

Abstract
Objective
Cellular senescence influences organismal aging and increases predisposition to age-related diseases, in particular cardiovascular disease, a leading cause of death and disability worldwide. PGC-1α is a master regulator of mitochondrial biogenesis and function, oxidative stress and insulin resistance. Senescence is associated with telomere and mitochondrial dysfunction and oxidative stress, inferring a potential causal role of PGC-1α in senescence pathogenesis.
Methods and Results
We generated a PGC-1α+/−/ApoE−/− mouse model and show that PGC-1α deficiency promotes a vascular senescence phenotype that is associated with increased oxidative stress, mitochondrial abnormalities, and reduced telomerase activity. PGC-1α disruption results in reduced expression of the longevity-related deacetylase sirtuin 1 (SIRT1) and the antioxidant catalase, and increased expression of the senescence marker p53 in aortas. Further, angiotensin II (Ang II), a major hormonal inducer of vascular senescence, induces prolonged lysine acetylation of PGC-1α and releases the PGC-1α·FoxO1 complex from the SIRT1 promoter, thus reducing SIRT1 expression. The phosphorylation defective mutant PGC-1α S570A is not acetylated, is constitutively active for FoxO1-dependent SIRT1 transcription and prevents Ang II-induced senescence. Acetylation of PGC-1α by Ang II interrupts the PGC-1α-FoxO1-SIRT1 feed-forward signaling circuit leading to SIRT1 and catalase downregulation and vascular senescence.
Conclusions
PGC-1α is a primary negative regulator of vascular senescence. Moreover, the central role of post-translational modification of PGC-1α in regulating Ang II-induced vascular senescence may inform development of novel therapeutic strategies for mitigating age-associated diseases such as atherosclerosis.
Keywords: Vascular senescence, PGC-1α, Angiotensin II, Signal transduction
Introduction
Age is a major risk factor for many chronic conditions including cardiovascular diseases such as hypertension and atherosclerosis, which lead to heart failure, myocardial infarction, and stroke. 1 These age-related diseases account for over 40 percent of the mortality in individuals 65 years and above in the United States. 1, 2 Cellular senescence, a hallmark of organismal aging, is a permanent non-replicating state characterized by telomere and mitochondrial dysfunction, elevated oxidative stress, 3 and increased expression of β-galactosidase (SA-βG), 4 tumor suppressor protein p53 and cyclin-dependent kinase inhibitors such as p21(CIP1/WAF1) and p16 (INK4a). 5 Vascular smooth muscle cell (VSMC) senescence in atherosclerotic plaques is a characteristic feature of atherosclerosis and is associated with increased levels of reactive oxygen species (ROS). 6 Understanding the mechanistic relationships among senescence and age-related disease phenotypes in general is currently a focus of great scientific interest. 7
Angiotensin II (Ang II) is a pluripotent hormone and an important mediator of hypertension and atherosclerosis. 8 Ang II exposure increases ROS production as well as VSMC senescence and acceleration of atherosclerosis.9 These effects are mediated importantly by elevated ROS generation through the G protein coupled Ang II type 1 receptor (AT1R). 10 AT1R deficiency in mice extends life span possibly by attenuation of oxidative stress. 11 The molecular mechanisms are incompletely understood.
Peroxisome proliferator-activated receptor γ coactivator-1α PGC-1α) enables transcriptional coactivation functions for multiple metabolic and stress response pathways, including carbohydrate and lipid metabolism, oxidative stress resistance, angiogenesis, and mitochondrial biogenesis and function. 12, 13 Regulation of PGC-1α activity is fine-tuned by several interacting post-translational modifications (including phosphorylation and acetylation) to integrate its multiple physiologic functions.14 Acetylation of PGC-1α by the histone acetyltransferase GCN5 (general control nonderepressible 5) inactivates PGC-1α, while deacetylation of PGC-1α by SIRT1 increases its activity. SIRT1 is a negative regulator of senescence.15 We showed recently that Ang II-stimulated phosphorylation of PGC-1α Ser570 increases the GCN5-mediated acetylation of PGC-1α, leading to reduced catalase expression, increased ROS levels and hypertrophy in VSMCs. 16 Reduction in catalase expression results in VSMC senescence.17 Overexpression of human catalase targeted to mitochondria promotes longevity in mice. 18, 19 We found that Ang II decreases catalase expression by releasing FoxO1 from the catalase promoter.16 Further, we found that FoxO1 binds to two cis-acting elements, IRS-1 and FKHD-L, in the SIRT1 promoter and directly activates SIRT1 transcription through a feed-forward, self-amplifying loop. 20 Collectively, these data infer a potentially important role in the pathogenesis of senescence for PGC-1α and its post-translational modification by phosphorylation and acetylation.
Until recently no specific role had been identified for PGC-1α in modulating the pathogenesis of senescence. Reduced telomerase activity and telomere length are well known to be associated with cellular senescence. 21 Genetically manipulated mice null for telomerase reverse transcriptase (Tert) exhibited accelerated DNA damage and aging that was associated with mitochondrial dysfunction and reduction in PGC-1α expression. 22 PGC-1α downregulation was mediated by the binding to and blocking of the PGC-1α promoter by the tumor suppressor p53, a mediator of senescence, as noted. A pivotal role of PGC-1α expression in creating the mitochondrial phenotype was established by rescue of compromised mitochondrial function upon PGC-1α transfection. 22 These findings inferred a potential role for PGC-1α in senescence and chronic diseases of aging. 23, 24
A causal relationship between cell senescence and organismal aging has been inferred also by observations that senescent cells accumulate in aging organs and disrupt structure and function by paracrine or endocrine secretion of substances inducing pathophysiologic responses. 25 Direct evidence was presented recently that senescent cells may harm nonsenescent cells and cause organ dysfunction and disease. 7 The reduction in PGC-1α expression observed in skeletal muscle during aging is consistent with this concept. 26 Furthermore, ectopic expression of PGC-1α in skeletal muscle prevents age-associated muscle wasting in mice. 27 Together, these data provide inferential evidence that the level of PGC-1α expression is an important determinant of cellular senescence, organismal aging, and related chronic diseases. The roles of post-translational modification and regulation of PGC-1α coactivator function in senescence pathogenesis are largely unknown.
Here we demonstrate that primary PGC-1α disruption in ApoE knockout mice (PGC-1α+/−/ApoE−/−) promotes vascular senescence that is associated with elevated ROS levels, mitochondrial abnormalities, and reduced telomerase activity. Further, PGC-1α deficiency decreases SIRT1 and catalase expression and augments p53 levels in aortas and mouse aortic smooth muscle cells (MASMs). These findings substantiate an important role for PGC-1α in vascular senescence. In primary rat aortic VSMCs (RASMs), Ang II induces prolonged lysine acetylation, suppresses PGC-1α coactivation function, and releases the PGC-1α·FoxO1 complex from the SIRT1 promoter, thus decreasing SIRT1 expression. This downregulation in turn enables the sustained acetylation of PGC-1α. Our results provide novel evidence that PGC-1α functions as an important negative regulator of vascular senescence and provide new insights into the role of PGC-1α post-translational modifications in the process. Further, the dependency upon PGC-1α cotranscriptional activity of the function of the FoxO1-SIRT1 transcriptional control loop informs a novel role for the coactivator in mitigating senescence. These observations substantiate the notion that PGC-1α might be a promising target in the development of interventions for age-related chronic diseases.
Results
Primary disruption of PGC-1α promotes vascular senescence that is associated with ROS production, mitochondrial abnormalities, and decreased telomerase activity in PGC-1α+/−/ApoE−/− mice
To investigate the role of PGC-1α in vascular senescence, we generated PGC-1α−/− and +/− mice in the ApoE−/− background. ApoE is a multifunctional protein that, in addition to its effects on lipoprotein metabolism, has intrinsic activities that enhance inflammation and oxidative stress. 28 The proinflammatory/prooxidation phenotype of the ApoE knockout enables Ang II-induced senescence in vivo. 6 Similar to PGC-1α−/− homozygotes, 29 PGC-1α−/−/ApoE−/− double knockout mice displayed small size, disordered control of thermogenesis, and increased perinatal mortality. We have, however, successfully nurtured a number of the compound homozygotes into old age (18 months) and used them to inform the role of PGC-1α in vascular senescence. The PGC-1α+/−/ApoE−/− mice, in contrast, showed no visually obvious phenotype and matured normally. Thus, we used these animals to evaluate in detail whether and how PGC-1α deficiency informs development of vascular senescence. Aortas of PGC-1α+/−/ApoE−/− animals exhibited increased SA-βG activity basally (Figure 1A, 1.47 ± 0.33-fold) and after Ang II infusion (Figure 1B, 5.11 ± 2.08-fold) compared with the PGC-1α+/+/ApoE−/− control mice. In addition, aortas of the younger mice (about 6 months) and older mice (about 18 months) from both PGC-1α+/−/ApoE−/− and PGC-1α−/−/ApoE−/− animals exhibit increased senescence activity compared with controls (Figure I in the online-only Data Supplement). No significant differences in blood pressure were observed between PGC-1α+/−/ApoE−/− and PGC-1α+/+/ApoE−/− mice after Ang II treatment (the online-only Data Supplemental Table). Histological examination showed SA-βG staining in VSMCs in the media and cells in the adventitia in aortas from PGC-1α+/−/ApoE−/− animals (Figure II in the online-only Data Supplement). To gain further insights into whether primary PGC-1α deficiency informs development of the senescence phenotype, we isolated and cultured MASMs from aortas of both genotypes. MASMs from PGC-1α+/−/ApoE−/− aortas showed increased SA-βG activity basally (1.38 ± 0.03-fold compared with control and after Ang II treatment (1.66 ± 0.04-fold compared with control 1.34 ± 0.05-fold) (Figure 1C–D). Consistent with the known role of PGC-1α in mitigating oxidative stress, we showed that its deficiency in MASMs (PGC-1α+/−/ApoE−/−) was associated with increased ROS levels basally (3.7 ± 0.2-fold compared with control) and after Ang II treatment (4.8 ± 0.2-fold compared with control 2.5 ± 0.3 -fold) (Figure 1E).
Figure 1. PGC-1α deficiency increases vascular senescence and ROS levels.
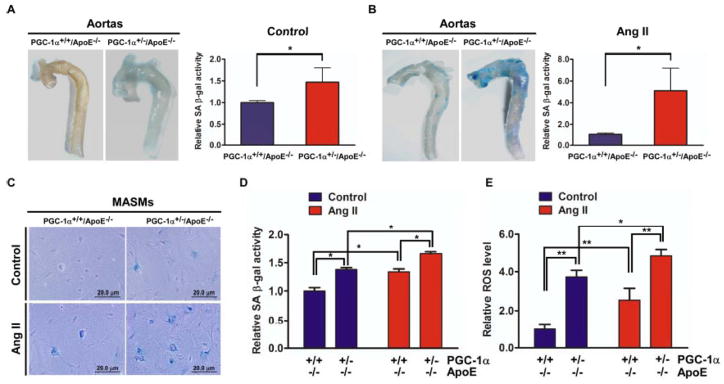
(A and B) The aortas from PGC-1α+/+/ApoE−/− and PGC-1α+/−/ApoE−/− mice at 6–7 months of age treated without (A) or with Ang II (B) via osmotic pump were excised for SA-βG staining as described in Methods. Quantitative analysis of SA-βG staining using fluorescein di-beta-D-galactopyranoside (FDG) was performed as described in Methods. Bar graphs represent relative SA-βG activity. Data (mean ± S.D, n=14) are expressed as fold change over wild type, *P<0.05 between the indicated conditions. (C, D and E) Mouse aortic smooth muscle cells (MASMs) isolated from aortas were quiesced and stimulated with or without 100 nM Ang II for 3 days. SA-βG staining (C) and quantitative analysis of SA-βG staining using FDG (D) and intracellular ROS levels (E) were determined as described in Methods, respectively. Scale bar=20 μm. *P<0.05, **P<0.01 between the indicated conditions.
PGC-1α plays an important role in mitochondrial biogenesis and function in multiple systems.14 Mitochondrial dysfunction has been associated frequently with mitochondrial structural abnormalities including mitochondrial fragmentation.30 We therefore examined the impact of PGC-1α deficiency on mitochondrial structure in MASMs. PGC-1α deficiency promotes accumulation of mitochondrial fragments (Figure 2A). Average mitochondrial length for PGC-1α wild type and deficiency was 2.57 and 2.14 μm, respectively (Figure 2B). Moreover, PGC-1α deficiency decreased expression of nuclear respiratory factor-1 (NRF-1) and mitochondrial transcription factor A (TFAM), determinants of mitochondrial biogenesis (Figure 2C). As expected, PGC-1α deficiency in MASMs (PGC-1α+/− and PGC-1α−/−/ApoE−/−) caused a significant increase in mitochondrial ROS generation, measured by MitoSOX fluorescence intensity (1.9 and 2.4-fold versus control, respectively, Figure III in the online-only Data Supplement). Mitochondria were visualized with MitoTracker Green. Image merging of MitoSOX Red with MitoTracker Green indicates that the enhanced superoxide production by PGC-1α deficiency was generated from mitochondria. Thus, these results are consistent with the notion that primary PGC-1α deficiency impairs mitochondrial structure and increases mitochondrial ROS levels.
Figure 2. PGC-1α disruption is associated with mitochondrial abnormalities and decreased telomerase activity.
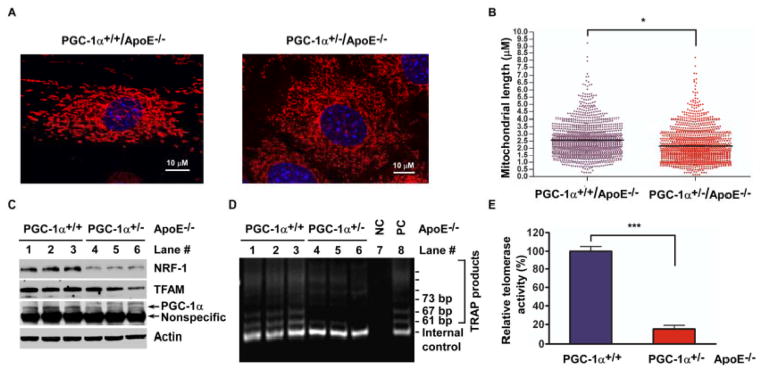
(A and B) PGC-1α-deficiency is associated with mitochondrial abnormalities. Representative images for mouse aortic smooth muscle cells (MASMs) isolated from PGC-1α+/+/ApoE−/− and PGC-1α+/−/ApoE−/− mice aortas. Cells were labeled with MitoTracker® Orange CMTMRos (A). Average mitochondrial tubule length for wild type and PGC-1α+/− MASMs was 2.57 and 2.14 μm, respectively (B). *P<0.05. (C) PGC-1α-deficiency impacts downstream expression networks. PGC-1α downstream targets expression was analyzed by immunoblotting with indicated antibody. (D and E) PGC-1α-deficiency decreases telomerase activity in MASMs. (D) Amplification products of telomeric repeat were subjected to electrophoresis on a non-denaturing 10% polyacrylamide gel. After ethidium bromide staining, the image was recorded with a CCD camera imaging system. Lane 7: Negative control (minus Taq polymerase); Lane 8: Positive control. (E) TRAP from the same experiment (D) was analyzed for quantification of telomerase activity by measuring fluorescence on a fluorescence plate reader. ***P<0.001.
Telomere dysfunction, an inducer of senescence, is associated with p53-dependent downregulation of PGC-1α and PGC-1α-dependent genes regulating mitochondrial biogenesis and function.22 It is unknown, however, whether primary PGC-1α disruption may affect telomere function/activities. We investigated the effect of PGC-1α deficiency on telomerase activity and found that telomerase activity was reduced 85% in MASMs from PGC-1α+/−/ApoE−/− compared with wild type mice (Figure 2D–E). Collectively, these data illustrate that PGC-1α gene disruption per se promotes vascular senescence that is associated with elevated oxidative stress, mitochondrial abnormalities, and decreased telomerase activity.
PGC-1α deficiency decreases SIRT1 and catalase expression, and increases p53 levels in PGC-1α+/−/ApoE−/− mice
To gain mechanistic insights into pathways by which PGC-1α deficiency mediates senescence and oxidative stress, we assessed the effects of this disruption on the expression levels of SIRT1, a negative regulator of senescence, the antioxidant catalase, and the senescence marker p53 in aortas of mice of both genotypes with and without Ang II infusion. The level of PGC-1α expression in the aortas of the heterozygote was decreased by 93% and was not affected significantly by Ang II infusion (Figure 3A–B). The reduction in PGC-1α expression in these mice was associated with a marked decrease in SIRT1 (93%) and catalase (79%) levels, and an increase in p53 expression (2.4 ± 0.4-fold compared with control 1.0 ± 0.3-fold), inferring their regulation by PGC-1α. In the PGC-1α+/+/ApoE−/− mice, Ang II infusion mimics the effects of PGC-1α deficiency in reducing SIRT1 and catalase expression and increasing p53 levels. Further reductions in SIRT1 and catalase expression and increase in p53 expression were observed in PGC-1α+/−/ApoE−/− after Ang II infusion (Figure 3A–B). Interestingly, in the PGC-1α+/+/ApoE−/− mice, prolonged Ang II exposure elicited these signals associated with senescence without significant downregulation of PGC-1α expression. These findings led us to posit that the Ang II-induced acetylation and loss of PGC-1α cotranscriptional function that we previously described might be important in inducing senescence. 16 The unanticipated downregulation of SIRT1 could play a pivotal role in this process.
Figure 3. PGC-1α-deficiency decreases SIRT1 and catalase expression, and augments p53 levels in PGC-1α+/−/ApoE−/− mice.

(A and B) PGC-1α+/+/ApoE−/− and PGC-1α+/−/ApoE−/− mice were given Ang II via osmotic pump for 4 weeks. Tissue lysates from aortas were analyzed for SIRT1, catalase, p53, and PGC-1α protein expression by Western blot (A). Bar graphs represent averaged data (mean ± S.D, n=4–6), expressed as fold change over PGC-1α wild type in sham control group (B). **P<0.01, vs. control or as indicated, ns denotes no significant difference.
PGC-1α acetylation results in reductions in SIRT1 and catalase expression
We showed that Ang II acetylates PGC-1α and down-regulates catalase expression in VSMCs in vitro, as noted. 16 Here we demonstrate that Ang II infusion in vivo is associated with increased serine phosphorylation and lysine acetylation of PGC-1α in the aortas of wild type C57BL/6J mice (Figure 4A–B). These modifications of PGC-1α were associated with decreased expression of SIRT1 and catalase, and increased p53 expression (Figure 4C). There was no change in the expression of PGC-1α. In order to provide insights and causal relationships among these signaling events, we utilized primary cultures of rat aortic smooth muscle cells (RASMs). 31 Similar to our observations in aortas, Ang II treatment in RASMs significantly decreased SIRT1 expression over time (Figure IV-A in the online-only Data Supplement), while no significant changes were observed in PGC-1α expression (Figure IV-B in the online-only Data Supplement). Taken together, these data suggest that acetylation of PGC-1α by Ang II may link to reduced SIRT1 and catalase expression.
Figure 4. SIRT1 downregulation by Ang II promotes PGC-1α acetylation.

(A and B) Mice were infused with Ang II (0.75 mg/kg/day) for 14 days. Nuclear extracts from aortas of Ang II-infused and control mice (3 animals in each group) were prepared and immunoprecipitated with PGC-1α antibody followed by blotting with phosphoserine (A) and acetylated lysine antibodies (B), respectively. Immunoblotting with PGC-1α is shown for total PGC-1α loading. (C) Mice were treated with Ang II (0.75 mg/kg/day) for 28 days. Tissue extracts from aortas of Ang II-infused and control mice were prepared and analyzed by immunoblotting with indicated antibody (C). Data (mean ± S.D, n=3) are quantified and expressed as fold change over control. *P<0.05, **P<0.01. (D and E) RASMs were infected with Ad.SIRT1 wild type (WT) or H363Y (DN) or Ad.Lac Z (as a control) for 24 hours (D), or transfected with 50 nM SIRT1 or control scrambled siRNA for 48 hours (E); cells were then treated with 100 nM Ang II for 30 minutes. PGC-1α acetylation was determined. (F) RASMs were transfected with 10, 30, 50 nM SIRT1 or control scrambled siRNA for 48 hours, and then infected with Ad.PGC-1α (S570A) or Ad.Lac Z for 24 hours. PGC-1α acetylation was determined and protein expression was analyzed with indicated antibodies.
We next determined whether the decrease in SIRT1 expression contributes to the acetylation and inactivation of PGC-1α by Ang II. Prolonged treatment with Ang II in RASMs induces sustained acetylation of PGC-1α Figure IV-B, in the online-only Data Supplement), which was decreased by overexpression of wild type SIRT1 but not SIRT1-H363Y, a catalytically inactive dominant negative mutant Figure 4D). Conversely, SIRT1 depletion by siRNA increases basal and Ang II-induced PGC-1α acetylation, which is associated with reduced expression of catalase (Figure 4E), and which is dependent upon PGC-1α and FoxO1. 16 To evaluate the role of PGC-1α deacetylation by SIRT1 in enabling catalase protein levels, we overexpressed PGC-1α S570A, a phosphorylation defective mutant that also blocks acetylation. 16 Overexpression of the PGC-1α S570A mutant increases catalase expression and prevents its downregulation by SIRT1 siRNA (Figure 4F). Thus, these data indicate that reduction in SIRT1 expression by Ang II leads to elevated and sustained PGC-1α acetylation, which represses PGC-1α co-transcriptional activity and thus results in decreased catalase expression.
FoxO1 phosphorylation is required for Ang II-induced SIRT1 downregulation
We showed recently that FoxO1 directly activates SIRT1 transcription through binding to the IRS-1 and FKHD-like response elements within the SIRT1 promoter. 20 The observation that Ang II reduces SIRT1 expression in vivo and in vitro prompted us to investigate if inactivation of FoxO1 could mediate Ang II-induced SIRT1 downregulation at the transcriptional level. To test this hypothesis, we conducted one-step RT-PCR and quantitative real-time PCR in RASMs to determine whether Ang II might impact SIRT1 mRNA abundance. Ang II significantly decreases SIRT1 mRNA after 2 hours of treatment and the levels were barely detectable by 8 hours (Figure V in the online-only Data Supplement). We next examined the impact of Ang II on FoxO1 phosphorylation that inhibits FoxO1 transcriptional activity. We found that Ang II (100 nM) stimulates phosphorylation of FoxO1 at both Ser256 and Thr24 sites, which peaks at 5 minutes (Figure 5A). We investigated the effect of Ang II on FoxO1 binding to the SIRT1 promoter. ChIP assays using a specific FoxO1 antibody revealed that Ang II stimulation decreases FoxO1 binding to IRS-1 and FKHD-L DNA-binding elements (DBE) of the SIRT1 promoter in a time-dependent manner (Figure 5B). The impaired binding of FoxO1 to the SIRT1 promoter by Ang II was prevented by overexpression of the FoxO1 phosphorylation mutant (TM, S256A/T24A/S319A) (Figure 5C, compare lane 12 with 8), but not FoxO1 wild type (WT) (Figure 5C, compare lane 10 with 8). We evaluated the effect of Ang II on FoxO1-dependent SIRT1 transcriptional activities using a luciferase reporter construct containing the 1.5 kb SIRT1 promoter. 20 Ang II stimulation suppresses in a dose-dependent manner FoxO1 (WT)- but not FoxO1 (TM)- mediated SIRT1 transcriptional activity. FoxO1 (TM) shows higher SIRT1 promoter activity than FoxO1 (WT) (Figure 5D). Moreover, overexpression of FoxO1 (TM), but not FoxO1 (WT), substantially inhibited Ang II-induced SIRT1 downregulation (Figure 5E). Thus, FoxO1 phosphorylation is an early, essential signal for transcriptional downregulation of SIRT1 expression in response to Ang II.
Figure 5. FoxO1 phosphorylation is required for SIRT1 downregulation by Ang II.

(A and B) RASMs were stimulated with 100 nM Ang II for the indicated times. FoxO1 phosphorylation was detected (A). Binding of FoxO1 to SIRT1 promoter was detected by ChIP assay (B). (C–E) RASMs were infected with Ad.FoxO1 wild type (WT) or its mutant (TM) or Ad.GFP (as a control) for 24 hours, and treated with Ang II for 4 hours for ChIP assay (C), or stimulated with increasing dose of Ang II for 16 hours for luciferase assay (D) or for 24 hours for SIRT1 expression (E). The relative activity was normalized and calculated as the folds to control or basal group. (*P<0.05, **P<0.01, n=6).
PGC-1α S570A coactivates FoxO1-dependent SIRT1 transcription
PGC-1α coactivates FoxO1-mediated catalase transcription. 16 The observation that PGC-1α disruption in ApoE knockout mice is associated with SIRT1 downregulation prompted us to posit that PGC-1α also regulates FoxO1-dependent SIRT1 transcription. Using reporter transfection assays, we demonstrated that overexpression of the PGC-1α S570A mutant elevates basal and FoxO1-mediated SIRT1 transcriptional activities in RASMs, inferring that PGC-1α coactivates SIRT1 transcription through FoxO1 (Figure VI-A in the online-only Data Supplement). Overexpression of PGC-1α S570A increases SIRT1 protein expression in a dose-dependent manner (Figure VI-B in the online-only Data Supplement). To investigate whether FoxO1 is required for PGC-1α-coactivated SIRT1 expression, we examined the effect of FoxO1 depletion using siRNA. ChIP assays showed that endogenous PGC-1α associates with the SIRT1 promoter regions encompassing IRS-1 and FKHD-L elements basally in a FoxO1-dependent manner (Figure VI-C in the online-only Data Supplement). We further determined the impact of FoxO1 depletion on PGC-1α-mediated SIRT1 expression (Figure VI-D in the online-only Data Supplement). Overexpression of the PGC-1α S570A mutant enhanced SIRT1 expression, which was abolished by FoxO1 depletion. FoxO1 siRNA also decreases basal SIRT1 expression as we previously reported. 20 Association of PGC-1α with FoxO1 in regulation of FoxO1-dependent gene expression in multiple systems is well established. 32, 33 These data suggest that PGC-1α S570A constitutively activates SIRT1 expression, which is mediated by the transcription factor FoxO1. These findings inform a previously unappreciated role for PGC-1α activity in controlling SIRT1 expression.
Inhibition of PGC-1α acetylation prevents Ang II-induced SIRT1 downregulation
Ang II induces PGC-1α acetylation as well as SIRT1 downregulation. In order to provide mechanistic understanding into the relationship of these events, we overexpressed the acetylation defective mutant PGC-1α S570A and determined the effects of Ang II on FoxO1-dependent SIRT1 expression. ChIP assays showed that Ang II stimulation inhibited binding of endogenous PGC-1α (Figure 6A) but not of the overexpressed PGC-1α S570A mutant (Figure 6B) to the SIRT1 promoter after 2 hours. Similarly, the PGC-1α mutant blocked Ang II-induced release of FoxO1 from the SIRT1 promoter (Figure 6C). Moreover, reporter assays indicated that the PGC-1α mutant blocked Ang II-induced reduction of SIRT1 promoter activity (Figure 6D). FoxO1 phosphorylation is required for Ang II-induced SIRT1 downregulation (Figure 5). We thus assessed whether PGC-1α S570A inhibits Ang II-stimulated FoxO1 phosphorylation. Overexpression of the PGC-1α mutant blocks basal and Ang II-stimulated FoxO1 phosphorylation at both sites (Figure 6E) as well as SIRT1 downregulation by Ang II (Figure 6F). Reciprocally, a constitutive active FoxO1 phosphorylation mutant prevents Ang II-induced PGC-1α acetylation (Figure VII in the online-only Data Supplement). Thus, the interdependent posttranslational modifications of FoxO1 and PGC-1α modulate feed-forward, self-amplifying SIRT1 transcription.
Figure 6. PGC-1α S570A prevents Ang II-reduced SIRT1 expression.

(A) Ang II impairs PGC-1α binding to the SIRT1 promoter. RASMs were stimulated with 100 nM Ang II for the indicated times (A). (B and C) PGC-1α S570A prevents dissociation of PGC-1α·FoxO1 complex from the SIRT1 promoter. RASMs were infected with Ad.PGC-1α S570A or Ad.Lac Z (control), and stimulated with Ang II for 2 hours. ChIP assay was performed. (D–F) PGC-1α S570A prevents reduced SIRT1 promoter activity, FoxO1 phosphorylation and decreased SIRT1 expression by Ang II. VSMCs were infected with Ad.PGC-1α S570A or Ad.Lac Z for 24 hours, and then treated with Ang II either for 16 hours for SIRT1 promoter activity by luciferase assay (D) (**P<0.01, vs. control or as indicated), or for 5 minutes for FoxO1 phosphorylation (E), or for 24 hours for SIRT1 expression (F).
PGC-1α is a negative regulator of VSMC senescence
PGC-1α gene deficiency displayed enhanced senescence both in aortas (Figures 1A-B and S1) and in cultured MASMs (Figure 1C–D) from ApoE−/− mice. We investigated the role of PGC-1α in Ang II-induced VSMC senescence. We overexpressed PGC-1α S570A in RASMs and determined senescence by SA-βG staining (Figures 7 and VIII in the online-only Data Supplement). Absence of PGC-1α acetylation inhibited Ang II-induced cellular senescence (Figure 7A). Similarly, overexpression of the FoxO1 constitutively active form (TM) inhibited Ang II-induced senescence (Figure 7B). PGC-1α coactivates FoxO1 to regulate SIRT1 expression. We next asked whether SIRT1 impacts Ang II-induced cellular senescence. Overexpression of SIRT1 wild type (WT) prevents Ang II-induced senescence (Figure 7C). In contrast, depletion of either PGC-1α or FoxO1 or SIRT1 by siRNA significantly enhances basal senescence (Figure 7D). These results, together with the previous in vivo observations, demonstrate that PGC-1α functions as a primary negative regulator of VSMC senescence. Together, these data are consistent with the notion that the inactivation of PGC-1α transcriptional coactivation capabilities by acetylation is sufficient to inhibit the FoxO1-SIRT1 signaling axis and enable Ang II-induced cellular senescence (Figure 7E). We cannot exclude the possibility that Ang II might target alternative pathways to induce vascular senescence. Thus, in the context of the development of the senescence phenotype, PGC-1α primary gene deficiency and inactivation of its cotranscriptional capability by posttranslational acetylation appear to act similarly in vivo and in vitro.
Figure 7. PGC-1α-FoxO1-SIRT1 signaling inhibits Ang II-induced VSMC senescence.
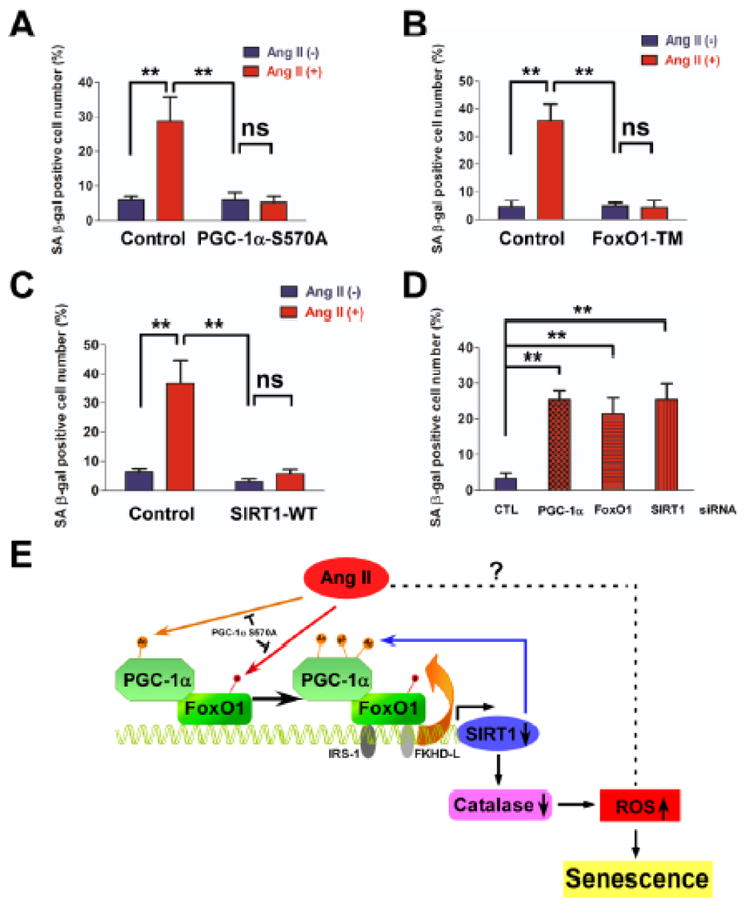
(A, B and C) Overexpression of constitutive active forms of PGC-1α, FoxO1, and SIRT1 wild type protects against Ang II-induced senescence. RASMs in serum-free DMEM were infected with either Ad.PGC-1α S570A (A), or FoxO1 constitutively active form-FoxO1(TM) (B), or SIRT1 wild type (WT) (C), or control vector for 24 hours. (D) Knock-down of either PGC-1α, or FoxO1, or SIRT1 promotes cellular senescence. RASMs were transfected with 100 nM PGC-1α or FoxO1 or SIRT1 or control scrambled siRNA for 5 days. SA-βG staining was performed, **P<0.01 between the indicated conditions, n=3–5. (E) Proposed model: PGC-1α acetylation by Ang II represses FoxO1-SIRT1 signaling to promote vascular senescence. PGC-1α coactivates FoxO1-mediated SIRT1 transcription. Ang II stimulates sustained acetylation of PGC-1α and suppresses PGC-1α co-transcriptional activity, thereby decreasing SIRT1 expression. Downregulation of SIRT1 expression in turn promotes PGC-1α acetylation, which further represses its coactivation ability on FoxO1-mediated SIRT1 transcription and decreases catalase expression, thus increasing ROS levels and vascular senescence. It is possible that Ang II might induce vascular senescence via alternative pathways.
Discussion
Cellular senescence, a hallmark of organismal aging, is an important risk factor for multiple age-associated diseases including neurodegenerative, cardiovascular, and metabolic disorders. 34 In particular, senescence has been inferentially implicated in atherosclerotic arterial disease clinically and in animals. 2, 35 PGC-1α, an essential regulator of mitochondrial biogenesis and function, is a putative inhibitor of aging-associated systemic conditions including telomere dysfunction, DNA damage, and elevated oxidative stress. 34 Deregulation of mitochondria with excessive production of ROS is a common unifying feature of diseases and conditions associated with aging. Decreased expression of PGC-1α has been implicated in the onset and progression of age-related neurodegenerative disorders such as Huntington, Parkinson, and Alzheimer diseases. 36–38 Overexpression of PGC-1 Drosophila homologue in fruit flies delays aging and prolongs lifespan. 39 The role of PGC-1α in mitigating senescence and the mechanisms involved have been largely unexplored.
Using genetic, transcriptional, molecular and functional assays, we demonstrate that both PGC-1α deficiency and inactivation of PGC-1α by acetylation induces vascular senescence. Cellular senescence triggered by primary PGC-1α disruption is associated with elevated ROS levels, abnormalities of mitochondrial structure, and reduced telomerase activity coinciding with increased p53 levels and reductions in SIRT1 and catalase expression (Figures 1–3). Moreover, Ang II-induced acetylation of PGC-1α decreases the binding of the PGC-1α·FoxO1 transcriptional complex to the SIRT1 and catalase promoters and downregulates their expression. Downregulation of SIRT1 expression in turn promotes PGC-1α acetylation, which further represses its coactivation ability on FoxO1-mediated SIRT1 and catalase transcription through a feed-forward inhibitory mechanism (Figure 7E). Depletion of PGC-1α, FoxO1, or SIRT1 induces senescence, while the overexpression of any one of them prevents it (Figure 7), suggesting that the feed-forward signaling loop among PGC-1α, FoxO1, and SIRT1 is required to prevent senescence. Roles for FoxO1 and SIRT1 in development of senescence have been demonstrated.40, 41 The findings here provide novel evidence that PGC-1α co-transcriptional activity is also essential in this regard. Taken together, the data provide novel mechanistic understanding of the generally important consequences of PGC-1α downregulation in senescence (Figure 7E). 22
PGC-1α co-transcriptional activity is fine-tuned by reversible posttranslational modifications including phosphorylation and acetylation. 14 Phosphorylation of PGC-1α Ser570 by insulin was identified originally as an inhibitory signal for PGC-1α coactivation of genes involved in gluconeogenesis and fatty acid oxidation. 42 Ser570 phosphorylation of PGC-1α by Ang II triggers enhanced GCN5 recruitment to and lysine acetylation of PGC-1α in vitro 16 and in vivo (Figures 4 and IX in the online-only Data Supplement). Acetylation of PGC-1α in RASMs releases the PGC-1α·FoxO1 complex from the catalase promoter, thereby inhibiting catalase expression and increasing hypertrophy. 16 We show that the acetylation-defective PGC-1α S570A mutant coactivates FoxO1-dependent SIRT1 expression and prevents Ang II-induced senescence (Figures 7 and VI in the online-only Data Supplement). In contrast, increased PGC-1α acetylation by SIRT1 downregulation induces senescence (Figures 4 and 7). Thus, Ang II enhances PGC-1α acetylation by both increasing its association with GCN5 and by decreasing SIRT1 expression. Moreover, disruption of the PGC-1α gene in mice is associated with reduction in SIRT1 expression (Figure 3). Reciprocally, the elevated SIRT1 expression levels observed in PGC-1α transgenic mice 27 support the notion that PGC-1α regulates SIRT1 expression.
FoxO1 and SIRT1 are evolutionarily conserved longevity genes involved in a broad spectrum of processes, many of which are salutary for health and development. They act synergistically to modulate aging, oxidative stress, and insulin resistance. 43 We recently showed that FoxO1 directly activates SIRT1 transcription through its association with the DNA-binding elements in the SIRT1 promoter. 20 Ang II treatment stimulates FoxO1 phosphorylation at both Ser256 and Thr24 sites, releases FoxO1 from the SIRT1 promoter, and represses FoxO1 transcriptional activity, thereby decreasing SIRT1 expression (Figure 5). PGC-1α S570A prevents Ang II-induced SIRT1 downregulation through repression of FoxO1 phosphorylation and chromatin occupancy in the SIRT1 promoter (Figure 6). Reciprocally, a constitutive active FoxO1 phosphorylation mutant prevents Ang II-induced PGC-1α acetylation (Figure VII in the online-only Data Supplement). These results indicate that the interdependent posttranslational modifications of FoxO1 and PGC-1α by Ang II are required for the coordinated regulation of SIRT1 expression. Ang II treatment does not alter SIRT1 mRNA half-life (t½ >8h, data not shown), suggesting that Ang II-induced SIRT1 downregulation is not mediated by impaired SIRT1 mRNA stability.
Telomere dysfunction is involved in replicative and stress-induced senescence. 24 Telomere dysfunction upregulates the senescence marker p53 and induces mitochondrial compromise through downregulation of the PGC-1α network. 22 We showed that both PGC-1α deficiency and inactivation by Ang II are associated with upregulation of p53 expression (Figures 3 and 4). Thus, vascular senescence may be induced and potentially linked to impaired telomere function by either PGC-1α deficiency or inhibitory posttranslational acetylation in the setting of preserved expression of PGC-1α protein (Figures 2–4). Interestingly, we observed that primary PGC-1α deficiency decreases telomerase activity contemporaneously with SIRT1 downregulation (Figures 2 and 3). SIRT1 deficiency leads to telomere shortening and genomic instability. 44 Increased SIRT1 expression in SIRT1super mice preserves telomere length, function, and genome stability with aging. Our findings not only broaden understanding of the scope of influence of SIRT1 on aging-related processes but also embellish the importance of the novel findings that PGC-1α is a major modulator of SIRT1 expression and cellular senescence.
The additive effect of PGC-1α deficiency and Ang II in vascular senescence (Figures 1B and D) suggests that Ang II-induced cell senescence is mediated by PGC-1α/FoxO1/SIRT1-dependent and independent mechanisms. Consistent with this notion, we observed an additive effect in ROS levels in PGC-1α deficient cells treated with Ang II (Figure 1E), suggesting that additional pathways mediated by Ang II could also contribute to development of the senescence phenotype (Figure. 7E).
PGC-1α plays a pivotal role in improving mitochondrial function and ameliorating oxidative stress via upregulation of genes improving mitochondrial biogenesis such as TFAM and expression of multiple antioxidant enzymes such as catalase.14, 45 Increased oxidative stress is an important stimulus of cell growth, hypertrophy and senescence in VSMCs.46 Overexpression of catalase in mitochondria extends murine life span. 18, 19 We showed previously that PGC-1α acetylation by Ang II downregulates catalase expression, which contributes, at least in part, to elevated ROS levels and VSMC hypertrophy. 16 Here we demonstrate that PGC-1α deficiency (PGC-1α+/−/ApoE−/−) in MASMs promotes cellular senescence that is associated with mitochondrial structure abnormalities (Figure 2A–B), augmented mitochondrial superoxide production (Figure III in the online-only Data Supplement), and increased intracellular ROS levels (Figure 1E) concomitant with reductions in TFAM, NRF-1, and catalase expression (Figure 2C). Furthermore, inactivation of PGC-1α by Ang II-induced acetylation mimics the role of PGC-1α disruption in downregulation of SIRT1 and catalase expression (Figure 3). Thus, acetylation of PGC-1α appears to be a central regulatory mechanism by which Ang II targets and represses the ROS defense mechanisms to mediate vascular senescence. Our data support the concept that reduced PGC-1α expression and inactivation of PGC-1α by acetylation enable development of cellular senescence, which is associated with elevated oxidative stress and mitochondrial abnormalities.
Recent evidence is consistent with the notion that linkage among oxidative stress, deterioration of telomere function, and mitochondrial dysfunction is mediated by the p53-PGC-1 axis, as noted. Thus, inferentially, PGC-1α likely plays a central role in the pathogenesis of multiple diseases of aging including atherosclerotic vascular disease. Stein et al. 47 reported that PGC-1α disruption does not alter atherogenesis in young PGC-1α−/−ApoE−/−, compared with PGC-1α+/+ApoE−/− controls. However, our preliminary observations suggest that elderly PGC-1α−/−ApoE−/− mice on a standard chow diet are more susceptible to age-related atherosclerosis than are PGC-1α+/+ApoE−/− mice of similar age, inferring a protective role for PGC-1α in the pathogenesis of the disease. An anti-aging role of PGC-1α in a lower organism was recently reported. 39 Additional studies are needed that incorporate age as a variable to clarify definitively the role of PGC-1α in atherosclerosis pathogenesis.
In conclusion, we describe novel functions for PGC-1α as a negative regulator of vascular senescence in vivo and in vitro. Oxidative stress and impaired mitochondrial and telomere function/activities are associated in the development of vascular senescence driven by PGC-1α gene deficiency. Deacetylated PGC-1α prevents senescence by enabling FoxO1-dependent SIRT1 transcription. PGC-1α acetylation by Ang II interrupts FoxO1-dependent SIRT1 transcription. This interactive relationship may result in a self sustaining feedback mechanism perpetuating PGC-1α inactivation (Figure 7E). These results provide unique mechanistic insights into the pivotal role for PGC-1α in linking diverse elements of cell senescence induction including telomere dysfunction, mitochondrial abnormalities, and elevated oxidative stress. Importantly, persistent inhibition of PGC-1α cotranscriptional function by environmentally-induced posttranslational modifications such as acetylation, as modeled here, could inform the pathogenesis of cardiovascular and other age-related chronic diseases. The findings should also guide new therapeutic strategies.
Supplementary Material
Significance.
Vascular senescence has been implicated in atherosclerotic arterial disease clinically and in animals. PGC-1α functions as an important inhibitor of senescence-associated conditions including mitochondrial dysfunction, increased oxidative stress, and insulin resistance. However, the role of PGC-1α in mitigating vascular senescence and the mechanisms involved have been largely unexplored. In the present study, we describe novel functions for PGC-1α as a central negative regulator of vascular senescence. Deacetylated PGC-1α prevents senescence by enabling FoxO1-dependent SIRT1 transcription. PGC-1α acetylation by Ang II interrupts FoxO1-dependent SIRT1 transcription. This interactive relationship may result in a self-sustaining feedback mechanism perpetuating PGC-1α inactivation. These results provide unique mechanistic insights into the pivotal role for PGC-1α in linking diverse elements of cell senescence induction. Importantly, persistent inhibition of PGC-1α cotranscriptional function by environmentally-induced post-translational modifications such as acetylation could inform the pathogenesis of cardiovascular and other age-related chronic diseases. The findings should also guide new therapeutic strategies.
Acknowledgments
We are grateful to Dr. Zolt Arany for providing PGC-1α knockout mice, Dr. J.A. Hill for AdFoxO1 (WT) and CA mutant, and Dr. Daniel P. Kelly for PGC-1α antibody.
Sources of Funding
This work was funded by grants from NIH UO1 HL80711 and NIH HL60728.
Non-standard Abbreviations
- Ang II
Angiotensin II
- AT1R
Ang II type 1 receptor
- ROS
Reactive oxygen species
- Tert
Telomerase reverse transcriptase
- PGC-1α
Peroxisome proliferator-activated receptor γ coactivator-1α
- GCN5
General control nonderepressible 5
- TFAM
Mitochondrial transcription factor A
- VSMC
Vascular smooth muscle cell
- MASMs
Mouse aortic smooth muscle cells
- RASMs
Rat aortic smooth muscle cells
- SiRNA
Small interference RNA
- Scr-siRNA
Scrambled small interference RNA
- TRAP
Telomeric Repeat Amplification Protocol
- ChIP
Chromatin Immunoprecipitation
- SA-βG
Senescence associated β-galactosidase
- FDG
Fluorescein di-β-D-galactopyranoside
Footnotes
No conflicts of interest exist.
Conflict of Interest Disclosures: None
References
- 1.Lakatta EG. Age-associated cardiovascular changes in health: impact on cardiovascular disease in older persons. Heart Fail Rev. 2002;7:29–49. doi: 10.1023/a:1013797722156. [DOI] [PubMed] [Google Scholar]
- 2.Kovacic JC, Moreno P, Nabel EG, Hachinski V, Fuster V. Cellular senescence, vascular disease, and aging: part 2 of a 2-part review: clinical vascular disease in the elderly. Circulation. 2011;123:1900–1910. doi: 10.1161/CIRCULATIONAHA.110.009118. [DOI] [PubMed] [Google Scholar]
- 3.Jeyapalan JC, Sedivy JM. Cellular senescence and organismal aging. Mechanisms of ageing and development. 2008;129:467–474. doi: 10.1016/j.mad.2008.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dimri GP, Lee X, Basile G, Acosta M, Scott G, Roskelley C, Medrano EE, Linskens M, Rubelj I, Pereira-Smith O, Peacocke M, Campsi J. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc Natl Acad Sci U S A. 1995;92:9363–9367. doi: 10.1073/pnas.92.20.9363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bringold F, Serrano M. Tumor suppressors and oncogenes in cellular senescence. Experimental gerontology. 2000;35:317–329. doi: 10.1016/s0531-5565(00)00083-8. [DOI] [PubMed] [Google Scholar]
- 6.Kunieda T, Minamino T, Nishi J, Tateno K, Oyama T, Katsuno T, Miyauchi H, Orimo M, Okada S, Takamura M, Nagai T, Kaneko S, Komuro I. Angiotensin II induces premature senescence of vascular smooth muscle cells and accelerates the development of atherosclerosis via a p21-dependent pathway. Circulation. 2006;114:953–960. doi: 10.1161/CIRCULATIONAHA.106.626606. [DOI] [PubMed] [Google Scholar]
- 7.Baker DJ, Wijshake T, Tchkonia T, LeBrasseur NK, Childs BG, van de Sluis B, Kirkland JL, van Deursen JM. Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature. 2011;479:232–236. doi: 10.1038/nature10600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Berk BC, Haendeler J, Sottile J. Angiotensin II, atherosclerosis, and aortic aneurysms. J Clin Invest. 2000;105:1525–1526. doi: 10.1172/JCI9820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Minamino T, Komuro I. Vascular cell senescence: contribution to atherosclerosis. Circ Res. 2007;100:15–26. doi: 10.1161/01.RES.0000256837.40544.4a. [DOI] [PubMed] [Google Scholar]
- 10.Griendling KK, Ushio-Fukai M, Lassegue B, Alexander RW. Angiotensin II signaling in vascular smooth muscle. New concepts. Hypertension. 1997;29:366–373. doi: 10.1161/01.hyp.29.1.366. [DOI] [PubMed] [Google Scholar]
- 11.Benigni A, Corna D, Zoja C, Sonzogni A, Latini R, Salio M, Conti S, Rottoli D, Longaretti L, Cassis P, Morigi M, Coffman TM, Remuzzi G. Disruption of the Ang II type 1 receptor promotes longevity in mice. J Clin Invest. 2009;119:524–530. doi: 10.1172/JCI36703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Puigserver P, Spiegelman BM. Peroxisome proliferator-activated receptor-gamma coactivator 1 alpha (PGC-1 alpha): transcriptional coactivator and metabolic regulator. Endocr Rev. 2003;24:78–90. doi: 10.1210/er.2002-0012. [DOI] [PubMed] [Google Scholar]
- 13.Arany Z, Foo SY, Ma Y, Ruas JL, Bommi-Reddy A, Girnun G, Cooper M, Laznik D, Chinsomboon J, Rangwala SM, Baek KH, Rosenzweig A, Spiegelman BM. HIF-independent regulation of VEGF and angiogenesis by the transcriptional coactivator PGC-1alpha. Nature. 2008;451:1008–1012. doi: 10.1038/nature06613. [DOI] [PubMed] [Google Scholar]
- 14.Fernandez-Marcos PJ, Auwerx J. Regulation of PGC-1alpha, a nodal regulator of mitochondrial biogenesis. Am J Clin Nutr. 2011;93:884S–890. doi: 10.3945/ajcn.110.001917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yao H, Chung S, Hwang JW, Rajendrasozhan S, Sundar IK, Dean DA, McBurney MW, Guarente L, Gu W, Ronty M, Kinnula VL, Rahman I. SIRT1 protects against emphysema via FOXO3-mediated reduction of premature senescence in mice. J Clin Invest. 2012;122:2032–2045. doi: 10.1172/JCI60132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xiong S, Salazar G, San Martin A, Ahmad M, Patrushev N, Hilenski L, Nazarewicz RR, Ma M, Ushio-Fukai M, Alexander RW. PGC-1 alpha serine 570 phosphorylation and GCN5-mediated acetylation by angiotensin II drive catalase down-regulation and vascular hypertrophy. J Biol Chem. 2010;285:2474–2487. doi: 10.1074/jbc.M109.065235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Patrushev N, Seidel-Rogol B, Salazar G. Angiotensin II requires zinc and downregulation of the zinc transporters ZnT3 and ZnT10 to induce senescence of vascular smooth muscle cells. PloS one. 2012;7:e33211. doi: 10.1371/journal.pone.0033211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schriner SE, Linford NJ, Martin GM, Treuting P, Ogburn CE, Emond M, Coskun PE, Ladiges W, Wolf N, Van Remmen H, Wallace DC, Rabinovitch PS. Extension of murine life span by overexpression of catalase targeted to mitochondria. Science. 2005;308:1909–1911. doi: 10.1126/science.1106653. [DOI] [PubMed] [Google Scholar]
- 19.Dai DF, Santana LF, Vermulst M, Tomazela DM, Emond MJ, MacCoss MJ, Gollahon K, Martin GM, Loeb LA, Ladiges WC, Rabinovitch PS. Overexpression of catalase targeted to mitochondria attenuates murine cardiac aging. Circulation. 2009;119:2789–2797. doi: 10.1161/CIRCULATIONAHA.108.822403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xiong S, Salazar G, Patrushev N, Alexander RW. FoxO1 mediates an autofeedback loop regulating SIRT1 expression. J Biol Chem. 2011;286:5289–5299. doi: 10.1074/jbc.M110.163667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bodnar AG, Ouellette M, Frolkis M, Holt SE, Chiu CP, Morin GB, Harley CB, Shay JW, Lichtsteiner S, Wright WE. Extension of life-span by introduction of telomerase into normal human cells. Science. 1998;279:349–352. doi: 10.1126/science.279.5349.349. [DOI] [PubMed] [Google Scholar]
- 22.Sahin E, Colla S, Liesa M, Moslehi J, Muller FL, Guo M, Cooper M, Kotton D, Fabian AJ, Walkey C, Maser RS, Tonon G, Foerster F, Xiong R, Wang YA, Shukla SA, Jaskelioff M, Martin ES, Heffernan TP, Protopopov A, Ivanova E, Mahoney JE, Kost-Alimova M, Perry SR, Bronson R, Liao R, Mulligan R, Shirihai OS, Chin L, DePinho RA. Telomere dysfunction induces metabolic and mitochondrial compromise. Nature. 2011;470:359–365. doi: 10.1038/nature09787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kelly DP. Cell biology: Ageing theories unified. Nature. 2011;470:342–343. doi: 10.1038/nature09896. [DOI] [PubMed] [Google Scholar]
- 24.Finkel T. Telomeres and mitochondrial function. Circ Res. 2011;108:903–904. doi: 10.1161/RES.0b013e31821bc2d8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Coppe JP, Patil CK, Rodier F, Sun Y, Munoz DP, Goldstein J, Nelson PS, Desprez PY, Campisi J. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. 2008;6:2853–2868. doi: 10.1371/journal.pbio.0060301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Anderson R, Prolla T. PGC-1alpha in aging and anti-aging interventions. Biochim Biophys Acta. 2009;1790:1059–1066. doi: 10.1016/j.bbagen.2009.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wenz T, Rossi SG, Rotundo RL, Spiegelman BM, Moraes CT. Increased muscle PGC-1alpha expression protects from sarcopenia and metabolic disease during aging. Proc Natl Acad Sci U S A. 2009;106:20405–20410. doi: 10.1073/pnas.0911570106. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 28.Jofre-Monseny L, Minihane AM, Rimbach G. Impact of apoE genotype on oxidative stress, inflammation and disease risk. Mol Nutr Food Res. 2008;52:131–145. doi: 10.1002/mnfr.200700322. [DOI] [PubMed] [Google Scholar]
- 29.Lin J, Wu PH, Tarr PT, Lindenberg KS, St-Pierre J, Zhang CY, Mootha VK, Jager S, Vianna CR, Reznick RM, Cui L, Manieri M, Donovan MX, Wu Z, Cooper MP, Fan MC, Rohas LM, Zavacki AM, Cinti S, Shulman GI, Lowell BB, Krainc D, Spiegelman BM. Defects in adaptive energy metabolism with CNS-linked hyperactivity in PGC-1alpha null mice. Cell. 2004;119:121–135. doi: 10.1016/j.cell.2004.09.013. [DOI] [PubMed] [Google Scholar]
- 30.Yu T, Robotham JL, Yoon Y. Increased production of reactive oxygen species in hyperglycemic conditions requires dynamic change of mitochondrial morphology. Proc Natl Acad Sci U S A. 2006;103:2653–2658. doi: 10.1073/pnas.0511154103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Griendling KK, Taubman MB, Akers M, Mendlowitz M, Alexander RW. Characterization of phosphatidylinositol-specific phospholipase C from cultured vascular smooth muscle cells. J Biol Chem. 1991;266:15498–15504. [PubMed] [Google Scholar]
- 32.Puigserver P, Rhee J, Donovan J, Walkey CJ, Yoon JC, Oriente F, Kitamura Y, Altomonte J, Dong H, Accili D, Spiegelman BM. Insulin-regulated hepatic gluconeogenesis through FOXO1-PGC-1alpha interaction. Nature. 2003;423:550–555. doi: 10.1038/nature01667. [DOI] [PubMed] [Google Scholar]
- 33.Handschin C, Lin J, Rhee J, Peyer AK, Chin S, Wu PH, Meyer UA, Spiegelman BM. Nutritional regulation of hepatic heme biosynthesis and porphyria through PGC-1alpha. Cell. 2005;122:505–515. doi: 10.1016/j.cell.2005.06.040. [DOI] [PubMed] [Google Scholar]
- 34.Wenz T. Mitochondria and PGC-1alpha in Aging and Age-Associated Diseases. Journal of aging research. 2011;2011:810619. doi: 10.4061/2011/810619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kovacic JC, Moreno P, Hachinski V, Nabel EG, Fuster V. Cellular senescence, vascular disease, and aging: part 1 of a 2-part review. Circulation. 2011;123:1650–1660. doi: 10.1161/CIRCULATIONAHA.110.007021. [DOI] [PubMed] [Google Scholar]
- 36.Cui L, Jeong H, Borovecki F, Parkhurst CN, Tanese N, Krainc D. Transcriptional repression of PGC-1alpha by mutant huntingtin leads to mitochondrial dysfunction and neurodegeneration. Cell. 2006;127:59–69. doi: 10.1016/j.cell.2006.09.015. [DOI] [PubMed] [Google Scholar]
- 37.Shin JH, Ko HS, Kang H, Lee Y, Lee YI, Pletinkova O, Troconso JC, Dawson VL, Dawson TM. PARIS (ZNF746) repression of PGC-1alpha contributes to neurodegeneration in Parkinson’s disease. Cell. 2011;144:689–702. doi: 10.1016/j.cell.2011.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Qin W, Haroutunian V, Katsel P, Cardozo CP, Ho L, Buxbaum JD, Pasinetti GM. PGC-1alpha expression decreases in the Alzheimer disease brain as a function of dementia. Archives of neurology. 2009;66:352–361. doi: 10.1001/archneurol.2008.588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rera M, Bahadorani S, Cho J, Koehler CL, Ulgherait M, Hur JH, Ansari WS, Lo T, Jr, Jones DL, Walker DW. Modulation of longevity and tissue homeostasis by the Drosophila PGC-1 homolog. Cell metabolism. 2011;14:623–634. doi: 10.1016/j.cmet.2011.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.van der Horst A, Burgering BM. Stressing the role of FoxO proteins in lifespan and disease. Nat Rev Mol Cell Biol. 2007;8:440–450. doi: 10.1038/nrm2190. [DOI] [PubMed] [Google Scholar]
- 41.Brooks CL, Gu W. How does SIRT1 affect metabolism, senescence and cancer? Nat Rev Cancer. 2009;9:123–128. doi: 10.1038/nrc2562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li X, Monks B, Ge Q, Birnbaum MJ. Akt/PKB regulates hepatic metabolism by directly inhibiting PGC-1alpha transcription coactivator. Nature. 2007;447:1012–1016. doi: 10.1038/nature05861. [DOI] [PubMed] [Google Scholar]
- 43.Banks AS, Kon N, Knight C, Matsumoto M, Gutierrez-Juarez R, Rossetti L, Gu W, Accili D. SirT1 gain of function increases energy efficiency and prevents diabetes in mice. Cell Metab. 2008;8:333–341. doi: 10.1016/j.cmet.2008.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Palacios JA, Herranz D, De Bonis ML, Velasco S, Serrano M, Blasco MA. SIRT1 contributes to telomere maintenance and augments global homologous recombination. J Cell Biol. 2010;191:1299–1313. doi: 10.1083/jcb.201005160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.St-Pierre J, Drori S, Uldry M, Silvaggi JM, Rhee J, Jager S, Handschin C, Zheng K, Lin J, Yang W, Simon DK, Bachoo R, Spiegelman BM. Suppression of reactive oxygen species and neurodegeneration by the PGC-1 transcriptional coactivators. Cell. 2006;127:397–408. doi: 10.1016/j.cell.2006.09.024. [DOI] [PubMed] [Google Scholar]
- 46.Matthews C, Gorenne I, Scott S, Figg N, Kirkpatrick P, Ritchie A, Goddard M, Bennett M. Vascular smooth muscle cells undergo telomere-based senescence in human atherosclerosis: effects of telomerase and oxidative stress. Circ Res. 2006;99:156–164. doi: 10.1161/01.RES.0000233315.38086.bc. [DOI] [PubMed] [Google Scholar]
- 47.Stein S, Lohmann C, Handschin C, Stenfeldt E, Boren J, Luscher TF, Matter CM. ApoE−/− PGC-1alpha−/− mice display reduced IL-18 levels and do not develop enhanced atherosclerosis. PloS one. 2010;5:e13539. doi: 10.1371/journal.pone.0013539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Roguska J, Simon NM, del Greco F. Pressor response to angiotensin II in hypertension. Correlation with plasma renin activity and response to norepinephrine and metaraminol. Am J Cardiol. 1968;21:705–713. doi: 10.1016/0002-9149(68)90269-5. [DOI] [PubMed] [Google Scholar]
- 49.Yang NC, Hu ML. A fluorimetric method using fluorescein di-beta-D-galactopyranoside for quantifying the senescence-associated beta-galactosidase activity in human foreskin fibroblast Hs68 cells. Anal Biochem. 2004;325:337–343. doi: 10.1016/j.ab.2003.11.012. [DOI] [PubMed] [Google Scholar]
- 50.Wright WE, Shay JW, Piatyszek MA. Modifications of a telomeric repeat amplification protocol (TRAP) result in increased reliability, linearity and sensitivity. Nucleic Acids Res. 1995;23:3794–3795. doi: 10.1093/nar/23.18.3794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nazarenko IA, Bhatnagar SK, Hohman RJ. A closed tube format for amplification and detection of DNA based on energy transfer. Nucleic Acids Res. 1997;25:2516–2521. doi: 10.1093/nar/25.12.2516. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.