

Abstract
The infectious agent of the transmissible spongiform encephalopathies, or prion diseases, has been the center of intense debate for decades. Years of studies have provided overwhelming evidence to support the prion hypothesis that posits a protein conformal infectious agent is responsible for the transmissibility of the disease. The recent studies that generate prion infectivity with purified bacterially expressed recombinant prion protein not only provides convincing evidence supporting the core of the prion hypothesis, that a pathogenic conformer of host prion protein is able to seed the conversion of its normal counterpart to the likeness of itself resulting in the replication of the pathogenic conformer and occurrence of disease, they also indicate the importance of cofactors, particularly lipid or lipid-like molecules, in forming the protein conformation-based infectious agent. This article reviews the literature regarding the chemical nature of the infectious agent and the potential contribution from lipid molecules to prion infectivity, and discusses the important remaining questions in this research area.
Keywords: prion, prion infectivity, prion protein conversion, lipid, recombinant prion
Introduction
Transmissible spongiform encephalopathies (TSEs), also known as prion diseases, are a group of neurodegenerative disorders that affect humans and a wide variety of animals [1–6]. TSE research started with searching for the infectious agent in scrapie, the prototypic prion disease in sheep and goats [1,7–12]. Decades of studies have provided unequivocal evidence that the ‘prion’, a proteinaceous infectious particle, is responsible for the infectivity (reviewed in [1,13–22]). According to the prion hypothesis, PrPSc, the pathogenic conformer of host prion protein (PrP) and the principle constituent of prion, is capable of imprinting its aberrant conformation to the normal cellular PrP (PrPC), resulting in the replication of PrPSc conformer and neurodegeneration [14,23]. The journey of exploring the chemical nature of the scrapie agent has been full of debates and controversies, along which numerous hypotheses have emerged and faded away [7,24–28]. The prion hypothesis, however, has withstood years of questioning and debate, and the latest studies have provided strong evidence supporting that the TSE agent is indeed a protein conformation-based infectious agent [29–41]. Despite these remarkable achievements, the molecular mechanism of prion infectivity, including the three-dimensional structure of the infectious PrPSc, the role of non-PrP cofactors in prion infectivity, and the molecular basis for PrPSc seeded conformational change of PrPC, remains unclear. Here, we review the literatures regarding the chemical nature of the infectious agent in TSEs with a special emphasis on the potential contribution from lipid to prion infectivity.
Early Studies on Scrapie
Scrapie is the prototypic TSE that affects sheep and goat. As early as in mid-18th century, scrapie has been suggested as an infectious disease and detailed clinical manifestations were described [42]. In 1898, scientists first reported neuronal vacuolation as the characteristic neuropathological change in the brain of scrapie affected sheep [43]. However, the same group of scientists reported in the following year their failed attempts to transmit scrapie to healthy sheep [44]. Those negative results were largely due to the short observation period whereas the incubation time of scrapie could be extraordinarily long. Indeed, the first successful transmission of scrapie to healthy sheep was achieved after long incubation time (>14 months) [45]. Later, it was also demonstrated that scrapie could be experimentally transmitted to goats despite long incubation time (>15 months) [46,47]. Those studies clearly established that scrapie is an infectious disease.
The observations that the scrapie agent was able to pass through antibacterial filter [48] and scrapie infected animals developed diseases only after long incubation periods [45,46] led to the belief that scrapie was caused by an unidentified ‘slow virus’ [24]. Efforts to isolate the scrapie agent failed to identify such ‘virus’, yet revealed the unusual physicochemical properties of the scrapie agent. In different laboratories, the scrapie agent was shown to survive diverse treatments that were used to inactivate viruses, including formalin treatment [49–51], boiling in water [52,53], extractions by low concentrations of phenol, chloroform [51] or ether [54,55], enzymatic degradation by nucleases [56–58], and ultraviolet (UV) and ionizing radiations [59,60]. Because of these unusual physicochemical characteristics, various speculations regarding the chemical nature of the scrapie agent emerged, including a protein [27], a polysaccharide [25,26], or a membrane fragment [7].
Inactivation of the Scrapie Agent by Ionizing and Ultraviolet Radiations
Protein-only theory
In an attempt to determine the size of the scrapie agent by ionizing radiation, Alper et al. [59] observed that an exceptionally high electron dose was required to inactivate the infectivity, leading to the prediction that the size of the scrapie agent was about 1/10 that of bacteriophages, the smallest virus at that time, and proposed for the first time that scrapie agent might replicate in the absence of nucleic acids. Based on the radiation inactivation result and other observations, Pattison and Jones [27] posited that the scrapie agent might just be a protein. To accommodate such self-propagating protein, Griffith [28] proposed three models to explain how a protein can replicate without nucleic acids. The second model, derived from a series of thermodynamic equations, predicts that the infectious agent (α2), a protein dimer, could convert the normal cellular protein bearing a different conformation (α′) into more protein dimers (α2). This hypothetical mechanism is very similar to the currently accepted replication mechanism of scrapie agent: misfolded PrPSc induces the conversion of normal cellular PrPC to PrPSc, a phenomenon now known as ‘prion replication’ [1,13,14,23].
Membrane hypothesis
In a follow-up study by Alper et al. [60], the scrapie agent was found to strongly resist inactivation by UV irradiation at the wavelength 254–265 nm, the wavelength where nucleic acids have the highest absorption and are most sensitive to inactivation. Inspired by the inactivation findings along with other distinct features of the agent, Gibbons and Hunter [7] proposed in the ‘membrane hypothesis’ that the scrapie agent might be a membrane fragment with a unique three-dimensional configuration. This unique membrane structure was hypothesized to self-replicate either by re-arrangement of the sugar or polysaccharide molecules attached to the naïve membranes or through the self-replication of polysaccharides [25,26] on the surface of the naïve membranes. The involvement of lipid membrane in scrapie agent is more convincingly supported by several later studies conducted by Alper et al. [61,62]. In an inactivation study using near monochromatic UV light, it was shown that 237 nm is the most effective wavelength to inactivate scrapie agent and the inactivation at this wavelength is six times more efficient compared with inactivation at 254 or 280 nm [61]. Of note, most DNA and RNA viruses are more susceptible to UV irradiation at 254 nm and the inactivation spectrum for scrapie agent is obviously different from that of viruses, but very similar to the absorbance spectrum of the purified bacterial endotoxin (lipopolysaccharide), a glycolipid molecule [61]. The notion that lipids might be essential to scrapie infectivity was strongly supported by another inactivation study using ionizing radiation in an oxygenated aqueous solution [62]. Oxygen in aqueous suspensions protects bacteriophages, DNA, RNA, or protein molecules from ionizing radiation, but is extremely effective in destroying lipid membranes and scrapie infectivity [62], suggesting that lipids might be an essential component of the scrapie agent. On the basis of these findings, Alper [63] proposed a modified membrane hypothesis that incorporates PrP as part of the infectious agent.
Prion Hypothesis
The difficulties encountered in early attempts to purify the scrapie agent [54,57,58,64–67] fostered the debates surrounding the chemical nature of the agent. Employing a series of differential centrifugations, detergent extractions, and enzymatic degradations, Prusiner et al. [68,69] were able to isolate and purify the scrapie infectivity-enriched, partially protease-resistant protein species of 27–30 kDa, and in 1982 Prusiner [13] coined the term ‘prion’ as the infectious agent in scrapie, which was originally defined as ‘small proteinaceous infectious particles that are resistant to inactivation by most procedures that modify nucleic acids’. The PrP of 27–30 kDa, or PrP27-30, was later identified as the N-terminus truncated, protease resistant core of PrPSc, the disease-associated conformational isoform of the normal cellular PrPC [70–75]. The prion hypothesis postulates that the prion replication involves PrPSc-seeded conversion of PrPC to PrPSc along with propagation of scrapie infectivity [1,14,23].
Although the possibility of large membrane fragments being part of the scrapie agent attenuated after prion was confirmed as the major component of the scrapie agent, the possibility that lipid molecules, regardless of the form and amount, might be involved in prion infectivity still remains [76–81].
In vitro conversion of PrP with denaturant treatment
Biophysical studies have shown that PrPSc is highly β-sheeted while PrPC is mainly α-helical [82–85]. Owing to substantial conformational disparities between PrPC and PrPSc, a large energy barrier has to be overcome or lowered before the conversion could occur. Therefore, during the course of conversion to PrPSc, the folded PrPC needs to be at least partially unfolded to reach a PrP* conformational state to make it kinetically feasible for the conversion (Fig. 1). Low concentrations of denaturants, including denaturing detergents or chaotropic agents, have been commonly used to partially denature PrP in vitro. In the first successful attempt to demonstrate PrPSc-seeded PrPC conversion in a cell-free system, Kocisko et al. [86] showed that mixing partially denatured PrPSc and PrPC together allowed the S35-labeled PrPC to gain the signature PK-resistant conformation and co-aggregate with the unlabeled PrPSc seed. This cell-free conversion system not only for the first time demonstrated the seeding capability of PrPSc, but also further recapitulated the species barrier and prion strain phenomena in vitro [87,88]. However, the low efficiency of this system made it impossible to estimate the infectivity of newly formed S35-labeled PrPSc.
Figure 1.
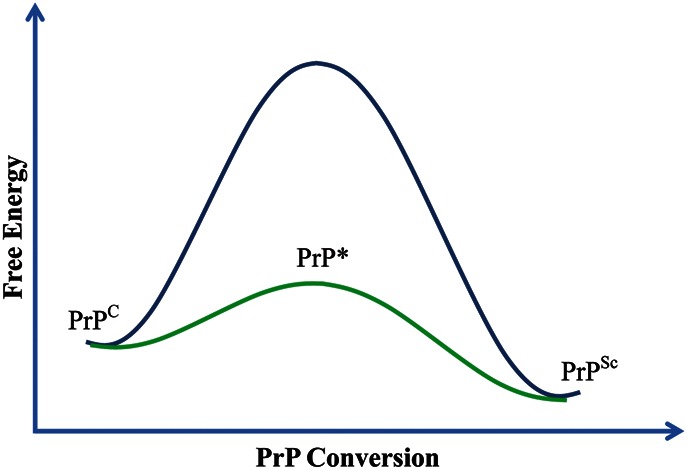
Diagram for PrP conversion In the presence of denaturants, lipid, or other co-factors, PrPC can reach the PrP* state and further convert to PrPSc.
The successful purification of fully folded bacterially expressed recombinant PrP (recPrP) [89–92] and the remarkable structural similarity between recPrP and purified native PrPC [93] make recPrP a legitimate substitute for PrPC in cell-free conversion studies. In a low pH solution containing 1 M GdnHCl, Swietnicki et al. [94] converted the α-helical recPrP into high β-sheeted conformers, some of which could aggregate and form fibrils. Other studies revealed that the recPrP amyloid fibers could be formed much more efficiently with higher concentrations of denaturants and constant shaking [33,95,96]. The recPrP in the form of amyloid fibers, however, does not have the typical biochemical property of most PrPSc conformer, a strong C-terminal PK-resistance. Moreover, when the recPrP amyloid fibers formed by the latter approach were tested for prion infectivity in mouse bioassay, they were only able to induce disease in PrP overexpressing transgenic mice after a prolonged incubation period, but not in wild-type mice [33,97].
In vitro conversion of PrP with lipids
The low infectivity associated with recPrP amyloid fibers indicates that the denaturant treatment induced recPrP amyloid conformation is most likely quite different from the infectious PrPSc conformation. A better approach to unfold PrP and lower the energy barrier is probably required to generate the infectious PrPSc conformer. The observations from early studies mentioned above and the studies described in the following paragraph suggest to us that lipids may play such a role to facilitate PrP conversion.
The normal PrPC is a cell surface glycoprotein attached to cell membrane via a glycosylphosphatidylinositol anchor and is released from lipid membranes after phosphoinositide phospholipase C (PI-PLC) digestion [98–101]. In contrast, PrPSc was tightly associated with lipid membranes and resistant to PI-PLC release, suggesting that the conversion from PrPC to PrPSc might take place on the cell surface or endocytic pathway [100–102] and involves interactions between PrP and lipid membranes. In an improved cell-free conversion system that excludes the usage of denaturant and detergent, a direct interaction between PrP and lipid membranes was found to be essential for PrP conversion [103,104]. Using human recPrP and liposomes consisting of synthetic lipids, Morillas et al. [105] showed that the interaction between recPrP and anionic lipids destabilizes the C-terminal structured domain of PrP, which indicates that lipid interaction may unfold the PrP to reach PrP* state and facilitate its conversion to the infectious PrPSc conformation. Studies from Pinheiro group [106–109] also showed that binding to lipid membrane could lead to secondary structural changes, aggregation and fibrillization of hamster recPrP (90–231).
We carried out detailed studies on the interactions between full-length, α-helical-rich mouse recPrP and anionic lipids under physiologically relevant conditions (150 mM NaCl, pH 7.4) [110]. Following our finding that recPrP could hydrophobically interact with liposomes containing lipids extracted from mouse brain [111], we further showed that the recPrP–lipid interaction was initiated by the electrostatic binding between positively charged amino acid residues and negatively charged anionic lipid headgroups, which was followed by the hydrophobic interactions between recPrP hydrophobic domain and lipid acyl chains [110]. Such recPrP–lipid interactions could lead to a striking conformational change of recPrP, converting from the α-helical structure to a β-sheeted conformation accompanied by gaining of a C-terminal PK-resistance [110], both of which are the signature biochemical properties of PrPSc conformer [82–84]. Furthermore, we also showed that the recPrP–anionic lipid interaction does not ubiquitously lead to the PK-resistant recPrP conformations, and the resulting recPrP conformations were greatly influenced by the structure of phospholipid headgroups and the distribution of these headgroups on the surface of lipid membranes. Another interesting observation from our study was that, depending on the composition of lipid membranes and the recPrP aggregation status, recPrP may also gain the N-terminal PK-resistance upon binding to lipid membranes [110]. Those results suggest that lipid membranes have a profound influence on PrP structure and PrP–lipid interactions may result in a variety of PrP conformations.
In the follow-up investigation, a series of recPrP mutations, including deletion of the N-terminal positively charged region, deletion of the highly conserved hydrophobic region, and point mutations in the middle charged regions, were employed to dissect the recPrP–lipid interactions [112]. Deletion of the conserved middle hydrophobic region completely demolished the lipid–recPrP hydrophobic interaction and resulted in an absolute loss of the PK-resistance, supporting a key role of hydrophobic PrP–lipid interaction in generating PK-resistant conformation. Deletion of the N-terminal positively charged region was found to reduce electrostatic interaction between recPrP mutant and anionic lipid, but the effect is not strong enough to alter the hydrophobic interaction and consequent PK-resistance. In contrast, mutations that eliminate the positive charges in front of the highly conserved hydrophobic region in the middle part of PrP appeared to affect neither electrostatic nor hydrophobic recPrP–lipid interaction, but markedly reduced the PK-resistance, suggesting a role of the middle charged residues in orienting recPrP and assisting the formation of PK-resistance. More interestingly, despite the similarity in location and amino acid substitution, the two human prion disease-associated mutations, P102L and P105L, exhibited dramatically different effects on recPrP–lipid interactions. The P102L mutant did not have an apparent effect on either electrostatic or hydrophobic PrP–lipid interaction, but completely abolished the PK-resistance. The P105L mutant, however, had a more complicated influence on recPrP–lipid interactions. Although the P105L mutation did not change the net charge of PrP, it significantly reduced the electrostatic PrP–lipid interaction. Furthermore, it had no apparent effect on the hydrophobic PrP–lipid interaction, but markedly changed the PK-resistance pattern. Since both mutations are located in the middle charged region and flanked by charged lysine residues, these results support a role of the middle charged region in the formation of PK-resistant PrP conformations. Moreover, the dramatic influence of both disease-associated mutations on PrP–lipid interaction and the resulting PrP conformations supports a role of lipid–PrP interaction in the pathogenesis of prion disease.
These results clearly showed that the recPrP–lipid interaction is sufficient to convert α-helical-rich recPrP to the β-sheeted, PK-resistant PrPSc-like conformations under physiologically relevant conditions, which supports that, similar to denaturant treatment, the lipid interaction is able to unfold recPrP and lower the energy barrier for converting recPrP to a conformation similar to PrPSc (Fig. 1).
In vitro generation of infectious prions
Although most of the foregoing conversion products recapitulate biochemical features of PrPSc, such as aggregation, β-sheeted secondary structure and protease resistance, none of which has been shown to induce prion disease in wild-type animals [33,97,113]. To support the prion hypothesis, demonstrating bona fide prion infectivity with converted PrP species is critically needed.
On the basis of the idea that fragmentation of protein aggregates by sonication increases the seeding capability, Soto et al. [114–116] developed a new technique termed Protein Misfolding Cylic Amplification (PMCA), in which the PrPSc-containing diseased brain homogenate and normal hamster brain homogenate with excessive PrPC were mixed in a testing tube and subjected to rounds of alternating sonication and incubation. The development of PMCA technique represents a landmark advance in prion research, which has been demonstrated to efficiently propagate PrPSc derived from various sources [35,117–119]. More importantly, the prion infectivity was concomitantly propagated with the PrPSc conformation and resulted in bona fide prion disease in wild-type hamsters [35]. Further studies have revealed that prion species barrier and prion strain phenomena could also be recapitulated by PMCA in a cell-free environment [120–122]. A major difference between PMCA and other cell-free conversion systems is the usage of brain homogenates and the non-PrP cofactors in the brain homogenates may facilitate the PrP conversion. Indeed, many studies have argued for the involvement of cofactors in PrP conversion, which includes polyanions such as proteoglycan [123] or RNA [124], metal ions [125–127] and lipids [103–110,112].
Proteoglycan was the first polyanion to show an effect in promoting the PrPSc propagation in a cell-free conversion system [123]. Nucleic acids, including both DNA and RNA, are able to interact with PrP and induce conformational changes of PrP [128,129] and RNA molecules have been found to be a potent cofactor fostering PrPSc-seeded in vitro prion propagation [124,130]. Using serial PMCA technique, Deleault et al. [36] successfully generated infectious hamster prion from purified native hamster PrPC with co-purified lipid molecules and additional RNA molecules, further supporting that RNA is potent factor promoting the formation of PrPSc conformer.
Early inactivation studies by Alper have implicated lipid as an essential component of prion infectivity [61,62], which was also supported by several lines of evidence from other laboratories. It has been shown that even the purest preparations of prion rod contain at least two different types of lipid molecules albeit in a very small amount [79]. Eluting solubilized prion rod through sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE) dissociates PrP27-30 from lipids and the eluted PrP27-30 completely lost prion infectivity [80]. Altered PrP27-30 conformation resulting from SDS-PAGE may explain the loss of infectivity in the frame of ‘protein only’ hypothesis, but an alternative interpretation that lipid–PrP interaction is essential for maintaining the infectious conformation could stand up by its own as well. Consistent with the latter explanation, purified prion rods could be dissolved into liposomes without losing infectivity; indeed, the infectivity titer associated with those prion liposomes increased 10–100 folds as evaluated by either incubation time or end-point titration bioassays [76,77]. In accord with the finding is a recent study showing that microsome associated PrPSc could infect SN56 cells with an efficiency higher than purified PrPSc [81]. The increased infectivity has also been suggested to result from the disassociation of large PrP aggregates, which could produce higher concentration of smaller PrP oligomers [77,81], which have been shown to possess the highest infectivity [131]. Also being suggested is that the lipid membrane may mediate a faster distribution of PrPSc or PrP27-30 onto cell plasma membranes resulting in faster prion propagations [77,81]. Nonetheless, the enhanced infectivity of lipid associated PrPSc or PrP27-30 supports a role of lipid in prion infectivity.
Inspired by the foregoing findings and our observations that recPrP–lipid interaction induces recPrP conformational change and lead to a PK-resistant conformation similar to PrPSc [110], we tested the possibility of generating infectious prion via serial PMCA with bacterially expressed recPrP in the presence of two cofactors, the synthetic phospholipid POPG (1-palmitoyl-2-oleoyl-sn-glycero-3-phospho-(1′-rac-glycerol)) and total RNA isolated from normal mouse liver [37]. The generation of a PK-resistant recPrP conformer (denoted as rPrP-res) was achieved after rounds of PMCA and the in vitro generated rPrP-res possesses all the hallmarks of diseased brain derived PrPSc: aggregated, C-terminal PK-resistant, capable of seeding the native PrPC conversion by PMCA, and able to infect cultured cells [37]. Most importantly, the newly generated rPrP-res induced prion disease in wild-type mice after a relatively short incubation period and with a quite synchronized onset, characteristics of a highly infectious prion [37]. We have shown that highly infectious rPrP-res can be generated when total mouse liver RNA was replaced by synthetic polyriboadenylic acid [poly(rA)] in a later study [40], ruling out the possibility that RNA carries the genetic information necessary for infection [36,40].
Prion infectivity has also been generated with bacterially expressed recPrP using PrPSc-seeded PMCA in the presence SDS and triton [38]. The recPrP conformer produced by this approach is able to cause prion disease in wild-type hamsters, but with a rather large variability in the incubation time and attack rate. In addition, the recPrP amyloid fibers going through an ‘annealing’ step (five cycles of incubations at 80°C and 37°C in the presence of normal hamster brain homogenate or bovine serum albumin) induced prion infectivity in asymptomatic wild-type hamsters [39]. More recently, prion infectivity has been successfully propagated by PMCA using recPrP plus a sole cofactor, phosphoethanolamine (PE), and more interestingly, when three diverse prion strains were propagated to recPrP with PE as the sole cofactor, they appear to converge to a single new strain type [41,132], supporting the notion that polyanions are dispensable for prion infectivity [133] and suggesting a role of cofactor in modulating prion strain phenotype.
Collectively, all these studies have demonstrated that purified bacterially expressed recPrP is able to convert to the infectious conformers in various in vitro systems, providing unequivocal evidence supporting the prion hypothesis.
Closing Remarks
Significant progress has been made in understanding the chemical nature of the infectious agent in TSEs, but several important questions regarding the agent still remain to be answered. Although an important role of lipids in prion infectivity is supported by the early radiation inactivation studies [61,62] and recent recombinant prion studies [37,41,134], whether lipids or lipid-like molecules are essential component of the infectious agent remains unclear. Further studies are also needed to clarify whether different types or combinations of cofactors [135], including both lipids and polyanions, contribute to prion strain diversity [4,136–138], the formation of seemingly endless PrPSc conformers [4,139,140], and the species barrier in prion transmission [141]. Moreover, the ability to generate recombinant prion in a clean in vitro system with defined synthetic cofactors opens the avenue to solve the three-dimensional structure of infectious PrPSc conformation, which is essential for us to understand the molecular mechanism of prion propagation. For this purpose, further optimization of the in vitro recombinant prion propagation system is needed to increase the quantity and quality of recombinant prion in order to meet the requirement of structural studies. Further investigations in this area are not only critical for unraveling the molecular mechanism of this unorthodox infectious agent and developing novel strategies against these fatal neurodegenerative disorders, they may also shed light on the mechanism of recently discovered ‘prion-like’ propagation of protein aggregates in a variety of common neurodegenerative disorders.
Funding
This work was supported by NIH grants RO1 NS071035 and RO1 NS060729 to J.M.
References
- 1.Prusiner SB. Prions. Proc Natl Acad Sci USA. 1998;95:13363–13383. doi: 10.1073/pnas.95.23.13363. doi:10.1073/pnas.95.23.13363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Collinge J. Prion diseases of humans and animals: their causes and molecular basis. Annu Rev Neurosci. 2001;24:519–550. doi: 10.1146/annurev.neuro.24.1.519. doi:10.1146/annurev.neuro.24.1.519. [DOI] [PubMed] [Google Scholar]
- 3.Weissmann C. The state of the prion. Nat Rev Microbiol. 2004;2:861–871. doi: 10.1038/nrmicro1025. doi:10.1038/nrmicro1025. [DOI] [PubMed] [Google Scholar]
- 4.Collinge J, Clarke AR. A general model of prion strains and their pathogenicity. Science. 2007;318:930–936. doi: 10.1126/science.1138718. doi:10.1126/science.1138718. [DOI] [PubMed] [Google Scholar]
- 5.Aguzzi A, Baumann F, Bremer J. The prion's elusive reason for being. Annu Rev Neurosci. 2008;31:439–477. doi: 10.1146/annurev.neuro.31.060407.125620. doi:10.1146/annurev.neuro.31.060407.125620. [DOI] [PubMed] [Google Scholar]
- 6.Caughey B, Baron GS, Chesebro B, Jeffrey M. Getting a grip on prions: oligomers, amyloids, and pathological membrane interactions. Annu Rev Biochem. 2009;78:177–204. doi: 10.1146/annurev.biochem.78.082907.145410. doi:10.1146/annurev.biochem.78.082907.145410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gibbons RA, Hunter GD. Nature of the scrapie agent. Nature. 1967;215:1041–1043. doi: 10.1038/2151041a0. doi:10.1038/2151041a0. [DOI] [PubMed] [Google Scholar]
- 8.Millson GC, Hunter GD, Kimberlin RH. The physico-chemical nature of the scrapie agent. Front Biol. 1976;44:243–266. [PubMed] [Google Scholar]
- 9.Prusiner SB. Scrapie prions. Ann Rev Microbiol. 1989;43:345–374. doi: 10.1146/annurev.mi.43.100189.002021. doi:10.1146/annurev.mi.43.100189.002021. [DOI] [PubMed] [Google Scholar]
- 10.Weissmann CA. ‘Unified theory’ of prion propagation. Nature. 1991;352:679–683. doi: 10.1038/352679a0. doi:10.1038/352679a0. [DOI] [PubMed] [Google Scholar]
- 11.Brown P, Bradley R. 1755 and all that: a historical primer of transmissible spongiform encephalopathy. BMJ. 1998;317:1688–1692. doi: 10.1136/bmj.317.7174.1688. doi:10.1136/bmj.317.7174.1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Taylor DM. Inactivation of transmissible degenerative encephalopathy agents: a review. Vet J. 2000;159:10–17. doi: 10.1053/tvjl.1999.0406. doi:10.1053/tvjl.1999.0406. [DOI] [PubMed] [Google Scholar]
- 13.Prusiner SB. Novel proteinaceous infectious particles cause scrapie. Science. 1982;216:136–144. doi: 10.1126/science.6801762. doi:10.1126/science.6801762. [DOI] [PubMed] [Google Scholar]
- 14.Prusiner SB. Molecular biology of prion diseases. Science. 1991;252:1515–1522. doi: 10.1126/science.1675487. doi:10.1126/science.1675487. [DOI] [PubMed] [Google Scholar]
- 15.Weissmann C. Spongiform encephalopathies. The prion's progress. Nature. 1991;349:569–571. doi: 10.1038/349569a0. [DOI] [PubMed] [Google Scholar]
- 16.Weissmann C. Molecular biology of prion disease. Trends in Cell Biol. 1994;4:10. doi: 10.1016/0962-8924(94)90032-9. doi:10.1016/0962-8924(94)90032-9. [DOI] [PubMed] [Google Scholar]
- 17.Weissmann C, Flechsig E. PrP knock-out and PrP transgenic mice in prion research. Br Med Bull. 2003;66:43–60. doi: 10.1093/bmb/66.1.43. doi:10.1093/bmb/66.1.43. [DOI] [PubMed] [Google Scholar]
- 18.Aguzzi A, Polymenidou M. Mammalian prion biology: one century of evolving concepts. Cell. 2004;116:313–327. doi: 10.1016/s0092-8674(03)01031-6. doi:10.1016/S0092-8674(03)01031-6. [DOI] [PubMed] [Google Scholar]
- 19.Hill AF, Collinge J. Prion strains and species barriers. Contrib Microbiol. 2004;11:33–49. doi: 10.1159/000077061. doi:10.1159/000077061. [DOI] [PubMed] [Google Scholar]
- 20.Caughey B, Baron GS. Prions and their partners in crime. Nature. 2006;443:803–810. doi: 10.1038/nature05294. doi:10.1038/nature05294. [DOI] [PubMed] [Google Scholar]
- 21.Soto C. Prion hypothesis: the end of the controversy? Trends Biochem Sci. 2011;36:151–158. doi: 10.1016/j.tibs.2010.11.001. doi:10.1016/j.tibs.2010.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Aguzzi A, Falsig J. Prion propagation, toxicity and degradation. Nat Neurosci. 2012;15:936–939. doi: 10.1038/nn.3120. doi:10.1038/nn.3120. [DOI] [PubMed] [Google Scholar]
- 23.Cohen FE, Prusiner SB. Pathologic conformations of prion proteins. Annu Rev Biochem. 1998;67:793–819. doi: 10.1146/annurev.biochem.67.1.793. doi:10.1146/annurev.biochem.67.1.793. [DOI] [PubMed] [Google Scholar]
- 24.Sigurdsson B. Rida, a chronic encephalitis of sheep. Br Vet J. 1954;110:341–354. [Google Scholar]
- 25.Field EJ. Transmission experiments with multiple sclerosis: an interim report. Br Med J. 1966;2:564–565. doi: 10.1136/bmj.2.5513.564. doi:10.1136/bmj.2.5513.564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Field EJ. The significance of astroglial hypertrophy in Scrapie, Kuru, Multiple Sclerosis and old age together with a note on the possible nature of the scrapie agent. Deutsche Zeitschrift für Nervenheilkunde. 1967;192:265–274. [Google Scholar]
- 27.Pattison IH, Jones KM. The possible nature of the transmissible agent of scrapie. Vet Rec. 1967;80:2–9. doi: 10.1136/vr.80.1.2. doi:10.1136/vr.80.1.2. [DOI] [PubMed] [Google Scholar]
- 28.Griffith JS. Self-replication and scrapie. Nature. 1967;215:1043–1044. doi: 10.1038/2151043a0. doi:10.1038/2151043a0. [DOI] [PubMed] [Google Scholar]
- 29.Bessen RA, Marsh RF. Biochemical and physical properties of the prion protein from two strains of the transmissible mink encephalopathy agent. J Virol. 1992;66:2096–2101. doi: 10.1128/jvi.66.4.2096-2101.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Collinge J, Sidle KC, Meads J, Ironside J, Hill AF. Molecular analysis of prion strain variation and the aetiology of ‘new variant’ CJD [see comments] Nature. 1996;383:685–690. doi: 10.1038/383685a0. doi:10.1038/383685a0. [DOI] [PubMed] [Google Scholar]
- 31.Telling GC, Parchi P, DeArmond SJ, Cortelli P, Montagna P, Gabizon R, Mastrianni J, et al. Evidence for the conformation of the pathologic isoform of the prion protein enciphering and propagating prion diversity. Science. 1996;274:2079–2082. doi: 10.1126/science.274.5295.2079. doi:10.1126/science.274.5295.2079. [DOI] [PubMed] [Google Scholar]
- 32.Safar J, Wille H, Itri V, Groth D, Serban H, Torchia M, Cohen FE, et al. Eight prion strains have PrP(Sc) molecules with different conformations. Nat Med. 1998;4:1157–1165. doi: 10.1038/2654. doi:10.1038/2654. [DOI] [PubMed] [Google Scholar]
- 33.Legname G, Baskakov IV, Nguyen HO, Riesner D, Cohen FE, DeArmond SJ, Prusiner SB. Synthetic mammalian prions. Science. 2004;305:673–676. doi: 10.1126/science.1100195. doi:10.1126/science.1100195. [DOI] [PubMed] [Google Scholar]
- 34.Jones EM, Surewicz WK. Fibril conformation as the basis of species- and strain-dependent seeding specificity of mammalian prion amyloids. Cell. 2005;121:63–72. doi: 10.1016/j.cell.2005.01.034. doi:10.1016/j.cell.2005.01.034. [DOI] [PubMed] [Google Scholar]
- 35.Castilla J, Saa P, Hetz C, Soto C. In vitro generation of infectious scrapie prions. Cell. 2005;121:195–206. doi: 10.1016/j.cell.2005.02.011. doi:10.1016/j.cell.2005.02.011. [DOI] [PubMed] [Google Scholar]
- 36.Deleault NR, Harris BT, Rees JR, Supattapone S. Formation of native prions from minimal components in vitro. Proc Natl Acad Sci USA. 2007;104:9741–9746. doi: 10.1073/pnas.0702662104. doi:10.1073/pnas.0702662104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang F, Wang X, Yuan CG, Ma J. Generating a prion with bacterially expressed recombinant prion protein. Science. 2010;327:1132–1135. doi: 10.1126/science.1183748. doi:10.1126/science.1183748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kim JI, Cali I, Surewicz K, Kong Q, Raymond GJ, Atarashi R, Race B, et al. Mammalian prions generated from bacterially expressed prion protein in the absence of any mammalian cofactors. J Biol Chem. 2010;285:14083–14087. doi: 10.1074/jbc.C110.113464. doi:10.1074/jbc.C110.113464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Makarava N, Kovacs GG, Bocharova O, Savtchenko R, Alexeeva I, Budka H, Rohwer RG, et al. Recombinant prion protein induces a new transmissible prion disease in wild-type animals. Acta Neuropathol. 2010;119:177–187. doi: 10.1007/s00401-009-0633-x. doi:10.1007/s00401-009-0633-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang F, Zhang Z, Wang X, Li J, Zha L, Yuan CG, Weissmann C, et al. Genetic informational RNA is not required for recombinant prion infectivity. J Virol. 2012;86:1874–1876. doi: 10.1128/JVI.06216-11. doi:10.1128/JVI.06216-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Deleault NR, Walsh DJ, Piro JR, Wang F, Wang X, Ma J, Rees JR, et al. Cofactor molecules maintain infectious conformation and restrict strain properties in purified prions. Proc Natl Acad Sci USA. 2012;109:E1938–E1946. doi: 10.1073/pnas.1206999109. doi:10.1073/pnas.1206999109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Leopoldt JG. Einleitung zu der landwirthschaft. Berlin: Christian Friedrich Günthern; 1759. Nützliche und auf die Erfahrung Gegründete. [Google Scholar]
- 43.Besnoit C, Morel C. Note sur les lésions nerveuses de la tremblante du mouton. Rev Vet. 1898;23:397–400. [Google Scholar]
- 44.Besnoit C. La tremblante ou névrite périphérique enzootique du mouton. VI. Etiologie Rev Vét. 1899;23:307–343. [Google Scholar]
- 45.Curril J, Chelle PL. Is the disease of scrapie inoculable? Cr Hebd Acad Sci. 1936;203:1552–1554. [Google Scholar]
- 46.Cuille J, Chelle PL. Experimental transmission of trembling to the goat. Cr Seances Acad Sci. 1939;208:1058–1160. [Google Scholar]
- 47.Pattison IH, Gordon WS, Millson GC. Experimental production of scrapie in goats. J Comp Pathol. 1959;69:300–312. doi: 10.1016/s0368-1742(59)80029-1. [DOI] [PubMed] [Google Scholar]
- 48.Cuille J, Chelle PL. Is the trembling of sheep detemined by a filtrable virus? Cr Hebd Acad Sci. 1938;206:1687–1688. [Google Scholar]
- 49.Gordon WS. Advances in veterinary research-looping ill, tick-borne fever and scrapie. Vet Rec. 1946;58:516–525. [PubMed] [Google Scholar]
- 50.Greig JR. Scrapie in sheep. J Comp Pathol. 1950;60:263–266. doi: 10.1016/s0368-1742(50)80024-3. [DOI] [PubMed] [Google Scholar]
- 51.Pattison IH. Resistance of the scrapie agent to formalin. J Comp Pathol. 1959;75:159–164. doi: 10.1016/0021-9975(65)90006-x. doi:10.1016/0021-9975(65)90006-X. [DOI] [PubMed] [Google Scholar]
- 52.Stamp JT, Brotherston JG, Zlotnik I, Mackay JM, Smith W. Further studies on scrapie. J Comp Pathol. 1959;69:268–280. doi: 10.1016/s0368-1742(59)80026-6. [DOI] [PubMed] [Google Scholar]
- 53.Pattison IH, Millson GC. Further experimental observations on scrapie. J Comp Pathol. 1961;71:350–359. doi: 10.1016/s0368-1742(61)80040-4. [DOI] [PubMed] [Google Scholar]
- 54.Butler EJ, Smith W. An attempt to separate the scrapie agent from brain tissue. Vet Rec. 1960;72:417–418. [Google Scholar]
- 55.Pattison IH, Millson GC. Scrapie produced experimentally in goats with special reference to the clinical syndrome. J Comp Pathol. 1961;71:101–109. doi: 10.1016/s0368-1742(61)80013-1. [DOI] [PubMed] [Google Scholar]
- 56.Pattison IH, Millson GC. Further observations on the experimental production of scrapie in goats and sheep. J Comp Pathol. 1960;70:182–193. doi: 10.1016/s0368-1742(60)80018-5. [DOI] [PubMed] [Google Scholar]
- 57.Mould DL, Smith W. The causal agent of scrapie. I. Extraction of the agent from infected sheep tissue. J Comp Pathol. 1962;72:97–105. doi: 10.1016/s0368-1742(62)80011-3. [DOI] [PubMed] [Google Scholar]
- 58.Hunter GD, Millson GC. Attempts to release the scrapie agent from tissue debris. J Comp Pathol. 1967;77:301–307. doi: 10.1016/0021-9975(67)90039-4. doi:10.1016/0021-9975(67)90039-4. [DOI] [PubMed] [Google Scholar]
- 59.Alper T, Haig DA, Clarke MC. The exceptionally small size of the scrapie agent. Biochem Biophys Res Commun. 1966;22:278–284. doi: 10.1016/0006-291x(66)90478-5. doi:10.1016/0006-291X(66)90478-5. [DOI] [PubMed] [Google Scholar]
- 60.Alper T, Cramp WA, Haig DA, Clarke MC. Does the agent of scrapie replicate without nucleic acid? Nature. 1967;214:764–766. doi: 10.1038/214764a0. doi:10.1038/214764a0. [DOI] [PubMed] [Google Scholar]
- 61.Latarjet R, Muel B, Haig DA, Clarke MC, Alper T. Inactivation of the scrapie agent by near monochromatic ultraviolet light. Nature. 1970;227:1341–1343. doi: 10.1038/2271341a0. doi:10.1038/2271341a0. [DOI] [PubMed] [Google Scholar]
- 62.Alper T, Haig DA, Clarke MC. The scrapie agent: evidence against its dependence for replication on intrinsic nucleic acid. J Gen Virol. 1978;41:503–516. doi: 10.1099/0022-1317-41-3-503. doi:10.1099/0022-1317-41-3-503. [DOI] [PubMed] [Google Scholar]
- 63.Alper T. The scrapie enigma: insights from radiation experiments. Radiat Res. 1993;135:283–292. doi:10.2307/3578866. [PubMed] [Google Scholar]
- 64.Mould DL, Smith W. The causal agent of scrapie. II. Extraction of the agent from infected goat tissue. J Comp Pathol. 1962;72:106–112. doi: 10.1016/s0368-1742(62)80012-5. [DOI] [PubMed] [Google Scholar]
- 65.Hunter GD, Millson GC, Meek G. The intracellular location of the agent of mouse scrapie. J Gen Microbiol. 1964;34:319–325. doi: 10.1099/00221287-34-2-319. [DOI] [PubMed] [Google Scholar]
- 66.Hunter GD, Millson GC. Studies on the heat stability and chromatographic behaviour of the scrapie agent. J Gen Microbiol. 1964;37:251–258. doi: 10.1099/00221287-37-2-251. [DOI] [PubMed] [Google Scholar]
- 67.Mould DL, Smith W, Dawson AM. Centrifugation studies on the infectivities of cellular fractions derived from mouse brain infected with scrapie (Suffolk strain) J Gen Microbiol. 1965;40:71–79. doi: 10.1099/00221287-40-1-71. [DOI] [PubMed] [Google Scholar]
- 68.Bolton DC, McKinley MP, Prusiner SB. Identification of a protein that purifies with the scrapie prion. Science. 1982;218:1309–1311. doi: 10.1126/science.6815801. doi:10.1126/science.6815801. [DOI] [PubMed] [Google Scholar]
- 69.Prusiner SB, Bolton DC, Groth DF, Bowman KA, Cochran SP, McKinley MP. Further purification and characterization of scrapie prions. Biochemistry. 1982;21:6942–6950. doi: 10.1021/bi00269a050. doi:10.1021/bi00269a050. [DOI] [PubMed] [Google Scholar]
- 70.McKinley MP, Bolton DC, Prusiner SB. A protease-resistant protein is a structural component of the scrapie prion. Cell. 1983;35:57–62. doi: 10.1016/0092-8674(83)90207-6. doi:10.1016/0092-8674(83)90207-6. [DOI] [PubMed] [Google Scholar]
- 71.Prusiner SB, Groth DF, Bolton DC, Kent SB, Hood LE. Purification and structural studies of a major scrapie prion protein. Cell. 1984;38:127–134. doi: 10.1016/0092-8674(84)90533-6. doi:10.1016/0092-8674(84)90533-6. [DOI] [PubMed] [Google Scholar]
- 72.Chesebro B, Race R, Wehrly K, Nishio J, Bloom M, Lechner D, Bergstrom S, et al. Identification of scrapie prion protein-specific mRNA in scrapie-infected and uninfected brain. Nature. 1985;315:331–333. doi: 10.1038/315331a0. doi:10.1038/315331a0. [DOI] [PubMed] [Google Scholar]
- 73.Oesch B, Westaway D, Walchli M, McKinley MP, Kent SB, Aebersold R, Barry RA, et al. A cellular gene encodes scrapie PrP 27–30 protein. Cell. 1985;40:735–746. doi: 10.1016/0092-8674(85)90333-2. doi:10.1016/0092-8674(85)90333-2. [DOI] [PubMed] [Google Scholar]
- 74.Basler K, Oesch B, Scott M, Westaway D, Walchli M, Groth DF, McKinley MP, et al. Scrapie and cellular PrP isoforms are encoded by the same chromosomal gene. Cell. 1986;46:417–428. doi: 10.1016/0092-8674(86)90662-8. doi:10.1016/0092-8674(86)90662-8. [DOI] [PubMed] [Google Scholar]
- 75.Meyer RK, McKinley MP, Bowman KA, Braunfeld MB, Barry RA, Prusiner SB. Separation and properties of cellular and scrapie prion proteins. Proc Natl Acad Sci USA. 1986;83:2310–2314. doi: 10.1073/pnas.83.8.2310. doi:10.1073/pnas.83.8.2310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Gabizon R, McKinley MP, Prusiner SB. Purified prion proteins and scrapie infectivity copartition into liposomes. Proc Natl Acad Sci USA. 1987;84:4017–4021. doi: 10.1073/pnas.84.12.4017. doi:10.1073/pnas.84.12.4017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Gabizon R, McKinley MP, Groth DF, Kenaga L, Prusiner SB. Properties of scrapie prion protein liposomes. J Biol Chem. 1988;263:4950–4955. [PubMed] [Google Scholar]
- 78.Stahl N, Baldwin MA, Teplow DB, Hood L, Gibson BW, Burlingame AL, Prusiner SB. Structural studies of the scrapie prion protein using mass spectrometry and amino acid sequencing. Biochemistry. 1993;32:1991–2002. doi: 10.1021/bi00059a016. doi:10.1021/bi00059a016. [DOI] [PubMed] [Google Scholar]
- 79.Klein TR, Kirsch D, Kaufmann R, Riesner D. Prion rods contain small amounts of two host sphingolipids as revealed by thin-layer chromatography and mass spectrometry. Biol Chem. 1998;379:655–666. doi: 10.1515/bchm.1998.379.6.655. [DOI] [PubMed] [Google Scholar]
- 80.Leffers KW, Wille H, Stohr J, Junger E, Prusiner SB, Riesner D. Assembly of natural and recombinant prion protein into fibrils. Biol Chem. 2005;386:569–580. doi: 10.1515/BC.2005.067. [DOI] [PubMed] [Google Scholar]
- 81.Baron GS, Magalhaes AC, Prado MA, Caughey B. Mouse-adapted scrapie infection of SN56 cells: greater efficiency with microsome-associated versus purified PrP-res. J Virol. 2006;80:2106–2117. doi: 10.1128/JVI.80.5.2106-2117.2006. doi:10.1128/JVI.80.5.2106-2117.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Caughey BW, Dong A, Bhat KS, Ernst D, Hayes SF, Caughey WS. Secondary structure analysis of the scrapie-associated protein PrP 27–30 in water by infrared spectroscopy. Biochemistry. 1991;30:7672–7680. doi: 10.1021/bi00245a003. doi:10.1021/bi00245a003. [DOI] [PubMed] [Google Scholar]
- 83.Pan KM, Baldwin M, Nguyen J, Gasset M, Serban A, Groth D, Mehlhorn I, et al. Conversion of alpha-helices into beta-sheets features in the formation of the scrapie prion proteins. Proc Natl Acad Sci USA. 1993;90:10962–10966. doi: 10.1073/pnas.90.23.10962. doi:10.1073/pnas.90.23.10962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Gasset M, Baldwin MA, Fletterick RJ, Prusiner SB. Perturbation of the secondary structure of the scrapie prion protein under conditions that alter infectivity. Proc Natl Acad Sci USA. 1993;90:1–5. doi: 10.1073/pnas.90.1.1. doi:10.1073/pnas.90.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Smirnovas V, Baron GS, Offerdahl DK, Raymond GJ, Caughey B, Surewicz WK. Structural organization of brain-derived mammalian prions examined by hydrogen-deuterium exchange. Nat Struct Mol Biol. 2011;18:504–506. doi: 10.1038/nsmb.2035. doi:10.1038/nsmb.2035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Kocisko DA, Come JH, Priola SA, Chesebro B, Raymond GJ, Lansbury PT, Caughey B. Cell-free formation of protease-resistant prion protein. Nature. 1994;370:471–474. doi: 10.1038/370471a0. doi:10.1038/370471a0. [DOI] [PubMed] [Google Scholar]
- 87.Kocisko DA, Priola SA, Raymond GJ, Chesebro B, Lansbury PT, Jr, Caughey B. Species specificity in the cell-free conversion of prion protein to protease-resistant forms: a model for the scrapie species barrier. Proc Natl Acad Sci USA. 1995;92:3923–3927. doi: 10.1073/pnas.92.9.3923. doi:10.1073/pnas.92.9.3923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Bessen RA, Kocisko DA, Raymond GJ, Nandan S, Lansbury PT, Caughey B. Non-genetic propagation of strain-specific properties of scrapie prion protein. Nature. 1995;375:698–700. doi: 10.1038/375698a0. doi:10.1038/375698a0. [DOI] [PubMed] [Google Scholar]
- 89.Mehlhorn I, Groth D, Stockel J, Moffat B, Reilly D, Yansura D, Willett WS, et al. High-level expression and characterization of a purified 142-residue polypeptide of the prion protein. Biochemistry. 1996;35:5528–5537. doi: 10.1021/bi952965e. doi:10.1021/bi952965e. [DOI] [PubMed] [Google Scholar]
- 90.Hornemann S, Korth C, Oesch B, Riek R, Wider G, Wuthrich K, Glockshuber R. Recombinant full-length murine prion protein, mPrP(23–231): purification and spectroscopic characterization. FEBS Lett. 1997;413:277–281. doi: 10.1016/s0014-5793(97)00921-6. doi:10.1016/S0014-5793(97)00921-6. [DOI] [PubMed] [Google Scholar]
- 91.Riek R, Hornemann S, Wider G, Glockshuber R, Wuthrich K. NMR characterization of the full-length recombinant murine prion protein, mPrP(23–231) FEBS Lett. 1997;413:282–288. doi: 10.1016/s0014-5793(97)00920-4. doi:10.1016/S0014-5793(97)00920-4. [DOI] [PubMed] [Google Scholar]
- 92.Zahn R, von Schroetter C, Wuthrich K. Human prion proteins expressed in Escherichia coli and purified by high-affinity column refolding. FEBS Lett. 1997;417:400–404. doi: 10.1016/s0014-5793(97)01330-6. doi:10.1016/S0014-5793(97)01330-6. [DOI] [PubMed] [Google Scholar]
- 93.Hornemann S, Schorn C, Wuthrich K. NMR structure of the bovine prion protein isolated from healthy calf brains. EMBO Rep. 2004;5:1159–1164. doi: 10.1038/sj.embor.7400297. doi:10.1038/sj.embor.7400297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Swietnicki W, Petersen R, Gambetti P, Surewicz WK. pH-dependent stability and conformation of the recombinant human prion protein PrP(90–231) J Biol Chem. 1997;272:27517–27520. doi: 10.1074/jbc.272.44.27517. doi:10.1074/jbc.272.44.27517. [DOI] [PubMed] [Google Scholar]
- 95.Baskakov IV, Legname G, Baldwin MA, Prusiner SB, Cohen FE. Pathway complexity of prion protein assembly into amyloid. J Biol Chem. 2002;277:21140–21148. doi: 10.1074/jbc.M111402200. doi:10.1074/jbc.M111402200. [DOI] [PubMed] [Google Scholar]
- 96.Bocharova OV, Breydo L, Parfenov AS, Salnikov VV, Baskakov IV. In vitro conversion of full-length mammalian prion protein produces amyloid form with physical properties of PrP(Sc) J Mol Biol. 2005;346:645–659. doi: 10.1016/j.jmb.2004.11.068. doi:10.1016/j.jmb.2004.11.068. [DOI] [PubMed] [Google Scholar]
- 97.Colby DW, Wain R, Baskakov IV, Legname G, Palmer CG, Nguyen HO, Lemus A, et al. Protease-sensitive synthetic prions. PLoS Pathog. 2010;6:e1000736. doi: 10.1371/journal.ppat.1000736. doi:10.1371/journal.ppat.1000736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Caughey B, Neary K, Buller R, Ernst D, Perry LL, Chesebro B, Race RE. Normal and scrapie-associated forms of prion protein differ in their sensitivities to phospholipase and proteases in intact neuroblastoma cells. J Virol. 1990;64:1093–1101. doi: 10.1128/jvi.64.3.1093-1101.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Stahl N, Borchelt DR, Prusiner SB. Differential release of cellular and scrapie prion proteins from cellular membranes by phosphatidylinositol-specific phospholipase C. Biochemistry. 1990;29:5405–5412. doi: 10.1021/bi00474a028. doi:10.1021/bi00474a028. [DOI] [PubMed] [Google Scholar]
- 100.Borchelt DR, Scott M, Taraboulos A, Stahl N, Prusiner SB. Scrapie and cellular prion proteins differ in their kinetics of synthesis and topology in cultured cells. J Cell Biol. 1990;110:743–752. doi: 10.1083/jcb.110.3.743. doi:10.1083/jcb.110.3.743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Caughey B, Raymond GJ. The scrapie-associated form of PrP is made from a cell surface precursor that is both protease- and phospholipase-sensitive. J Biol Chem. 1991;266:18217–18223. [PubMed] [Google Scholar]
- 102.Caughey B, Raymond GJ, Ernst D, Race RE. N-terminal truncation of the scrapie-associated form of PrP by lysosomal protease(s): implications regarding the site of conversion of PrP to the protease-resistant state. J Virol. 1991;65:6597–6603. doi: 10.1128/jvi.65.12.6597-6603.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Baron GS, Wehrly K, Dorward DW, Chesebro B, Caughey B. Conversion of raft associated prion protein to the protease-resistant state requires insertion of PrP-res [PrP(Sc)] into contiguous membranes. EMBO J. 2002;21:1031–1040. doi: 10.1093/emboj/21.5.1031. doi:10.1093/emboj/21.5.1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Baron GS, Caughey B. Effect of glycosylphosphatidylinositol anchor-dependent and -independent prion protein association with model raft membranes on conversion to the protease-resistant isoform. J Biol Chem. 2003;278:14883–14892. doi: 10.1074/jbc.M210840200. doi:10.1074/jbc.M210840200. [DOI] [PubMed] [Google Scholar]
- 105.Morillas M, Swietnicki W, Gambetti P, Surewicz WK. Membrane environment alters the conformational structure of the recombinant human prion protein. J Biol Chem. 1999;274:36859–36865. doi: 10.1074/jbc.274.52.36859. doi:10.1074/jbc.274.52.36859. [DOI] [PubMed] [Google Scholar]
- 106.Sanghera N, Pinheiro TJ. Binding of prion protein to lipid membranes and implications for prion conversion. J Mol Biol. 2002;315:1241–1256. doi: 10.1006/jmbi.2001.5322. doi:10.1006/jmbi.2001.5322. [DOI] [PubMed] [Google Scholar]
- 107.Kazlauskaite J, Sanghera N, Sylvester I, Venien-Bryan C, Pinheiro TJ. Structural changes of the prion protein in lipid membranes leading to aggregation and fibrillization. Biochemistry. 2003;42:3295–3304. doi: 10.1021/bi026872q. doi:10.1021/bi026872q. [DOI] [PubMed] [Google Scholar]
- 108.Critchley P, Kazlauskaite J, Eason R, Pinheiro TJ. Binding of prion proteins to lipid membranes. Biochem Biophys Res Commun. 2004;313:559–567. doi: 10.1016/j.bbrc.2003.12.004. doi:10.1016/j.bbrc.2003.12.004. [DOI] [PubMed] [Google Scholar]
- 109.Kazlauskaite J, Pinheiro TJ. Aggregation and fibrillization of prions in lipid membranes. Biochem Soc Symp. 2005;72:211–222. doi: 10.1042/bss0720211. [DOI] [PubMed] [Google Scholar]
- 110.Wang F, Yang F, Hu Y, Wang X, Jin C, Ma J. Lipid interaction converts prion protein to a PrPSc-like proteinase K-resistant conformation under physiological conditions. Biochemistry. 2007;46:7045–7053. doi: 10.1021/bi700299h. doi:10.1021/bi700299h. [DOI] [PubMed] [Google Scholar]
- 111.Wang X, Wang F, Arterburn L, Wollmann R, Ma J. The interaction between cytoplasmic prion protein and the hydrophobic lipid core of membrane correlates with neurotoxicity. J Biol Chem. 2006;281:13559–13565. doi: 10.1074/jbc.M512306200. doi:10.1074/jbc.M512306200. [DOI] [PubMed] [Google Scholar]
- 112.Wang F, Yin S, Wang X, Zha L, Sy MS, Ma J. Role of the highly conserved middle region of prion protein (PrP) in PrP-lipid interaction. Biochemistry. 2010;49:8169–8176. doi: 10.1021/bi101146v. doi:10.1021/bi101146v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Hill AF, Antoniou M, Collinge J. Protease-resistant prion protein produced in vitro lacks detectable infectivity. J Gen Virol. 1999;80:11–14. doi: 10.1099/0022-1317-80-1-11. [DOI] [PubMed] [Google Scholar]
- 114.Saborio GP, Permanne B, Soto C. Sensitive detection of pathological prion protein by cyclic amplification of protein misfolding. Nature. 2001;411:810–813. doi: 10.1038/35081095. doi:10.1038/35081095. [DOI] [PubMed] [Google Scholar]
- 115.Saa P, Castilla J, Soto C. Cyclic amplification of protein misfolding and aggregation. Methods Mol Biol. 2005;299:53–65. doi: 10.1385/1-59259-874-9:053. [DOI] [PubMed] [Google Scholar]
- 116.Castilla J, Saa P, Morales R, Abid K, Maundrell K, Soto C. Protein misfolding cyclic amplification for diagnosis and prion propagation studies. Methods Enzymol. 2006;412:3–21. doi: 10.1016/S0076-6879(06)12001-7. doi:10.1016/S0076-6879(06)12001-7. [DOI] [PubMed] [Google Scholar]
- 117.Castilla J, Saa P, Soto C. Detection of prions in blood. Nat Med. 2005;11:982–985. doi: 10.1038/nm1286. [DOI] [PubMed] [Google Scholar]
- 118.Saa P, Castilla J, Soto C. Ultra-efficient replication of infectious prions by automated protein misfolding cyclic amplification. J Biol Chem. 2006;281:35245–35252. doi: 10.1074/jbc.M603964200. doi:10.1074/jbc.M603964200. [DOI] [PubMed] [Google Scholar]
- 119.Gonzalez-Romero D, Barria MA, Leon P, Morales R, Soto C. Detection of infectious prions in urine. FEBS Lett. 2008;582:3161–3166. doi: 10.1016/j.febslet.2008.08.003. doi:10.1016/j.febslet.2008.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Green KM, Castilla J, Seward TS, Napier DL, Jewell JE, Soto C, Telling GC. Accelerated high fidelity prion amplification within and across prion species barriers. PLoS Pathog. 2008;4:e1000139. doi: 10.1371/journal.ppat.1000139. doi:10.1371/journal.ppat.1000139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Castilla J, Gonzalez-Romero D, Saa P, Morales R, De Castro J, Soto C. Crossing the species barrier by PrP(Sc) replication in vitro generates unique infectious prions. Cell. 2008;134:757–768. doi: 10.1016/j.cell.2008.07.030. doi:10.1016/j.cell.2008.07.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Castilla J, Morales R, Saa P, Barria M, Gambetti P, Soto C. Cell-free propagation of prion strains. EMBO J. 2008;27:2557–2566. doi: 10.1038/emboj.2008.181. doi:10.1038/emboj.2008.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Wong C, Xiong LW, Horiuchi M, Raymond L, Wehrly K, Chesebro B, Caughey B. Sulfated glycans and elevated temperature stimulate PrP(Sc)-dependent cell-free formation of protease-resistant prion protein. EMBO J. 2001;20:377–386. doi: 10.1093/emboj/20.3.377. doi:10.1093/emboj/20.3.377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Deleault NR, Lucassen RW, Supattapone S. RNA molecules stimulate prion protein conversion. Nature. 2003;425:717–720. doi: 10.1038/nature01979. doi:10.1038/nature01979. [DOI] [PubMed] [Google Scholar]
- 125.Qin K, Yang DS, Yang Y, Chishti MA, Meng LJ, Kretzschmar HA, Yip CM, et al. Copper(II)-induced conformational changes and protease resistance in recombinant and cellular PrP. Effect of protein age and deamidation. J Biol Chem. 2000;275:19121–19131. doi: 10.1074/jbc.275.25.19121. [DOI] [PubMed] [Google Scholar]
- 126.Quaglio E, Chiesa R, Harris DA. Copper converts the cellular prion protein into a protease-resistant species that is distinct from the scrapie isoform. J Biol Chem. 2001;276:11432–11438. doi: 10.1074/jbc.M009666200. doi:10.1074/jbc.M009666200. [DOI] [PubMed] [Google Scholar]
- 127.Basu S, Mohan ML, Luo X, Kundu B, Kong Q, Singh N. Modulation of proteinase K-resistant prion protein in cells and infectious brain homogenate by redox iron: implications for prion replication and disease pathogenesis. Mol Biol Cell. 2007;18:3302–3312. doi: 10.1091/mbc.E07-04-0317. doi:10.1091/mbc.E07-04-0317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Cordeiro Y, Machado F, Juliano L, Juliano MA, Brentani RR, Foguel D, Silva JL. DNA converts cellular prion protein into the beta-sheet conformation and inhibits prion peptide aggregation. J Biol Chem. 2001;276:49400–49409. doi: 10.1074/jbc.M106707200. doi:10.1074/jbc.M106707200. [DOI] [PubMed] [Google Scholar]
- 129.Adler V, Zeiler B, Kryukov V, Kascsak R, Rubenstein R, Grossman A. Small, highly structured RNAs participate in the conversion of human recombinant PrP(Sen) to PrP(Res) in vitro. J Mol Biol. 2003;332:47–57. doi: 10.1016/s0022-2836(03)00919-7. doi:10.1016/S0022-2836(03)00919-7. [DOI] [PubMed] [Google Scholar]
- 130.Deleault NR, Geoghegan JC, Nishina K, Kascsak R, Williamson RA, Supattapone S. Protease-resistant prion protein amplification reconstituted with partially purified substrates and synthetic polyanions. J Biol Chem. 2005;280:26873–26879. doi: 10.1074/jbc.M503973200. doi:10.1074/jbc.M503973200. [DOI] [PubMed] [Google Scholar]
- 131.Silveira JR, Raymond GJ, Hughson AG, Race RE, Sim VL, Hayes SF, Caughey B. The most infectious prion protein particles. Nature. 2005;437:257–261. doi: 10.1038/nature03989. doi:10.1038/nature03989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Deleault NR, Piro JR, Walsh DJ, Wang F, Ma J, Geoghegan JC, Supattapone S. Isolation of phosphatidylethanolamine as a solitary cofactor for prion formation in the absence of nucleic acids. Proc Natl Acad Sci USA. 2012;109:8546–8551. doi: 10.1073/pnas.1204498109. doi:10.1073/pnas.1204498109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Piro JR, Harris BT, Supattapone S. In situ photodegradation of incorporated polyanion does not alter prion infectivity. PLoS Pathog. 2011;7:e1002001. doi: 10.1371/journal.ppat.1002001. doi:10.1371/journal.ppat.1002001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Wang F, Wang X, Ma J. Conversion of bacterially expressed recombinant prion protein. Methods. 2011;53:208–213. doi: 10.1016/j.ymeth.2010.12.013. doi:10.1016/j.ymeth.2010.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Ma J. The role of cofactors in prion propagation and infectivity. PLoS Pathog. 2012;8:e1002589. doi: 10.1371/journal.ppat.1002589. doi:10.1371/journal.ppat.1002589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Aguzzi A, Heikenwalder M, Polymenidou M. Insights into prion strains and neurotoxicity. Nat Rev Mol Cell Biol. 2007;8:552–561. doi: 10.1038/nrm2204. doi:10.1038/nrm2204. [DOI] [PubMed] [Google Scholar]
- 137.Aguzzi A. Unraveling prion strains with cell biology and organic chemistry. Proc Natl Acad Sci USA. 2008;105:11–12. doi: 10.1073/pnas.0710824105. doi:10.1073/pnas.0710824105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Weissmann C. Thoughts on mammalian prion strains. Folia neuropathologica/Association of Polish Neuropathologists and Medical Research Centre. Polish Acad Sci. 2009;47:104–113. [PubMed] [Google Scholar]
- 139.Li J, Browning S, Mahal SP, Oelschlegel AM, Weissmann C. Darwinian evolution of prions in cell culture. Science. 2010;327:869–872. doi: 10.1126/science.1183218. doi:10.1126/science.1183218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Weissmann C, Li J, Mahal SP, Browning S. Prions on the move. EMBO Rep. 2011;12:1109–1117. doi: 10.1038/embor.2011.192. doi:10.1038/embor.2011.192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Beringue V, Herzog L, Jaumain E, Reine F, Sibille P, Le Dur A, Vilotte JL, Laude H. Facilitated cross-species transmission of prions in extraneural tissue. Science. 2012;335:472–475. doi: 10.1126/science.1215659. doi:10.1126/science.1215659. [DOI] [PubMed] [Google Scholar]