

Abstract
AIM: To examine whether the heteroplasmy level for 15059G>A mutation in the mitochondrial genome might be associated with essential hypertension.
METHODS: This cross-sectional study involved 196 unrelated participants randomly selected from general population (90 males and 106 females) who underwent a regular medical check-up at the Institute for Atherosclerosis Research (Moscow, Russia). One hundred and twenty of them (61%) had essential hypertension, and 76 (39%) were apparently healthy normotensive persons. The level of heteroplasmy for 15059G>A mutation occurring in the coding region of cytochrome b gene (MT-CYB) of mtDNA isolated from the blood leukocytes, was quantified using DNA pyrosequencing method.
RESULTS: The 15059G>A heteroplasmy level ranged between 4% and 83%, with a median level of 31%. Between the upper and lower quartiles of 15059G>A heteroplasmy distribution, significant differences were observed for patients’ age, systolic blood pressure, and triglyceride levels. 15059G>A heteroplasmy correlated both with age (r = 0.331, P < 0.001) and the presence of hypertension (r = 0.228, P = 0.002). Regression analysis revealed that the age explains 12% variability of 15059G>A heteroplasmy, and hypertension independently explains more 5% variability. The 15059G>A heteroplasmy exceeding 31% was found to be significantly associated with a higher risk of essential hypertension (odds ratio 2.76; P (Fisher) 0.019]. The study participants with high 15059G>A heteroplasmy level were found to have significantly higher age (P < 0.001) and the prevalence of essential hypertension (P = 0.033), as compared to those with low 15059G>A heteroplasmy level. These observations suggested a positive correlation between the level of 15059G>A heteroplasmy and essential hypertension.
CONCLUSION: This study provides the evidence of association of mtDNA 15059G>A mutation heteroplasmy with essential hypertension.
Keywords: Essential hypertension, Heteroplasmy, Mitochondrial DNA, 15059G>A mutation
Core tip: The pathophysiology of essential hypertension (EH) is insufficiently understood; in particular, the impact of mitochondrial DNA mutations on the development of EH is poorly investigated. We undertook this study in order to see whether the level of heteroplasmy for the 15059G>A mutation in the mitochondrial cytochrome b gene might be associated with EH. The 15059G>A heteroplasmy level in mtDNA in blood leukocytes obtained from 196 study participants, randomly selected from general population (120 of whom had EH), exceeding 31%, was found to be significantly associated with a higher risk of EH.
INTRODUCTION
It is known that mutations in mitochondrial DNA (mtDNA) cause a variety of hereditary disorders with complex phenotypes including those that have hypertension as one of their clinical outcomes (such as the HUPRA syndrome comprising hyperuricemia, metabolic alkalosis, pulmonary hypertension, and progressive renal failure in infancy)[1].
Essential hypertension (EH), that represents a common form of hypertension, is a highly polygenic pathological condition which is caused by a combination of small-scale changes in the expression of many genes, in conjunction with a variable collection of environmental factors[2-4]. To the date, in total, 14 independent chromosome loci have been recognized for blood pressure traits that reached genome-wide significance including replication in independent cohorts[2]. Nevertheless, these variants explain just a very small fraction of the heritability of blood pressure traits[2]. Because chromosomal DNA variants exhibit only a modest effect in EH[2], it is impossible to exclude that, in contrast, somatic mtDNA mutations might importantly contribute to the development of hypertension and that a genetic predisposition to EH may be influenced by a ratio between mutated and wild-type mtDNA, e.g., by heteroplasmy level. In support of this possibility, a non-redundant role of mtDNA heteroplasmy has been reported in human aging[5] and several age-related pathologic conditions including atherosclerosis[6,7], Alzheimer’s disease[8], and diabetes[9]. It has been also reported that the entire mtDNA sequencing in United States pedigrees of African and European descent allowed to identify significant changes in the mtDNA sequence of hypertensive probands, which implies a potential role of mtDNA mutations in EH[10]. To the date, the role of somatic mtDNA mutations in EH is poorly studied and poorly understood. Therefore, it is obvious that any report dealing with the consideration of the involvement of mtDNA sequence alterations in hypertension may represent interest for further understanding of “genetic roots” and the mechanisms of the development of EH.
Initially, a G-to-A mutation at nucleotide 15059 of the mtDNA sequence was described in a patient with mitochondrial myopathy[11]. It has been established that G-to-A mutation occurs as a result of replacement of glycine at amino acid position 190 of mitochondrial cytochrome b with a stop codon leading to a truncated protein that misses 244 amino acids at the C-terminus of cytochrome b[12]. Earlier it was shown that 15059G>A heteroplasmy is associated with fibro-fatty atherosclerotic plaques which suggests a potential involvement of 15059G>A heteroplasmy in atherosclerosis[6,7]. In the present report, we proved the results of a study that involved an analysis of 196 randomly selected individuals which indicate an association of this mtDNA mutation with EH.
MATERIALS AND METHODS
Patients
This study was conducted in accordance with the Helsinki Declaration of 1975 as revised in 1983. All participants gave their written informed consent prior to their inclusion in the study, and the protocol was approved by the ethics committee of the Institute for Atherosclerosis Research, Moscow, Russia.
The study involved 196 unrelated patients (90 males and 106 females) who underwent a regular medical check-up at the Institute for Atherosclerosis Research, Moscow. On admission, a careful analysis of history was taken with special attention to cardiovascular risk factors, including a family history of cardiovascular diseases.
EH was diagnosed according to the European Society of Hypertension and the European Society of Cardiology classifications[13]. The presence of concomitant coronary heart disease (CHD) was evaluated according to American College of Physicians/American College of Cardiology Foundation/American Heart Association guidelines[14]. Standard 12-lead echocardiography was used for the diagnosis of left ventricular hypertrophy (LVH)[13]. Myocardial infarction (MI) was diagnosed according to the joint criteria of the Expert Consensus Document[15].
Biochemical measurements
The venous blood for lipid analysis was taken after overnight fasting. To obtain serum, the blood was incubated for 1 h at 37 °C and centrifuged for 15 min at 1500 g, and serum was stored at -70 °C. Serum concentrations of cholesterol and triglycerides were measured by enzymatic method using commercially available kits (Analyticon Biotechnologies AG, Germany)[16]. High density lipoprotein (HDL) cholesterol was measured enzymatically in the supernatant after the precipitation of apolipoprotein B-containing lipoproteins[17], and low density lipoprotein (LDL) cholesterol were calculated using the Friedewald formula[18].
DNA analysis
Mitochondrial DNA was isolated with the Aquapure Genomic Tissue Kit (Bio-Rad Laboratories, Hercules, CA, United States) according to the manufacturer’s protocols. The heteroplasmy level of the mtDNA mutation 15059G>A was quantified by the pyrosequencing method using the automated pyrosequencing machine PSQ HS96MA (Pyrosequencing AB, Uppsala, Sweden). Briefly, a 450-bp polymerase chain reaction (PCR) fragment of mtDNA was amplified using forward primer 5’-Bio-CATTATTCTCGCACGGACT-3’ and reverse primer 5’-GCTATAGTTGCAAGCAGGAG-3’ and then sequenced using the primer 5’-TTTCTGAGTAGAGAAATGAT-3’. The quantitative assay of the mutant allele 15059A was performed by peak height analysis of the pyrogram in the studied domain of a single strand PCR fragment of the mitochondrial genome as previously described[6]. Primers were synthesized by Syntol (Moscow, Russia).
Statistical analysis
Data were analyzed using a software package SPSS 14.0 (SPSS Inc., Chicago, IL, United States). The comparisons of mean values were performed by the Mann-Whitney U-test for continuous variables, and by chi-square Pearson’s test for categorical variables. The data are presented in terms of mean and SD. The normality of the 15059G>A heteroplasmy distribution was estimated from normal probability plots and by the Shapiro–Wilk W-test[19]. Quartiles with their confidence intervals (CI) were computed according to Aczel[20] and Conover[21] and analyzed by t-test. Odds ratios (OR) and their 95%CI were calculated using the Calculator for Confidence Intervals of OR[22]. Two-tailed Fisher’s exact test was used to examine whether the 15059G>A heteroplasmy level is associated with EH. The significance of differences was defined at the 0.05 level of confidence.
RESULTS
The characteristics of study participants are presented in Table 1. Of 196 participants, 120 (61%) and 45 (23%) had clinically manifested EH and CHD, respectively. Compared to 76 normotensive subjects, hypertensive patients were significantly older [66.3 (SD 8.7) vs 62.1 (SD 9.0) years, P < 0.001], had higher systolic blood pressure [systolic blood pressure (SBP) 147 (SD 16) vs 127 (SD 13) mmHg, P < 0.001], elevated plasma triglycerides [127 (SD 55) vs 112 (SD 47) mg/dL, P < 0 .001], and had a more frequent family history of EH (45% vs 28%, P = 0.034). Compared to 151 CHD-free study participants, CHD patients exhibited no significant differences in clinical characteristics, except for age [69.9 (SD 8.6) vs 63.4 (SD 9.1) years, respectively, P < 0.001].
Table 1.
Antropometric, clinical and biochemical characteristics of study participants (mean ± SD) n (%)
| Characteristics | |
| Age, yr | 65.1 ± 9.8 |
| BMI, kg/m2 | 26.2 ± 4.7 |
| SBP, mm/Hg | 138 ± 19 |
| DBP, mm/Hg | 83 ± 9 |
| Cholesterol, mg/dL | 234 ± 49 |
| TG, mg/dL | 125 ± 70 |
| HDL cholesterol, mg/dL | 66 ± 11 |
| LDL cholesterol, mg/dL | 143 ± 47 |
| EH | 120 (61) |
| LVH | 53 (27) |
| CHD | 45 (23) |
| Type 2 diabetes | 23 (12) |
| Myocardial infarction | 8 (4) |
| Family history of EH | 75 (38) |
| Family history of myocardial infarction | 51 (26) |
BMI: Body mass index; SBP: Systolic blood pressure; DBP: Diastolic blood pressure; EH: Essential hypertension; LVH: Left ventricular hypertrophy; CHD: Coronary heart disease; TG: Triglycerides; HDL: High density lipoproteins; LDL: Low density lipoproteins.
The distribution histogram of the 15059G>A heteroplasmy level in 196 study participants is presented in Figure 1. The heteroplasmy percentage ranged between 4% and 83%, with a median level of 31%. Except for three samples, the 15059G>A heteroplasmy level fitted the normal distribution (Shapiro-Wilk W-test; P = 0.18), with a mean level of 30.4% (SD 17.9%).
Figure 1.
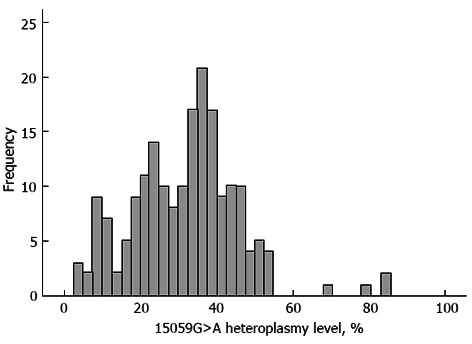
A frequency distribution histogram of the mtDNA 15059 G>A mutation heteroplasmy level in 191 studied individuals. The bell-shaped curve represents the expected normal frequency distribution of the mutated allele.
Clinical characteristics of patients were compared using a quartile scale of the 15059G>A heteroplasmy level distribution, with the first quartile being the lowest, and the fourth quartile being the highest. Between the upper and lower quartiles, significant differences were observed for patients’ age, SBP, and triglycerides (TG) levels (Table 2). However, there was no significant correlation between age and the level of SBP in the given sample (r = 0.108, P = 0.2). On the other side, 15059G>A heteroplasmy level correlated both with age (r = 0.331, P < 0.001) and the presence of hypertension (r = 0.228, P = 0.002). Regression analysis revealed that the age explains 12% variability of 15059G>A heteroplasmy level, and hypertension independently explains more 5% variability.
Table 2.
Antropometric, clinical and biochemical characteristics of study participants from the 1st and 2nd quartiles of distribution of 15059G>A heteroplasmy level n (%)
| Characteristics |
15059G>A heteroplasmy level1 |
P value | |
| Quartile 1 (n = 49) | Quartile 4 (n = 48) | ||
| Age, yr | 60.1 ± 7.1 | 66.2 ± 9.9 | 0.001 |
| BMI, kg/m2 | 26.4 ± 4.5 | 27.1 ± 5.1 | 0.41 |
| SBP, mm/Hg | 132 ± 18 | 143 ± 22 | 0.022 |
| DBP, mm/Hg | 83 ± 12 | 84 ± 12 | 0.50 |
| Cholesterol, mg/dL | 228 ± 42 | 243 ± 43 | 0.10 |
| TG, mg/dL | 112 ± 57 | 141 ± 61 | 0.021 |
| HDL cholesterol, mg/dL | 66 ± 14 | 68 ± 19 | 0.69 |
| LDL cholesterol, mg/dL | 140 ± 40 | 147 ± 44 | 0.44 |
| EH | 22 (45) | 30 (63) | 0.17 |
| LVH | 13 (27) | 13 (27) | 0.97 |
Data are mean ± SD. BMI: Body mass index; SBP: Systolic blood pressure; DBP: Diastolic blood pressure; EH: Essential hypertension; LVH: Left ventricular hypertrophy; TG: Triglycerides; HDL: High density lipoproteins; LDL: Low density lipoproteins.
In the subgroup of CHD patients, significant differences between the upper and lower quartiles of 15059G>A heteroplasmy level were found for patients’ age and SBP (Table 3). In CHD-free patients, significant differences between the upper and lower quartiles of 15059G>A heteroplasmy level remained for patients’ age and TG levels (Table 4). Thus, in comparison with study participants who had a lower level of the 15059G>A heteroplasmy, the presence of CHD in those with higher heteroplasmy positively correlated with increased SBP but not with elevated serum TG.
Table 3.
Antropometric, clinical and biochemical characteristics of coronary heart disease patients from the 1st and 2nd quartiles of distribution of 15059G>A heteroplasmy level n (%)
| Characteristics |
15059G>A heteroplasmy level1 |
P value | |
| Quartile 1 (n = 11) | Quartile 4 (n = 10) | ||
| Age, yr | 63.2 ± 7.0 | 72.9 ± 8.7 | 0.011 |
| BMI, kg/m2 | 27.2 ± 6.2 | 26.1 ± 5.9 | 0.69 |
| SBP, mmHg | 129 ± 22 | 153 ± 22 | 0.03 |
| DBP, mmHg | 80 ± 12 | 85 ± 13 | 0.32 |
| Cholesterol, mg/dL | 224 ± 54 | 236 ± 49 | 0.59 |
| TG, mg/dL | 130 ± 77 | 121 ± 56 | 0.78 |
| HDL cholesterol, mg/dL | 62 ± 15 | 69 ± 15 | 0.39 |
| LDL cholesterol, mg/dL | 136 ± 47 | 143 ± 42 | 0.69 |
| EH | 7 (64) | 7 (70) | 0.86 |
| LVH | 4 (36) | 4 (40) | 0.68 |
Data are mean ± SD. BMI: Body mass index; SBP: Systolic blood pressure; DBP: Diastolic blood pressure; EH: Essential hypertension; LVH: Left ventricular hypertrophy; TG: Triglycerides; HDL: High density lipoproteins; LDL: Low density lipoproteins.
Table 4.
Antropometric, clinical and biochemical characteristics of CHD-free study participants from the 1st and 2nd quartiles of distribution of 15059G>A heteroplasmy level n (%)
| Characteristics |
15059G>A heteroplasmy level1 |
P value | |
| Quartile 1 (n = 38) | Quartile 4 (n = 37) | ||
| Age, yr | 59.4 ± 7.2 | 64.7 ± 8.8 | 0.005 |
| BMI, kg/m2 | 26.0 ± 4.0 | 27.4 ± 5.3 | 0.2 |
| SBP, mmHg | 132 ± 14 | 141 ± 21 | 0.17 |
| DBP, mmHg | 84 ± 11 | 84 ± 10 | 0.99 |
| Cholesterol, mg/dL | 231 ± 41 | 246 ± 47 | 0.14 |
| TG, mg/dL | 106 ± 46 | 147 ± 61 | 0.003 |
| HDL cholesterol, mg/dL | 67 ± 14 | 67 ± 18 | 0.99 |
| LDL cholesterol, mg/dL | 143 ± 37 | 150 ± 42 | 0.44 |
| EH | 15 (39) | 23 (62) | 0.19 |
| LVH | 10 (26) | 10 (27) | 0.98 |
Data are mean ± SD. BMI: Body mass index; SBP: Systolic blood pressure; DBP: Diastolic blood pressure; EH: Essential hypertension; LVH: Left ventricular hypertrophy; TG: Triglycerides; HDL: High density lipoproteins; LDL: Low density lipoproteins.
Using a two-step cluster analysis, the 15059G>A heteroplasmy level was classified as “low heteroplasmy” and “high heteroplasmy”. In the “high heteroplasmy” group, study participants were found to have significantly higher age (P < 0.001) and EH prevalence then those from “low heteroplasmy” group (Table 5). These observations suggest an association between the level of 15059G>A heteroplasmy and EH. The 15059G>A heteroplasmy level exceeding 31% was associated with increased risk of EH [OR =2.76, P (Fisher) = 0.019] (Table 6). The relative risk accounted for 1.47 (95%CI: 1.15-1.84; P = 0.002). The presence of CHD in study participants with high 15059G>A heteroplasmy seemed to further increase the risk for EH by -1.2-fold but this association did not reach statistical significance [OR = 3.31, P (Fisher) = 0.18], obviously due to insufficient sample size.
Table 5.
Comparison of antropometric, clinical and biochemical characteristics of study participants from “low heteroplasmy” and “high heteroplasmy” groups n (%)
| Characteristics |
15059G>A heteroplasmy level1 |
P value | |
| Low (n = 99) | High (n = 97) | ||
| Age, yr | 60.2 ± 8.4 | 68.6 ± 8.4 | < 0.001 |
| BMI, kg/m2 | 26.3 ± 4.1 | 27.0 ± 5.0 | 0.52 |
| SBP, mmHg | 133 ± 16 | 142 ± 18 | 0.024 |
| DBP, mmHg | 82 ± 11 | 84 ± 11 | 0.39 |
| Cholesterol, mg/dL | 232 ± 48 | 241 ± 48 | 0.27 |
| TG, mg/dL | 118 ± 56 | 135 ± 62 | 0.065 |
| HDL cholesterol, mg/dL | 67 ± 14 | 66 ± 15 | 0.92 |
| LDL cholesterol, mg/dL | 141 ± 42 | 148 ± 44 | 0.49 |
| EH | 51 (52) | 69 (71) | 0.033 |
| LVH | 20 (20) | 33 (34) | 0.1 |
Data are mean ± SD. BMI: Body mass index; SBP: Systolic blood pressure; DBP: Diastolic blood pressure; EH: Essential hypertension; LVH: Left ventricular hypertrophy; TG: Triglycerides; HDL: High density lipoproteins; LDL: Low density lipoproteins.
Table 6.
Association between the 15059G>A heteroplasmy level and essential hypertension prevalence
| 15059G>A heteroplasmy level | No hypertension, n | Hypertension, n | Odds ratio (95%CI) | P value |
| Low (< 31%) | 51 | 51 | ||
| High (> 31%) | 25 | 69 | 2.76 (1.45-5.27) | 0.019 |
| Total | 76 | 120 |
DISCUSSION
The pathophysiology of essential hypertension (EH) is insufficiently understood; in particular, the impact of mitochondrial DNA mutations on the development of EH is poorly investigated. We undertook this study in order to see whether the level of heteroplasmy for the 15059G>A mutation in the mitochondrial cytochrome b gene might be associated with EH. The 15059G>A heteroplasmy level in mtDNA in blood leukocytes obtained from 196 study participants, randomly selected from general population (120 of whom had EH), exceeding 31%, was found to be significantly associated with a higher risk of EH.
Compared to the nuclear DNA, mitochondria are known to lack the efficient DNA repair and protection systems[23]. Because of the large number of mitochondrial genome copies exist within each cell, a ratio of mutated to wild-type mtDNA that represents significant determinant of phenotype[24]. In a number of studies, including the analysis of a large Han Chinese pedigree with suggestively maternally transmitted hypertension, the role of homoplasmic, inherited mtDNA mutations in etiology of familial, maternally inherited forms of hypertension (MIH) has been acknowledged[3,25-29]. It is known that all homoplasmic mtDNA mutations which are associated with MIH cause functional defects[3,25-29]. The 4435A>G mutation which is located at 3’ end to the anticodon (corresponding to the conventional position 37 of tRNAMet) affects the fidelity of codon recognition, structural formation, and stabilization of functional tRNAs[27]. The 4263A>G mutation resided at the processing site for the tRNAIle 5’-end precursor results in reduced efficiency of the tRNAIle precursor 5’-end cleavage catalyzed by RNase P[28]. The 4401A>G mutation that is situated at the spacer immediately to the 5’ end of tRNAMet and tRNAGln genes causes a reduction in the steady-state levels of both mitochondrial tRNAs[25]. The 4295 A>G mutation, which is located at immediately 3’ end to the anticodon, corresponding to conventional position 37 of tRNAIle, has a functional effect similar to that of the 4435A>G mutation[3]. The mitochondrial hypertension-associated ND1 T3308C mutation that locates in two nucleotides which are located to be adjacent to the 3’ end of mitochondrial tRNALeu UUR has been shown to result in a change of the H-strand polycistronic RNA precursor processing as well as in the destabilization of ND1 mRNA[26]. Despite a high penetrance, these mutations are thought to be infrequent as such mutations were identified in just a few families. In relation to the 4263A>G mutation. This mutation was identified only in one family and was not detected in 49 other families with matrilineal hypertension[28].
The fact that we observed higher levels of the 15059G>A mutation heteroplasmy in the elderly is not unexpected and is formally consistent with a theory of aging purports[30]. According to this theory, reactive oxygen species (ROS), normally produced by mitochondrial respiration, affect mitochondria by causing oxidative damage to the mitochondrion membrane components and cytosolic elements[30-34]. This eventually leads to dysfunction and further production of ROS and an increase in mtDNA mutation[30-34]. An increase in the ratio of mutated to wild-type mtDNA in mitochondrial genes encoding the respiratory chain subunits might thus lead to reduced steady-state levels of respiratory chain proteins and respiratory chain deficiency.
It has been reported that the nonsense 15059G>A mutation affecting the mitochondrial cytochrome b results in the formation of an inactive truncated product lacking a pair of ubiquinol/ubiquinone-binding sites that is likely to uncouple the mitochondrial respiratory chain[11]. Practically all homoplastic mutations found in the MT-CYB gene have been found to lead to deleterious effects associated with the respiratory chain complex III deficiency in muscles and clinical presentation involving exercise intolerance[31]. For example, truncating mutations 15242G>A and 15761G>A in MT-CYB, which, similar to 15059G>A, result in loss of the last N-terminal amino acids of cytochrome b, were heteroplasmic and abundant (87% and 73% respectively) in affected tissue (skeletal muscle) but were rare (0.7%) or absent in unaffected tissue (blood) of patients with symptoms of mitochondrial myopathy[32,33]. The 15059G>A homoplasmy might lead to pathological consequences, and the severity of clinical outcomes caused by this mutation should correlate with the percentage of the mutated mtDNA[30-34]. However, compared to blood cells, effects of truncating heteroplasmic mutations in MT-CYB are likely to be more harmful in tissues involved in active mitochondrial glucose oxidation and high energy consumption such as skeletal muscle[30-34]. The information about the dynamic nature of mitochondria has been outlined in large number original studies and reviews[35-43]. The dynamic nature of mitochondria is a concept that includes the movement of mitochondria along the cytoskeleton, the regulation of mitochondrial architecture (morphology and distribution), and connectivity mediated by tethering and fusion/fission events[35]. This dynamic networks are essential in order to maintain normal mitochondrial functions and participate in key functional processes including development, metabolic efficiency, apoptosis, and aging[36].
One cannot exclude that a positive correlation between the high 15059G>A heteroplasmy and increased plasma TG levels in non-CHD patients, found in our study, may reflect an insufficient lipid intake in individuals with increased levels of the mutant allele 15059A. This may result from the reduced capacity of mutant mitochondria to metabolize fatty acids. Elevated plasma TG itself and a high TG/HDL-cholesterol ratio indicate an atherogenic lipid profile that predisposes to atherosclerosis and CHD[42,43]. Increased plasma TG were shown to predispose to CHD more strongly in the subsets of hypertensive patients[44,45]. This is in accordance with our observations that showed a high frequency of EH subjects in CHD-free patients who had the highest 15059G>A heteroplasmy levels.
There are no doubts that the present study has limitations. First of all, the sample size was rather small in order to detect significant differences in the level of 15059G>A heteroplasmy between EH-free individuals and EH patients. Secondly, the study participants with known EH were on treatment; and thirdly, although not all of them reached treatment goals, blood pressure levels were affected anyway. We have found an association of heteroplasmy both with the prevalence of EH and SBP, but not DBP. This difference was observed for the whole group of study participants; on the subdivision into CHD patients and CHD-free subjects, the difference in EH prevalence was not significant. The observed findings are not extremely big, and reliable statistical hints were applied: two-step cluster analysis was able to demonstrate an association of EH and G15059A heteroplasmy. It should also be noted that the sample was taken from ethnically heterogeneous population of Moscow inhabitants of senior and elderly ages. Therefore, at present there is insufficient evidence to interpolate the results of this study to other populations and age groups. Finally, the given study was cross-sectional, and the assessment of actual risk of EH due to the presence of a high level of 15059G>A heteroplasmy requires further prospective studies.
The precise mechanism by which 15059G>A mutation might affect in the development of EH is currently unknown. Earlier, Wang et al[28] showed that the homoplasmic 4263 A>G mutation in the MT-T1 gene associated with familial MIH also involved in changes of codon AGA to AGG in MT-ND1 gene coding for NADH dehydrogenase subunit 1 of the respiratory chain complex I. Functional assays have revealed that this mutation results in a marked reduction in substrate-dependent oxygen consumption reflective of complexes I, III, and IV by 70%-80% and increased ROS levels in the lymphoblastoid cell lines derived from mutation carriers[28]. The 15059G>A mutation associated with a deficiency in the production and activity of mitochondrial cytochrome b may contribute to EH involving a similar mechanism associated with defects in oxidative phosphorylation, reduced mitochondrial-dependent oxygen consumption, and increased ROS generation[28]. Elevated ROS levels may induce oxidative stress, which represents a ubiquitous risk factor for a variety of vascular diseases including EH[46]. There is a strong possibility that ROS may directly alter vascular function as well as may be responsible for changes in vascular tone by several actions, for example, altering nitric oxide (NO) bioavailability or signaling[46]. It is well known that a reduced bioavailability of NO represents one of the key processes by which endothelial dysfunction is manifested in hypertension[46]. As a result, an imbalance of counteracting mechanisms, designed to maintain vascular homeostasis, occurs and this leads to vasoconstriction, impaired vascular function, and chronic hypertension[47].
It is well known that a variety of cell types, including endothelial cells, smooth muscle cells, pericytes and dendritic cells reside in the intact vascular wall[48,49]. However, it is currently unknown in which cell type(s) of the vessel wall the mitochondrial 15059G>A mutation may exert its effects associated with EH. A quantification of the 15059G>A heteroplasmy in different vascular cells in autopsy samples derived from patients with chronically manifested EH should help with unraveling this puzzle. It is worth to noting here that an ultrastructural examination of arterial cells in a variety of vascular pathologies allowed to reveal that marked alterations in the structural appearance of mitochondria occur[50-52]. These alterations include a reduction in number of mitochondrial cristae and changes in electron density of mitochondrial matrix[50-52]. However, the question whether the structural alterations of mitochondria might reflect the presence of mitochondrial mutation(s) in these organelles requires further investigation.
In conclusion, the present study provides the evidence of mtDNA 15059G>A mutation heteroplasmy association with EH.
Footnotes
Supported by The Russian Ministry of Science and Education
P- Reviewers Erne P, Figueroa X S- Editor Song XX L- Editor A E- Editor Lu YJ
References
- 1.Belostotsky R, Ben-Shalom E, Rinat C, Becker-Cohen R, Feinstein S, Zeligson S, Segel R, Elpeleg O, Nassar S, Frishberg Y. Mutations in the mitochondrial seryl-tRNA synthetase cause hyperuricemia, pulmonary hypertension, renal failure in infancy and alkalosis, HUPRA syndrome. Am J Hum Genet. 2011;88:193–200. doi: 10.1016/j.ajhg.2010.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ehret GB. Genome-wide association studies: contribution of genomics to understanding blood pressure and essential hypertension. Curr Hypertens Rep. 2010;12:17–25. doi: 10.1007/s11906-009-0086-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Li Z, Liu Y, Yang L, Wang S, Guan MX. Maternally inherited hypertension is associated with the mitochondrial tRNA(Ile) A4295G mutation in a Chinese family. Biochem Biophys Res Commun. 2008;367:906–911. doi: 10.1016/j.bbrc.2007.12.150. [DOI] [PubMed] [Google Scholar]
- 4.Manolio TA, Collins FS, Cox NJ, Goldstein DB, Hindorff LA, Hunter DJ, McCarthy MI, Ramos EM, Cardon LR, Chakravarti A, et al. Finding the missing heritability of complex diseases. Nature. 2009;461:747–753. doi: 10.1038/nature08494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Salvioli S, Capri M, Santoro A, Raule N, Sevini F, Lukas S, Lanzarini C, Monti D, Passarino G, Rose G, et al. The impact of mitochondrial DNA on human lifespan: a view from studies on centenarians. Biotechnol J. 2008;3:740–749. doi: 10.1002/biot.200800046. [DOI] [PubMed] [Google Scholar]
- 6.Sazonova M, Budnikov E, Khasanova Z, Sobenin I, Postnov A, Orekhov A. Studies of the human aortic intima by a direct quantitative assay of mutant alleles in the mitochondrial genome. Atherosclerosis. 2009;204:184–190. doi: 10.1016/j.atherosclerosis.2008.09.001. [DOI] [PubMed] [Google Scholar]
- 7.Sobenin IA, Sazonova MA, Postnov AY, Bobryshev YV, Orekhov AN. Mitochondrial mutations are associated with atherosclerotic lesions in the human aorta. Clin Dev Immunol. 2012;2012:832464. doi: 10.1155/2012/832464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Coon KD, Valla J, Szelinger S, Schneider LE, Niedzielko TL, Brown KM, Pearson JV, Halperin R, Dunckley T, Papassotiropoulos A, et al. Quantitation of heteroplasmy of mtDNA sequence variants identified in a population of AD patients and controls by array-based resequencing. Mitochondrion. 2006;6:194–210. doi: 10.1016/j.mito.2006.07.002. [DOI] [PubMed] [Google Scholar]
- 9.Laloi-Michelin M, Meas T, Ambonville C, Bellanné-Chantelot C, Beaufils S, Massin P, Vialettes B, Gin H, Timsit J, Bauduceau B, et al. The clinical variability of maternally inherited diabetes and deafness is associated with the degree of heteroplasmy in blood leukocytes. J Clin Endocrinol Metab. 2009;94:3025–3030. doi: 10.1210/jc.2008-2680. [DOI] [PubMed] [Google Scholar]
- 10.Andreu AL, Hanna MG, Reichmann H, Bruno C, Penn AS, Tanji K, Pallotti F, Iwata S, Bonilla E, Lach B, et al. Exercise intolerance due to mutations in the cytochrome b gene of mitochondrial DNA. N Engl J Med. 1999;341:1037–1044. doi: 10.1056/NEJM199909303411404. [DOI] [PubMed] [Google Scholar]
- 11.Andreu AL, Bruno C, Dunne TC, Tanji K, Shanske S, Sue CM, Krishna S, Hadjigeorgiou GM, Shtilbans A, Bonilla E, et al. A nonsense mutation (G15059A) in the cytochrome b gene in a patient with exercise intolerance and myoglobinuria. Ann Neurol. 1999;45:127–130. doi: 10.1002/1531-8249(199901)45:1<127::aid-art20>3.0.co;2-y. [DOI] [PubMed] [Google Scholar]
- 12.Mancia G, De Backer G, Dominiczak A, Cifkova R, Fagard R, Germano G, Grassi G, Heagerty AM, Kjeldsen SE, Laurent S, et al. 2007 Guidelines for the Management of Arterial Hypertension: The Task Force for the Management of Arterial Hypertension of the European Society of Hypertension (ESH) and of the European Society of Cardiology (ESC) J Hypertens. 2007;25:1105–1187. doi: 10.1097/HJH.0b013e3281fc975a. [DOI] [PubMed] [Google Scholar]
- 13.Chobanian AV, Bakris GL, Black HR, Cushman WC, Green LA, Izzo JL, Jones DW, Materson BJ, Oparil S, Wright JT, et al. The Seventh Report of the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure: the JNC 7 report. JAMA. 2003;289:2560–2572. doi: 10.1001/jama.289.19.2560. [DOI] [PubMed] [Google Scholar]
- 14.Qaseem A, Fihn SD, Williams S, Dallas P, Owens DK, Shekelle P. Diagnosis of stable ischemic heart disease: summary of a clinical practice guideline from the American College of Physicians/American College of Cardiology Foundation/American Heart Association/American Association for Thoracic Surgery/Preventive Cardiovascular Nurses Association/Society of Thoracic Surgeons. Ann Intern Med. 2012;157:729–734. doi: 10.7326/0003-4819-157-10-201211200-00010. [DOI] [PubMed] [Google Scholar]
- 15.Thygesen K, Alpert JS, White HD, Jaffe AS, Apple FS, Galvani M, Katus HA, Newby LK, Ravkilde J, Chaitman B, et al. Universal definition of myocardial infarction. Circulation. 2007;116:2634–2653. doi: 10.1161/CIRCULATIONAHA.107.187397. [DOI] [PubMed] [Google Scholar]
- 16.Siedel J, Hägele EO, Ziegenhorn J, Wahlefeld AW. Reagent for the enzymatic determination of serum total cholesterol with improved lipolytic efficiency. Clin Chem. 1983;29:1075–1080. [PubMed] [Google Scholar]
- 17.Burstein M, Scholnick HR, Morfin R. Rapid method for the isolation of lipoproteins from human serum by precipitation with polyanions. J Lipid Res. 1970;11:583–595. [PubMed] [Google Scholar]
- 18.Friedewald WT, Levy RI, Fredrickson DS. Estimation of the concentration of low-density lipoprotein cholesterol in plasma, without use of the preparative ultracentrifuge. Clin Chem. 1972;18:499–502. [PubMed] [Google Scholar]
- 19.Shapiro SS, Wilk MB. An analysis of variance test for normality. Biometrika. 1965;52:591–611. [Google Scholar]
- 20.Aczel AD. Complete business statistics. 2nd ed. Burr Ridge, IL: Irwin; 1993. [Google Scholar]
- 21.Conover WJ. Practical nonparametric statistics. 3rd ed. New York, NY: John Wiley & Sons; 1999. [Google Scholar]
- 22.Bland JM, Altman DG. Statistics notes. The odds ratio. BMJ. 2000;320:1468. doi: 10.1136/bmj.320.7247.1468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu P, Demple B. DNA repair in mammalian mitochondria: Much more than we thought? Environ Mol Mutagen. 2010;51:417–426. doi: 10.1002/em.20576. [DOI] [PubMed] [Google Scholar]
- 24.Krishnan KJ, Reeve AK, Samuels DC, Chinnery PF, Blackwood JK, Taylor RW, Wanrooij S, Spelbrink JN, Lightowlers RN, Turnbull DM. What causes mitochondrial DNA deletions in human cells? Nat Genet. 2008;40:275–279. doi: 10.1038/ng.f.94. [DOI] [PubMed] [Google Scholar]
- 25.Li R, Liu Y, Li Z, Yang L, Wang S, Guan MX. Failures in mitochondrial tRNAMet and tRNAGln metabolism caused by the novel 4401A& gt; G mutation are involved in essential hypertension in a Han Chinese Family. Hypertension. 2009;54:329–337. doi: 10.1161/HYPERTENSIONAHA.109.129270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liu Y, Li Z, Yang L, Wang S, Guan MX. The mitochondrial ND1 T3308C mutation in a Chinese family with the secondary hypertension. Biochem Biophys Res Commun. 2008;368:18–22. doi: 10.1016/j.bbrc.2007.12.193. [DOI] [PubMed] [Google Scholar]
- 27.Liu Y, Li R, Li Z, Wang XJ, Yang L, Wang S, Guan MX. Mitochondrial transfer RNAMet 4435A& gt; G mutation is associated with maternally inherited hypertension in a Chinese pedigree. Hypertension. 2009;53:1083–1090. doi: 10.1161/HYPERTENSIONAHA.109.128702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang S, Li R, Fettermann A, Li Z, Qian Y, Liu Y, Wang X, Zhou A, Mo JQ, Yang L, et al. Maternally inherited essential hypertension is associated with the novel 4263A& gt; G mutation in the mitochondrial tRNAIle gene in a large Han Chinese family. Circ Res. 2011;108:862–870. doi: 10.1161/CIRCRESAHA.110.231811. [DOI] [PubMed] [Google Scholar]
- 29.Lu Z, Chen H, Meng Y, Wang Y, Xue L, Zhi S, Qiu Q, Yang L, Mo JQ, Guan MX. The tRNAMet 4435A& gt; G mutation in the mitochondrial haplogroup G2a1 is responsible for maternally inherited hypertension in a Chinese pedigree. Eur J Hum Genet. 2011;19:1181–1186. doi: 10.1038/ejhg.2011.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Madamanchi NR, Runge MS. Mitochondrial dysfunction in atherosclerosis. Circ Res. 2007;100:460–473. doi: 10.1161/01.RES.0000258450.44413.96. [DOI] [PubMed] [Google Scholar]
- 31.Blakely EL, Mitchell AL, Fisher N, Meunier B, Nijtmans LG, Schaefer AM, Jackson MJ, Turnbull DM, Taylor RW. A mitochondrial cytochrome b mutation causing severe respiratory chain enzyme deficiency in humans and yeast. FEBS J. 2005;272:3583–3592. doi: 10.1111/j.1742-4658.2005.04779.x. [DOI] [PubMed] [Google Scholar]
- 32.Keightley JA, Anitori R, Burton MD, Quan F, Buist NR, Kennaway NG. Mitochondrial encephalomyopathy and complex III deficiency associated with a stop-codon mutation in the cytochrome b gene. Am J Hum Genet. 2000;67:1400–1410. doi: 10.1086/316900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mancuso M, Filosto M, Stevens JC, Patterson M, Shanske S, Krishna S, DiMauro S. Mitochondrial myopathy and complex III deficiency in a patient with a new stop-codon mutation (G339X) in the cytochrome b gene. J Neurol Sci. 2003;209:61–63. doi: 10.1016/s0022-510x(02)00462-8. [DOI] [PubMed] [Google Scholar]
- 34.Davidson SM, Duchen MR. Endothelial mitochondria: contributing to vascular function and disease. Circ Res. 2007;100:1128–1141. doi: 10.1161/01.RES.0000261970.18328.1d. [DOI] [PubMed] [Google Scholar]
- 35.Palmer CS, Osellame LD, Stojanovski D, Ryan MT. The regulation of mitochondrial morphology: intricate mechanisms and dynamic machinery. Cell Signal. 2011;23:1534–1545. doi: 10.1016/j.cellsig.2011.05.021. [DOI] [PubMed] [Google Scholar]
- 36.Chan DC. Mitochondria: dynamic organelles in disease, aging, and development. Cell. 2006;125:1241–1252. doi: 10.1016/j.cell.2006.06.010. [DOI] [PubMed] [Google Scholar]
- 37.Duvezin-Caubet S, Jagasia R, Wagener J, Hofmann S, Trifunovic A, Hansson A, Chomyn A, Bauer MF, Attardi G, Larsson NG, et al. Proteolytic processing of OPA1 links mitochondrial dysfunction to alterations in mitochondrial morphology. J Biol Chem. 2006;281:37972–37979. doi: 10.1074/jbc.M606059200. [DOI] [PubMed] [Google Scholar]
- 38.Song Z, Chen H, Fiket M, Alexander C, Chan DC. OPA1 processing controls mitochondrial fusion and is regulated by mRNA splicing, membrane potential, and Yme1L. J Cell Biol. 2007;178:749–755. doi: 10.1083/jcb.200704110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ishihara N, Fujita Y, Oka T, Mihara K. Regulation of mitochondrial morphology through proteolytic cleavage of OPA1. EMBO J. 2006;25:2966–2977. doi: 10.1038/sj.emboj.7601184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yaffe MP. The machinery of mitochondrial inheritance and behavior. Science. 1999;283:1493–1497. doi: 10.1126/science.283.5407.1493. [DOI] [PubMed] [Google Scholar]
- 41.Wu S, Zhou F, Zhang Z, Xing D. Mitochondrial oxidative stress causes mitochondrial fragmentation via differential modulation of mitochondrial fission-fusion proteins. FEBS J. 2011;278:941–954. doi: 10.1111/j.1742-4658.2011.08010.x. [DOI] [PubMed] [Google Scholar]
- 42.Hopkins PN, Wu LL, Hunt SC, Brinton EA. Plasma triglycerides and type III hyperlipidemia are independently associated with premature familial coronary artery disease. J Am Coll Cardiol. 2005;45:1003–1012. doi: 10.1016/j.jacc.2004.11.062. [DOI] [PubMed] [Google Scholar]
- 43.da Luz PL, Favarato D, Faria-Neto JR, Lemos P, Chagas AC. High ratio of triglycerides to HDL-cholesterol predicts extensive coronary disease. Clinics (Sao Paulo) 2008;63:427–432. doi: 10.1590/S1807-59322008000400003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Connelly PW, Petrasovits A, Stachenko S, MacLean DR, Little JA, Chockalingam A. Prevalence of high plasma triglyceride combined with low HDL-C levels and its association with smoking, hypertension, obesity, diabetes, sedentariness and LDL-C levels in the Canadian population. Canadian Heart Health Surveys Research Group. Can J Cardiol. 1999;15:428–433. [PubMed] [Google Scholar]
- 45.Chen CY, Hwu CM, Lin MW, Tsai CH, Yeh HI. High triglyceride level is associated with severe coronary artery disease in hypertensive subjects. Scand Cardiovasc J. 2008;42:146–152. doi: 10.1080/14017430701840325. [DOI] [PubMed] [Google Scholar]
- 46.Schulz E, Gori T, Münzel T. Oxidative stress and endothelial dysfunction in hypertension. Hypertens Res. 2011;34:665–673. doi: 10.1038/hr.2011.39. [DOI] [PubMed] [Google Scholar]
- 47.Watson T, Goon PK, Lip GY. Endothelial progenitor cells, endothelial dysfunction, inflammation, and oxidative stress in hypertension. Antioxid Redox Signal. 2008;10:1079–1088. doi: 10.1089/ars.2007.1998. [DOI] [PubMed] [Google Scholar]
- 48.Davies PF. Vascular cell interactions with special reference to the pathogenesis of atherosclerosis. Lab Invest. 1986;55:5–24. [PubMed] [Google Scholar]
- 49.Orekhov AN, Andreeva ER, Andrianova IV, Bobryshev YV. Peculiarities of cell composition and cell proliferation in different type atherosclerotic lesions in carotid and coronary arteries. Atherosclerosis. 2010;212:436–443. doi: 10.1016/j.atherosclerosis.2010.07.009. [DOI] [PubMed] [Google Scholar]
- 50.Bobryshev YV, Andreeva ER, Mikhailova IA, Andrianova IV, Moisenovich MM, Khapchaev S, Agapov II, Sobenin IA, Lusta KA, Orekhov AN. Correlation between lipid deposition, immune-inflammatory cell content and MHC class II expression in diffuse intimal thickening of the human aorta. Atherosclerosis. 2011;219:171–183. doi: 10.1016/j.atherosclerosis.2011.07.016. [DOI] [PubMed] [Google Scholar]
- 51.Sobenin IA, Sazonova MA, Ivanova MM, Zhelankin AV, Myasoedova VA, Postnov AY, Nurbaev SD, Yuri V. Bobryshev YV, Orekhov AN. Mutation C3256T of Mitochondrial Genome in White Blood Cells: Novel Genetic Marker of Atherosclerosis and Coronary Heart Disease. PLoS ONE. 2012;7:e46573. doi: 10.1371/journal.pone.0046573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chistiakov DA, Sobenin IA, Bobryshev YV, Orekhov AN. Mitochondrial dysfunction and mitochondrial DNA mutations in atherosclerotic complications in diabetes. World J Cardiol. 2012;4:148–156. doi: 10.4330/wjc.v4.i5.148. [DOI] [PMC free article] [PubMed] [Google Scholar]