

Background: Dbf4/Cdc7 triggers DNA replication by phosphorylating Mcm2–7 helicase.
Results: Disruption of both the Dbf4-Mcm2 and Cdc7-Mcm4 interactions results in growth inhibition and sensitivity to genotoxic stress.
Conclusion: Functionally overlapping Dbf4-Mcm2 and Cdc7-Mcm4 interactions promote DNA replication and resistance to fork inhibition.
Significance: Characterizing how Dbf4/Cdc7 interacts with the Mcm2–7 ring is crucial to understanding the regulation of DNA replication.
Keywords: Cell Cycle, Checkpoint Control, DNA Helicase, DNA Replication, Yeast, Cdc7, Dbf4, MCM
Abstract
The essential cell cycle target of the Dbf4/Cdc7 kinase (DDK) is the Mcm2–7 helicase complex. Although Mcm4 has been identified as the critical DDK phosphorylation target for DNA replication, it is not well understood which of the six Mcm2–7 subunits actually mediate(s) docking of this kinase complex. We systematically examined the interaction between each Mcm2–7 subunit with Dbf4 and Cdc7 through two-hybrid and co-immunoprecipitation analyses. Strikingly different binding patterns were observed, as Dbf4 interacted most strongly with Mcm2, whereas Cdc7 displayed association with both Mcm4 and Mcm5. We identified an N-terminal Mcm2 region required for interaction with Dbf4. Cells expressing either an Mcm2 mutant lacking this docking domain (Mcm2ΔDDD) or an Mcm4 mutant lacking a previously identified DDK docking domain (Mcm4ΔDDD) displayed modest DNA replication and growth defects. In contrast, combining these two mutations resulted in synthetic lethality, suggesting that Mcm2 and Mcm4 play overlapping roles in the association of DDK with MCM rings at replication origins. Consistent with this model, growth inhibition could be induced in Mcm4ΔDDD cells through Mcm2 overexpression as a means of titrating the Dbf4-MCM ring interaction. This growth inhibition was exacerbated by exposing the cells to either hydroxyurea or methyl methanesulfonate, lending support for a DDK role in stabilizing or restarting replication forks under S phase checkpoint conditions. Finally, constitutive overexpression of each individual MCM subunit was examined, and genotoxic sensitivity was found to be specific to Mcm2 or Mcm4 overexpression, further pointing to the importance of the DDK-MCM ring interaction.
Introduction
The minichromosome maintenance (MCM)2 complex is composed of six distinct subunits (Mcm2–7) that function together as an essential helicase required for DNA replication. The heterohexamer first assembles in the cytoplasm and is co-imported into the nucleus with Cdt1 (1). It is then targeted to and maintained at origins of DNA replication through an interaction between Cdt1 and the Orc6 subunit of the origin recognition complex (ORC) (2, 3). The tight association of two MCM heterohexamers with individual origins is brought about by the sequential hydrolysis of Cdc6- and ORC-bound ATP (4–6). Several MCM subunits then undergo priming phosphorylation by multiple kinases, including Mec1 (7). In late G1 phase, levels of Dbf4 rise, activating the Dbf4-dependent kinase (DDK) Cdc7 (for review, see Ref. 8), which then phosphorylates primed MCM subunits, thereby stimulating DNA replication.
Several lines of evidence indicate that Dbf4/Cdc7 activates Mcm2–7 by bringing about a conformational change to the complex. The essential function of DDK can be bypassed by the mcm5-bob1 allele, even though it appears that Mcm5 is not itself phosphorylated by this kinase complex (7, 9). Structural analysis has suggested a model in which the mutant form of Mcm5 may render the MCM complex permissive for DNA replication (10, 11). Viability can similarly be rescued in cells lacking DDK activity through phosphomimetic mutations of DDK target sites in an N-terminal region of Mcm4 or by removal of this domain altogether, indicating that it plays an inhibitory role that can be altered by the kinase (12). Although the consequences of a DDK-dependent change in MCM complex conformation have not been fully characterized, there is evidence to suggest that it may stimulate association with two proteins required for attracting DNA polymerases to origins, Sld3 and Cdc45 (13, 14).
Misregulation of Mcm2–7 function has been implicated as a cause of genomic instability and mammalian cancer phenotypes. Altered levels of MCM subunits have been associated with numerous human cancer types (for review, see Refs. 15, 16), and mice that are hypomorphic for MCM activity have demonstrated chromosomal abnormalities and a dramatic increase in cancer susceptibility (17, 18). Interestingly, although DDK phosphorylation of budding yeast Mcm2 is not required for normal growth, mutation of the two DDK target sites (Ser-164, Ser-170) to alanine rendered cells sensitive to the DNA-damaging agent methyl methanesulfonate (MMS), the ribonucleotide reductase inhibitor hydroxyurea (HU), the base analog 5-fluorouracil, and caffeine, which inhibits the S phase checkpoint kinases Tel1 and Mec1 (19, 20).
Despite the precise mapping of amino acid residues phosphorylated by Dbf4/Cdc7 (7, 19, 21), the way in which this kinase complex is targeted to the Mcm2–7 heterohexamer is still not well understood. We have previously shown that two conserved regions of Dbf4 mediate interactions with the MCM complex (motifs C and M) (22, 23). Mutation of these Dbf4 domains compromises cell growth, DNA replication, and MCM phosphorylation (22–25). We have also recently determined that a short region at the C terminus of Cdc7 is required for MCM binding.3 Similarly, Sheu and Stillman have identified a region of Mcm4 that binds to the Dbf4/Cdc7 complex, and mutation of this domain reduces the level of Mcm4 phosphorylation by DDK (13).
In the present report, we show that Dbf4 and Cdc7 interact with distinct MCM subunits and that deletion of the MCM association region of either protein results in only modest DNA replication and growth defects. In contrast, simultaneous impairment of Dbf4- and Cdc7-MCM interactions strongly inhibits growth and contributes to increased sensitivity to DNA replication stress.
EXPERIMENTAL PROCEDURES
Yeast Strains
DY-1 (MATa, ade2–1, can1–100, trp1–1, his3–11,-15, ura3–1, leu2–3,-112, pep4:LEU2) was used for the two-hybrid analyses and co-immunoprecipitations. DY-262 (MATα, leu2Δ0, met15Δ, ura3Δ0, lys2Δ0, mcm2::HIS3) and DY-263 (MATα his3Δ1 leu2Δ0 met15Δ ura3Δ0 mcm4::KanMX), supported for growth with MCM2 or MCM4 on a URA3 CEN vector, respectively (26), were used for plasmid shuffling. MCM2 and MCM4 plasmid shuffle strains were transformed with YCplac111-Mcm2WT, -Mcm2ΔDDD, -Mcm4WT, or -Mcm4ΔDDD and grown on synthetic complete (SC)-selective medium lacking uracil and leucine. Colonies from these transformation plates were streaked on SC plates lacking leucine, with added 5-fluoro-orotic acid to select for cells that had lost the URA3 support plasmid. This resulted in the only copy of MCM2 or MCM4 being on the YCplac111 LEU2, CEN plasmid. These shuffle strains were then mated to DY-196 (MATa, his3Δ1, leu2Δ0, ura3Δ0), and the resulting diploids were sporulated and dissected to generate the haploid MATa shuffle strains. DY-228 (MATa, his3Δ200, met15Δ0, trp1Δ63, ura3Δ0) was used as the parental strain to generate GAL1-MCM2 (DY-215), GAL1-MCM3 (DY-216), GAL1-MCM4 (DY-217), GAL1-MCM5 (DY-218), GAL1-MCM6 (DY-219), and GAL1-MCM7 (DY-220) strains, using TRP1 as a selectable marker.
Plasmids
pEG-Dbf4-FL and pJG-Mcm2 have been described previously (22). pJG-Mcm3, pJG-Mcm5, and pJG-Mcm6 were generated by PCR amplification of genomic MCM3, MCM5, and MCM6, respectively, from strain DY-26 (MATa, his3Δ200, met15Δ0, trp1Δ63, ura3Δ0), with the forward and reverse primers containing ApaI and XhoI restriction sites, respectively. pJG-Mcm4 was generated by PCR amplification of genomic MCM4 from DY-26 with the forward and reverse primers containing NcoI and XhoI restriction sites, respectively. pJG-Mcm7 was generated by PCR amplification of genomic MCM7 from DY-26 with the forward and reverse primers containing NcoI and EcoRI sites, respectively. pJG-Mcm2Δ63 was generated by PCR amplification of genomic MCM2 from DY-26 with the forward and reverse primers corresponding to sequence encoding amino acids 64–868, containing NcoI and XhoI sites, respectively. Both pJG-Mcm2(505–868) and pJG-Mcm2(1–504) were generated by PCR amplification of genomic MCM2 from DY-1 with the forward and reverse primers corresponding to DNA encoding either amino acids 1–504 or 505–868, containing NcoI and XhoI sites, respectively. In all cases, the PCR products were kit-purified (GE Healthcare) and then ligated into the appropriately digested vector. pEG-Cdc7 was generated by PCR amplification of genomic CDC7 from DY-26 genomic DNA with the forward and reverse primers containing EcoRI and BglII sites, respectively. pEG-202 (27) was then cut with EcoRI and BamHI with the fragment and vector then ligated, thus generating an in-frame fusion with the LexA coding sequence. pCM190-Mcm2WT and pCM190-Mcm2Δ63 were generated by PCR amplification of MCM2 and mcm2Δ63 from pJG-Mcm2WT and pJG-Mcm2Δ63, respectively, with the forward and reverse primers containing NotI and BamHI respectively, in both cases, and cloned into corresponding sites in pCM190. pCM190-Mcm4WT was generated by PCR amplification of MCM4 using pJG-Mcm4 as template, with forward and reverse primers including NotI and BglII sites, respectively, followed by ligation into pCM190 digested with NotI and BamHI. pJG-Mcm2Δ2–4,10–63 was generated by PCR amplification of genomic MCM2 from DY-26 with a forward primer containing both NcoI and NdeI sites followed by the sequence encoding amino acids 5–9 and 64–75. The reverse primer contained BamHI and XhoI sites, followed by sequence corresponding to the C-terminal coding region of MCM2. Following PCR amplification and purification, the insert was cut with NcoI and XhoI and cloned into equivalent sites in pJG4–6. pJG-Mcm4ΔDDD was generated by PCR amplification of two fragments of MCM4 from DY-26 genomic DNA (encoding amino acids 1–174 and 334–878). An NcoI site was incorporated into the reverse primer of the first fragment and the forward primer of the second fragment, thus creating a junction for the two fragments. These two cut and purified fragments were then cloned together into the pJG4–6 vector using ApaI and XhoI. The plasmid shuffle vector (YCplac111) is a CEN vector with a LEU2 selectable marker. The YCplac111-Mcm2WT and YCplac111-Mcm4WT plasmid shuffle vectors have been previously described (26). The YCplac111-Mcm2WT vector along with pJG-Mcm2Δ2–4,10–63 was cut with NdeI and BamHI, and the resulting Mcm2ΔDDD fragment was cloned into the cut YCplac111 vector to generate YCplac111-Mcm2ΔDDD. Mcm4ΔDDD was PCR-amplified using pJG-Mcm4ΔDDD as template with a forward primer corresponding to the MCM4 N-terminal coding sequence containing NdeI and a reverse primer corresponding to the C-terminal coding region of MCM4 downstream of an internal MluI site. This PCR product was then cut with NdeI and MluI and cloned into the Mcm4WT plasmid shuffle vector, which was also cut with NdeI and MluI to generate YCplac111-Mcm4ΔDDD. pCM190-Mcm2ΔDDD and pCM190-Mcm4ΔDDD were generated by PCR amplification of pJG-Mcm2Δ2–4,10–63 and pJG-Mcm4ΔDDD, respectively, with forward and reverse primers containing NheI and BglII sites, respectively, to allow ligation into pCM190 at the equivalent sites. All plasmid constructs were verified by DNA sequencing.
Two-hybrid Analysis
Liquid culture two-hybrid assays were performed as described previously (27). The lacZ reporter plasmid pSH18–34, pEG-202-derived bait, and pJG4–6-derived prey plasmids were transformed into DY-1 cells. Cultures were grown to an initial concentration of 1 × 107 cells/ml in 10 ml of SC medium lacking uracil, histidine, and tryptophan. Cells were then washed in water and induced for 6 h in 20 ml of 2% galactose-1% raffinose medium lacking uracil, histidine, and tryptophan. The interactions between the fusion proteins were detected by the quantitative β-galactosidase assay on 5 × 106 permeabilized cells. The β-galactosidase activity was determined by the following formula: β-galactosidase activity = 1000 × A420/(t × V × A600), where t = time of the reaction (in minutes) and V = volume of culture used in the assay (in milliliters).
Co-immunoprecipitation
DY-1 cells were transformed with pCM190- and pJG4–6-derived expression vectors and were initially grown to 1 × 107 cells/ml in 10 ml of SC medium lacking uracil and tryptophan. Cells were then centrifuged (2000 × g, 3.5 min), after washing with 20 ml of dH2O, the pellet was resuspended in 20 ml of 2% galactose-1% raffinose medium (Sigma) lacking uracil and tryptophan, grown for 6 h, and centrifuged (2000 × g, 3.5 min). All subsequent steps were carried out at 4 °C. Pellets were resuspended in 400 μl of ice-cold lysis buffer (50 mm Hepes, pH 7.5, 140 mm NaCl, 1 mm Na-EDTA, 1% Triton X-100) supplemented with protease inhibitor mixture (Roche Applied Science) and lysed with a bead beater by using 0.5 g of glass beads/sample. The lysate was centrifuged (13,000 × g, 30 s) and the supernatant collected. Supernatant was incubated on a rotating wheel (overnight, 4 °C) with 15 μl of protein A-Sepharose beads saturated with rabbit-α-Myc monoclonal antibody (Sigma). Following incubation, the unbound supernatant was removed and saved, and the beads were washed twice in 600 μl of lysis buffer with a final resuspension in 30 μl of lysis buffer.
Whole Cell Extract Preparation and Western Blotting
Whole cell extracts were prepared as described previously (22). Protein concentrations were assayed immediately (Bio-Rad Protein Assay), followed by the addition of a half-volume of sample buffer (60% 4× buffer (15% SDS; 40% glycerol, 166 mm Tris); 0.26 m DTT; 7% bromphenol blue) to the sample and boiling for 7 min. The sample was then stored at −20 °C until it was run on a 7.5% SDS-polyacrylamide gel. Following transfer, nitrocellulose membranes were stained with 0.1% Ponceau S and imaged, then destained with TEN+T (20 mm Tris-HCl, 1 mm EDTA, 0.14 m NaCl, 0.05% Tween 20). Detections were carried out with mouse anti-HA (1:1000; Sigma), mouse anti-Myc (1:5000; Sigma), and rabbit anti-LexA (1:3000; ABR) primary antibodies, in conjunction with Alexa Fluor 488 goat anti-mouse IgG and Alexa Fluor 647 goat anti-rabbit IgG (1:2500; Invitrogen) secondary antibodies.
Growth Assays
Spot plate growth assays involving plasmid shuffle strains were performed by growing cells to a concentration of 2 × 107 cells/ml. Cultures were then serially diluted, and 5-μl aliquots were spotted onto SC plates lacking uracil and leucine (to select for plasmid maintenance), which were then incubated at 30 °C for 2–4 days. Spotting assays for genotoxic sensitivity were performed in the same manner on plates that were untreated or supplemented with either HU or MMS (Sigma), except that for MCM subunit genomic overexpression assays the aliquots were spotted on YPD plates, either with or without HU or MMS added. In some cases, doxycycline (6 μg/ml) was added to the plates to repress expression from pCM190-derived constructs.
RESULTS
To determine the way in which DDK associates with the Mcm2–7 helicase, we systematically examined the extent to which Dbf4 and Cdc7 are able to interact with each one of the MCM subunits. In the case of Dbf4, two-hybrid analysis revealed a robust interaction with Mcm2, and a much weaker, but reproducible, association with Mcm6 (Fig. 1A). To confirm these results, we carried out co-immunoprecipitation trials using extracts from a series of budding yeast transformants overexpressing Myc-tagged Dbf4 and one each of the MCM subunits that had been tagged with HA epitopes. In good agreement with the two-hybrid analysis, only Mcm2 and Mcm6 were pulled down with Dbf4, although in this case the extent of interaction with these two MCM subunits appeared similar (Fig. 1B). Contrasting these findings, similar analyses with Cdc7 indicated an interaction with multiple MCM subunits, but, notably, Mcm2 and Mcm6 were not among these (Fig. 1, C and D). When comparing the two-hybrid and co-immunoprecipitation results for Cdc7, Mcm4 and Mcm5 were found to interact in both cases.
FIGURE 1.

Dbf4 and Cdc7 interact with mutually exclusive subsets of Mcm2–7 subunits. A and C, two-hybrid assays were carried out using either bait construct pEG-Dbf4 (A) or pEG-Cdc7 (C), whereas pJG-Mcm2, -Mcm3, -Mcm4, -Mcm5, -Mcm6, and -Mcm7 were used as prey constructs, along with pJG4–6 as an empty vector control. To confirm that all baits and preys were expressed properly, culture aliquots were removed just prior to the measurement of β-galactosidase activity, and whole cell extracts were prepared and subjected to immunoblot analysis. The average of three replicates is shown ± S.D. (error bars). Bait proteins were detected with anti-LexA antibodies, and prey proteins were detected with anti-HA antibodies. Ponceau S staining of the membrane was carried out to determine relative sample loading. B and D, immunoprecipitation (IP) of Myc-tagged Dbf4 (B) or Cdc7 (D) was carried out in strains overexpressing HA-tagged Mcm2, Mcm3, Mcm4, Mcm5, Mcm6, or Mcm7. Shown are immunoblots of IP and supernatant (S) fractions detected with anti-HA or anti-Myc antibodies. 20 μg of input and one-fourth of the final bead suspension were loaded for the Dbf4 IP immunoblot, whereas 50 μg of input and one-fourth of the final bead suspension were loaded for the Cdc7 IP immunoblot. Ponceau S staining of the membrane was carried out to determine relative sample loading.
Because our analysis pointed to Mcm2 being the major MCM subunit bound by Dbf4, we decided to examine this interaction in greater detail. A series of MCM2 truncation mutants was constructed (Fig. 2A), and we assessed the ability of the corresponding proteins to interact with Dbf4, as above. Separating Mcm2 into N-terminal(1–504) and C-terminal(505–868) fragments demonstrated that neither was sufficient to mediate normal levels of binding to Dbf4 (Fig. 2B). Given that removal of the N-terminal region reduced association with Dbf4 to a greater extent, we further dissected this part of the protein and determined that removal of the N-terminal 63 amino acids resulted in an abrogation of the Dbf4-Mcm2 interaction (Fig. 2, C and D, and results not shown). It has been shown previously that one of two nuclear localization signals (NLSs) for Mcm2–7 resides very near to the N terminus of Mcm2 (amino acids 5–9) (28). Even though the expression constructs we had used for the interaction assays above included additional NLS sequences as part of the protein fusions, we investigated the effect of the original nuclear localization signal by restoring this sequence (Δ2–4,10–63, Fig. 2A). As with the original N-terminal truncation, association with Dbf4 was disrupted with this mutant (Fig. 2, E and F). In contrast, the Δ2–4,10–63 mutation had little effect on the interaction between Mcm2 and neighboring MCM ring subunit Mcm6 (Fig. 3).
FIGURE 2.

An N-terminal Mcm2 region mediates interaction with Dbf4. A, Mcm2WT and mutant cassettes used in this study. The location of the conserved MCM box, including Walker A, Walker B, and arginine finger motifs, is indicated. B, C, and E, two-hybrid assays carried out using bait construct pEG-Dbf4. pJG-Mcm2 (WT), -Mcm2(1–504), -Mcm2(505–868), -Mcm2Δ63, and -Mcm2Δ2–4,10–63 were used as prey. The average of three replicates is shown ± S.D. (error bars). Immunoblot analyses to verify bait and prey expression were carried out as described for Fig. 1. D and F, immunoprecipitation (IP) of Myc-tagged Dbf4. Shown are immunoblots of IP and supernatant (S) fractions detected with monoclonal anti-HA (Mcm2 detection) and anti-Myc antibodies (Dbf4 detection). 20 μg of input and one-fourth of the final bead suspension were loaded.
FIGURE 3.

Mcm2Δ2–4,10–63 maintains an interaction with Mcm6. A, two-hybrid assays were carried out using bait construct pEG-Mcm2. pJG-Mcm3, -Mcm4, -Mcm5, Mcm6, -Mcm7, and pJG-4–6 (empty) were used as prey. The average of three replicates is shown ± S.D. (error bars). Immunoblot analysis to verify bait and prey expression was carried out as described for Fig. 1. B, two-hybrid assays were carried out using prey constructs pJG-Mcm6 and pJG-4–6 (Empty). pEG-Mcm2 and pEG-Mcm2Δ2–4,10–63 were used as bait. The average of three replicates is shown ± S.D. Immunoblot analysis to verify bait and prey expression was carried out as described in the legend for Fig. 1.
Consistent with our findings, previous work from the Stillman laboratory has identified a region of Mcm4 (amino acids 175–333) that interacts with the Dbf4/Cdc7 complex which they referred to as the DDK docking domain (13). We therefore proceeded to assess the relative importance of the Dbf4-Mcm2 and Cdc7-Mcm4 interactions for cell proliferation and DNA replication using the Mcm2Δ2–4,10–63 (henceforth referred to as Mcm2ΔDDD) and Mcm4ΔDDD mutants. Strains were established for which the single genomic copy of either MCM2 or MCM4 was deleted, and growth was supported by CEN (single copy per cell) plasmid-based expression of either wild-type or mutant Mcm2 or Mcm4. When Mcm2ΔDDD and Mcm4ΔDDD were used to support growth in mcm2 and mcm4 deletion strains, respectively, modest growth impairment was observed relative to what was seen when their wild-type counterparts were used (Fig. 4A). We assessed these same strains for DNA replication by first arresting cultures in late G1 phase using the mating pheromone α-factor and then removing the α-factor to allow for a synchronous release into the cell cycle. Both the Mcm2ΔDDD and Mcm4ΔDDD strains showed slight, but reproducible, defects traversing S phase compared with their wild-type counterparts (Fig. 4B).
FIGURE 4.
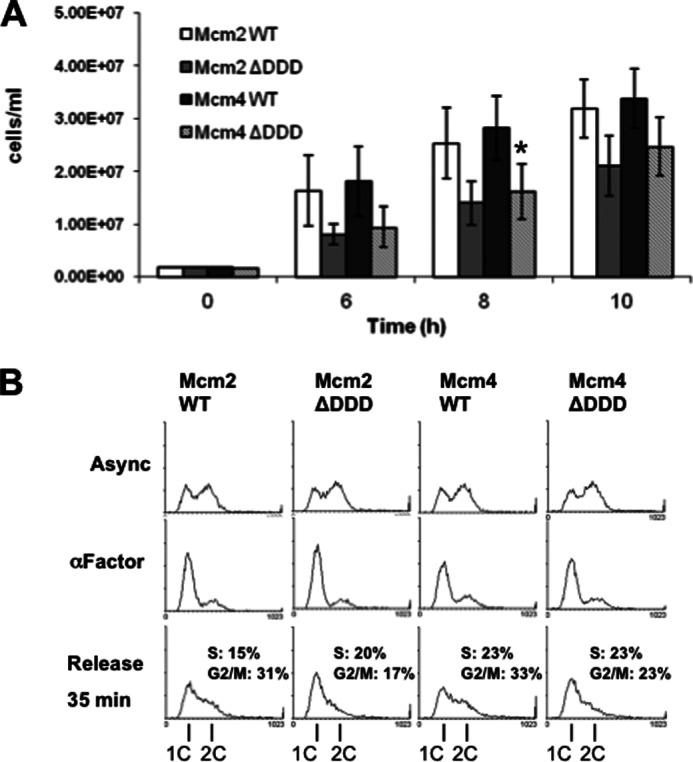
Mcm2 and Mcm4ΔDDD mutants have modest growth defects. CEN/ARS plasmid constructs YCplac111-Mcm2WT and -Mcm2ΔDDD were used to support growth in mcm2::HIS3 background, whereasYCplac111-Mcm4WT and -Mcm4ΔDDD were used to support growth in a mcm4::KanMX background. A, cultures were grown in selective media and cell concentrations determined at the indicated time points. The average of three replicates is shown ± S.D. (error bars). The asterisk indicates a significant difference between Mcm4ΔDDD and Mcm4WT at the 8 h time point (paired Student's t test, p < 0.05). B, asynchronous (Async) cultures were arrested in α-factor (30 μg/ml) for 2.5 h followed by release into pheromone-free medium containing 50 μg/ml Pronase E (Sigma) with samples taken for FACS analysis. Areas representing S and G2/M phase cells for the released cells were determined using WinMDI and then converted to percentages of total cells in each sample as indicated.
The above results indicated that abrogation of either the Dbf4-Mcm2 or Cdc7-Mcm4 interaction had only minor consequences for DNA replication and cell cycle progression. This suggests a functional overlap, such that either one of these interactions is sufficient to target the DDK complex to Mcm2–7. To investigate whether the Dbf4-Mcm2 and Cdc7-Mcm4 associations represent redundant interaction mechanisms, the Mcm2ΔDDD and Mcm4ΔDDD strains were crossed, sporulation was induced in the diploids, and the resultant tetrads were dissected. Of 55 spores analyzed, none was mcm2Δ, mcm4Δ supported by episomal Mcm2ΔDDD, and Mcm4ΔDDD, whereas in a control cross of the Mcm2 and Mcm4 wild-type plasmid shuffle strains, 10 of 36 spores analyzed were both mcm2Δ and mcm4Δ supported by episomal Mcm2WT and Mcm4WT. These results suggest that the combination of Mcm2ΔDDD and Mcm4ΔDDD is synthetic lethal, consistent with a model whereby disruption of redundant Dbf4-Mcm2 and Cdc7-Mcm4 interactions simultaneously prevents proper association of the DDK complex with the Mcm2–7 ring.
Given the lethality of the Mcm2ΔDDD and Mcm4ΔDDD combination, we examined an induced disruption of Dbf4 interaction with MCM rings in cells where the Cdc7-Mcm4 association had already been compromised. This was done by transforming the Mcm4ΔDDD and Mcm4WT control strains with either a doxycycline (Dox)-repressible Mcm2 expression vector or an empty vector control. When Dox was present, all four transformants demonstrated comparable growth (Fig. 5A). In the Mcm4WT strain, the absence of Dox and consequent overexpression of Mcm2 resulted in mild growth inhibition (Fig. 5, A and B), consistent with the notion that surplus Mcm2 is able to partially titrate the DDK complex from the Mcm2–7 ring through its interaction with Dbf4. Strikingly, when Mcm2 was overexpressed in the Mcm4ΔDDD strain, the growth defect was much more severe, supporting a model whereby simultaneous disruption of the Dbf4-Mcm2 and Cdc7-Mcm4 interactions compromises the ability of the DDK complex to associate with Mcm2–7. When we overexpressed Mcm4 in the Mcm2WT and Mcm2ΔDDD strains, we similarly saw growth inhibition that was exacerbated in the mutant background. In this case, however, the difference was very slight (Fig. 5A, see highest dilutions) and variable from trial to trial (compare Fig. 5, A and C). To examine whether the growth inhibition observed when Mcm2 was overexpressed in the Mcm4ΔDDD strain was due to a DDK titration effect, we performed another plate growth assay, this time adding Mcm4ΔDDD and Mcm4WT cells transformed with a Dox-repressible Mcm2ΔDDD expression vector as additional controls. In marked contrast to what was observed with Mcm2, Mcm2ΔDDD overexpression had only a minor effect on growth in Mcm4ΔDDD cells (Fig. 5C), likely due to incorporation of the mutant Mcm2 in some Mcm2–7 rings.
FIGURE 5.

Mcm4ΔDDD cells are sensitive to Mcm2 overexpression. Mcm4WT, Mcm4ΔDDD, Mcm2WT, and Mcm2ΔDDD plasmid-supported strains were transformed with either empty pCM190, pCM190-Mcm2WT, or pCM190-Mcm4WT for which expression is under the control of a doxycycline (DOX)-repressible promoter. A, 10-fold serial dilutions of each transformant were spotted on selective media with or without Dox at a starting concentration of 1 × 107 cells/ml and grown for 2 days. B, the same Mcm4WT and Mcm4ΔDDD transformants were grown in selective medium without Dox and the cell concentration determined at the indicated time points. The average of three replicates is shown ± S.D. (error bars). C, serial dilutions were carried as in A, with the addition of strains transformed with pCM190-Mcm2ΔDDD or pCM190-Mcm4ΔDDD, as indicated.
Previously, we reported that Dbf4 motif C mutants weakened for their interaction with Mcm2 are hypersensitive to genotoxic stress (23). This is consistent with the idea that Dbf4/Cdc7 may help to stabilize and/or restart replication forks under checkpoint conditions by associating with Mcm2–7 and phosphorylating the helicase or another fork component. To determine the sensitivity of cells that have compromised Dbf4/Cdc7 association with MCM rings, we repeated the previous experiment for Mcm2 overexpression, but this time we also examined growth on media plates supplemented with different levels of either the DNA-alkylating agent MMS or the ribonucleotide reductase inhibitor HU, both of which impede fork progression. When comparing relative growth between the Mcm4ΔDDD/Mcm2 overexpression and Mcm4ΔDDD/empty vector strains on -Dox alone plates with those on -Dox with either HU or MMS added, the contrast was clearly greater in the presence of these genotoxic agents (Fig. 6; note that images are for plates grown 3 days, as opposed to 2 in Fig. 5).
FIGURE 6.
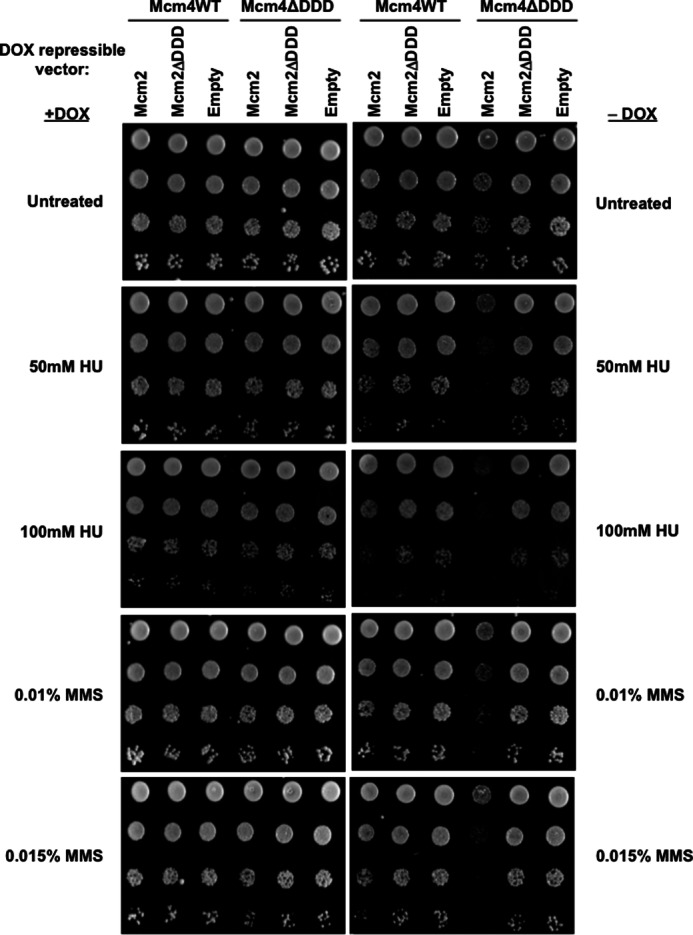
Exposure to genotoxic agents exacerbates growth defects in Mcm4ΔDDD cells overexpressing Mcm2. Mcm4WT and Mcm4ΔDDD plasmid-supported strains transformed with pCM190-Mcm2WT, pCM190-Mcm2ΔDDD, or empty pCM190 were tested for sensitivity to genotoxic agents. 10-fold serial dilutions of each transformed strain were plated on selective media containing the indicated concentrations of HU or MMS, with or without added Dox, at a starting concentration of 1 × 107 cells/ml, and grown for 3 days.
The notion that altered activity or stoichiometry of individual Mcm2–7 subunits can contribute to genomic instability has been previously highlighted by murine studies in which hypomorphic mutants or reduced levels of specific MCM proteins resulted in cancer phenotypes (17, 18). To investigate the consequences of constitutive Mcm2–7 subunit overexpression in Saccharomyces cerevisiae, yeast strains were generated in which the genomic promoters controlling expression of individual MCM genes were replaced with a strong GAL1 promoter. Strikingly, when these strains were exposed to MMS or HU, those overexpressing Mcm2 or Mcm4, but not the other subunits, demonstrated hypersensitivity (Fig. 7). These results support the idea that excess Mcm2 or Mcm4 can sequester Dbf4 and Cdc7 from origin bound Mcm2–7 rings and that this reduction of the Dbf4/Cdc7 association with the MCM helicase has more severe consequences under conditions of replication stress.
FIGURE 7.
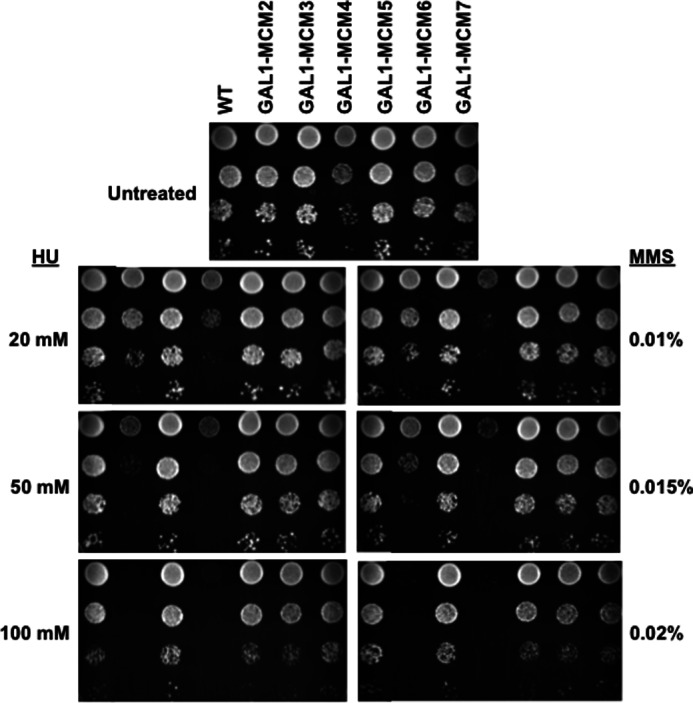
Constitutive overexpression of Mcm2 or Mcm4 imparts sensitivity to genotoxic agents. Yeast strains were generated in which the endogenous promoter for genes encoding each of the Mcm2–7 subunits were individually replaced with the GAL1 promoter. A, 10-fold serial dilutions of each GAL1-MCM overexpression strain were plated on YPG/R (2% galactose, 1% raffinose) containing the indicated concentrations of HU or MMS, at a starting concentration of 1 × 107 cells/ml, and grown for 4 days.
DISCUSSION
Previous studies of DDK have shown that it acts locally throughout S phase to bring about the sequential activation of early, middle, and late firing origins of DNA replication (for review, see Ref. 29). Although it had been well established that the critical physiological targets of DDK are Mcm2–7 subunits, little was known about the way in which this essential replicative kinase initially associates with the MCM complex.
The present study reports the first systematic examination of the way in which the two DDK complex subunits, Dbf4 and Cdc7, each contribute to interaction with the MCM helicase. In the case of Dbf4, our results suggest that the major interaction is with Mcm2, whereas an association with Mcm6, which lies adjacent to Mcm2 in the MCM ring (30), was also observed. Strikingly, these two MCM subunits were not among those we found to associate with Cdc7. These observations are consistent with previous work indicating that Dbf4, but not Cdc7, binds tightly to Mcm2 (21). Examination of various Mcm2 domains revealed that residues in both the N- and C-terminal halves of the protein participate in the Dbf4 interaction, with the N terminus appearing to make the larger contribution. Our identification of a region encompassing most of the N-terminal 63 amino acids being required for interaction with Dbf4 represents a heretofore uncharacterized functional domain in the protein, which resides well away from the conserved C-terminal MCM box that mediates Mcm2 association with other Mcm2–7 subunits (for review, see Ref. 31) and distinct from the actual Mcm2 DDK phosphorylation sites (19, 21). Interestingly, a second Mcm2 region spanning amino acids 204–278 has also been reported to mediate interaction with Dbf4, although the effect of removing this region from full-length Mcm2 was not evaluated (21). Along with our observations for Dbf4, the identification of Mcm4 and Mcm5 as interaction partners for Cdc7 indicates that the two DDK complex components interact with mutually exclusive subsets of the MCM subunits. This suggests at least two mechanisms for DDK complex interaction with the MCM ring. In the first scenario, both Dbf4-MCM and Cdc7-MCM interactions are required, ensuring that monomeric Dbf4 or Cdc7 subunit binding does not interfere with DDK complex MCM targeting. A second possibility is that the interactions are largely redundant, promoting a more efficient DDK complex-MCM association and minimizing the consequences of mutations that interfere with either the Dbf4-MCM or Cdc7-MCM interactions. Our results clearly support the latter model. The identification of a short N-terminal Mcm2 region necessary for interaction with Dbf4, along with the previous identification of a region of Mcm4 that docks with Cdc7, allowed us to contrast the relatively minor effects of disrupting the Dbf4-Mcm2 or Cdc7-Mcm4 interactions on their own, with the severe consequences of breaking both tethers simultaneously. Although we identified Mcm5 and Mcm6 as additional binding partners for Cdc7 and Dbf4, respectively, the synthetic lethality we observed when combining Mcm2ΔDDD and Mcm4ΔDDD mutations suggests that the effect of Cdc7-Mcm5 and Dbf4-Mcm6 interactions is not sufficient for proper DDK association with Mcm2–7. Nevertheless, it will be of interest to evaluate the relative importance of these additional interactions for cell cycle progression in future studies.
Relative to the impaired growth we observed when overexpressing Mcm2 in the presence of Mcm4ΔDDD, the effect of extra Mcm4 in an Mcm2ΔDDD background was modest. This suggests that there are differences in the abilities of soluble Mcm2 and Mcm4 to compete with their origin-bound counterparts for interaction with DDK components. Mcm4 may effectively sequester some free Cdc7 but fail to displace Cdc7 that is already associated with loaded MCM rings. This is likely due to a less efficient targeting of Mcm4 to the nucleus. Unlike Mcm2, Mcm4 has no NLS, and its nuclear import therefore relies on forming complexes that include Mcm2 or Mcm3, which also has an NLS.
The growth defects caused by Mcm2 overexpression in Mcm4ΔDDD cells were exacerbated in the presence of genotoxic compounds known to impede replication fork progression. This is reminiscent of what we saw with Dbf4 motif C mutants that are compromised for interaction with Mcm2 but maintain the region required for interaction with the checkpoint kinase Rad53 (23, 32). Furthermore, recent data from the Davey laboratory demonstrates similar sensitivity when the two Mcm2 DDK target sites (serines 164 and 170) are mutated to alanine (19, 20), suggesting that efficiency of DDK complex association with the MCM ring may be particularly important during conditions of replication stress. An intriguing possibility is that DDK phosphorylation of one or more MCM subunits may help to stabilize and/or restart stalled or blocked replication forks. Consistent with this notion, a genetic screen with the aforementioned Mcm2 DDK target site mutant revealed synthetic lethal interactions with factors implicated in fork progression (20). Another possible scenario is that interaction with the MCM ring serves to direct DDK to other targets at or near the forks. Candidates include Cdc45 and the polα-primase complex, both of which are DDK substrates (9, 33), as well as histone H3, because its phosphorylation by DDK has been shown to play a role in maintaining genomic integrity (34).
Intriguingly, a number of the phenomena we have observed with budding yeast mirror findings in higher eukaryotes. For example, roles for both Mcm2–7 and Dbf4/Cdc7 during replication stress have been identified in Xenopus (35, 36), and altered abundance of both MCM and DDK subunits have been implicated in human cancers (37, 38). The extents to which Dbf4-Mcm2 and Cdc7-Mcm4 interactions are conserved, influence control of DNA replication, and help preserve genome integrity in metazoan organisms are important subjects for future study.
Acknowledgments
We thank Megan Davey and Brent Stead (Western University) for yeast strains and plasmid constructs and Darryl Jones for critical reading of the manuscript.
This work was supported by Natural Sciences and Engineering Research Council of Canada Grant RGPIN 238392.
E. S. Suman and B. P. Duncker, unpublished results.
- MCM
- minichromosome maintenance
- DDD
- DDK docking domain
- DDK
- Dbf4-dependent kinase
- Dox
- doxycycline
- HU
- hydroxyurea
- MMS
- methyl methanesulfonate
- NLS
- nuclear localization signal
- ORC
- origin recognition complex
- SC
- synthetic complete
- CEN
- centromeric.
REFERENCES
- 1. Tanaka S., Diffley J. F. (2002) Interdependent nuclear accumulation of budding yeast Cdt1 and Mcm2–7 during G1 phase. Nat. Cell Biol. 4, 198–207 [DOI] [PubMed] [Google Scholar]
- 2. Semple J. W., Da-Silva L. F., Jervis E. J., Ah-Kee J., Al-Attar H., Kummer L., Heikkila J. J., Pasero P., Duncker B. P. (2006) An essential role for Orc6 in DNA replication through maintenance of pre-replicative complexes. EMBO J. 25, 5150–5158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Chen S., de Vries M. A., Bell S. P. (2007) Orc6 is required for dynamic recruitment of Cdt1 during repeated Mcm2–7 loading. Genes Dev. 21, 2897–2907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Randell J. C., Bowers J. L., Rodríguez H. K., Bell S. P. (2006) Sequential ATP hydrolysis by Cdc6 and ORC directs loading of the Mcm2–7 helicase. Mol. Cell 21, 29–39 [DOI] [PubMed] [Google Scholar]
- 5. Evrin C., Clarke P., Zech J., Lurz R., Sun J., Uhle S., Li H., Stillman B., Speck C. (2009) A double-hexameric MCM2–7 complex is loaded onto origin DNA during licensing of eukaryotic DNA replication. Proc. Natl. Acad. Sci. U.S.A. 106, 20240–20245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Remus D., Beuron F., Tolun G., Griffith J. D., Morris E. P., Diffley J. F. (2009) Concerted loading of Mcm2–7 double hexamers around DNA during DNA replication origin licensing. Cell 139, 719–730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Randell J. C., Fan A., Chan C., Francis L. I., Heller R. C., Galani K., Bell S. P. (2010) Mec1 is one of multiple kinases that prime the Mcm2–7 helicase for phosphorylation by Cdc7. Mol. Cell 40, 353–363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sclafani R. A., Holzen T. M. (2007) Cell cycle regulation of DNA replication. Annu. Rev. Genet. 41, 237–280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Weinreich M., Stillman B. (1999) Cdc7p-Dbf4p kinase binds to chromatin during S phase and is regulated by both the APC and the RAD53 checkpoint pathway. EMBO J. 18, 5334–5346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hardy C. F., Dryga O., Seematter S., Pahl P. M., Sclafani R. A. (1997) Mcm5/Cdc46-Bob1 bypasses the requirement for the S phase activator Cdc7p. Proc. Natl. Acad. Sci. U.S.A. 94, 3151–3155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hoang M. L., Leon R. P., Pessoa-Brandao L., Hunt S., Raghuraman M. K., Fangman W. L., Brewer B. J., Sclafani R. A. (2007) Structural changes in Mcm5 protein bypass Cdc7-Dbf4 function and reduce replication origin efficiency in Saccharomyces cerevisiae. Mol. Cell. Biol. 27, 7594–7602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sheu Y. J., Stillman B. (2010) The Dbf4-Cdc7 kinase promotes S phase by alleviating an inhibitory activity in Mcm4. Nature 463, 113–117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sheu Y. J., Stillman B. (2006) Cdc7-Dbf4 phosphorylates MCM proteins via a docking site-mediated mechanism to promote S phase progression. Mol. Cell 24, 101–113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Heller R. C., Kang S., Lam W. M., Chen S., Chan C. S., Bell S. P. (2011) Eukaryotic origin-dependent DNA replication in vitro reveals sequential action of DDK and S-CDK kinases. Cell 146, 80–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Semple J. W., Duncker B. P. (2004) ORC-associated replication factors as biomarkers for cancer. Biotechnol. Adv. 22, 621–631 [DOI] [PubMed] [Google Scholar]
- 16. Gonzalez M. A., Tachibana K. E., Laskey R. A., Coleman N. (2005) Control of DNA replication and its potential clinical exploitation. Nat. Rev. Cancer 5, 135–141 [DOI] [PubMed] [Google Scholar]
- 17. Shima N., Alcaraz A., Liachko I., Buske T. R., Andrews C. A., Munroe R. J., Hartford S. A., Tye B. K., Schimenti J. C. (2007) A viable allele of Mcm4 causes chromosome instability and mammary adenocarcinomas in mice. Nat. Genet. 39, 93–98 [DOI] [PubMed] [Google Scholar]
- 18. Chuang C. H., Wallace M. D., Abratte C., Southard T., Schimenti J. C. (2010) Incremental genetic perturbations to MCM2–7 expression and subcellular distribution reveal exquisite sensitivity of mice to DNA replication stress. PLoS Genet. 6, e1001110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Stead B. E., Brandl C. J., Davey M. J. (2011) Phosphorylation of Mcm2 modulates Mcm2–7 activity and affects the cell's response to DNA damage. Nucleic Acids Res. 39, 6998–7008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Stead B. E., Brandl C. J., Sandre M. K., Davey M. J. (2012) Mcm2 phosphorylation and the response to replicative stress. BMC Genet. 13, 36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bruck I., Kaplan D. (2009) Dbf4-Cdc7 phosphorylation of Mcm2 is required for cell growth. J. Biol. Chem. 284, 28823–28831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Varrin A. E., Prasad A. A., Scholz R. P., Ramer M. D., Duncker B. P. (2005) A mutation in Dbf4 motif M impairs interactions with DNA replication factors and confers increased resistance to genotoxic agents. Mol. Cell. Biol. 25, 7494–7504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Jones D. R., Prasad A. A., Chan P. K., Duncker B. P. (2010) The Dbf4 motif C zinc finger promotes DNA replication and mediates resistance to genotoxic stress. Cell Cycle 9, 2018–2026 [DOI] [PubMed] [Google Scholar]
- 24. Francis L. I., Randell J. C., Takara T. J., Uchima L., Bell S. P. (2009) Incorporation into the prereplicative complex activates the Mcm2–7 helicase for Cdc7-Dbf4 phosphorylation. Genes Dev. 23, 643–654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Harkins V., Gabrielse C., Haste L., Weinreich M. (2009) Budding yeast Dbf4 sequences required for Cdc7 kinase activation and identification of a functional relationship between the Dbf4 and Rev1 BRCT domains. Genetics 183, 1269–1282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Stead B. E., Sorbara C. D., Brandl C. J., Davey M. J. (2009) ATP binding and hydrolysis by Mcm2 regulate DNA binding by Mcm complexes. J. Mol. Biol. 391, 301–313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ausubel F., Brent R., Kingston R. E., Moore D. D., Seidman J. G., Smith J. A., Struhl K. (1995) Short Protocols in Molecular Biology, 3rd Ed., John Wiley & Sons, New York [Google Scholar]
- 28. Liku M. E., Nguyen V. Q., Rosales A. W., Irie K., Li J. J. (2005) CDK phosphorylation of a novel NLS-NES module distributed between two subunits of the Mcm2–7 complex prevents chromosomal rereplication. Mol. Biol. Cell 16, 5026–5039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Pasero P., Schwob E. (2000) Think global, act local: how to regulate S phase from individual replication origins. Curr. Opin. Genet. Dev. 10, 178–186 [DOI] [PubMed] [Google Scholar]
- 30. Davey M. J., Indiani C., O'Donnell M. (2003) Reconstitution of the Mcm2–7p heterohexamer, subunit arrangement, and ATP site architecture. J. Biol. Chem. 278, 4491–4499 [DOI] [PubMed] [Google Scholar]
- 31. Forsburg S. L. (2004) Eukaryotic MCM proteins: beyond replication initiation. Microbiol. Mol. Biol. Rev. 68, 109–131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Matthews L. A., Jones D. R., Prasad A. A., Duncker B. P., Guarné A. (2012) Saccharomyces cerevisiae Dbf4 has unique fold necessary for interaction with Rad53 kinase. J. Biol. Chem. 287, 2378–2387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Nougarède R., Della Seta F., Zarzov P., Schwob E. (2000) Hierarchy of S-phase-promoting factors: yeast Dbf4-Cdc7 kinase requires prior S phase cyclin-dependent kinase activation. Mol. Cell. Biol. 20, 3795–3806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Baker S. P., Phillips J., Anderson S., Qiu Q., Shabanowitz J., Smith M. M., Yates J. R., 3rd, Hunt D. F., Grant P. A. (2010) Histone H3 Thr-45 phosphorylation is a replication-associated post-translational modification in S. cerevisiae. Nat. Cell Biol. 12, 294–298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Woodward A. M., Göhler T., Luciani M. G., Oehlmann M., Ge X., Gartner A., Jackson D. A., Blow J. J. (2006) Excess Mcm2–7 license dormant origins of replication that can be used under conditions of replicative stress. J. Cell Biol. 173, 673–683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Tsuji T., Lau E., Chiang G. G., Jiang W. (2008) The role of Dbf4/Drf1-dependent kinase Cdc7 in DNA-damage checkpoint control. Mol. Cell 32, 862–869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Bonte D., Lindvall C., Liu H., Dykema K., Furge K., Weinreich M. (2008) Cdc7-Dbf4 kinase overexpression in multiple cancers and tumor cell lines is correlated with p53 inactivation. Neoplasia 10, 920–931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lau K. M., Chan Q. K., Pang J. C., Li K. K., Yeung W. W., Chung N. Y., Lui P. C., Tam Y. S., Li H. M., Zhou L., Wang Y., Mao Y., Ng H. K. (2010) Minichromosome maintenance proteins 2, 3, and 7 in medulloblastoma: overexpression and involvement in regulation of cell migration and invasion. Oncogene 29, 5475–5489 [DOI] [PubMed] [Google Scholar]