

Abstract
Longevity is regulated by a network of intimately linked metabolic systems. We used a combination of mouse population genetics and RNAi in C. elegans to identify mitochondrial ribosomal protein S5 (Mrps5) and other mitochondrial ribosomal proteins (MRPs) as metabolic and longevity regulators. MRP knockdown triggers mitonuclear protein imbalance, reducing mitochondrial respiration and activating the mitochondrial unfolded protein response (UPRmt). Specific antibiotics targeting mitochondrial translation and ethidium bromide, which impairs mitochondrial DNA transcription, pharmacologically mimic mrp knockdown and extend lifespan by inducing mitonuclear protein imbalance, also in mammalian cells. In addition, resveratrol and rapamycin, longevity compounds acting on different molecular targets, similarly induced mitonuclear protein imbalance, UPRmt and lifespan extention in C. elegans. Collectively these data demonstrate that MRPs represent an evolutionary conserved protein family that ties the mitochondrial ribosome and mitonuclear protein imbalance to UPRmt, an overarching longevity pathway across multiple species.
Longevity is coordinated by intersecting pathways, often converging on metabolic networks1–4. A key player in lifespan regulation is the mitochondrion. Over a thousand proteins encoded by nuclear DNA (nDNA) translocate to and function in mitochondria5, in synchrony with 13 proteins encoded by the mitochondrial DNA (mtDNA) that require a separate translation machinery, including mitochondrial ribosomal proteins (MRPs)6,7. Many molecular studies of longevity have exploited simple organisms and loss- or gain-of-function mutations, but the complex connectedness of mitochondrial and metabolic longevity networks benefits from an integrative cross-species approach2.
Here we pioneered such a strategy and used the BXD reference population of mice2,8–10 to identify mitochondrial ribosomal protein S5 (Mrps5) and other members of the MRP family as longevity genes. In C. elegans, we confirmed this role of MRPs and demonstrated that they induce a stoichiometric imbalance between nDNA- and mtDNA-encoded OXPHOS proteins, hereafter termed “mitonuclear protein imbalance”, which activates the mitochondrial unfolded protein response (UPRmt). Our conclusions were corroborated using specific antibiotics targeting bacterial/mitochondrial translation, and ethidium bromide, which inhibits mtDNA transcription. This mechanism is shared with pathways that induce mitonuclear protein imbalance from a nuclear perspective, such as the UPRmt and lifespan effects of rapamycin and resveratrol. Our data hence tie mitochondrial translation and metabolism to natural lifespan regulation across species.
A QTL for mouse longevity
The BXD family consists of fully inbred progeny of a cross between C57BL/6J and DBA/2J mice, with a complexity that matches many human populations11. Both parental strains have been sequenced enabling analysis of sequence variants linked to phenotypes12. We used new genomic and genetic resources to reanalyze longevity data for BXD lines13 using forward and reverse genetic methods9.
The forward strategy exploits longevity data and updated high-density SNP genotypes14 archived in www.GeneNetwork.org. As reported13, lifespan of BXDs varies from ~365d for the shortest lived strain to ~900d for the longest lived (Fig. 1a). We remapped longevity using the new genotypes and detected one genome-wide significant locus on chromosome 2 with a peak at 124–129Mb (Fig. 1b, LOD=4.0). Two additional loci, on chromosomes 4 and 7, were not significant, but suggestive (LOD=2.8 and 3.0, respectively). However, neither was suggestive after controlling for SNP rs6374387 on chromosome 2 using composite interval mapping.
Figure 1. Lifespan regulation in BXD recombinant inbred mice.
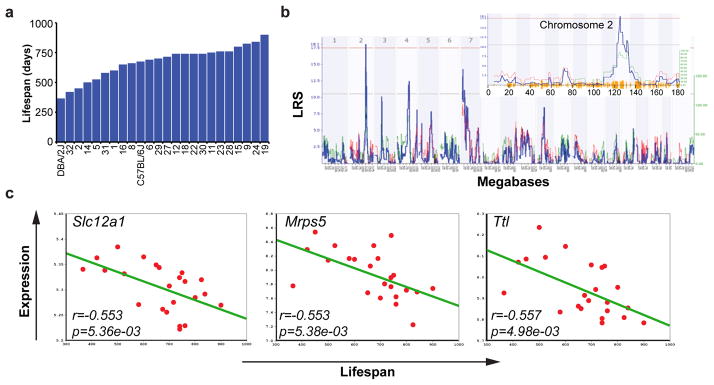
a, Life span in different BXD strains. b, Interval mapping using the BXD lifespan data reveals a strong QTL on chromosome 2, between 124–129 Mb. The red line depicts cutoff for statistical significance (p genome-wide <0.05), while the grey line represents the limit for suggestive QTLs. See also Supplementary Table 1. c, Pearson’s r correlation coefficient with corresponding p values for the covariation between BXD lifespan (x-axis) and mRNA expression of the indicated gene in the BXD eye microarrays (y-axis). Expression of Slc12a1, Mrps5 and Ttl robustly correlates with longevity (p<0.01).
The chromosome 2 locus contains ~70 genes (Supplementary Table 1), none of which previously linked to longevity. To evaluate and rank candidates, we correlated lifespan with multiple gene expression data sets. Only three genes in the locus correlate strongly with lifespan (Fig. 1c, p<0.01; Supplementary Fig. 1), i.e. solute carrier family 12 member 1 (Slc12a1), mitochondrial ribosomal protein S5 (Mrps5), and tubulin-tyrosine ligase (Ttl). From the natural variation in expression of these genes we deduced that 50% reduction of expression corresponds to a ~250d lifespan difference.
Conservation of longevity in C. elegans
We identified Y37A1C.1/nkcc-1, E02A10.1/mrps-5, and F25C8.5/ttll-9 as worm homologs of Slc12a1, Mrps5, and Ttl, respectively. RNAi-mediated knockdown of nkcc-1 and mrps-5, but not of ttll-9, extended lifespan (Fig. 2a).
Figure 2. Validation of Mrps5 as a candidate longevity gene.

a, Knockdown during entire life of mrps-5, nkcc-1 or ttll-9 in C. elegans increased lifespan by 60%, 23% or 3%, respectively. See also Supplementary Table 2. b, One-way hierarchical clustering showing expression differences in gastrocnemius between young (5mo), old (25mo) and caloric restricted C57BL/6J mice15. Expression of mouse Mrp’s decreases upon aging, and reverts with CR, whereas Slc12a1 and Ttl do not change. c, Pearson’s r correlation coefficient with corresponding p values for covariation between BXD lifespan (x-axis) and mRNA expression of eleven other Mrp’s (y-axis) indicates robust correlation. Principle component analysis (PCA) reveals a highly significant correlation between the Mrp gene family and BXD lifespan. d, Mrps5 strongly correlates with genes involved in OXPHOS. Red lines indicate a positive Pearson correlation coefficient of 0.7–1.0, and blue lines of 0.5–0.7.
Next, we compared expression of Mrps5 and other Mrp family members in a muscle microarray of aging and CR in C57BL/6J15. Mrp expression decreased with age, an effect rescued by CR; in contrast, expression of Slc12a1 and Ttl was unaffected (Fig. 2b). Linkage of MRPs with lifespan is strengthened as many other Mrp family members also correlate with longevity (Fig. 2c). We extended our analyses to the DNA level using sequence data for Mrps5 in both parental strains and identified missense variants in exon 3 (rs29667217 and rs13471334; V60A and V67I, respectively). Other sequence variants in Mrps5 contribute to variation in transcript abundance; Mrps5 mRNA levels among the BXDs are associated with a strong QTL superimposed over the gene itself—a cis expression QTL.
Using a reverse genetics approach, we studied the Mrps5-associated network. Mrps5 expression covaries with genes involved in oxidative phosphorylation (OXPHOS). Considering that oxidative metabolism is involved in known longevity pathways2, the set of transcripts that covary with Mrps5 qualified as an appealing longevity network. OXPHOS was the most enriched network of Mrps5 covariates in both BXDs16 and a conventional F2 intercross17 (p=1.53e-21, p=5.78e-10, respectively). Finally, we generated an interaction network of OXPHOS genes with Mrps5 (Fig. 2d), in which Ndufb7 provides the hinge that links Mrps5 to OXPHOS. Interestingly, knockdown of the worm homologs for the network components Ndufb7 and Ndufa6 robustly extended lifespan18–20. Mrps5 hence emerged as a strong longevity candidate, integrating protein synthesis and mitochondrial metabolism—both important longevity modulators.
Mitonuclear protein imbalance and aging
To define causality of the MRPs in determining lifespan, we knocked down mrp genes during the entire life of the worm and robustly increased lifespan (Fig. 3a, Supplementary Table 2). Similarly to well-characterized mitochondrial mutants that live longer, larval development was delayed (Supplementary Fig. 2a)21. Knockdown during development proved crucial and sufficient to extend lifespan, while RNAi during adulthood alone did not (Fig. 3b, Supplementary Fig. 2b and Table 2), as reported in other long-lived mitochondrial mutants22. Increased lifespan was not due to effects on feeding, as pharyngeal pumping rates were normal (Supplementary Fig. 2c). mrps-5 RNAi also delayed physiological decline with age. Even though they moved slightly less in early adulthood (d3), mrps-5 RNAi worms move twice as much as controls at d13, and this effect becomes more pronounced at d20 (Supplementary Fig. 2d–e and Movies 1–4). This difference was accompanied by a delay in decline of pharyngeal pumping (Supplementary Fig. 2c) and in muscle fiber disorganization (Fig. 3c), hallmarks of fitness of aged mrps-5 RNAi worms.
Figure 3. mrps-5 RNAi prevents aging-associated functional decline and alters mitochondrial function.

a, Knockdown of mrpl-1, mrpl-2 or mrpl-37 increased lifespan by 57%, 54%, or 41%, respectively. b, When RNAi of mrps-5 was performed during the larval stages only, lifespan increased by 48%, while RNAi started from the L4 stage had no effect. p≤0.001 is for larval-only versus either vector control or adult-only. c, mrps-5 or cco-1 RNAi prevented age-related changes in muscle morphology as evidenced by a pmyo-3MYO-3::GFP reporter worm highlighting myosin heavy chain. d, mrp RNAi in C. elegans decreased respiration. Respiration/worm is shown here, but respiration was similarly decreased when corrected for protein. FCCP was added at the indicated time. Values are mean±SEM (n=10), *** p≤0.001. e–g, mrps-5 RNAi decreased ATP levels (e, n=3), citrate synthase activity (f, n=3), and altered the ratio between nDNA (ATP5A) versus mtDNA-encoded (MTCO1) OXPHOS proteins, similar to cco-1, but not mev-1 (g, n=4). * p≤0.05. h, mrps-5 RNAi resulted in fragmented mitochondria, as visualized using the pmyo-3::mito::GFP reporter, which expresses mitochondria-targeted GFP driven by the muscle-specific myo-3 promoter. i, mrps-5 RNAi increased mean lifespan by 40%. j-m, mrps-5 RNAi extends lifespan of daf-16(mu86) (j), sir-2.1(ok434) (k), aak-2(ok524) (l), mev-1(kn1) (m) mutants by 37%, 40%, 69%, 112%. n, Knockdown of cco-1 does not extend lifespan of mrps-5 RNAi worms. See Supplementary Table 2 and Fig. 4.
In line with the mitochondrial connection of Mrps5, basal respiration was reduced upon mrp knockdown and unresponsive to the uncoupler FCCP (Fig. 3d). As a consequence, mrps-5 RNAi worms displayed reduced ATP levels and citrate synthase activity (Fig. 3e–f), indicative for reduced mitochondrial abundance or activity. Consistent with its role in mitochondrial translation, mrps-5 RNAi induced a stoichiometric imbalance between nDNA- and mtDNA-encoded OXPHOS subunits, termed mitonuclear protein imbalance, visualized by selective reduction in MTCE.26 (MTCO-1 homolog; from mtDNA) relative to H28O16.1 (ATP5A homolog; from nDNA) expression (Fig. 3g). The mitonuclear protein imbalance and impact on mitochondrial function was similar to the long-lived cco-1 mutant—deficient for the nDNA-encoded worm homolog of complex IV, subunit Vb/COX4—but not observed in the short-lived complex II SDHC mutant mev-1 (Fig. 3g). Furthermore, mitochondria had a more punctuate globular pattern instead of the regular reticular/tubular appearance in both muscle (Fig. 3h) and intestine, a finding confirmed by electron microscopy (Supplementary Fig. 3a–b).
To identify which “longevity pathways”—insulin/IGF-1 signaling23, CR24, and mitochondrial dysfunction22—are required for the lifespan phenotype, we reduced mrps-5 expression in worms carrying mutations in these pathways. mrps-5 RNAi increases lifespan by ~40% in wild types (Fig. 3i), similar to the effect in daf-2, daf-16, eat-2, sir-2.1, and aak-2 mutants (Fig. 3j–l, Supplementary Fig. 4a–e) indicating that mrps-5 regulates longevity independently of insulin/IGF-1 (daf-16/daf-2) and CR (eat-2/sir-2.1) and acts downstream of mitochondrial regulator aak-2.
We focused on the mitochondrial pathway, since: (1) it robustly impacts longevity22; (2) MRPs function in the translation of mtDNA-encoded OXPHOS subunits6,7; and (3) in the BXDs, Mrps5 networked with several OXPHOS components (Fig 2d). Interestingly, mrps-5 RNAi reverts the short-lived phenotype of mev-1 mutants, with a dramatic 112% lifespan extension (Fig. 3m, Supplementary Fig. 4f). mrps-5 RNAi in the cco-1 mutants did not extend lifespan compared to mrps-5 RNAi alone, suggesting that cco-1 and mrps-5 act in a similar fashion (Fig. 3n, Supplementary Fig. 4g). The same is true for mrps-5 RNAi in the mitochondrial clk-1(e2519) mutant, confirming the link with mitochondrial longevity pathways (Supplementary Fig. 4h).
Mitochondrial unfolded protein response
The mitochondrial unfolded protein response (UPRmt) accounts for longevity upon cco-1 loss-of-function and is selective for the mitochondrial pathway and not involved in the CR or insulin/IGF-1 pathways25. UPRmt is induced by mitochondrial stress, subsequently activating a nuclear transcriptional response, inducing the chaperones HSP-6 (HSP-70 in mammals) and HSP-60 to restore mitochondrial proteostasis26,27. We monitored UPRmt using hsp-6::GFP and hsp-60::GFP reporter worms with reduced mrp expression. Similar to the cco-1 mutant25, hsp-6 and hsp-60 were induced in worms with reduced mrp (Fig. 4a–c, Supplementary Fig. 5a–b). This was specific for UPRmt, as mrps-5 RNAi did not affect UPR in the ER (UPRER) and cytosolic heat shock response (HSR) (Supplementary Fig. 5c). As for lifespan, UPRmt was not induced when mrp expression was only inhibited during adulthood (Supplementary Fig. 5d). We then measured UPRmt upon combined mrps-5 and mev-1 inactivation. Whereas mev-1 RNAi alone did not induce UPRmt (Fig 4c–d), combined inactivation induced mitonuclear protein imbalance (Supplementary Fig. 5e) and synergistically induced UPRmt (Fig. 4d), accounting for the amplified lifespan. Double inactivation of mrps-5 and cco-1 did not further enhance UPRmt compared to mrps-5 alone (Fig. 4d), in line with the similar lifespan.
Figure 4. mrp genes confer longevity effects through UPRmt.

a, RNAi of mrp genes induced UPRmt (hsp-6::GFP reporter), similar to cco-1 knockdown. Worms are synchronized at day 1 of adulthood. b, Quantification of UPRmt upon knockdown of mrp or cco-1. (n=4) c, mrps-5 and cco-1, but not mev-1, RNAi, induce UPRmt as reflected by the induction of HSP-6-GFP protein. d, Combined RNAi of mrps-5 and mev-1 synergistically increased UPRmt, while combined cco-1/mrps-5 RNAi did not further increase UPRmt (n=6). e, Knockdown of different mrp genes results in different levels of UPRmt, which correlates with mean lifespan (n=33–61 worms for lifespan, n=3 for GFP). f–h, Epistasis with UPRmt regulator haf-1. Double RNAi of mrps-5 and haf-1 partially prevented lifespan extension (f), UPRmt (g, n=5), and reduction in respiration (h, n=10), compared to mrps-5 RNAi alone. i, In various tissues of mouse crosses, Ubl5, Abcb10 and Hspd1 expression correlated with Mrp expression. j, Hspd1 (HSP60) ties in a correlation network with Mrp and OXPHOS genes. Connecting lines indicate a Pearson correlation coefficient of 0.75–1.0. Bar graphs are mean±SEM, * p≤0.05; ** p≤0.01; *** p≤0.001. See also Supplementary Fig. 5–7 and Table 3.
There are individual differences in the degree of UPRmt within the mrps-5 RNAi worm population, which tightly correlate with lifespan extension (Supplementary Fig. 5f–h). Importantly, GFP expression stayed similar throughout life, demonstrating that this is not an artifact of transiently reduced food intake (Supplementary Fig. 5i). To further link the level of UPRmt to longevity, we measured UPRmt in worms treated with RNAi against mrp genes28. Reduced expression of each mrp gene activated UPRmt to a different degree (Supplementary Fig. 6a). The level of UPRmt again correlated significantly with lifespan extension (Fig. 4e, Supplementary Fig. 6a–b and Table 3).
Two downstream effectors of UPRmt are HAF1, a mitochondrial peptide transporter29, and UBL5, a ubiquitin-like protein that regulates the transcriptional activation of mitochondrial chaperones30. Knockdown of haf-1 in the face of mrps-5 RNAi reduced lifespan extension, UPRmt and increased respiration (Fig. 4f–h). Similarly, when both ubl-5 and mrps-5 were knocked down, lifespan extension, the respiration phenotype and UPRmt were partially lost, in line with double cco-1 and ubl-5 RNAi25 (Supplementary Fig. 7a–f).
This network could be traced back to mice, as Ubl5 and the most likely mouse haf-1 homolog—Abcb10—correlated tightly with Mrp genes, for instance in hippocampus of the BXDs31 and in adipose tissue of F2-intercrossed mice17 (Fig. 4i, Supplementary Fig. 7g). Additionally, Hspd1—encoding HSP60—correlated with multiple Mrp genes (Fig. 4i, data not shown). Gene ontology analysis showed strong connectivity between Ubl5 and OXPHOS genes (p=9e-4 in eye; p=8.62e-10 in hippocampus), the translation process or ribosome (p=6e-4 eye; p=6.03e-10 hippocampus), and the mitochondrial inner membrane (p=1e-4 eye; p=3.31e-27 hippocampus) in the BXDs. Finally, we tied Hspd1 in a close correlation network with various Mrp and OXPHOS genes (Fig. 4j).
Pharmacological mitonuclear protein imbalance
Many mitochondrial functions can be traced back to their endosymbiotic “bacterial” origin. Consequently, antibiotics that target bacterial translation also inhibit mitochondrial translation. We therefore used doxycycline to confirm the role of mitochondrial translation in longevity, while using carbenicillin—targeting the bacterial cell wall—as control. We used heat-killed OP50 or live HT115 bacteria—the latter insensitive to low concentrations of doxycycline (not shown)—to feed worms, to prevent antibiotic effects on bacteria. Doxycycline, given throughout life, dose-dependently extended lifespan, induced UPRmt, not UPRER, and reduced oxygen consumption, without affecting ATP levels or citrate synthase activity (Fig. 5a–e, Supplementary Fig. 8a–b). Doxycycline at 60μg/mL caused developmental delay, like mrps-5 RNAi, but no abnormalities were apparent at lower concentrations (not shown). A low concentration of doxycycline (6μg/mL), given only during development, also increased lifespan and UPRmt, and attenuated respiration (Fig. 5f–h). Chloramphenicol—belonging to a different class of antibiotics targeting translation—also increased lifespan and UPRmt, while decreasing respiration (Fig. 5i–k), when administered during development. Similar to mrps-5 RNAi, doxycycline increased the ratio of nDNA- (ATP5A) over mtDNA-encoded (MTCO1) OXPHOS proteins (Fig. 5l).
Figure 5. Specific antibiotics extend lifespan by phenocopying mrps-5 knockdown.

a, Effects on worm lifespan of doxycycline (30μg/mL), compared to carbenicilin (30μg/mL) or vehicle. p≤0.001 refers to statistical significance of doxycycline compared to either vehicle or the carbenicilin control. Antibiotics were given throughout life in panels a–e. b, The effects of doxycycline on lifespan are dose-dependent. c–e, Doxycycline induced UPRmt (c, n=5) and reduced respiration (d, n=10), without changing citrate synthase activity (e, n=3). f–k, When treated only during larval development, doxycycline (6μg/mL; f–h) and chloramphenicol (100μg/mL; i–k) extend lifespan (f, i), induced UPRmt (g, j, n=5) and reduced respiration (h, k, n=6). l, Doxycycline alters the ratio between nDNA- (ATP5A) and mtDNA-encoded (MTCO1) OXPHOS proteins in worms. m, Doxycycline decreased respiration in a cultured hepatocyte cell line (n=5), n, induced Hsp60 transcription, as measured using an Hsp60 promoter reporter (n=8), o, increased HSP60 protein expression and altered the ratio of nDNA- (UQCRC2) versus mtDNA- (MTCOI) encoded proteins (n=2). p, Doxycycline increased HSP60 protein and altered the ratio of nDNA- versus mtDNA-encoded proteins in primary murine hepatocytes. q, Doxycycline (50 mpkd) for 10 days in C57BL/6N mice decreased oxygen consumption (n=10). See also Supplementary Table 2.
Linking back to mammals, doxycycline decreased respiration in a mouse hepatocyte cell line (Fig. 5m). Doxycycline also induced UPRmt, as evidenced by induction of Hsp60 (Fig. 5n) and the UPRmt protease ClpP (Supplementary Fig. 8c), and increased HSP60 protein expression, in hepatocyte cell lines and primary murine hepatocytes (Fig. 5o–p). Doxycycline induced a striking mitonuclear protein imbalance in hepatocytes (Fig. 5o–p). Finally, feeding mice with doxycycline for 10d lowered oxygen consumption in vivo, indicative for attenuated mitochondrial function (Fig. 5q).
Similar effects on mitonuclear protein imbalance, UPRmt, respiration and lifespan, without affecting mitochondrial morphology, were also observed in worms exposed to low concentrations ethidium bromide, which inhibits mtDNA transcription specifically32 (Supplementary Fig. 8d–h). This suggests that mitonuclear protein imbalance is the common underlying mechanism that links basic mitochondrial function to lifespan regulation.
A conserved longevity mechanism
To define how intricately mitonuclear protein imbalance and UPRmt are involved in longevity, we analyzed its activation in worms exposed to rapamycin33,34. Rapamycin inhibits TOR signaling to alter nDNA translation, inducing mitonuclear protein imbalance35, and increases lifespan in various species, including mice33. Rapamycin also increased mean worm lifespan (by 16%)34 in a ubl-5-dependent manner, induced UPRmt, but not UPRER or HSR, and increased respiration (Fig. 6a–c, Supplementary Fig. 9a). This was associated with increased ATP levels, equal citrate synthase activity, and altered nDNA/mtDNA OXPHOS protein ratio (Fig. 6d–e). Additionally, rapamycin dose-dependently changed the balance between nDNA and mtDNA-encoded OXPHOS subunits in mouse hepatocytes (Fig. 6f–g). This mitonuclear protein imbalance induced HSP60 and ClpP (Fig. 6f–h). Similarly, the acclaimed lifespan enhancer resveratrol induced mitonuclear protein imbalance in hepatocytes (Fig. 6i) and ubl-5-dependently increased worm lifespan and UPRmt, but not UPRER or HSR, while increasing respiration and maintaining ATP levels and citrate synthase activity (Supplementary Fig. 9b–f). Mitonuclear protein imbalance and UPRmt hence represent an overarching mechanism of longevity that also can be engaged by pathways that signal mainly through the nucleus. Finally, we tested whether reactive oxygen species (ROS) and mitohormesis, which poses that an initial ROS burst (after 24h) induces adaptive long-term protection36, could explain our worm phenotypes. However, no ROS was produced after 24h mrps-5 RNAi or doxycycline, rapamycin or ethidium bromide (Supplementary Fig. 10a). In addition, the mitohormesis regulators daf-16 and aak-236,37 were not involved in UPRmt induction (Supplementary Fig. 10b) or lifespan extension (Fig. 3k, m) following mrps-5 RNAi. Finally, the ROS scavenger N-acetylcysteine (NAC) did not abrogate the mrps-5 RNAi- or rapamycin-induced UPRmt, nor did it suppress longevity (Supplementary Fig. 10c–f), similar to NAC-treated cco-1 RNAi worms25. Together, these data demonstrate that, even though mitohormesis is important for insulin/IGF1-dependent aging37, UPRmt-mediated longevity is independent of ROS.
Figure 6. Conserved function of mitonuclear protein imbalance and UPRmt in longevity.

a, Rapamycin (1nM) extends worm lifespan in a ubl-5-dependent manner, and b, ubl-5-dependently induced UPRmt (hsp-6::GFP) but not UPRER (hsp-4::GFP) (n=4). c–e, Rapamycin increased respiration (c, n=10) and ATP content but not citrate synthase activity (d, n=3) and induced mitonuclear protein imbalance (e). f–h, In mouse hepatocytes, rapamycin induces mitonuclear protein imbalance (f–g) and induces UPRmt as shown at the protein (f–g, n=3), and transcriptional (h, n=8) level. i, Resveratrol (25 μM) induced mitonuclear protein imbalance in mouse hepatocytes (n=4). j, Hypothetical scheme of the mechanism by which reduced Mrp expression (during aging or genetic inactivation), specific antibiotics, ethidium bromide and rapamycin and resveratrol extend lifespan by inducing UPRmt. Bar graphs are expressed as mean±SEM, * p≤0.05; *** p≤0.001. See also Supplementary Table 2.
Discussion
Using the BXD inbred wild type mouse panel, we identified a chromosome 2 QTL that is responsible for longevity, with Slc12a1, Mrps5 and Ttl showing strong correlation with lifespan. A holistic approach, involving bioinformatics, and genetic and pharmacological strategies in both worms and mammals, established that Mrps5 and the Mrp protein family are the main actors in metabolic lifespan regulation. The MRPs constitute the mitoribosome that regulates translation of 13 mtDNA-encoded proteins, underscoring the vital importance of mitochondrial protein production6,7.
Inhibiting mitochondrial translation reduced respiration and extended lifespan. There is an apparent dichotomy, however, as rapamycin (this study) as well as NAD+ boosters—resveratrol (this study), nicotinamide riboside, nicotinamide and PARP inhibitors (LM, RHH, JA, unpublished data)—couple longevity to increased respiration. Abnormal mitochondrial proteostasis could reconcile these disparate observations. Knockdown of mrps-5 or cco-1 affect proteostasis and activate UPRmt25,28. From the cco-1 study25 it was, however, unclear if there was a direct connection between the level of UPRmt and the lifespan extension. Our data unequivocally demonstrate a positive correlation between the level of UPRmt and lifespan. Moreover, UPRmt seems to result from an imbalance between mtDNA- and nDNA-encoded proteins and is a common feature linking mitochondrial longevity pathways. Genetic defects in mrp’s or respiratory chain genes, antibiotics that inhibit translation, or moderate mtDNA transcription inhibition, induce such a mitonuclear protein imbalance from within mitochondria. Conversely, resveratrol and rapamycin change the production of nDNA-encoded mitochondrial proteins and if this is not matched with the levels of mtDNA-encoded proteins, mitonuclear protein imbalance and UPRmt will also ensue, which favors longevity (Fig. 6j). The reason why mev-1 mutants do not display UPRmt is consistent with the fact that complex II is entirely nDNA-encoded and therefore does not require a balanced production of proteins from the nDNA and mtDNA. Additionally, complex II can be bypassed for mitochondrial ATP generation and is not part of OXPHOS supercomplexes38. Although further work to validate this hypothesis is warranted, this could explain apparent contradictions such as why mutations that decrease and increase respiration both can induce longevity.
In summary, our data implicate MRPs as a novel longevity protein family, conserved from worms to mammals. The identification of these genes was triggered by analysis of murine reference populations. Hence, it is natural variation in Mrp expression, not artificial loss- or gain-of-function, that translates to longevity. In worms, longevity involves enhanced fitness and UPRmt, and correlates tightly with levels of mrp knockdown. Our data suggest that stoichiometric imbalance between nDNA- and mtDNA-encoded OXPHOS proteins, or mitonuclear protein imbalance, is at the core of UPRmt activation, both in worms and mammals. The apparent conservation of mitonuclear protein imbalance and UPRmt as a general longevity mechanism should incite studies to explore whether targeting UPRmt can prevent aging-associated functional decline and treat diseases linked with aging.
Methods
Methods and any associated references are available in the online version of the paper at http://www.nature.com/nature/.
Supplementary Material
Acknowledgments
We thank Pierre Gönczy and the Caenorhabditis Genetics Center for sharing/providing reagents. RHH is supported by fellowships of NWO-Rubicon and AMC, and LM by an FRM fellowship. JA is the Nestlé Chair in Energy Metabolism and supported by EPFL, ERC (2008-AdG-23138), Velux Stiftung, and SNSF. RWW and GeneNetwork are supported by NIH (P20-DA 21131, UO1AA13499 and U01AA14425), and the UT Center for Integrative and Translational Genomics. RWW and JA are supported by NIH (R01AG043930).
Footnotes
Supplementary Information is linked to the online version of the paper at www.nature.com/nature
Author Contributions RHH, LM and JA conceived and designed the project. RHH and RWW performed QTL mapping and sequence analyses. RHH, LM, EK, DR, NM, GK performed experiments. RHH and JA wrote the manuscript, with contributions from all other authors.
Author Information Reprints and permissions information is available at www.nature.com/reprints. The authors declare no competing financial interests.
References
- 1.Fontana L, Partridge L, Longo VD. Extending healthy life span--from yeast to humans. Science. 2010;328:321–326. doi: 10.1126/science.1172539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Houtkooper RH, Williams RW, Auwerx J. Metabolic networks of longevity. Cell. 2010;142:9–14. doi: 10.1016/j.cell.2010.06.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kenyon CJ. The genetics of ageing. Nature. 2010;464:504–512. doi: 10.1038/nature08980. [DOI] [PubMed] [Google Scholar]
- 4.Mair W, Dillin A. Aging and survival: the genetics of life span extension by dietary restriction. Annu Rev Biochem. 2008;77:727–754. doi: 10.1146/annurev.biochem.77.061206.171059. [DOI] [PubMed] [Google Scholar]
- 5.Pagliarini DJ, et al. A mitochondrial protein compendium elucidates complex I disease biology. Cell. 2008;134:112–123. doi: 10.1016/j.cell.2008.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sharma MR, et al. Structure of the mammalian mitochondrial ribosome reveals an expanded functional role for its component proteins. Cell. 2003;115:97–108. doi: 10.1016/s0092-8674(03)00762-1. [DOI] [PubMed] [Google Scholar]
- 7.Anderson S, et al. Sequence and organization of the human mitochondrial genome. Nature. 1981;290:457–465. doi: 10.1038/290457a0. [DOI] [PubMed] [Google Scholar]
- 8.Liao CY, Rikke BA, Johnson TE, Diaz V, Nelson JF. Genetic variation in the murine lifespan response to dietary restriction: from life extension to life shortening. Aging Cell. 2010;9:92–95. doi: 10.1111/j.1474-9726.2009.00533.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Argmann CA, Chambon P, Auwerx J. Mouse phenogenomics: the fast track to “systems metabolism”. Cell Metab. 2005;2:349–360. doi: 10.1016/j.cmet.2005.11.002. [DOI] [PubMed] [Google Scholar]
- 10.Andreux PA, et al. Systems Genetics of Metabolism: The Use of the BXD Murine Reference Panel for Multiscalar Integration of Traits. Cell. 2012;150:1287–1299. doi: 10.1016/j.cell.2012.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Peirce JL, Lu L, Gu J, Silver LM, Williams RW. A new set of BXD recombinant inbred lines from advanced intercross populations in mice. BMC Genet. 2004;5:7. doi: 10.1186/1471-2156-5-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Keane TM, et al. Mouse genomic variation and its effect on phenotypes and gene regulation. Nature. 2011;477:289–294. doi: 10.1038/nature10413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.De Haan G, Van Zant G. Genetic analysis of hemopoietic cell cycling in mice suggests its involvement in organismal life span. FASEB J. 1999;13:707–713. doi: 10.1096/fasebj.13.6.707. [DOI] [PubMed] [Google Scholar]
- 14.Shifman S, et al. A high-resolution single nucleotide polymorphism genetic map of the mouse genome. PLoS Biol. 2006;4:e395. doi: 10.1371/journal.pbio.0040395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Edwards MG, et al. Gene expression profiling of aging reveals activation of a p53-mediated transcriptional program. BMC Genomics. 2007;8:80. doi: 10.1186/1471-2164-8-80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Geisert EE, et al. Gene expression in the mouse eye: an online resource for genetics using 103 strains of mice. Mol Vis. 2009;15:1730–1763. [PMC free article] [PubMed] [Google Scholar]
- 17.Chen Y, et al. Variations in DNA elucidate molecular networks that cause disease. Nature. 2008;452:429–435. doi: 10.1038/nature06757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lee SS, et al. A systematic RNAi screen identifies a critical role for mitochondria in C. elegans longevity. Nat Genet. 2003;33:40–48. doi: 10.1038/ng1056. [DOI] [PubMed] [Google Scholar]
- 19.Hamilton B, et al. A systematic RNAi screen for longevity genes in C. elegans. Genes Dev. 2005;19:1544–1555. doi: 10.1101/gad.1308205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hansen M, Hsu AL, Dillin A, Kenyon C. New genes tied to endocrine, metabolic, and dietary regulation of lifespan from a Caenorhabditis elegans genomic RNAi screen. PLoS Genet. 2005;1:119–128. doi: 10.1371/journal.pgen.0010017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wong A, Boutis P, Hekimi S. Mutations in the clk-1 gene of Caenorhabditis elegans affect developmental and behavioral timing. Genetics. 1995;139:1247–1259. doi: 10.1093/genetics/139.3.1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dillin A, et al. Rates of behavior and aging specified by mitochondrial function during development. Science. 2002;298:2398–2401. doi: 10.1126/science.1077780. [DOI] [PubMed] [Google Scholar]
- 23.Kenyon C, Chang J, Gensch E, Rudner A, Tabtiang R. A C. elegans mutant that lives twice as long as wild type. Nature. 1993;366:461–464. doi: 10.1038/366461a0. [DOI] [PubMed] [Google Scholar]
- 24.Lakowski B, Hekimi S. The genetics of caloric restriction in Caenorhabditis elegans. Proc Natl Acad Sci USA. 1998;95:13091–13096. doi: 10.1073/pnas.95.22.13091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Durieux J, Wolff S, Dillin A. The cell-non-autonomous nature of electron transport chain-mediated longevity. Cell. 2011;144:79–91. doi: 10.1016/j.cell.2010.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Haynes CM, Ron D. The mitochondrial UPR - protecting organelle protein homeostasis. J Cell Sci. 2010;123:3849–3855. doi: 10.1242/jcs.075119. [DOI] [PubMed] [Google Scholar]
- 27.Zhao Q, et al. A mitochondrial specific stress response in mammalian cells. EMBO J. 2002;21:4411–4419. doi: 10.1093/emboj/cdf445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yoneda T, et al. Compartment-specific perturbation of protein handling activates genes encoding mitochondrial chaperones. J Cell Sci. 2004;117:4055–4066. doi: 10.1242/jcs.01275. [DOI] [PubMed] [Google Scholar]
- 29.Haynes CM, Yang Y, Blais SP, Neubert TA, Ron D. The matrix peptide exporter HAF-1 signals a mitochondrial UPR by activating the transcription factor ZC376.7 in C. elegans. Mol Cell. 2010;37:529–540. doi: 10.1016/j.molcel.2010.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Benedetti C, Haynes CM, Yang Y, Harding HP, Ron D. Ubiquitin-like protein 5 positively regulates chaperone gene expression in the mitochondrial unfolded protein response. Genetics. 2006;174:229–239. doi: 10.1534/genetics.106.061580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Overall RW, et al. Genetics of the hippocampal transcriptome in mouse: a systematic survey and online neurogenomics resource. Front Neurosci. 2009;3:55. doi: 10.3389/neuro.15.003.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zylber E, Vesco C, Penman S. Selective inhibition of the synthesis of mitochondria-associated RNA by ethidium bromide. J Mol Biol. 1969;44:195–204. doi: 10.1016/0022-2836(69)90414-8. [DOI] [PubMed] [Google Scholar]
- 33.Harrison DE, et al. Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature. 2009;460:392–395. doi: 10.1038/nature08221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Robida-Stubbs S, et al. TOR Signaling and Rapamycin Influence Longevity by Regulating SKN-1/Nrf and DAF-16/FoxO. Cell Metab. 2012;15:713–724. doi: 10.1016/j.cmet.2012.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zid BM, et al. 4E-BP extends lifespan upon dietary restriction by enhancing mitochondrial activity in Drosophila. Cell. 2009;139:149–160. doi: 10.1016/j.cell.2009.07.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schulz TJ, et al. Glucose restriction extends Caenorhabditis elegans life span by inducing mitochondrial respiration and increasing oxidative stress. Cell Metab. 2007;6:280–293. doi: 10.1016/j.cmet.2007.08.011. [DOI] [PubMed] [Google Scholar]
- 37.Zarse K, et al. Impaired insulin/IGF1 signaling extends life span by promoting mitochondrial L-proline catabolism to induce a transient ROS signal. Cell Metab. 2012;15:451–465. doi: 10.1016/j.cmet.2012.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schagger H, Pfeiffer K. Supercomplexes in the respiratory chains of yeast and mammalian mitochondria. EMBO J. 2000;19:1777–1783. doi: 10.1093/emboj/19.8.1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.