

Background: The role of RRM1 in the pathogenesis of TDP-43 proteinopathy is unclear.
Results: RRM1 was aggregate-prone, mediated by a self-assembly at newly identified amyloidogenic β-strands containing cysteines; cysteine substitution(s) replicated diverse cytopathologies of TDP-43 in ALS.
Conclusion: RRM1 misfolding may underlie TDP-43 proteinopathy.
Significance: This study proposes a novel mechanism and a new in vitro model for TDP-43 proteinopathy.
Keywords: Amyotrophic Lateral Sclerosis (Lou Gehrig's Disease), Cell Biology, Protein Chemical Modification, Protein Misfolding, Structural Biology, TAR DNA-binding Protein 43 kDa
Abstract
Aggregation of TAR DNA-binding protein of 43 kDa (TDP-43) is a pathological signature of amyotrophic lateral sclerosis (ALS). Although accumulating evidence suggests the involvement of RNA recognition motifs (RRMs) in TDP-43 proteinopathy, it remains unclear how native TDP-43 is converted to pathogenic forms. To elucidate the role of homeostasis of RRM1 structure in ALS pathogenesis, conformations of RRM1 under high pressure were monitored by NMR. We first found that RRM1 was prone to aggregation and had three regions showing stable chemical shifts during misfolding. Moreover, mass spectrometric analysis of aggregated RRM1 revealed that one of the regions was located on protease-resistant β-strands containing two cysteines (Cys-173 and Cys-175), indicating that this region served as a core assembly interface in RRM1 aggregation. Although a fraction of RRM1 aggregates comprised disulfide-bonded oligomers, the substitution of cysteine(s) to serine(s) (C/S) resulted in unexpected acceleration of amyloid fibrils of RRM1 and disulfide-independent aggregate formation of full-length TDP-43. Notably, TDP-43 aggregates with RRM1-C/S required the C terminus, and replicated cytopathologies of ALS, including mislocalization, impaired RNA splicing, ubiquitination, phosphorylation, and motor neuron toxicity. Furthermore, RRM1-C/S accentuated inclusions of familial ALS-linked TDP-43 mutants in the C terminus. The relevance of RRM1-C/S-induced TDP-43 aggregates in ALS pathogenesis was verified by immunolabeling of inclusions of ALS patients and cultured cells overexpressing the RRM1-C/S TDP-43 with antibody targeting misfolding-relevant regions. Our results indicate that cysteines in RRM1 crucially govern the conformation of TDP-43, and aberrant self-assembly of RRM1 at amyloidogenic regions contributes to pathogenic conversion of TDP-43 in ALS.
Introduction
TAR DNA-binding protein of 43 kDa (TDP-43)3 is a DNA- and RNA-binding nuclear protein that has been identified as a core component of ubiquitinated inclusions in frontotemporal lobar degeneration (FTLD) and amyotrophic lateral sclerosis (ALS) (1, 2). Accumulating evidence suggests that TDP-43 proteinopathy mediates motor neuron degeneration in familial ALS caused by diverse genetic defects in ubiquilin 2, profilin 1, optineurin, valosin-containing protein, and C9ORF72 (reviewed in Ref. 3). Moreover, the presence of mutations in TDP-43 in a subpopulation of familial ALS indicates its primary role in ALS pathogenesis (4). Biochemical signatures of TDP-43 proteinopathy include cytosolic redistribution accompanied by loss of nuclear TDP-43 and the formation of 25- or 35-kDa C-terminal fragments, which are often phosphorylated or ubiquitinated (5, 6). However, the molecular basis of how native TDP-43 is converted to pathogenic species remains elusive.
TDP-43 consists of several functional domains, including a bipartite nuclear localizing signal (NLS; aa 82–98); two RNA-recognition motifs, RRM1 (aa 103–183) and RRM2 (aa 190–260); and a glycine-rich C-terminal region (7, 8). Genetic mutations of TDP-43 in familial ALS patients are concentrated in the C-terminal region, suggesting that structural alteration of this region is closely linked to ALS pathology (4). In agreement with this theory, analysis of recombinant TDP-43 protein from Escherichia coli revealed that the C-terminal tail is extremely insoluble in Sarkosyl buffer (9). Recent work shows that residues 321–366 in the C-terminal region are responsible for TDP-43 aggregate formation (10). However, despite a large body of in vitro evidence regarding the role of C-terminal fragments in TDP-43 aggregation, the existing in vivo evidence is insufficient to support an initial contribution of these fragments in the pathogenesis of ALS or FTLD. Full-length TDP-43, as well as the C-terminal fragment, is reportedly phosphorylated or ubiquitinated in the affected regions in ALS and FTLD (1, 2, 11); hence, a more intensive analysis of other domains is required to elucidate the chronological structural changes of full-length TDP-43 proteins in ALS and FTLD.
The role of the two TDP-43 RRMs, particularly in protein folding, is unclear; however, both RRMs contribute to both cytosolic aggregate formation and phenotypic deterioration, including growth defects in yeast, neurite outgrowth inhibition, and motor disturbance in Caenorhabditis elegans (12). We previously reported that Asp-246 and Glu-247 in the RRM2 domain play important roles to preserve the function and conformation of TDP-43 (13). A recent study showed that stress granule formation is linked to cytoplasmic TDP-43 inclusions, in which RRM1 interacts with RNA (14). On the other hand, it is reported that TDP-43 aggregates under extremely toxic conditions are distinct from stress granules, with which TDP-43 associates under non-lethal stresses (15). Considering that the predominant role of RRM1 is RNA processing (16), structural damage to this domain may cause serious defects in neuronal development and neurological diseases (17–19). However, there are few investigations of the relationship between RRM1 conformation and TDP-43 proteinopathy. Recent advances in structural biology indicate that conformational fluctuations between the basic folded and disordered states are important for protein misfolding and the formation of amyloid fibrils (20). Nuclear magnetic resonance (NMR) spectroscopy is a useful tool to characterize conformational changes of proteins at the atomic level; stresses such as pressure and temperature help to elucidate the intermediate structure of unfolded or misfolded species, as described previously for prion disease (21, 22). A recent NMR study documented that N-terminal fragments of TDP-43 form oligomers in solution via self-assembly (23).
In the present study, we first demonstrated that RRM1 readily acquires amyloidogenicity under physical stresses, in which three misfolding-relevant regions are involved, using a combination of NMR and mass spectrometry. More specifically, analyses using biochemical, cell biological, and immunohistochemical investigations showed that two cysteine residues located in one of the core regions play crucial roles to maintain the conformation and function of RRM1. Finally, RRM1 self-assembly at this core may contribute to ALS-linked pathogenic conversion of full-length TDP-43.
EXPERIMENTAL PROCEDURES
Plasmid Construction and Protein Purification
cDNA for RRM1 (aa 103–108), N-terminal RRM1 (aa 1–183), and N-terminal RRM2 (aa 1–265) of human TDP-43 was cloned by PCR using a previously reported construct (pcDNA3-TDP-43-FLAG) as a template (24), using the following primers: RRM1, 5′-GGGATCCCCGGAATTCACATCCGATTTAATAGTGT-3′ and 5′-GTCGACCCGGGAATTCTTAGCTTTGCTTAGAATTAGGA-3′; RRM2, 5′-GGGATCCCCGGAATTCAGCAGAAAAGTGTTTGTGG-3′ and 5′-GTCGACCCGGGAATTCTTAATTGTGCTTAGGTTCGGCA-3′; N-terminal RRM1, 5′-CGCGGGCCCGGGATCCATGTCTGAATATATTCGG-3′ and 5′-GTCGACCCGGGAATTCTTAGCTTTGCTTAGAATTAGGA-3′; N-terminal RRM2, 5′-CGCGGGCCCGGGATCCATGTCTGAATATATTCGG-3′ and 5′-GTCGACCCGGGAATTCTTAATTGTGCTTAGGTTCGGCA-3′. Substitution mutants for cysteine with serine (C/S) or alanine (C/A), sporadic or familial ALS-linked mutant (D169G, A315T, and Q331K), and NLS mutation (mNLS) were generated using site-directed mutagenesis as described previously (24). The sequences of the mutagenized oligonucleotides were as follows: C173S, 5′-TGATAGATGGACGATGGAGTGACTGCAAACTTCCT-3′; C175S, 5′-GATGGACGATGGTGTGACAGCAAACTTCCTAATTCTA-3′; C173S/C175S (DCS), 5′-ATATGATAGATGGACGATGGAGTGACAGCAAACTTCCTAATTCTAAG-3′; C173A, 5′-CATATGATAGATGGACGATGGGCTGACTGCAAACTTCCTAATTC-3′; C173A/C175A, 5′-CGACATATGATAGATGGACGATGGGCTGACGCCAAACTTCCTAATTCTAAGCAAAG-3′; C198S, 5′-GTGTTTGTGGGGCGCAGTACAGAGGACATGA-3′; C244S, 5′-TGATCAGATTGCGCAGTCTCTTAGTGGAGAGGACT-3′; D169G, 5′-ACAGCGACATATGATAGGTGGACGATGGTGTGAC-3′; A315T, 5′-GAACTTTGGTACGTTCAGCATTAATCCAGC-3′; G331K, 5′-TGGCTGCCGCCAAGGCAGCACTACAGAGCA-3′. Deletion mutants of human TDP-43 for RRM1 (ΔRRM1), or glycine-rich C terminus (aa 266–414) were generated by PCR by designing primers to eliminate the deletion site as follows: ΔRRM1, 5′-CAAGATGAGCCTTTGAGAA-3′ and 5′-TTTCTGGACTGCTCTTTTC-3′; ΔC-term, 5′-GGATCCATCGCCACCATGG-3′ and 5′-ATTGTGCTTAGGTTCGGCA-3′. These cDNAs were subcloned into pcDNA3 (Invitrogen) or pEGFP-N2 (Clontech, Palo Alto, CA) for culture cell experiments at BamHI/XhoI, XhoI/BamHI, or pGEX-6p-1 to generate recombinant proteins at BamHI/XhoI sites, respectively (24). To generate expression vectors for WT or RRM1-C/S TDP-43 fused with mCherry and EGFP at the N and C terminus, respectively, PCR products of TDP-43 were subcloned into mCherry-C1 vector (Clontech), using pcDNA3-TDP-43-FLAG of WT or various RRM1-C/S mutants as templates. Primer pairs used were 5′-GGGCTCGAGCTATGTCTGAATATATTCGGGTAACC-3′ and 5′-CCCGGATCCACATTCCCCAGCCAGAAGAC-3′. Then the NheI-EcoRI fragment from mCherry-TDP-43 encoding mCherry-fused TDP-43 was further subcloned into the same sites of pEGFP-N2 vectors. Recombinant proteins were generated and purified in E. coli as described previously (24). Renilla luciferase reporter plasmid pGL4.74 was purchased from Promega (Madison, WI).
Antibodies
A rabbit polyclonal anti-TDP-43 antibody (Sigma) was used for Western blot analysis of the RRM1 domain (1:1000 dilution). Another rabbit polyclonal anti-TDP-43 antibody (Proteintech, Chicago, IL) was used for immunofluorescence (1:1000). Mouse monoclonal anti-FLAG (M2) (Sigma) was used at a 1:500 dilution for immunofluorescence and 1:1000 for Western blotting. Rabbit polyclonal anti-phospho-TDP-43 antibody (Wako, Tokyo, Japan) was used at 1:500 for immunofluorescence and 1:1000 for Western blotting; rabbit monoclonal anti-Lys-48-specific ubiquitin (Millipore, Temecula, CA) was used at 1:500 for immunofluorescence. Methods for generating the antisera against misfolding-relevant cores of RRM1 domain are described below, separately.
NMR Measurements
Protein samples were prepared in 20 mm Tris-HCl buffer at 1.1 mm and pH 7.0. 1H NMR and 15N/1H HSQC measurements were collected every 500 bars from 30 to 2000 bars at 25 °C. Analyses were performed using a DRX600 spectrometer (Bruker BiosSpin Co., Fällanden, Switzerland) combined with the on-line high pressure NMR cell, as described previously (25). The three-dimensional structure of RRM1 in solution was determined by conventional double and triple resonance NMR techniques (Protein Data Bank code 2CQG). 1H chemical shifts were directly referenced to the methyl resonance of 4,4-dimethyl-4-silapentane-1-sulfonic acid, whereas 15N chemical shifts were indirectly referenced to the 1H chemical shift of 4,4-dimethyl-4-silapentane-1-sulfonic acid.
Biochemical Analysis of Recombinant Proteins and Culture Cell Lysates
Recombinant proteins were agitated in a multivortex mixer at 800 rpm for 16 h at 22 °C or heated for 10 min at 70 °C, followed by postincubation at 4 °C for the indicated time. In several experiments, the mixtures were separated into PBS-soluble and -insoluble fractions after centrifugation for 5 min at 17,000 × g at 4 °C. For SDS-PAGE analyses of denatured proteins, samples were denatured with LDS sampling buffer (Invitrogen) with or without 100 mm dithiothreitol (DTT) at 70 °C for 20 min. Perfuoro-octanoic acid (PFO, Fluorochem Ltd., Derbyshire, UK) is a non-dissociative detergent that preserves the protein-protein interactions of both cytosolic and membranous proteins (26). Protein samples were mixed with the same amount of PFO sampling buffer (100 mm Tris base, 2% NaPFO, 20% glycerol, 0.005% bromphenol blue, pH 8.0), incubated for 1 h at 22 °C, and separated on a 4–12% gradient polyacrylamide gel (Novex, BisTris gel, Invitrogen) with prechilled running buffer (0.5% NaPFO, 25 mm Tris-HCl, 192 mm glycine, pH 8.5) at 140 V. After SDS-PAGE or PFO-PAGE, gels were analyzed by Coomassie Brilliant Blue staining or by Western blotting. For Western blotting, samples were denatured in 2% SDS-sample buffer containing protease inhibitor mixture (Roche Applied Science) and/or phosphatase inhibitor mixture (Nacalai Tesque, Kyoto, Japan) with or without DTT. The samples were separated by SDS-PAGE and then transferred to polyvinylidene difluoride (PVDF) membranes. Antibody detection was performed using enhanced chemiluminescence (Nacalai Tesque).
Molecular Size Filtration Chromatography
The molecular size of the PBS-soluble RRM1 domain with or without agitation was estimated by size exclusion chromatography. Before and after agitation, RRM1 samples were incubated for 7 days at 4 °C, centrifuged for 20 min at 15,000 × g and 4 °C, and then subjected to molecular size filtration HPLC (AKTA Explorer 10S, GE Healthcare) at a flow rate of 0.5 ml/min on a Superdex 75 10/300 column (GE Healthcare) equilibrated with PBS. In the experiment for the interaction of RRM1 and RNA, soluble RRM1 proteins (2 mg/ml) in PBS with or without (TG)12 oligonucleotides were applied to a Superdex 75 10/300 column equilibrated with PBS. Recombinant proteins and (TG)12 oligonucleotides were detected by absorbance with 215 and 254 nm, respectively. Calibration of the columns for molecular weight was performed using ovalbumin (43 kDa; GE Healthcare), copper zinc superoxide dismutase (32 kDa; Sigma), myoglobin (17.6 kDa; Sigma), and aprotinin (6.5 kDa; Sigma) as protein standards.
Atomic Force Microscope Imaging
Samples for atomic force microscope imaging were placed on a muscovite mica surface for 1 min, rinsed with Milli-Q water, and then dried at room temperature for at least 12 h. Imaging was performed with a Nanoscope 3 microscope (Veeco, Plainview, NY) for tapping mode imaging in air. Images were collected with amplitude and height modes. Images of the protein aggregates were analyzed using Nanoscope software (Veeco).
Thioflavin T Assay
Amyloid formation by agitated or heat-denatured RRM1 was estimated using the thioflavin T assay. After postincubation at 4 °C for the indicated times, the protein solution (0.1 mm in PBS) was reacted with an equimolar amount of BTA-1 (Sigma), a derivative of thioflavin T. Fluorescence was measured at 440 nm (excitation) and 480 nm (emission) using a multiplate reader (Tecan, Männedorf, Switzerland).
LC-MS/MS Analysis
LC-MS/MS analysis of Pronase-digested RRM1 protein aggregates was performed as described previously (9), with minor modifications. RRM1 protein aggregates (25 μg) were resuspended in 100 μl of digestion buffer (50 mm Tris, 100 mm NaCl, 5 mm CaCl2, pH 8.0), mixed with 5 μg of Pronase (Millipore), and incubated for 1 h at 37 °C. The proteins in the reaction mixture were precipitated by ultracentrifugation for 30 min at 100,000 × g and 4 °C. The pellet was recovered in 20 μl of buffer (50 mm Tris, 500 mm NaCl, 6 m guanidine HCl, 5 mm EDTA, 5 mm DTT, pH 8.0) and desalted using a NuTip C-18 cartridge (Glygen Co., Columbia, MD) according to the manufacturer's protocol. The buffer was replaced with 2% acetonitrile and 0.1% trifluoroacetate (Nacalai Tesque) in distilled water. Digested fragments were analyzed by LC-MS/MS (Paradigm MS4, AMR, Kyoto, Japan; Finigan LCQ Advantage MAX, Thermo Electron, Waltham, MA).
Thiol Detection in the RRM1 Protein by Ellman's Test
RRM1 protein (1 mg/ml) was incubated with 0.67 mm 5,5′-dithiobis(2-nitrobenzoic) acid (Dojindo, Kumamoto, Japan) for 1 h at room temperature. The reaction was measured by photometry at 412 nm.
Cell Culture, Transfection
All culture cells were at 37 °C with 5% CO2 and 100% humidity. HEK 293A cells (Invitrogen) were maintained in Dulbecco's modified Eagle's medium containing 10% fetal bovine serum and penicillin/streptomycin, whereas the human neuroblastoma cell line SHSY-5Y (24) was cultured in DMEM/F-12-Ham medium containing 15% fetal bovine serum, non-essential amino acids (Invitrogen), and penicillin/streptomycin. Murine motor neuron cell line NSC34 cells (Cellutions Biosystems, Vancouver, Canada) were maintained in DMEM containing 10% FBS for growth. For differentiation, medium was changed to differentiation medium comprising 3% FBS in DMEM/F-12-Ham with 1% non-essential amino acids (27). A FuGene HD transfection kit (Roche Applied Science) was used for the plasmid transfection, according to the manufacturer's protocol. 48 h after transfection, cells were processed for various analyses.
Immunocytochemistry and Confocal Microscopy
After fixation with 4% PFA, cells were incubated in PBS containing 5% goat serum and 0.2% Triton X-100 for 1 h at room temperature. Cells were reacted with various primary antibodies in PBS containing 0.1% Triton X-100 for 1 h at room temperature and then with the fluorescent secondary antibody (Alexa 488, Invitrogen; CF 568, Biotium, Hayward, CA) in the same buffer. After washing three times in PBS, nuclei were stained by incubating cells in PBS containing 2 μg/ml DAPI. Stained cells were analyzed by confocal laser microscopy (Nikon, C1 si, Tokyo, Japan). For the cell counting experiments, seven or eight photographs (20–50 GFP-positive cells/image) were randomly taken. Cell counting for total fluorescent cells and inclusion-positive cells was performed in a blind fashion using ImageJ software. For quantification of the aggregate-specific reactivity of antibodies raised against misfolding-relevant regions in comparison with that of a commercially available anti-TDP-43 antibody (Proteintech), fluorescence of each channel and the area of stained cells were obtained as pixel values using the operating EZ-C1 software (Nikon). Fluorescence per area was calculated, and the red/green ratio of each cell was obtained. 11–34 spots from 4–9 photos/antibody were analyzed to be averaged and were standardized to the value for C173S/C175S (DCS) staining.
Exon 9 Skipping Assay
RNA splicing of TDP-43 was investigated using an exon 9 skipping assay with a TG13T5 minigene reporter plasmid as described previously with some minor modifications (16, 28). The TDP-43-EGFP (0.5 μg/well) and pTG12 (3 μg/well; a generous gift from Dr. E. Buratti and Dr. F. E. Baralle, International Centre for Genetic Engineering and Biotechnology) plasmids were simultaneously introduced into HEK293A cells in 6-well dishes using the FuGeneHD reagent (Roche Applied Science). After harvesting the mRNA using the Purelink RNA purification kit (Invitrogen), reverse transcription was performed using 2 μg of total RNA, primer mixtures of random hexamer and oligo(dT), and a RiverTra Ace qPCR RT kit (Toyobo, Osaka, Japan). Exon 9 skipping was estimated according to the density of the PCR products of the unspliced or spliced species. The primer sequences were 5′-TAGGATCCGGTCACCAGGAAGTTGGTTAAATCA-3′ and 5′-CAACTTCAAGCTCCTAAGCCACTGC-3′.
Cell Toxicity Assay
We estimated the viability and toxicity of the transfected cells, using a commercially available double fluorescent assay, which can estimate live cells and dead cells simultaneously by measuring the activity of proteases in the cytosol of live cells and in the extracellular space released from dead cells (MultiTox-Fluor multiplex cytotoxicity assay, Promega). NSC34 cells were plated onto a 96-well black culture plate at a density of 1 × 104 cells/well a day before the transfection. Plasmids for FLAG-tagged TDP-43 of WT or various mutants were introduced at 0.2 μg/well using the FuGene HD transfection kit, and the media were changed to differentiation medium on the next day. 48 h after the transfection, cell-permeant GF-AFC and cell-impermeant bis-AAF-R110, fluorescent indicators for live cells and dead cells, respectively, were applied. After a 2-h incubation, relative fluorescence units were obtained in the multiplate reader (Infinite 200, Tecan) with excitation/emission of 400/505 nm for live cells and 485/520 nm for dead cells, respectively. The toxicity ratio was obtained by the division of bis-AAF-R110 by GF-AFC fluorescence, which enables us to estimate the cell viability regardless of the well-to-well difference in the transfection efficiency or cell density. In separate experiments, 0.02 μg/well of Renilla luciferase reporter plasmid was co-transfected with TDP-43-FLAG, to normalize bis-AAF-R110 fluorescence to luciferase activity, for the standardization of the transfection efficiency using the Renilla luciferase assay system (Promega). The cell toxicity ratio was expressed as -fold increase compared with the vector control.
Generation of Antibodies Targeting Aggregation-linked RRM1
Rabbits were immunized with keyhole limpet hemocyanin-conjugated peptides, identified as aggregation-relevant sequences by LC-MS/MS and high pressure NMR analysis (CVLGLPWKTT, CQVKKDLKTG, and SQRHMIDGRWC; designated as RRM1-a, RRM1-b, and RRM1-c, respectively). Antisera obtained after immunization with RRM1-a and RRM1-c peptides showed specific and high reactivity against recombinant RRM1 and full-length TDP-43 protein by Western blotting and ELISA. IgG purified using protein A was used for Western blotting (1:1000) and ELISA as well as for immunofluorescence and immunohistochemistry (1:500).
ELISA
Target proteins in coating buffer (Roche Applied Science) were coated onto an ELISA plate (Nunc, Rochester, NY) at a concentration of 1 g/ml. Coated proteins were then reacted with primary antibodies and subsequently with peroxidase-conjugated secondary antibody (Jackson Immunoresearch). Finally, the reaction buffer containing 2,2′-azino-bis(3-ethylbenzothiazoline-6-sulfonate) (Roche Applied Science) was applied, and the antibody titer was measured by a spectrometer at 405 nm with a reference of 490 nm.
Immunohistochemistry of Specimens from ALS Patients
Experimental protocols were approved by and performed under the guidelines of our institutional ethics committee. Informed consent was obtained from all individuals or their guardians before the analysis. Post-mortem lumbar spinal cords from three patients with a definite diagnosis of sporadic ALS, from three age-matched neurological disease controls other than ALS, and from three age-matched neurologically normal subjects were analyzed. At autopsy, spinal cords were removed, and tissue blocks from the lumbar level were immediately placed in 10% buffered formalin and embedded in paraffin. Sections (6 μm) were treated by autoclaving (10 min at 121 °C in 10 mm sodium citrate buffer) for antigen retrieval and then incubated overnight with primary antibodies against RRM1-a or RRM1-c in PBS containing 3% BSA at 4 °C. The antibody reaction was visualized using a Vectastain Elite ABC kit (Vector Laboratories, Burlingame, CA), with 3,3′-diaminobenzidine tetrahydrochloride as the chromogen. The staining specificity was assessed by replacing the primary antibodies with the appropriate amounts of PBS containing 3% BSA. No reaction product deposits were detected.
Statistics
Multiple comparisons were analyzed by one-way ANOVA of Bonferroni's or Dunnett's multiple-comparison test using Prism software (GraphPad, La Jolla, CA).
RESULTS
Identification of Core Regions Involved in RRM1 Misfolding
Based on a previous report that RRM1 is a predominant domain for RNA processing (16), we hypothesized that structural alteration of RRM1 underlies TDP-43 proteinopathy. To test our hypothesis, a site-specific conformational fluctuation of the RRM1 domain was investigated using high pressure NMR spectroscopy; pressure perturbation was used to increase the population of partially disordered conformations, which have a potential risk of molecular association (29). Superposition of the 15N/1H HSQC spectra of the domain at 30 bars, before and after pressure treatment at 2000 bars, which usually induces transient conformational change in many proteins, revealed stable changes in the chemical shifts (Fig. 1A), particularly at residues in the aa 113–122, 133–147, and 166–173 regions (Fig. 1, B and C; designated as a, b, and c, respectively). Extended exposure to high pressure for 2 weeks resulted in marked precipitation of RRM1 protein. SDS-PAGE analysis revealed that the precipitates comprised monomers and DTT-sensitive dimers, indicating that an intermolecular disulfide bond was formed by high pressure stress (Fig. 1D). Because chemical shifts are sensitive to the nuclear microenvironment, the stable changes in 1H and 15N chemical shifts may indicate irreversible changes in the RRM1 structure or in the molecular association between the a, b, and c regions.
FIGURE 1.

Pressure-induced structural changes in RRM1. A, overlaid 15N/1H HSQC spectra for the stable isotope-labeled RRM1 domain at 30 bars and 25 °C before (blue) and after (red) pressure treatment. Residues showing large chemical shift changes are indicated by the residue number. B, deviations of 1H chemical shifts in RRM1. ΔδNH was calculated by the equation, ((ΔδH)2 + (ΔδN/5)2)0.5. The α-helix and β-sheet are indicated at the top by lines and arrows, respectively. C, three-dimensional structure of RRM1 in solution as determined by NMR. Residues showing large chemical shifts are indicated by arrowheads; colors indicate the degree of shift (0.02 < Δδ < 0.03 ppm (orange) and 0.03 < Δδ ppm (red)). Three clusters of amino acid residues showing irreversible chemical shifts on NMR are indicated by arrowheads: core-a (a, black), core-b (b, gray), and core-c (c, white). D, SDS-polyacrylamide gel showing the SDS-resistant dimer of the a-, b-, and c-domains following exposure to 2000-bar pressure. Bands (kDa) corresponding to the monomer and dimer proteins are indicated by arrows.
RRM1 Is Prone to Aggregation during Physical Stress and Serves as Seeds
We also examined an effect of transient agitation stress on the aggregate formation of RRM1. Non-reducing SDS-PAGE analysis of aggregated RRM1 displayed increasing amounts of SDS-resistant dimer along with postincubation time, a trend that was clearly expressed as the dimer/monomer ratio of the total protein (Fig. 2A). This result indicates that early RRM1 precipitates served as seeds for development of the aggregation. Molecular size filtration chromatography of protein samples incubated in PBS for 7 days at 4 °C after agitation showed a single peak representing the monomer. RRM1 formed a precipitate that mounted to 95% of total protein after agitation, indicating that RRM1 is aggregation-prone and exists as a monomer in solution in the absence of DNA/RNA and that the oligomeric species is exclusively hydrophobic (Fig. 2B). PFO-PAGE, which preserves protein-protein interactions, displayed oligomers and aggregates of RRM1 by heat or agitation stresses (Fig. 2C). The results obtained from analyses using pressure, agitation, and heat stresses indicate that these perturbations may cause the common aberrant assembly of RRM1 during the aggregation process. Interestingly, the protein samples after agitation and heat stresses showed a small increase in Thioflavin-T fluorescence (Fig. 2D), suggesting a formation of amyloid fibrils during the postincubation. Indeed, atomic force microscope analysis displayed a time-dependent formation of short fibrils in the samples (Fig. 2E). To study whether RRM2 has features similar to those of RRM1, we analyzed truncated TDP-43 proteins containing RRM1, RRM2, the N-terminal fragment spanning RRM1 (aa 1–183), or both RRMs (aa 1–265). Non-reducing SDS-PAGE revealed that all RRM1-containing fragments precipitated with the aberrant dimer in the pellet fractions (Fig. 2F, b, lanes 1, 2, and 5–8), whereas the RRM2 fragment remained as a hydrophilic monomer upon agitation (Fig. 2F, b, lanes 3 and 4). RRM2 is also resistant to aggregation by physical stresses (13). It should be noted that the N terminus did not alter the aggregation propensity of RRM1-containing proteins. To investigate the effect of DNA/RNA interaction on the RRM1 misfolding, we first confirmed the function of our recombinant RRM1 proteins. The gel filtration assay revealed that RRM1 interacts with TG-repeated oligonucleotide (TG)12 and remained as a monomer (Fig. 3A). Moreover, the aggregation propensity estimated by the dimer/monomer ratio was not altered by the simultaneous incubation with (TG)12 nucleotides during agitation (Fig. 3B).
FIGURE 2.

Native RRM1 is a soluble monomer that is prone to aggregation. A, seeding effect of RRM1 aggregates assessed with disulfide-mediated dimers. The RRM1 proteins were agitated for 16 h or non-treated (ctrl), followed by postincubation at 4 °C for 1, 5, 7, or 14 days, and total protein mixture was separated by non-reducing SDS-PAGE. a, Coomassie Brilliant Blue staining; b, a densitometry for the ratio of the dimer to the monomer. B, gel filtration analysis showing the absorbance at 215 nm of RRM1 with (closed circles) or without (open circles) agitation. Agitation markedly reduces the amount of monomeric RRM1 in solution with no emergence of oligomers. Molecular size markers are ovalbumin (43 kDa), copper zinc superoxide dismutase (32 kDa), myoglobin (17.6 kDa), and aprotinin (6.5 kDa). C, Coomassie Brilliant Blue staining of a PFO-polyacrylamide gel (see “Experimental Procedures”) for the RRM1 domain after transient agitation or the application of heat stress. Single, double, and triple arrowheads indicate the monomer, dimer, and trimer sizes of RRM1, respectively. Asterisks indicate the high molecular weight species of RRM1. D, thioflavin T (ThT) fluorescence assay showing time-dependent amyloid formation of agitated RRM1. Recombinant WT RRM1 was incubated with BTA-1 at either 24 h or 7 days after 16-h agitation or static incubation at room temperature (RT) as a control. *, p < 0.05 versus control sample by one-way ANOVA of Bonferroni's test. Each value is expressed as ThT ratio to room temperature treatment with a 24-h postincubation (n = 3; ±S.E. (error bars)). E, atomic force microscope analysis of RRM1 aggregation after a 16-h agitation. Images show proteins on days 0 (a), 5 (b), and 7 (c and d). Large globular particles and fibrillar aggregates, indicated by arrowheads were present after agitation. F, a, schematic illustration of human TDP-43 with domain information. b, non-reducing SDS-PAGE analysis of RRM1, RRM2, and N-terminal fragments spanning RRM1 (aa 1–183) and RRM2 (aa 1–265), with or without overnight agitation. Protein solutions were separated into supernatant and pellet fractions after a 7-day incubation at 4 °C after the agitation. *, dimer forms of each protein (RRM1, aa 1–183 fragment, and aa 1–265 fragment).
FIGURE 3.

The interaction of RRM1 with TG-repeated oligonucleotides did not influence the aggregate formation of RRM1. A, size exclusion chromatography showing the interaction between RRM1 and oligonucleotides comprising 12 repeats of thymine-guanine, (TG)12. The figure shows that peak flows for the mixture of RRM1 and (TG)12 merged into a single fraction, corresponding to the combined molecular weight of both, whereas individual applications led to different and smaller fractions. B, SDS-polyacrylamide gels showing recombinant RRM1 proteins (40 μm) incubated with (TG)12, adenine-cytosine (AC)12 (40 μm), or PBS and subjected to 16-h agitation at room temperature. Samples with (+) or without (−) DTT were separated by SDS-PAGE and stained using Coomassie Brilliant Blue. No alteration of dimer formation was observed in the presence of (TG)12. Single and double asterisks indicate RRM1 monomer and dimer, respectively.
RRM-1 Displays Amyloidogenicity following Assembly of Specific β-Strands
To further investigate the role of the three RRM1 core regions identified by NMR, mass spectrometric analysis of agitation-induced RRM1 aggregates digested with the nonspecific protease, Pronase, was performed. Abundant undigested peptides were identified (Table 1), indicating that the soluble RRM1 domain was transformed to a protease-inaccessible conformer during physical stress. Interestingly, three clusters of protease-resistant fragments matched those identified by NMR (core-a, core-b, and core-c; Fig. 4A). In particular, core-c, located in the fourth and fifth β-strands, was the most protease-inaccessible (Figs. 1C and 4A and Table 1).
TABLE 1.
LC-MS/MS results showing the Pronase-resistant domain sequences of aggregated TDP-43
Shown are LC-MS/MS profiles with monoisotopic m/z values of the agitation-induced aggregates of recombinant RRM1 proteins treated with pronase. MH+, protonized molecules.
| m/z submitted | MH+ matched | Intensity | Δ ppm | Mass | Start | End | Sequence |
|---|---|---|---|---|---|---|---|
| ppm | % | ||||||
| 684.41 | 684.4079 | 100 | 3.03 | 7.28 | 6 | 11 | VLGLPW |
| 600.35 | 600.3504 | 100 | −0.681 | 6.39 | 8 | 12 | GLPWK |
| 1287.66 | 1287.658 | 100 | 1.6 | 13.70 | 8 | 18 | GLPWKTTEQDL |
| 962.52 | 962.5153 | 100 | 4.88 | 10.24 | 12 | 19 | KTTEQDLK |
| 733.36 | 733.3727 | 100 | −17.3 | 7.80 | 14 | 19 | TEQDLK |
| 503.32 | 503.3188 | 100 | 2.44 | 5.36 | 35 | 38 | KDLK |
| 657.37 | 657.3678 | 100 | 3.27 | 6.99 | 38 | 43 | KTGHSK |
| 529.27 | 529.2729 | 100 | −5.45 | 5.63 | 39 | 43 | TGHSK |
| 733.36 | 733.3628 | 100 | −3.77 | 7.80 | 39 | 45 | TGHSKGF |
| 937.45 | 937.4526 | 100 | −2.82 | 9.97 | 39 | 47 | TGHSKGFGF |
| 639.3 | 639.2984 | 100 | 2.45 | 6.80 | 53 | 57 | YETQV |
| 1001.5 | 1001.502 | 100 | −1.91 | 10.66 | 59 | 66 | VMSQRHMI |
| 902.43 | 902.4335 | 100 | −3.88 | 9.60 | 60 | 66 | MSQRHMI |
| 914.43 | 914.4301 | 100 | −0.145 | 9.73 | 64 | 70 | HMIDGRW |
| 654.24 | 654.2374 | 100 | 3.93 | 6.96 | 70 | 74 | WCDCK |
| 1193.54 | 1193.544 | 100 | −3.5 | 12.70 | 70 | 79 | WCDCKLPNSK |
| 1408.63 | 1408.635 | 100 | −3.39 | 14.99 | 70 | 81 | WCDCKLPNSKQS |
| 1007.46 | 1007.465 | 100 | −4.83 | 10.72 | 71 | 79 | CDCKLPNSK |
| 773.42 | 773.4152 | 100 | 6.21 | 8.23 | 75 | 81 | LPNSKQS |
FIGURE 4.

Amyloid formation by RRM1 via specific β-sheet assembly by cysteine substitutions. A, schematic illustration of RRM1 residues; protease-resistant fragments and secondary structures are indicated by solid bars and gray schematics, respectively. Pronase-resistant fragments of aggregated RRM1 were analyzed by LC-MS/MS; regions with monoisotopic m/z > 800 are shown. The amino acid residues indicated by asterisks showed irreversible changes in NMR chemical shift after 2000-bar pressure treatment, which were designated as misfolding-relevant core-a, -b, and -c. B, a, Western blot analysis of WT or single RRM1-C/S mutants (C173S or C175S) under reducing (DTT (+)) or non-reducing (DTT (−)) conditions. Proteins were incubated for 7 days at 4 °C after 16-h agitation (+) or non-treatment (−). b, densitometric analysis of the dimer/monomer ratio, indicating that the single C/S mutation accelerates aggregate formation containing an intermolecular disulfide bond. agi, agitation. C, ThT fluorescence assay showing that RRM1 aggregates form amyloid fibrils. Recombinant RRM1 proteins (0.03 mm) were reacted with a ThT derivative (BTA-1) after 24-h incubation at 4 or 37 °C. Data represent the -fold increase in the ThT ratio compared with the controls. Each value expresses mean ± S.E. (n = 3). *, p < 0.05 versus control sample by one way ANOVA of Bonferroni's test. D, free state of cysteine residues in the de novo RRM1 protein. Absorbance (412 nm) values obtained from the Ellman's test using 5,5′-dithiobis(2-nitrobenzoic acid). RRM1 proteins at the indicated concentrations were incubated with 5,5′-dithiobis(2-nitrobenzoic) acid for 20 min at 22 °C. Data are expressed as the average of duplicate values. RRM1 proteins either with (+) or without (−) DTT effectively reacted with 5,5′-dithiobis(2-nitrobenzoic) acid, indicating the free thiol moieties.
RRM1 aggregates contained a fraction of disulfide-bonded oligomers, which were mediated by Cys-173 and Cys-175 in the fifth β-strand (β5). Because a previous report (30) documented that Cys-173 and Cys-175 are among the targets for oxidation to form oligomers of TDP-43 in ALS, the effect of cysteine substitution of Cys-173 and Cys-175 with Ser (C/S mutants) on the RRM1 oligomerization was investigated. Surprisingly, the single C/S mutants (C173S and C175S) displayed accelerated formation of DTT-sensitive oligomers even without agitation (Fig. 4B, a and b). The double C/S mutant (C173S/C175S) was more unstable and formed irregular oligomers or aggregates (data not shown). Moreover, a thioflavin T assay also revealed that cysteine substitution increased β-sheet formation, and double C/S mutants displayed higher thioflavin T fluorescence than single C/S mutants (Fig. 4C). These results indicate that free thiol of RRM1 governs its conformation, which acquires amyloidogenicity upon modification. An intermolecular disulfide bond accelerates the β-sheet assembly but is not a prerequisite to the RRM1 aggregation. The possibility of intramolecular disulfide bonds between Cys-173 and Cys-175 for the preservation of RRM1 conformation was also eliminated, because Ellman's test indicated that both cysteine residues were free in the de novo condition (Fig. 4D).
Disruption of Free Cysteine Residues in RRM1 Induces Nuclear and Cytosolic TDP-43 Aggregates through Successive Participation of C-terminal Domain
We further investigated the role of Cys-173 and Cys-175 in the conformation of full-length TDP-43. C-terminally EGFP-fused TDP-43 constructs containing WT or C/S mutant (C173S, C175S, and C173S/C175S) RRM1 were overexpressed in HEK293A cells for confocal analysis. Consistent with the data obtained from RRM1 domain analysis, the single and double C/S mutations resulted in the formation of marked aggregates or inclusions of full-length TDP-43, which were chiefly in the nucleus and occasionally in the cytosol (Fig. 5A). Because nuclear inclusions of TDP-43 with RRM1-C/S mutations may simply reflect a chromosomal sequestration of TDP-43 due to a failure to interact with DNA/RNA (31), we generated cytosolic TDP-43 mutants with C173S and/or C175S by modifying the NLS (24). TDP-43 containing RRM1-C/S mutations and mNLS frequently formed cytosolic round inclusions (Fig. 5B). Although TDP-43 proteins lacking the RRM1 domain indeed form multiple round inclusions in the nucleus (Fig. 5C, a and b), as reported previously (31), those with modified NLS did not aggregate in the cytosol (Fig. 5C, c and d). This indicates that the loss of DNA/RNA interaction of TDP-43 by non-functional RRM1 is not responsible for cytosolic aggregation of the RRM1-C/S mutant; it rather indicates that the aberrant assembly or conformation of RRM1 induces TDP-43 aggregation in the cytosol. The role of two cysteines in RRM2 domain was also examined; double substitution of C198S and C244S in RRM2 induced only negligible nuclear aggregates (Fig. 5D), indicating the unique role of the RRM1 cysteines. Moreover, a TDP-43 mutant, in which all four cysteines in RRM1 (Cys-173 and Cys-175) and RRM2 (Cys-198 and Cys-244) were substituted with serine, formed the same inclusions as those without the Cys-198 and Cys-244 substitutions (Fig. 5E), which can exclude the possibility of the compensation by other conserved cysteines. Substitution with alanine (C173A or C173A/C175A) had the same effect (Fig. 5F), indicating that the aggregation propensity of the C/S mutants was caused by ablation of free cysteines, not by the introduction of serine.
FIGURE 5.
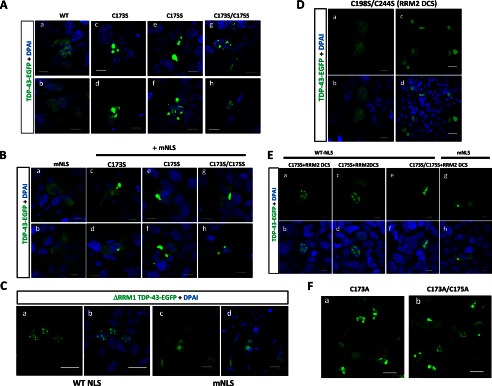
RRM1-C/S substitutions induced cytoplasmic and nuclear inclusions of TDP-43. Shown are confocal analyses of HEK293A cells transiently transfected with EGFP-fused TDP-43. A and B, RRM1-C/S mutations in TDP-43 induced aggregates in the nucleus and cytosol. Cells were transiently transfected with full-length WT or RRM1-C/S of TDP-43-EGFP (green) with WT (A) or modified (B) NLS. Scale bar, 10 μm. C, the RRM1 deletion mutant of TDP-43 formed marked nuclear inclusions, but RRM1-deleted TDP-43 with modified NLS did not. HEK293A cells were transiently transfected with EGFP-fused TDP-43 lacking RRM1 with WT (a and b) or modified (c and d) NLS. Scale bar, 10 μm. D, RRM2-C/S had no impact on the TDP-43 aggregation. Cells were transfected with full-length WT or RRM2-C/S double mutant (RRM2 DCS) TDP-43-EGFP (green). Scale bars, 10 μm (a and b) and 30 μm (c and d). E, disulfide bonding in RRM1 and RRM2 is not inevitable for TDP-43 aggregation. Cells were transfected with RRM1 single mutants and the RRM2-C/S double mutant (RRM2 DCS) of TDP-43-EGFP (green). Double C/S substitutions at Cys-198/Cys-244 (RRM2 DCS) did not affect nuclear or cytosolic TDP-43 inclusions caused by C/S mutations at RRM1. Scale bar, 10 μm. F, C/A mutants in Cys-173 and/or C175S showed the same aggregation as C/S mutants. Scale bar, 10 μm. Nuclei were stained by DAPI (blue).
The specific role of β4 and β5, especially cysteine residues in RRM1, in the preservation of TDP-43 conformation was suggested, because another sporadic ALS-linked mutant, D169G TDP-43 (32), which is located at the loop region between β4 and β5 of RRM1 core-c, had no impact on the aggregate formation (Fig. 6A). Furthermore, we investigated the effect of C terminus deletion on the aggregate formation of TDP-43 with RRM1-C/S, because the prion domain of the C terminus is implicated in TDP-43 proteinopathy. Confocal microscope analysis of transfected HEK293A cells revealed that TDP-43 aggregates either in the nucleus or the cytosol were markedly abolished by deletion of the C terminus (Fig. 6B, g). This finding indicates that RRM1 misfolding triggers C terminus assembly to form the cytosolic inclusions of full-length TDP-43. It should be noted that C-terminal deletion constructs still formed cytosolic and nuclear aggregates, indicating the presence of RRM1-mediated aggregation, as observed in vitro.
FIGURE 6.

Crucial roles of cysteines in RRM1 and C-terminal domain in the formation of cytosolic inclusions of TDP-43. A, sporadic ALS mutation D169G mutation in RRM1 domain caused no TDP-43 aggregation. Confocal analysis of HEK293A cells overexpressing nuclear (A) or mislocalized (mNLS) (B) TDP-43-EGFP (green), with C175S (c and d) or D169G (e and f) mutations. Scale bar, 10 mm (a, c, and e) or 50 μm (b, d, and f). B, C terminus mediates RRM1-C/S-induced aggregation of TDP-43. Confocal analysis of HEK293A cells transiently transfected with nuclear (a–c, WT NLS) or cytosolic (d–f, mNLS) TDP-43-EGFP carrying the double C173S/C175S mutation (DCS), either with (c and f) or without (b and e) the C terminus (aa 266–414, ΔCt). WT*, WT NLS. g, quantification of the effect of the C-terminal tail of TDP-43 on nuclear or cytosolic aggregates caused by RRM1-C/S mutation. The percentage of aggregate holding cells in the total transfected cells was obtained by counting. *, p < 0.05 versus nuclear or cytosolic TDP-43 without any C/S mutation (WT or mNLS). †, p < 0.05 between DCS mutants with and without C-terminal deletions by one-way ANOVA of Bonferroni's test. Data represent the mean ± S.E. (error bars) (n = 6).
TDP-43 with Misfolded RRM1 Results in Disulfide-independent Oligomerization, Ubiquitination, and Aberrant Phosphorylation
To assess the extent of pathogenic features of TDP-43 proteinopathy shared in our RRM1-C/S mutants, immunocytochemistry for ubiquitination or phosphorylation was performed. In human neuronal SHSY-5Y cells, most of the nuclear inclusions of RRM1-C/S mutant TDP-43 were not phosphorylated, whereas occasional cytosolic inclusions showed strong reactivity to antibodies targeting phospho-TDP-43 (Fig. 7A, d–i) or Lys-48-linked ubiquitin chains (Fig. 7A, m–o). Moreover, cytosolic TDP-43 inclusions caused by RRM1-C/S mutation with modified NLS signal were frequently phosphorylated at Ser-409 and Ser-410 (Fig. 7B, d–i) and ubiquitinated (Fig. 7B, m–o). It should be noted that not all cytosolic inclusions were modified by ubiquitin or phosphorylation, indicating that aggregation precedes such modifications in the cytosol. Cytosol-mislocalized TDP-43 without the RRM1-C/S substitutions showed no overt reactivity to anti-phospho-TDP-43 (Fig. 7B, a–c) or anti-ubiquitin (Fig. 7B, j–l). Western blot analysis of FLAG-tagged TDP-43 harboring RRM1-C/S substitutions, containing either a WT (Fig. 7C) or a mutant NLS (Fig. 7D), revealed that RRM1-C/S TDP-43 was readily oligomerized in the presence of a proteasome inhibitor. DTT treatment marginally reduced oligomer formation and increased the amount of dimeric single C/S mutant TDP-43 (Fig. 7, C and D). Moreover, TDP-43 proteins containing RRM1-C/S substitutions were more heavily phosphorylated at Ser-409/Ser-410 than WT TDP-43 proteins, especially in the presence of the proteasome inhibitor lactacystin (Fig. 7E, left), consistent with the previous report showing that proteasome inhibition induced cytosolic mislocalization and the formation of phosphorylated inclusions of TDP-43 (33). Furthermore, mislocalized forms of RRM1-C/S mutants were more susceptible to Ser-409 and Ser-410 phosphorylation, even without lactacystin (Fig. 7E, right). We also observed that RRM1-C/S-linked inclusions of TDP-43 were phosphorylated at Ser-403 and Ser-404 (data not shown).
FIGURE 7.
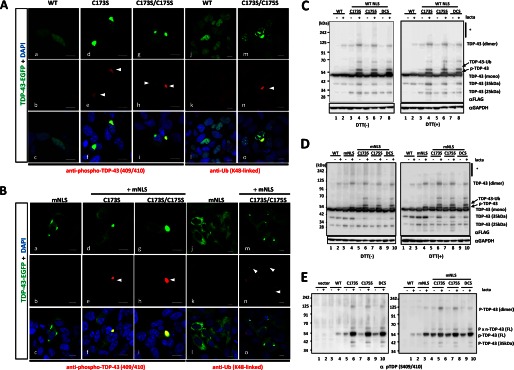
TDP-43 inclusions with RRM1-C/S mutation were phosphorylated, ubiquitinated, and disulfide-free. A and B, confocal analysis of SHSY-5Y cells transfected with full-length WT or RRM1-C/S mutants of TDP-43-EGFP (green). Cells were immunostained with antibodies (red) targeting phospho-TDP-43 at Ser-409/Ser-410 (a–i) or Lys-48-linked ubiquitin (j–o). In B, all constructs contained mNLS. Nuclei were stained by DAPI (blue). Phosphorylated or ubiquitinated inclusions in the cytoplasm are indicated by arrowheads. Note that cytoplasmic inclusions (green) are strongly immunoreactive to both antibodies compared with nuclear aggregates. Scale bar, 10 μm. C and D, Western blots showing that disulfide-irrelevant oligomers of TDP-43 harboring RRM1-C/S substitutions under the proteasome inhibitor lactacystin are more prominent in the cytosol (D) than in the nucleus (C). TDP-43-FLAG was overexpressed in HEK293A cells. Cell lysates were analyzed by SDS-PAGE and incubated with antibodies targeting FLAG and GAPDH. Note that DTT marginally reduced oligomerization (indicated by an asterisk) and increased dimer formation in C173S and C175S mutants. E, Western blot of TDP-43 with RRM1-C/S substitutions using an antibody targeting TDP-43 phosphorylated at Ser-409/Ser-410. TDP-43 with mNLS is phosphorylated to a greater extent than WT, which is enhanced by lactacystin (right, lanes 3 and 4). Conversely, TDP-43 harboring the RRM1-C/S substitution is more strongly phosphorylated, and modification of NLS or lactacystin treatment augmented the phosphorylated TDP-43 (both left and right, lanes 5–10).
RRM1-C/S Substitution Replicates and Enhances TDP-43 Proteinopathy
We further performed several functional assessments of the misfolded TDP-43 caused by RRM1-C/S mutation, focusing on the features of TDP-43 proteinopathy. An assay for exon 9 skipping in the cystic fibrosis transmembrane conductance regulator by misfolded TDP-43 with RRM1-C/S in HEK293A cells revealed that all of the RRM1-C/S mutants failed to augment the spliced fragments to the same extent as the RRM1 deletion mutant (Fig. 8A). Next, it was also shown that TDP-43 with RRM1-C/S, especially harboring nuclear aggregates, induced cytosolic mislocalization of TDP-43 by immunofluorescence using anti-TDP-43 antibody (Fig. 8B). Moreover, both nuclear and cytosolic inclusions of TDP-43 with RRM1-C/S were colocalized with co-transfected WT TDP-43-FLAG proteins (Fig. 8C, f and i), indicating that misfolded TDP-43 could sequester native nuclear TDP-43 in both nucleus and cytosol. Notably, cytosolic inclusions of TDP-43 with RRM1-C/S chiefly comprised full-length forms of TDP-43, but not cleaved species from NLS-containing N terminus, as demonstrated by confocal analysis of TDP-43 tagged with mCherry and EGFP at the N and C terminus, respectively, in which the co-existence of two fluorescences in both nuclear and cytosolic aggregates was shown (Fig. 8D).
FIGURE 8.

C/S substitution in RRM1 induces diverse dysfunctions of ALS-linked TDP-43 proteinopathies. A, the exon 9 skipping assay for cystic fibrosis transmembrane conductance regulator RNA. cDNA from HEK293A cells was analyzed 48 h after co-transfection with TDP-43-EGFP and TG13T5 minigene reporter plasmids. a and b, which show the agarose gel image and densitometric analysis, respectively, show the impaired RNA splicing effect of the WT and mutant TDP-43-EGFP proteins, and the ratio of sliced to unspliced fragments was obtained. Data represent the mean ± S.E. n = 4. *, p < 0.05 versus vector control; #, p < 0.05 versus WT TDP-43 by one-way ANOVA of Bonferroni's test. B, TDP-43 with RRM1-C/S substitutions induces cytosolic mislocalization of TDP-43. HEK293A cells were transiently transfected with WT (a–c) or RRM1-C/S mutant TDP-43-EGFP (d–i) and were analyzed using rabbit polyclonal anti-TDP-43 antibody. Scale bar, 50 μm. j, quantification of TDP-43-EGFP-expressing cells with cytosolic mislocalized TDP-43. Data represent the mean ± S.E. (n = 4). In each experiment, 21–70 cells were counted. *, p < 0.05 versus WT by one-way ANOVA of Bonferroni's test. C, immunofluorescence analysis showing that nuclear or cytosolic aggregates of TDP-43 with RRM1-C/S substitution recruit nuclear WT TDP-43. HEK293A cells were co-transfected with the nuclear (a–f) or cytosolic (g–l) forms of WT or C/S mutant TDP-43-EGFP (green) together with FLAG-tagged WT TDP-43. Cells were stained with an anti-FLAG antibody (red). Colocalized TDP-43-EGFP and TDP-43-FLAG are indicated by arrowheads. Scale bar, 10 μm. D, full-length TDP-43 is redistributed into the cytosol by RRM1-C/S substitution. a, schematic illustration of mCherry-TDP-43-GFP. 5′- and 3′- wild-type and mutant TDP-43 genes were fused with mCherry and GFP, respectively, in an in-frame manner. b–m, co-localization of both N and C termini of TDP-43 containing C173S and/or C175S substitutions. HEK-293A cells were transfected with the indicated constructs and were cultured for 32 h. The transfected cells were further treated with 10 mm lactacystin for 16 h, followed by observation with a confocal microscope. Scale bar, 10 μm.
Despite the potentially dominant roles of mutant TDP-43 in subpopulations of familial ALS patients, there is only a subtle difference in the aggregation propensity of mutant TDP-43 compared with the WT (Fig. 9, A (a–c) and B). Intriguingly, the introduction of RRM1-C/S into the familial ALS (FALS)-linked TDP-43 mutant A315T or Q331K clearly accelerated the aggregate formation of both FALS mutants compared with WT (Fig. 9, A (d–f) and B). Because the species of the TDP-43 conformer that links to neurotoxicity remains unclear, we investigated the toxicity of the TDP-43 with RRM1-C/S mutations using motor neuron cell line NSC34 cells. First, we confirmed that RRM1-C/S mutation in TDP-43-EGFP with modified NLS induced ubiquitinated inclusions in the cytosol of differentiated NSC-34 cells (Fig. 9C, a–l), as observed in other cell lines. Next, the toxic effect of RRM1-C/S mutants of TDP-43-FLAG was assessed by fluorometry after simultaneous treatment with cell-impermeable bis-AAF-R110 and cell-permeable GF-AFC that react with dead cell protease and with live cell protease, respectively. This method has an advantage of minimizing the well-to-well difference of transfection efficiency or cell density by normalization of bis-AAF-R100 to GF-AFC, designated as a toxicity ratio. The ratio revealed that RRM1-C/S mutants, especially those with modified NLS, significantly augmented the toxicity compared with the vector control (Fig. 9D, a). Moreover, we also obtained normalized bis-AAF-R100 fluorescence to co-transfected Renilla luciferase activity to validate that this effect is caused by the transgene. The normalized toxicity ratio also showed similar results, in which significant toxicity in C175S and C173S/C175S with mNLS was observed (Fig. 9D, b). C173S with mNLS showed a trend of higher toxicity, although it was not significant. Taken together, these results indicate that the aggregation propensity associated with the misfolding of RRM1 might underlie the toxicity of mislocalized TDP-43. It should be noted that the amount of carboxyl 25- or 35-kDa fragments (Fig. 7D) did not correlate with the toxicity of TDP-43.
FIGURE 9.

RRM1-C/S mutations unravel the pathogenic properties of TDP-43. A, RRM1-C/S substitution enhances the aggregation propensity of TDP-43 with familial ALS-linked mutations in the C-terminal domain. EGFP-fused WT or A315T and Q331K mutants of TDP-43, either with (d–f) or without (a–c) the C175S mutation, were transiently expressed in HEK293A cells. B, quantification of cells with aggregates. Data represent the mean percentages of cells harboring aggregates in total transfected cells ± S.E. (n = 8 images); 20–120 cells were counted in each image. *, p < 0.05 by one-way ANOVA of Bonferroni's test. C, cytosolic aggregates of TDP-43 with RRM1-C/S injure the motor neuron cell line. a–l, confocal micrographs showing subcellular distributions and inclusion formation of TDP-43-EGFP in NSC34. D, a and b, fluorometric assay for neurotoxicity of RRM1-C/S mutants in NSC-34 cells. Murine motor neuron NSC34 cells plated in a 96-well culture plate were transiently transfected with plasmids expressing TDP-43-FLAG (0.2 μg/well) of WT, ALS-linked mutants (D169G and A315T), or Cys-173/Cys-175 mutants (C173S, C175S, and C173S/C175S (DCS)) with or without mNLS. Data represent the mean ± S.E. (n = 12 wells from 4 independent experiments; each value is normalized to vector control in each experiment). *, p < 0.05 versus vector control by one-way ANOVA of Bonferroni's test. b, dead cell marker bis-AAF-R110 was also normalized to luciferase activity from co-transfected Renilla luciferase reporter plasmid. The Renilla luciferase reporter gene (0.02 μg/well) was co-transfected with pcDNA-TDP-43-FLAG. Data represent the mean ± S.E. (n = 11–12). Each value is normalized to vector control in each experiment). *, p < 0.05 versus vector control by one-way ANOVA of Dunnett's test.
Misfolding-relevant Cores in RRM1 Are Molecular Signatures of Pathological TDP-43 Deposition in ALS
To investigate if TDP-43 inclusions caused by the RRM1-C/S mutations link to ALS pathology, we generated rabbit polyclonal antibodies (pAbs) against the three RRM1 core regions (Fig. 10A). Two antisera that showed a high titration against both recombinant RRM1 and full-length TDP-43 were obtained for core-a and core-c (pAb RRM1-a and pAb RRM1-c) tested by Western blotting (Fig. 10B) and ELISA (Fig. 10C). In cultured cells, pAb RRM1-a and pAb RRM1-c showed a weak reactivity to endogenous or external WT TDP-43 (Fig. 11A, a–c and j–l), except for a few cases of concentrated expression in the nucleus. By contrast, both pAbs clearly recognized aggregation of TDP-43 with the RRM1-C/S mutations (Fig. 11A, d–i and m–r), whereas a commercially available polyclonal antibody raised against N-terminal 260 residues of TDP-43 recognized both TDP-43 of WT and C175S mutant (Fig. 11, A (s–x) and B). These results indicate that the aggregation-relevant cores were exposed in the TDP-43 RRM1-C/S mutants.
FIGURE 10.

Evaluation of polyclonal antibody for the detection of aggregation-relevant cores. A, schematic illustration showing the antigenic sequences in RRM1 used to generate the polyclonal antibodies raised against core-a (aa 108–116), core-b (aa 134–142), and core-c (aa 163–173), which are designated as pAb RRM1-a, -b, and -c, respectively. Asterisks indicate the residues that showed irreversible chemical shifts on NMR. B, Western blot analysis using antibody against three aggregation-relevant domains (Ab1, Ab2, and Ab3) in the presence (DTT (+)) and absence (DTT (−)) of DTT. The antibody 1 (Ab1; against aa 108–116) and the antibody 3 (Ab3; against aa 167–172) recognize RRM1 and full-length recombinant TDP-43 but not RRM2 or the RRM1 deletion mutant of TDP-43. C, ELISA study showing the immunoreactivity of pAb RRM1-a (a) and pAb RRM1-c (b) against recombinant human TDP-43 proteins of various types.
FIGURE 11.
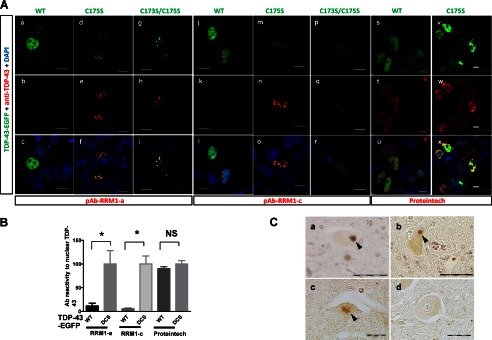
Misfolding-relevant core in RRM1 is an immunogenic marker of TDP-43 inclusions. A, confocal micrographs showing SHSY-5Y cells transiently expressing EGFP-fused WT or C/S mutant TDP-43. Cells were immunostained with the antibody targeting core-a (a–i; pAb RRM1-a) or core-c (j–r; pAb RRM1-c) or a commercially available anti-TDP-43 antibody (s–x; Proteintech). The bottom panels show overlaid images of the EGFP (green) and antibody (red) signals. Nuclei were stained with DAPI (blue). Scale bar, 10 μm. B, quantification of the aggregate-specific reactivity of the pAb RRM1-a and pAb RRM1-c antibodies, compared with that of a commercially available anti-TDP-43 antibody (Proteintech). The fluorescence of EGFP and the antibody was measured in each nucleus expressing WT or C173S/C175S mutant (DCS) TDP-43. The ratio of antibody to EGFP fluorescence was determined; WT data are normalized to the DCS data, which are expressed as Ab reactivity to nuclear TDP-43. Data represent the mean ± S.E. (n = 15–117 nuclei). *, p < 0.05 by one-way ANOVA of Bonferroni's test. C, representative immunohistochemical photographs of spinal cord sections from three sporadic ALS patients (a–c) and a subject with myasthenia gravis (d) were stained with antibodies targeting RRM1-a. Arrowheads indicate Lewy body-like hyaline inclusions (a and b) and a skein-like inclusion (c). Scale bar, 20 μm.
We then examined whether the pAbs recognized TDP-43 inclusions in ALS patients. Immunohistochemical analyses showed that pAb RRM1-a recognized Lewy body-like hyaline inclusions (Fig. 11C, a and b) and skein-like inclusions (Fig 11C, c) in the spinal motor neurons of sporadic ALS patients. Spinal cord sections from individuals with non-neurological disease or with other neurological diseases, including myasthenia gravis, Parkinson disease, and myotonic dystrophy, showed no positive staining (see Fig. 11C (d) for myasthenia gravis). Nuclear TDP-43 was not stained by pAb RRM1-a in motor neurons, regardless of the presence of cytosolic TDP-43 aggregates, which is consistent with the cell culture results. These results suggest that RRM1-C/S substitution-induced inclusions of TDP-43 share the common conformation change with those in ALS. RRM1-c staining was nonspecific in human sections (data not shown).
DISCUSSION
In this study, we have presented a novel finding, that the TDP-43 RRM1 domain crucially governs the conformation, subcellular distribution, and function of TDP-43. It was also shown that the RRM1 was aggregation-prone under physical stresses, such as high pressure or agitation, and served as seeds for further aggregation. Because the high pressure and aggregation allow molecular association in solution, regional increases or condensation of TDP-43 may underlie aberrant assembly of RRM1 domains as well as oxidative stress (30, 34). In agreement with this notion, higher concentrations of RRM1 were more susceptible to precipitation after shorter periods of mechanical stress and postincubation (data not shown). It is also reported that RRM1-RRM2 tandem fragments are readily precipitated at higher concentrations (23), and an effect of high expression levels of TDP-43 on aggregate formation has been also been reported (10).
As the initial step in the misfolding process, regional structural changes were observed at three regions in RRM1, designated as core-a, -b, and -c; these regions showed irreversible chemical shifts after transient high pressure treatment (Fig. 1C). Core-a and core-b are located in the loop between the β-strands, whereas core-c is located within the β4 and β5 strands. Moreover, core-c coincides with the protease-resistant regions of aggregated RRM1. Two cysteine residues, Cys-173 and Cys-175, are located in the core-c-containing β-strands (Fig. 4A); the presence of intermolecular disulfide bonds in RRM1 aggregates strongly suggests that core-c serves as the assembly interface for dimerization or oligomerization. Intriguingly, these core regions lie on the same face of the molecule in NMR analysis as shown in Fig. 1C, supporting our notion that they might serve as a dimer interface in solution under physical stresses.
RRM1 aggregation caused by physical stresses contained intermolecular disulfide bonds at Cys-173 and Cys-175, which are shown to be targets for oxidation in TDP-43 (30). Our in vitro data indicating that RRM1-C/S mutation accelerates intermolecular disulfide bonds and aggregation agree with this notion. However, double substitution of Cys-173 and Cys-175 did not reduce but rather promoted aggregation of both RRM1 and full-length TDP-43. From these results, it is suggested that free thiols of Cys-173 and Cys-175 crucially govern the conformation of RRM1, especially at β4 and β5. Intermolecular disulfide bonds might be promoted by β-sheet assembly through disordered conformation of β4 and β5, which, in turn, may further accelerate the oligomerization of RRM1. The unique feature that both Cys-173 and Cys-175 are closely located on the β-strands might link to the aggregation propensity of the RRM1 domain. In general, thiol modifications variously affect the protein conformation (35). In particular, the oxidative stress is implicated in ALS pathogenesis; indeed, nuclear factor κB, a redox-dependent transcription factor, is activated in ALS (36). In this sense, free cysteines relentlessly face risks of pathogenic modifications, such as intra- or intermolecular disulfides, sulfonic acids (35, 37), or S-nitrosothiol (38). It should be noted, however, that the disulfide bond formation might be a transient factor but not a prerequisite for ultimate aggregates of TDP-43 caused by RRM1-C/S mutations. Moreover, our finding that inclusion formation of full-length TDP-43 with RRM1-C/S required the C-terminal domain (Fig. 6B) suggests that the aberrant assembly of RRM1 may accelerate the turbulence of the aggregenic C terminus, resulting in the cytopathological features of TDP-43 proteinopathy (Fig. 12A). This could explain why the aggregation propensity of two FALS-linked TDP-43 mutants in the C terminus was aggravated by the RRM1-C/S mutation (Fig. 9A). A loss of RNA binding affinity of RRM1 should be considered, as previously documented in the case of RRM2 (39). Indeed, TDP-43 with RRM1-C/S was deprived of RNA processing (Fig. 8A), and we have confirmed that aggregated RRM1 remarkably lost the interaction with (TG)12 nucleotides (data not shown). However, aggregation propensity of WT RRM1 was not altered in the presence of (TG)12 nucleotides during the agitation (Fig. 3B), and TDP-43 with mNLS lacking the RRM1 domain did not form as overt aggregates as RRM1-C/S mutant (Fig. 5C). These suggest that DNA/RNA binding affinity cannot fully explain the aggregate formation of TDP-43 with RRM1 misfolding. The functional effect of RRM1 misfolding should also be considered. Because RRM1 is a dominant RNA-binding domain (40) and binds a large number of RNAs (17–19), the misfolding of RRM1 domain caused by various stresses might seriously disrupt cellular homeostasis. As expected, C/S mutations in RRM1 abolished RNA splicing activity (Fig. 8A).
FIGURE 12.

Hypothetical roles of misfolding-relevant cores in RRM1. A, ribbon structures showing the locations of misfolding-relevant RRM1 core-a, -b, and -c. B, hypothetical scheme of TDP-43 proteinopathy development. For clarity, RRM1 is indicated by a shaded area; core-a, -b, and -c are also indicated. These cores may not be exposed under physiological conditions. Various cellular stresses, such as oxidative stress or regional condensation of TDP-43, may alter the β-sheet assembly, which may induce transient disulfide bonds, ultimately resulting in pathological TDP-43 proteinopathies with the participation of the C terminus.
To our surprise, RRM1-C/S mutation recapitulates diverse phenomena observed in TDP-43 proteinopathies in ALS, including the induction of cytosolic mislocalization accompanied by the formation of ubiquitinated or phosphorylated inclusions, sequestration of otherwise native nuclear TDP-43, and the motor neuron toxicity in culture cells. We also confirmed that RRM1-C/S TDP-43 with no tag resulted in the same aberrant nuclear or cytosolic inclusions immunoreactive to pAb RRM1-a and antibodies against Lys-48 ubiquitin or phospho-TDP-43 but to a lesser extent than tagged proteins (data not shown). To date, limited information is available as to the mechanisms of toxicity of TDP-43 proteinopathy in mammalian cells. Previous work generating TDP-43 transgenic rodents shows that the overexpression of TDP-43 of WT or FALS-linked mutants (41) or the NLS-modified type (42) induces cytoplasmic ubiquitinated or phosphorylated inclusions of TDP-43. However, both animals show no overt neuronal loss. By simultaneous measurements of both indicators of dead cells and live cells, we demonstrated that RRM1 misfolding may induce toxic conversion of TDP-43 in a cell culture study. However, molecular mechanisms to account for the toxic effect of the RRM1-C/S remain unknown. The effect of dysfunctional RRM1 on cell death is higher in the cytosol than in the nucleus. This may suggest that the RNA mishandling of accumulated TDP-43 in the stress granules is linked to this toxicity. It is also possible that cytosolic TDP-43 inclusions sequester miscellaneous survival factors. The cell death appeared non-apoptotic, because we observed no activation of caspase-3/7 during the 48-h cell culture (not shown). It was reported that C-terminal 25- or 35-kDa fragments, not containing RRM1, replicate TDP-43 proteinopathy in cells (28) and in vivo models (43, 44). However, we found no relationship between the amount of these fragments (Fig. 7, C and D) and the severity of neurotoxicity of cytosolic TDP-43 fragments with RRM1-C/S (Fig. 9D). Notably, full-length TDP-43 preferentially accumulates in spinal cords of ALS patients (45), which fits our finding that cytosolic inclusions were full-length.
Last, a polyclonal antibody raised against a misfolding-relevant core in RRM1 stained Lewy body-like hyaline inclusions or skein-like inclusions in ALS sections, indicating that RRM1 misfolding is involved in inclusion formation in ALS. Although antibodies targeting core-a strongly recognized recombinant RRM1 proteins by ELISA (Fig. 10C), they did not react with WT nuclear TDP-43 proteins in cultured cells or spinal cord sections of ALS patients. This may indicate that the core regions in the normal full-length TDP-43 might face inside or be shielded by other molecules or domains; the structure of these domains might be deformed under toxic environments, such as oxidative stress, or after regional accumulation (Fig. 12A). Our results also suggest that core-c, consisting of tandem β-strands β4 and β5, may acquire amyloidogenicity during RRM1 misfolding, resulting in enhancement of β-sheet-mediated oligomer formation. It is also possible that deformity of core-b may affect access of RNA to the phenylalanine residues in the adjacent β-strand (Fig. 12B).
In conclusion, this study proposes a novel mechanism of TDP-43 proteinopathy in which ALS- and FTLD-relevant misfolding of TDP-43 is triggered by specific β-sheet assembly of the RRM1 domain, which is tightly regulated by free cysteine residues on the β-strands. Moreover, aggregated recombinant RRM1 proteins and cytosolic inclusions of TDP-43 with RRM1-C/S can serve for high throughput screening toward potential therapy for ALS/FTLD. Further investigation regarding how RRM1 misfolding induces pathogenic conversion of C terminus may also expand our understanding of the pathomechanism of TDP-43 proteinopathy.
Acknowledgments
We thank Dr. E. Buratti and Dr. F. Baralle for the generous gift of the TG13T5 minigene reporter plasmid. We thank the Central Research Laboratory, Shiga University of Medical Science, for technical support. We are grateful for Dr. Y. Furukawa for experimental advice regarding Pronase digestion. We thank Dr. T. Yokota for providing plasmids for mutant TDP-43 (A315T and Q331K). We also thank Dr. Y. Tomabechi, Dr. N. Iwasaki, Dr. T. Harada, Dr. Y. Tomo, Dr. T. Nagira, Dr. M. Ikari, Dr. S. Watanabe, Dr. F. He, and Dr. T. Terada for experimental support.
This work was supported by KAKENHI Grants 23300131 for Scientific Research (B) and 22659171 for Challenging Exploratory Research; Grant-in-aid for Scientific Research on Innovative Areas 23111002 from the Ministry of Education, Culture, Sports, Science, and Technology (MEXT); and a Grant for Research on Neurodegenerative Diseases from Japan Health and Labor Science.
- TDP-43
- TAR DNA-binding protein of 43 kDa
- ALS
- amyotrophic lateral sclerosis
- FALS
- familial ALS
- NLS
- nucleus localization signal
- mNLS
- NLS mutation
- RRM
- RNA-recognition motif
- PFO
- perfuoro-octanoic acid
- FTLD
- frontotemporal lobar degeneration
- aa
- amino acid(s)
- DCS
- C173S/C175S double cysteine to serine mutation
- ANOVA
- analysis of variance
- Ab
- antibody
- pAb
- polyclonal antibody
- EGFP
- enhanced green fluorescent protein
- ThT
- thioflavin T
- GF-AFC
- glycyl-phenylalanyl-aminofluorocoumarin
- bis-AAF-R110
- bis-alanyl-alanyl-phenylalanyl-rhodamine 110.
REFERENCES
- 1. Arai T., Hasegawa M., Akiyama H., Ikeda K., Nonaka T., Mori H., Mann D., Tsuchiya K., Yoshida M., Hashizume Y., Oda T. (2006) TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem. Biophys. Res. Commun. 351, 602–611 [DOI] [PubMed] [Google Scholar]
- 2. Neumann M., Sampathu D. M., Kwong L. K., Truax A. C., Micsenyi M. C., Chou T. T., Bruce J., Schuck T., Grossman M., Clark C. M., McCluskey L. F., Miller B. L., Masliah E., Mackenzie I. R., Feldman H., Feiden W., Kretzschmar H. A., Trojanowski J. Q., Lee V. M. (2006) Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 314, 130–133 [DOI] [PubMed] [Google Scholar]
- 3. Baloh R. H. (2012) How do the RNA-binding proteins TDP-43 and FUS relate to amyotrophic lateral sclerosis and frontotemporal degeneration, and to each other? Curr. Opin. Neurol. 25, 701–707 [DOI] [PubMed] [Google Scholar]
- 4. Pesiridis G. S., Lee V. M., Trojanowski J. Q. (2009) Mutations in TDP-43 link glycine-rich domain functions to amyotrophic lateral sclerosis. Hum. Mol. Genet. 18, R156–R162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Gendron T. F., Josephs K. A., Petrucelli L. (2010) Review. Transactive response DNA-binding protein 43 (TDP-43). Mechanisms of neurodegeneration. Neuropathol. Appl. Neurobiol. 36, 97–112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Cohen T. J., Lee V. M., Trojanowski J. Q. (2011) TDP-43 functions and pathogenic mechanisms implicated in TDP-43 proteinopathies. Trends Mol. Med. 17, 659–667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Winton M. J., Igaz L. M., Wong M. M., Kwong L. K., Trojanowski J. Q., Lee V. M. (2008) Disturbance of nuclear and cytoplasmic TAR DNA-binding protein (TDP-43) induces disease-like redistribution, sequestration, and aggregate formation. J. Biol. Chem. 283, 13302–13309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kuo P. H., Doudeva L. G., Wang Y. T., Shen C. K., Yuan H. S. (2009) Structural insights into TDP-43 in nucleic acid binding and domain interactions. Nucleic Acids Res. 37, 1799–1808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Furukawa Y., Kaneko K., Watanabe S., Yamanaka K., Nukina N. (2011) A seeding reaction recapitulates intracellular formation of Sarkosyl-insoluble transactivation response element (TAR) DNA-binding protein-43 inclusions. J. Biol. Chem. 286, 18664–18672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Budini M., Buratti E., Stuani C., Guarnaccia C., Romano V., De Conti L., Baralle F. E. (2012) Cellular model of TAR DNA-binding protein 43 (TDP-43) aggregation based on its C-terminal Gln/Asn-rich region. J. Biol. Chem. 287, 7512–7525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hasegawa M., Arai T., Nonaka T., Kametani F., Yoshida M., Hashizume Y., Beach T. G., Buratti E., Baralle F., Morita M., Nakano I., Oda T., Tsuchiya K., Akiyama H. (2008) Phosphorylated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Ann. Neurol. 64, 60–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ash P. E., Zhang Y. J., Roberts C. M., Saldi T., Hutter H., Buratti E., Petrucelli L., Link C. D. (2010) Neurotoxic effects of TDP-43 overexpression in C. elegans. Hum. Mol. Genet. 19, 3206–3218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Shodai A., Ido A., Fujiwara N., Ayaki T., Morimura T., Oono M., Uchida T., Takahashi R., Ito H., Urushitani M. (2012) Conserved acidic amino acid residues in a second RNA recognition motif regulate assembly and function of TDP-43. PloS One 7, e52776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bentmann E., Neumann M., Tahirovic S., Rodde R., Dormann D., Haass C. (2012) Requirements for stress granule recruitment of fused in sarcoma (FUS) and TAR DNA-binding protein of 43 kDa (TDP-43). J. Biol. Chem. 287, 23079–23094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Higashi S., Kabuta T., Nagai Y., Tsuchiya Y., Akiyama H., Wada K. (2013) TDP-43 associates with stalled ribosomes and contributes to cell survival during cellular stress. J. Neurochem. 10.1111/jnc.12194 [DOI] [PubMed] [Google Scholar]
- 16. Buratti E., Baralle F. E. (2001) Characterization and functional implications of the RNA binding properties of nuclear factor TDP-43, a novel splicing regulator of CFTR exon 9. J. Biol. Chem. 276, 36337–36343 [DOI] [PubMed] [Google Scholar]
- 17. Polymenidou M., Lagier-Tourenne C., Hutt K. R., Huelga S. C., Moran J., Liang T. Y., Ling S. C., Sun E., Wancewicz E., Mazur C., Kordasiewicz H., Sedaghat Y., Donohue J. P., Shiue L., Bennett C. F., Yeo G. W., Cleveland D. W. (2011) Long pre-mRNA depletion and RNA missplicing contribute to neuronal vulnerability from loss of TDP-43. Nat. Neurosci. 14, 459–468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Tollervey J. R., Curk T., Rogelj B., Briese M., Cereda M., Kayikci M., König J., Hortobágyi T., Nishimura A. L., Zupunski V., Patani R., Chandran S., Rot G., Zupan B., Shaw C. E., Ule J. (2011) Characterizing the RNA targets and position-dependent splicing regulation by TDP-43. Nat. Neurosci. 14, 452–458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Xiao S., Sanelli T., Dib S., Sheps D., Findlater J., Bilbao J., Keith J., Zinman L., Rogaeva E., Robertson J. (2011) RNA targets of TDP-43 identified by UV-CLIP are deregulated in ALS. Mol. Cell. Neurosci. 47, 167–180 [DOI] [PubMed] [Google Scholar]
- 20. Chiti F., Dobson C. M. (2006) Protein misfolding, functional amyloid, and human disease. Annu. Rev. Biochem. 75, 333–366 [DOI] [PubMed] [Google Scholar]
- 21. Kuwata K., Nishida N., Matsumoto T., Kamatari Y. O., Hosokawa-Muto J., Kodama K., Nakamura H. K., Kimura K., Kawasaki M., Takakura Y., Shirabe S., Takata J., Kataoka Y., Katamine S. (2007) Hot spots in prion protein for pathogenic conversion. Proc. Natl. Acad. Sci. U.S.A. 104, 11921–11926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Torrent J., Alvarez-Martinez M. T., Liautard J. P., Lange R. (2006) Modulation of prion protein structure by pressure and temperature. Biochim. Biophys. Acta 1764, 546–551 [DOI] [PubMed] [Google Scholar]
- 23. Chang C. K., Wu T. H., Wu C. Y., Chiang M. H., Toh E. K., Hsu Y. C., Lin K. F., Liao Y. H., Huang T. H., Huang J. J. (2012) The N-terminus of TDP-43 promotes its oligomerization and enhances DNA binding affinity. Biochem. Biophys. Res. Commun. 425, 219–224 [DOI] [PubMed] [Google Scholar]
- 24. Urushitani M., Sato T., Bamba H., Hisa Y., Tooyama I. (2010) Synergistic effect between proteasome and autophagosome in the clearance of polyubiquitinated TDP-43. J. Neurosci. Res. 88, 784–797 [DOI] [PubMed] [Google Scholar]
- 25. Akasaka K. (2006) Probing conformational fluctuation of proteins by pressure perturbation. Chem. Rev. 106, 1814–1835 [DOI] [PubMed] [Google Scholar]
- 26. Ramjeesingh M., Huan L. J., Garami E., Bear C. E. (1999) Novel method for evaluation of the oligomeric structure of membrane proteins. Biochem. J. 342, 119–123 [PMC free article] [PubMed] [Google Scholar]
- 27. Matusica D., Fenech M. P., Rogers M. L., Rush R. A. (2008) Characterization and use of the NSC-34 cell line for study of neurotrophin receptor trafficking. J. Neurosci. Res. 86, 553–565 [DOI] [PubMed] [Google Scholar]
- 28. Che M. X., Jiang Y. J., Xie Y. Y., Jiang L. L., Hu H. Y. (2011) Aggregation of the 35-kDa fragment of TDP-43 causes formation of cytoplasmic inclusions and alteration of RNA processing. FASEB J. 25, 2344–2353 [DOI] [PubMed] [Google Scholar]
- 29. Kitahara R., Yamaguchi Y., Sakata E., Kasuya T., Tanaka K., Kato K., Yokoyama S., Akasaka K. (2006) Evolutionally conserved intermediates between ubiquitin and NEDD8. J. Mol. Biol. 363, 395–404 [DOI] [PubMed] [Google Scholar]
- 30. Cohen T. J., Hwang A. W., Unger T., Trojanowski J. Q., Lee V. M. (2012) Redox signalling directly regulates TDP-43 via cysteine oxidation and disulphide cross-linking. EMBO J. 31, 1241–1252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ayala Y. M., Zago P., D'Ambrogio A., Xu Y. F., Petrucelli L., Buratti E., Baralle F. E. (2008) Structural determinants of the cellular localization and shuttling of TDP-43. J. Cell Sci. 121, 3778–3785 [DOI] [PubMed] [Google Scholar]
- 32. Kabashi E., Valdmanis P. N., Dion P., Spiegelman D., McConkey B. J., Vande Velde C., Bouchard J. P., Lacomblez L., Pochigaeva K., Salachas F., Pradat P. F., Camu W., Meininger V., Dupre N., Rouleau G. A. (2008) TARDBP mutations in individuals with sporadic and familial amyotrophic lateral sclerosis. Nat. Genet. 40, 572–574 [DOI] [PubMed] [Google Scholar]
- 33. Nonaka T., Arai T., Buratti E., Baralle F. E., Akiyama H., Hasegawa M. (2009) Phosphorylated and ubiquitinated TDP-43 pathological inclusions in ALS and FTLD-U are recapitulated in SH-SY5Y cells. FEBS Lett. 583, 394–400 [DOI] [PubMed] [Google Scholar]
- 34. Iguchi Y., Katsuno M., Takagi S., Ishigaki S., Niwa J., Hasegawa M., Tanaka F., Sobue G. (2012) Oxidative stress induced by glutathione depletion reproduces pathological modifications of TDP-43 linked to TDP-43 proteinopathies. Neurobiol. Dis. 45, 862–870 [DOI] [PubMed] [Google Scholar]
- 35. Biswas S., Chida A. S., Rahman I. (2006) Redox modifications of protein-thiols. emerging roles in cell signaling. Biochem. Pharmacol. 71, 551–564 [DOI] [PubMed] [Google Scholar]
- 36. Swarup V., Phaneuf D., Dupré N., Petri S., Strong M., Kriz J., Julien J. P. (2011) Deregulation of TDP-43 in amyotrophic lateral sclerosis triggers nuclear factor κB-mediated pathogenic pathways. J. Exp. Med. 208, 2429–2447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Fujiwara N., Nakano M., Kato S., Yoshihara D., Ookawara T., Eguchi H., Taniguchi N., Suzuki K. (2007) Oxidative modification to cysteine sulfonic acid of Cys111 in human copper-zinc superoxide dismutase. J. Biol. Chem. 282, 35933–35944 [DOI] [PubMed] [Google Scholar]
- 38. Akhtar M. W., Sunico C. R., Nakamura T., Lipton S. A. (2012) Redox regulation of protein function via cysteine S-nitrosylation and its relevance to neurodegenerative diseases. Int. J. Cell Biol. 2012, 463756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Pesiridis G. S., Tripathy K., Tanik S., Trojanowski J. Q., Lee V. M. (2011) A “two-hit” hypothesis for inclusion formation by carboxyl-terminal fragments of TDP-43 protein linked to RNA depletion and impaired microtubule-dependent transport. J. Biol. Chem. 286, 18845–18855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Buratti E., Brindisi A., Pagani F., Baralle F. E. (2004) Nuclear factor TDP-43 binds to the polymorphic TG repeats in CFTR intron 8 and causes skipping of exon 9. A functional link with disease penetrance. Am. J. Hum. Genet. 74, 1322–1325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Swarup V., Phaneuf D., Bareil C., Robertson J., Rouleau G. A., Kriz J., Julien J. P. (2011) Pathological hallmarks of amyotrophic lateral sclerosis/frontotemporal lobar degeneration in transgenic mice produced with TDP-43 genomic fragments. Brain 134, 2610–2626 [DOI] [PubMed] [Google Scholar]
- 42. Igaz L. M., Kwong L. K., Lee E. B., Chen-Plotkin A., Swanson E., Unger T., Malunda J., Xu Y., Winton M. J., Trojanowski J. Q., Lee V. M. (2011) Dysregulation of the ALS-associated gene TDP-43 leads to neuronal death and degeneration in mice. J. Clin. Invest. 121, 726–738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Zhang T., Mullane P. C., Periz G., Wang J. (2011) TDP-43 neurotoxicity and protein aggregation modulated by heat shock factor and insulin/IGF-1 signaling. Hum. Mol. Genet. 20, 1952–1965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Caccamo A., Majumder S., Oddo S. (2012) Cognitive decline typical of frontotemporal lobar degeneration in transgenic mice expressing the 25-kDa C-terminal fragment of TDP-43. Am. J. Pathol. 180, 293–302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Igaz L. M., Kwong L. K., Xu Y., Truax A. C., Uryu K., Neumann M., Clark C. M., Elman L. B., Miller B. L., Grossman M., McCluskey L. F., Trojanowski J. Q., Lee V. M. (2008) Enrichment of C-terminal fragments in TAR DNA-binding protein-43 cytoplasmic inclusions in brain but not in spinal cord of frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Am. J. Pathol. 173, 182–194 [DOI] [PMC free article] [PubMed] [Google Scholar]