonald & Thornton, 1994 ▶; Wallace et al., 1995 ▶).
A remarkable structural feature of phiYFPv responsible for its yellow fluorescence is a nearly perfect π-stacking (d ≃ 3.6 Å) between the antiparallel phenolic rings of the chromophore Tyr66 and Tyr203 (Fig. 4 ▶). This particular interaction has been observed previously in the crystal structures of several engineered YFPs (Griesbeck et al., 2001 ▶; Rekas et al., 2002 ▶; Wachter et al., 1998 ▶).
Figure 4.
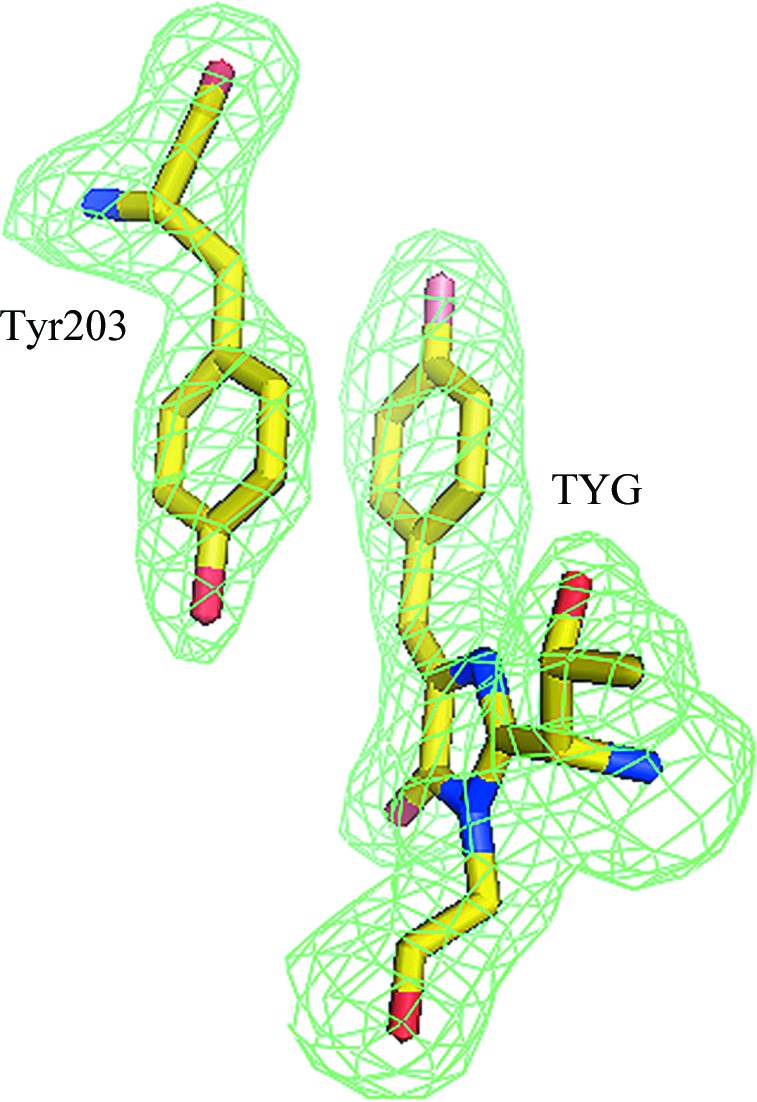
π-Stacking interaction of the chromophore Tyr66 with the proximal Tyr203 viewed in an F o − F c OMIT electron-density map (cutoff ρ = 3.0σ). This figure was produced using PyMOL (DeLano, 2002 ▶).
3.4. Structure-based site-directed mutagenesis
The availability of the crystal structure allowed us to perform site mapping in the neighbourhood of the chromophore aimed at identifying the key residues that affect the fluorescence characteristics of phiYFPv. The mapping results were subsequently tested by site-directed mutagenesis (Table 3 ▶). The functional roles of the sites which mostly account for the advanced spectral properties of dimeric phiYFPv have also been tested by point mutations in the monomeric yellow fluorescent biomarker tagYFP (λex max ≃ 508 nm, λem max ≃ 524 nm), which appears to share some important similarities with phiYFPv. Although the nearest chromophore environments of phiYFPv and tagYFP are not identical, both proteins possess the same chromophore-forming triad and have the π-stacking interaction of a tyrosine in position 203 with the chromophore phenolic ring in common. TagYFP belongs to a homologous group of genetically engineered monomeric/weak dimeric yellow biomarkers (here named groupYFP) consisting of mCitrine, Venus, YPet, EYFP and tagYFP (Evrogen_Moscow; http://www.evrogen.com; Griesbeck et al., 2001 ▶; Nagai et al., 2002 ▶; Nguyen & Daugherty, 2005 ▶; Tsien, 1998 ▶; Fig. 5 ▶). The biomarkers of this group exhibit similar fluorescence spectra and are used for protein labelling and FRET techniques (Chudakov et al., 2010 ▶; Rekas et al., 2002 ▶; Shaner et al., 2005 ▶, 2007 ▶).
Table 3. Spectral characteristics.
(a).
PhiYFP mutants.
| λex max/λem max † (nm) | ECmol ‡ (M −1 cm−1) | QY§ | B ¶ | B rel phiYFP †† (%) | |
|---|---|---|---|---|---|
| PhiYFPv | 524/537 | 101305 | 0.59 | 59.8 | 100 |
| Tyr203Ser | 495/515 | 62854 | 0.31 | 19.5 | 33 |
| Tyr203Thr‡‡ | 500/515 | 72919 | 0.28 | 20.4 | 34 |
| Tyr203Val‡‡ | 510/520 | 75944 | 0.39 | 29.6 | 49 |
| Tyr14Ile | 520/532 | n/d§§ | n/d | n/d | n/d |
| Val205Ser | 516/529 | 37340 | 0.32 | 11.9 | 20 |
| Ser161Ala | 524/537 | 103023 | 0.76 | 78.3 | 130 |
(b).
TagYFP mutants.
| λex max/λem max † (nm) | ECmol ‡ (M −1 cm−1) | QY§ | B ¶ | B rel tagYFP †† (%) | |
|---|---|---|---|---|---|
| TagYFP | 508/524 | 20056 | 0.74 | 14.8 | 100 |
| Val14Tyr/Leu68Ala | 505/520 | 32461 | 0.49 | 15.9 | 107 |
| Thr205Val | 515/530 | 45905 | 0.38 | 17.4 | 118 |
λex max and λem max are the excitation and emission maxima, respectively.
ECmol is the extinction coefficient.
QY is the fluorescence quantum yield.
B is the brightness calculated as (ECmol × QY)/1000.
B rel phiYFP = [B(mutant)/B(phiYFP)] × 100; B rel tagYFP = [B(mutant)/B(tagYFP)] × 100.
Pakhomov & Martynov (2011 ▶).
Not detectable.
Figure 5.

Sequence alignment of phiYFPv and the yellow variants of the YFP group (total identity 44.2%) showing the chromophore-forming sequence (highlighted in grey), residues from the environment nearest to the chromophore (in red; see also Fig. 3 ▶) and dimer-stabilizing residues from the intersubunit contact surface (highlighted in yellow; see also Table 1 ▶).
Site-directed mutagenesis confirmed that π-stacking interactions between the aromatic rings of the chromophore Tyr66 and the proximal Tyr203 is a crucial structural feature defining the spectral characteristics of phiYFPv and, obviously, its wild-type analogue phiYFP. A single replacement of Tyr203 by Ser/Thr/Val shifts the emission maximum from the yellow (∼537 nm) to the green spectral range (∼515–520 nm), with considerable dimming of the fluorescence (Pakhomov & Martynov, 2011 ▶; see also Table 3 ▶).
Our mutagenesis study has revealed the important functional role of sites 14 and 205 proximal to the chromophore. In phiYFPv these sites are occupied by Tyr14 and Val205 and in groupYFPs by residues of a substantially different nature: Val/Ile14 and Thr/Ser205 (Figs. 5 ▶ and 6 ▶; the numbering used here and subsequently corresponds to phiYFPv). The replacement of Tyr14 or Val205 in phiYFPv by Ile or Ser, respectively, strongly decreased the protein-maturation rate, resulting in very dim variants (Table 3 ▶). In contrast, the introduction of Tyr or Val at the respective positions 14 or 205 in the homologous monomeric biomarker tagYFP had a positive influence on the spectral properties. The most significant effect was achieved by the Thr205Val mutation, which increased the brightness of tagYFP by ∼18%, accompanied by a desirable ∼6 nm red shift of the emission band.
Figure 6.
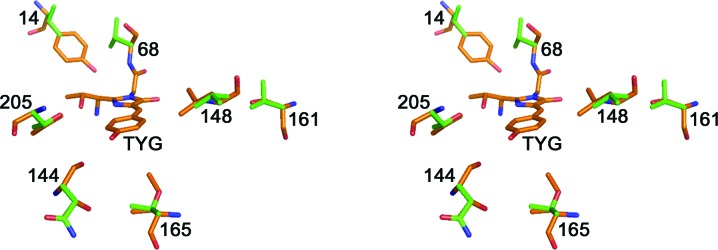
Stereoview showing the residue differences around the TYG chromophore in phiYFPv (orange) and tagYFP (green). Sites in phiYFPv/tagYFP: 14, Tyr/Val; 68, Ala/Val; 144, Thr/Asn; 148, Leu/Val; 161, Ser/Val; 165, Ile/Thr; 205, Val/Thr. This figure was produced using PyMOL (DeLano, 2002 ▶).
It has been suggested that Ser at position 161 in phiYFPv (represented by Ala/Val in groupYFP) plays the same role as Ser158 in mKate (Pletnev et al., 2008 ▶). The Ser158Ala mutation in mKate increased the brightness of fluorescence by almost a factor of two (Pletnev et al., 2008 ▶); similarly, the replacement of Ser161 by Ala in phiYFPv increased its brightness by 30%.
4. Discussion
The subfamily of yellow fluorescent proteins can be divided into two groups. The first group consists of two wild-type proteins, dimeric phiYFP (Phialidium) and tetrameric zYFP538 (Zoanthus sp.), both of which exhibit almost identical spectral characteristics (λex max ≃ 525 nm and λem max ≃ 537 nm; Matz et al., 1999 ▶; Shagin et al., 2004 ▶). The second group is represented by homologous monomeric/weak dimeric genetically engineered biomarkers (here named groupYFP): mCitrine, mVenus, YPet, EYFP and tagYFP (λex max ≃ 515 nm and λem max ≃ 528 nm; Griesbeck et al., 2001 ▶; Nagai et al., 2002 ▶; Nguyen & Daugherty, 2005 ▶; Tsien, 1998 ▶; see also Rekas et al., 2002 ▶; Shaner et al., 2005 ▶, 2007 ▶). All of these ‘yellow’ FPs, except for zYFP538, are characterized by a coplanar bicyclic structure of the chromophore which is typical of ‘green’ FPs (Fig. 2 ▶ a). In zYFP538, on the other hand, post-translational modification results in a tricyclic chromophore structure with an additional tetrahydropyridine ring (Fig. 2 ▶ b; Pletneva et al., 2007 ▶; Remington et al., 2005 ▶).
In an attempt to explain the spectral properties of phiYFPv, we performed structure-based site-directed mutagenesis. The most promising mutations were further transferred to the monomeric biomarker tagYFP, a homologous member of groupYFP. The observed ∼20–30 nm bathochromic shift of phiYFPv and groupYFP fluorescence relative to that of green FPs is apparently a consequence of the combined influence of several structural features. The π-stacking interactions between the highly polarizable aromatic rings of the chromophore Tyr66 and the proximal Tyr203 have been identified as a major structural feature responsible for this shift. The importance of such interactions has been demonstrated previously for a number of other designed variants of GFPs (Dickson et al., 1997 ▶; Ormo et al., 1996 ▶; Wachter et al., 1998 ▶). The introduction of similar interactions in the green mutant avGFP_S65T (by a Thr203Tyr mutation) with emission at ∼512 nm yielded a yellow variant with emission at ∼527 nm. It was suggested that the enhanced polarizability of groups adjacent to the chromophore, such as the phenyl of Tyr or Phe and the imidazole of His, is likely to be a major factor in the observed red shift (Wachter et al., 1998 ▶). These adjacent groups were proposed to increase the chromophore polarity with a greater dipole moment, resulting in longer wavelength fluorescence.
The functional importance of the observed interactions between the π-stacked aromatic rings of tyrosines in phiYFPv (similar to those present in wild-type phiYFP) has been tested by introducing a number of single-point mutations: Tyr203Thr, Tyr203Val (Pakhomov & Martynov, 2011 ▶) and Tyr203Ser (Table 3 ▶). The mutants are characterized by weak fluorescence, demonstrating a hypsochromic shift of the emission maximum from ∼537 to ∼515–520 nm. In phiYFPv the side chain of Tyr203 is located in a tightly packed environment created by the chromophore phenolic ring and the side chains of Gln69, Leu148, Met201, Val205, Glu222 and Val224. The replacement of Tyr203 by smaller residues (Val, Thr and Ser) creates an empty cavity of ∼4.4 Å within this environment, which presumably favours vibrational relaxation of the chromophore, causing the observed dimming of fluorescence. An additional contribution to the decrease in the emission intensity could also arise from partial protonation of the chromophore phenolate O atom. The latter arises from direct hydrogen bonding to the proton-donating hydroxyl groups of Ser203 or Thr203 in the corresponding mutants. The possibility of the formation of these hydrogen bonds has been proposed based on computer modelling. Ser and Thr at position 205 in the yellow FPs from the groupYFPs (Griesbeck et al., 2001 ▶; Rekas et al., 2002 ▶; Wachter et al., 1998 ▶) presumably demonstrate a similar protonation effect owing to participation in water-mediated hydrogen bonding with the chromophore tyrosine. In tagYFP, the replacement of Thr205 by Val resulted in a brightness increase of 18% (Table 3 ▶).
The observed π-stacking interaction is absent in zYFP538, which shows yellow fluorescence at ∼538 nm caused by an extra double Cα=Nζ bond of the additional tetrahydropyridine ring of the chromophore that extends its π-conjugated system (Fig. 2 ▶ b; Pletneva et al., 2007 ▶; Remington et al., 2005 ▶).
Our structure-based mutagenesis study of the nearest environment of the chromophore in phiYFPv has revealed the important functional roles of Tyr and Val at positions 14 and 205, respectively (Fig. 5 ▶). Tyr14 is positioned ∼4.2 Å from the chromophore. It forms two direct hydrogen bonds to the carbonyl of Leu64 preceding the chromophore and to the side chain of Asn119, which is involved in the hydrogen-bonded network around the chromophore. Disrupting these hydrogen bonds by the replacement of Tyr14 with a hydrophobic Ile resulted in a poorly matured dim variant (Table 3 ▶). The hydrophobic Val205 in phiYFPv mostly resides in a hydrophobic environment and presumably stabilizes the conformational state of Tyr203, which is involved in the key π-stacking interactions mentioned above. The replacement of Val205 in phiYFPv by the hydrophilic Ser results in 80% fluorescence quenching and in an ∼8 nm blue shift of the excitation/emission bands.
These results led to testing of the effect of Tyr and Val at the corresponding positions 14 and 205 on the spectral properties in the monomeric biomarker tagYFP. In this protein positions 14 and 205 are occupied by residues of a substantially different nature: Val and Thr, respectively. The mutation Val14Tyr in tagYFP (combined with Leu68Ala in order to remove steric hindrance) had only a minor effect, showing an unwanted ∼4 nm blue shift and an ∼7% increase in fluorescence brightness (Table 3 ▶). The replacement Thr205Val demonstrated a quite significant positive effect, exhibiting an ∼18% brightness increase of tagYFP and a desirable ∼6 nm red shift of the emission band maximum.
The Ser158Ala mutation in the far-red mKate breaks a hydrogen bond between Ser158 and the hydroxyl of the chromophore Tyr66 in a nonfluorescent trans state (Pletnev et al., 2008 ▶). Destabilization of the trans state in the corresponding mKate variant shifted the equilibrium to a fluorescent cis state, increasing the fluorescence brightness by 95%. In phiYFPv Ser161 occupies a position similar to that of Ser158 in mKate; thus, it could be assumed that it stabilizes a hypothetically possible nonfluorescent trans state in a similar manner (Fig. 7 ▶). A certain fraction of the trans isomer presumably appears owing to cis-to-trans isomerization of the chromophore (the kindling effect) upon irradiation at the excitation wavelength (Chudakov et al., 2003 ▶). Indeed, the replacement Ser161Ala in phiYFPv resulted in an increase in brightness of ∼30% (Table 3 ▶). It is worth noting that in all members of groupYFP position 161 is occupied by the hydrophobic residues Ala or Val that are incapable of forming hydrogen bonds.
Figure 7.
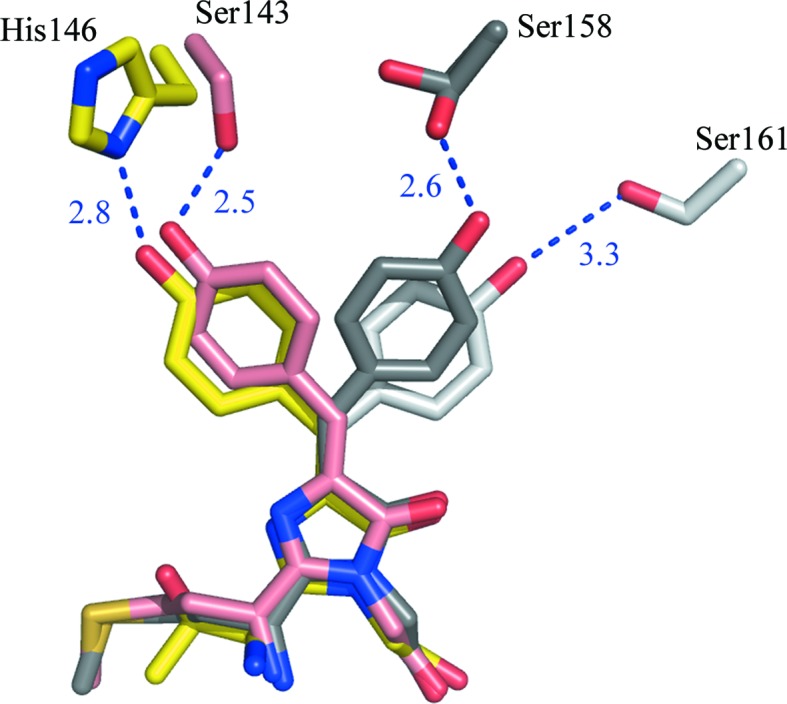
Structures of the superimposed chromophores of mKate (Pletnev et al., 2008 ▶) and phiYFPv with the isomer-stabilizing residues shown. mKate: fluorescent cis form at pH 7.0, pink; nonfluorescent trans form at pH 2.0, dark grey. phiYFPv: fluorescent cis form at pH 8.0, yellow; hypothetical modelled nonfluorescent trans form, light grey. This figure was produced using PyMOL (DeLano, 2002 ▶).
Interestingly, despite having identical chromophores and the same π-stacking between the two key tyrosines, the dimeric phiYFPv (λem max ≃ 537 nm) and the monomeric tagYFP (λem max ≃ 524 nm) exhibit an ∼13 nm difference in fluorescence-band position. We mostly attribute this difference to the combined effect of individual stereochemical features in the chromophore areas of both proteins. The largest contribution is from the hydrophobicity/hydrophilicity of the residue at position 205, which is a hydrophobic Val in phiYFPv and a similar-sized hydrophilic Thr in tagYFP. The replacement of Val205 by a hydrophilic Ser in phiYFPv resulted in an ∼8 nm blue shift of the emission band (Table 3 ▶). On the other hand, the replacement of the hydrophilic Thr205 by a hydrophobic Val in monomeric tagYFP resulted in an ∼6 nm red shift. An additional contribution to the observed spectral difference between the dimeric phiYFPv and the monomeric tagYFP could arise from differences in their oligomeric state. Extensive intersubunit interactions, represented by 12 hydrogen bonds and six salt bridges in the phiYFPv dimer (Table 2 ▶) could affect the electrostatic field of the protein matrix and, as a consequence, the spectral properties of the internal chromophore.
Supplementary Material
PDB reference: phiYFPv, 4he4
Acknowledgments
We acknowledge the use of beamline 22-ID of the Southeast Regional Collaborative Access Team (SER-CAT) located at the Advanced Photon Source, Argonne National Laboratory. Use of the APS was supported by the US Department of Energy, Office of Science, Office of Basic Energy Sciences under Contract No. W-31-109-Eng-38. This project has been supported in part by Federal funds from the National Cancer Institute, National Institutes of Health (NIH) contract No. HHSN261200800001E, the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research and by grants from the President of the Russian Federation MK-2382.2012.4, the Russian Foundation for Basic Research 11-04-00241, 11-04-92122 and 12-04-33023, MCB RAS and Measures to Attract Leading Scientists to Russian Educational Institutions (11.G34.31.0017). The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does the mention of trade names, commercial products or organizations imply endorsement by the US Government.
References
- Barondeau, D. P., Putnam, C. D., Kassmann, C. J., Tainer, J. A. & Getzoff, E. D. (2003). Proc. Natl Acad. Sci. USA, 100, 12111–12116. [DOI] [PMC free article] [PubMed]
- Carlson, H. J. & Campbell, R. E. (2009). Curr. Opin. Biotechnol. 20, 19–27. [DOI] [PubMed]
- Chudakov, D. M., Feofanov, A. V., Mudrik, N. N., Lukyanov, S. & Lukyanov, K. A. (2003). J. Biol. Chem. 278, 7215–7219. [DOI] [PubMed]
- Chudakov, D. M., Matz, M. V., Lukyanov, S. & Lukyanov, K. A. (2010). Physiol. Rev. 90, 1103–1163. [DOI] [PubMed]
- Davidson, M. W. & Campbell, R. E. (2009). Nature Methods, 6, 713–717. [DOI] [PubMed]
- DeLano, W. L. (2002). PyMOL http://www.pymol.org.
- Dickson, R. M., Cubitt, A. B., Tsien, R. Y. & Moerner, W. E. (1997). Nature (London), 388, 355–358. [DOI] [PubMed]
- Emsley, P. & Cowtan, K. (2004). Acta Cryst. D60, 2126–2132. [DOI] [PubMed]
- Evans, S. V. (1993). J. Mol. Graph. 11, 134–138. [DOI] [PubMed]
- Evdokimov, A. G., Pokross, M. E., Egorov, N. S., Zaraisky, A. G., Yampolsky, I. V., Merzlyak, E. M., Shkoporov, A. N., Sander, I., Lukyanov, K. A. & Chudakov, D. M. (2006). EMBO Rep. 7, 1006–1012. [DOI] [PMC free article] [PubMed]
- Griesbeck, O., Baird, G. S., Campbell, R. E., Zacharias, D. A. & Tsien, R. Y. (2001). J. Biol. Chem. 276, 29188–29194. [DOI] [PubMed]
- Ho, S. N., Hunt, H. D., Horton, R. M., Pullen, J. K. & Pease, L. R. (1989). Gene, 77, 51–59. [DOI] [PubMed]
- Lam, A. J., St-Pierre, F., Gong, Y., Marshall, J. D., Cranfill, P. J., Baird, M. A., McKeown, M. R., Wiedenmann, J., Davidson, M. W., Schnitzer, M. J., Tsien, R. Y. & Lin, M. Z. (2012). Nature Methods, 9, 1005–1012 [DOI] [PMC free article] [PubMed]
- Laskowski, R. A., MacArthur, M. W., Moss, D. S. & Thornton, J. M. (1993). J. Appl. Cryst. 26, 283–291.
- Matz, M. V., Fradkov, A. F., Labas, Y. A., Savitsky, A. P., Zaraisky, A. G., Markelov, M. L. & Lukyanov, S. A. (1999). Nature Biotechnol. 17, 969–973. [DOI] [PubMed]
- McDonald, I. K. & Thornton, J. M. (1994). J. Mol. Biol. 238, 777–793. [DOI] [PubMed]
- Miyawaki, A. (2011). Annu. Rev. Biochem. 80, 357–373. [DOI] [PubMed]
- Murshudov, G. N., Skubák, P., Lebedev, A. A., Pannu, N. S., Steiner, R. A., Nicholls, R. A., Winn, M. D., Long, F. & Vagin, A. A. (2011). Acta Cryst. D67, 355–367. [DOI] [PMC free article] [PubMed]
- Nagai, T., Ibata, K., Park, E. S., Kubota, M., Mikoshiba, K. & Miyawaki, A. (2002). Nature Biotechnol. 20, 87–90. [DOI] [PubMed]
- Nguyen, A. W. & Daugherty, P. S. (2005). Nature Biotechnol. 23, 355–360. [DOI] [PubMed]
- Ormo, M., Cubitt, A. B., Kallio, K., Gross, L. A., Tsien, R. Y. & Remington, S. J. (1996). Science, 273, 1392–1395. [DOI] [PubMed]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol. 276, 307–326. [DOI] [PubMed]
- Pakhomov, A. A. & Martynov, V. I. (2011). Biochem. Biophys. Res. Commun. 407, 230–235. [DOI] [PubMed]
- Passamaneck, Y. J., Di Gregorio, A., Papaioannou, V. E. & Hadjantonakis, A. K. (2006). Microsc. Res. Tech. 69, 160–167. [DOI] [PubMed]
- Piston, D. W. & Kremers, G. J. (2007). Trends Biochem. Sci. 32, 407–414. [DOI] [PubMed]
- Pletneva, N. V., Pletnev, S. V., Chudakov, D. M., Tikhonova, T. V., Popov, V. O., Martynov, V. I., Wlodawer, A., Dauter, Z. & Pletnev, V. Z. (2007). Bioorg. Khim. 33, 421–430. [DOI] [PubMed]
- Pletnev, S., Shcherbo, D., Chudakov, D. M., Pletneva, N., Merzlyak, E. M., Wlodawer, A., Dauter, Z. & Pletnev, V. (2008). J. Biol. Chem. 283, 28980–28987. [DOI] [PMC free article] [PubMed]
- Rekas, A., Alattia, J. R., Nagai, T., Miyawaki, A. & Ikura, M. (2002). J. Biol. Chem. 277, 50573–50578. [DOI] [PubMed]
- Remington, S. J. (2006). Curr. Opin. Struct. Biol. 16, 714–721. [DOI] [PubMed]
- Remington, S. J., Wachter, R. M., Yarbrough, D. K., Branchaud, B., Anderson, D. C., Kallio, K. & Lukyanov, K. A. (2005). Biochemistry, 44, 202–212. [DOI] [PubMed]
- Shagin, D. A., Barsova, E. V., Yanushevich, Y. G., Fradkov, A. F., Lukyanov, K. A., Labas, Y. A., Semenova, T. N., Ugalde, J. A., Meyers, A., Nunez, J. M., Widder, E. A., Lukyanov, S. A. & Matz, M. V. (2004). Mol. Biol. Evol. 21, 841–850. [DOI] [PubMed]
- Shaner, N. C., Patterson, G. H. & Davidson, M. W. (2007). J. Cell Sci. 120, 4247–4260. [DOI] [PubMed]
- Shaner, N. C., Steinbach, P. A. & Tsien, R. Y. (2005). Nature Methods, 2, 905–909. [DOI] [PubMed]
- Shcherbo, D., Merzlyak, E. M., Chepurnykh, T. V., Fradkov, A. F., Ermakova, G. V., Solovieva, E. A., Lukyanov, K. A., Bogdanova, E. A., Zaraisky, A. G., Lukyanov, S. & Chudakov, D. M. (2007). Nature Methods, 4, 741–746. [DOI] [PubMed]
- Shcherbo, D., Shemiakina, I. I., Ryabova, A. V., Luker, K. E., Schmidt, B. T., Souslova, E. A., Gorodnicheva, T. V., Strukova, L., Shidlovskiy, K. M., Britanova, O. V., Zaraisky, A. G., Lukyanov, K. A., Loschenov, V. B., Luker, G. D. & Chudakov, D. M. (2010). Nature Methods, 7, 827–829. [DOI] [PMC free article] [PubMed]
- Stepanenko, O. V., Stepanenko, O. V., Shcherbakova, D. M., Kuznetsova, I. M., Turoverov, K. K. & Verkhusha, V. V. (2011). Biotechniques, 51, 313–327. [DOI] [PMC free article] [PubMed]
- Stewart, C. N. Jr (2006). Trends Biotechnol. 24, 155–162. [DOI] [PubMed]
- Tsien, R. Y. (1998). Annu. Rev. Biochem. 67, 509–544. [DOI] [PubMed]
- Vagin, A. & Teplyakov, A. (2010). Acta Cryst. D66, 22–25. [DOI] [PubMed]
- Wachter, R. M., Elsliger, M. A., Kallio, K., Hanson, G. T. & Remington, S. J. (1998). Structure, 6, 1267–1277. [DOI] [PubMed]
- Wacker, S. A., Oswald, F., Wiedenmann, J. & Knöchel, W. (2007). Dev. Dyn. 236, 473–480. [DOI] [PubMed]
- Wallace, A. C., Laskowski, R. A. & Thornton, J. M. (1995). Protein Eng. 8, 127–134. [DOI] [PubMed]
- Wang, Y. S., Yoo, C. M. & Blancaflor, E. B. (2008). New Phytol. 177, 525–536. [DOI] [PubMed]
- Wiedenmann, J., Oswald, F. & Nienhaus, G. U. (2009). IUBMB Life, 61, 1029–1042. [DOI] [PubMed]
- Winn, M. D. et al. (2011). Acta Cryst. D67, 235–242.
- Wu, S., Koizumi, K., Macrae-Crerar, A. & Gallagher, K. L. (2011). PLoS One, 6, e27536. [DOI] [PMC free article] [PubMed]
- Zubova, N. N. & Savitsky, A. P. (2005). Usp. Biol. Khim. 45, 1–66.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: phiYFPv, 4he4