

Background: S-Nitrosation is an emerging post-translational modification that is not yet well understood on a molecular level.
Results: We propose a mechanism for S-nitrosation of glutathione transferase P1-1 by S-nitrosoglutathione.
Conclusion: Cys101 is nitrosated in a single step, but Cys47 nitrosation is limited by the rate of helix 2 opening.
Significance: Detailing the mechanism of spontaneous transnitrosation is crucial to understanding how protein S-nitrosation is controlled.
Keywords: Covalent Regulation, Nitric Oxide, Nitrosative Stress, Post-translational Modification, Protein Dynamics, S-Nitrosation, Glutathione Transferase, Transient Kinetics
Abstract
S-Nitrosation is a post-translational modification of protein cysteine residues, which occurs in response to cellular oxidative stress. Although it is increasingly being linked to physiologically important processes, the molecular basis for protein regulation by this modification remains poorly understood. We used transient kinetic methods to determine a minimal mechanism for spontaneous S-nitrosoglutathione (GSNO)-mediated transnitrosation of human glutathione transferase (GST) P1-1, a major detoxification enzyme and key regulator of cell proliferation. Cys47 of GSTP1-1 is S-nitrosated in two steps, with the chemical step limited by a pre-equilibrium between the open and closed conformations of helix α2 at the active site. Cys101, in contrast, is S-nitrosated in a single step but is subject to negative cooperativity due to steric hindrance at the dimer interface. Despite the presence of a GSNO binding site at the active site of GSTP1-1, isothermal titration calorimetry as well as nitrosation experiments using S-nitrosocysteine demonstrate that GSNO binding does not precede S-nitrosation of GSTP1-1. Kinetics experiments using the cellular reductant glutathione show that Cys101-NO is substantially more resistant to denitrosation than Cys47-NO, suggesting a potential role for Cys101 in long term nitric oxide storage or transfer. These results constitute the first report of the molecular mechanism of spontaneous protein transnitrosation, providing insight into the post-translational control of GSTP1-1 as well as the process of protein transnitrosation in general.
Introduction
S-Nitrosation is a post-translational modification resulting from the covalent linkage of nitric oxide (NO) to cysteine thiols. The experimentally verified S-nitrosoproteome is rapidly increasing (1) and S-nitrosation is recognized to control a number of important physiological processes such as vascular homeostasis (2), autophagy (3), the innate immune response (4), and a spectrum of other post-translational modifications (5). Unsurprisingly, dysregulation of protein S-nitrosation has been associated with several diseases, including neurodegenerative disorders, various cancers, and diabetes (reviewed in Ref. 6).
S-Nitrosothiols are formed either by the direct, diffusion controlled reaction between the thiyl radical and NO (7) or via the intermediate N2O3 (8). Several enzymes have been identified as catalysts of protein S-nitrosation reactions (reviewed in Ref. 9). However, the cellular abundance of GSH (10) means that S-nitrosoglutathione (GSNO)2 is the major S-nitrosothiol in vivo (11) and therefore spontaneous transnitrosation of protein thiols by GSNO is a significant route for protein S-nitrosation. In support of this, cellular GSNO appears to be in equilibrium with protein-SNOs, with an increase in GSNO levels leading to increased protein S-nitrosation (12). In addition, GSNO treatment causes widespread S-nitrosation of yeast proteins (13) and GSNO is commonly used to stimulate S-nitrosation of purified protein in vitro.
Rates of transnitrosation by GSNO or other NO donors have been measured for small molecule thiols (14), bovine serum albumin (15), hemoglobin (16), and thioredoxin (17). However, the kinetic mechanism of transnitrosation has yet to be reported for any protein. Because protein S-nitrosation is largely not under enzymatic control, elucidating possible mechanisms of spontaneous GSNO-mediated transnitrosation is crucial to understanding how S-nitrosation is controlled, as well as targeted to specific cysteines.
Human glutathione transferase P1-1 is a well characterized detoxification enzyme that also functions to regulate cell proliferation by inhibiting c-Jun N-terminal kinase (18). GSTP1-1 S-nitrosated at cysteines 47 and 101 has previously been discovered in vivo (19–21) and produced in vitro (22, 23). The modification is highly disruptive, significantly impairing the catalytic activity of the enzyme (22). Here, we establish a minimal mechanism for GSTP1-1 transnitrosation and denitrosation at both Cys47 and Cys101. This model gives new insight into the post-translational control of GSTP1-1 activity, as well as protein transnitrosation reactions in general.
EXPERIMENTAL PROCEDURES
Protein Expression, Purification, and S-Nitrosation
The pET-15b vector encoding N-terminal His6-tagged GSTP1-1 was a kind gift from S. Y. Blond (Centre for Pharmaceutical Biotechnology, University of Illinois, Chicago, IL). His6-GSTP1-1 was expressed in Escherichia coli T7 cells and purified by Co2+ affinity chromatography as described previously (24). C47S, C101S, and C47S/C101S mutant GSTP1-1 were prepared by site-directed mutagenesis and purified identically to the wild-type protein. S-Nitrosated GSTP1-1 was prepared by mixing fully reduced protein with a 50-fold molar excess of GSNO or CysNO at 37 °C for 1 h. Excess nitrosating agent was removed by dialysis or buffer exchange. S-Nitrosation stoichiometry was determined by UV-difference spectroscopy as described in Ref. 22 based on an S-NO extinction coefficient of 750 m−1 cm−1 at 330 nm.
Synthesis of GSNO and CysNO
GSNO and CysNO were synthesized as described previously (25) and frozen in aliquots at −80 °C. SNO concentrations were determined by UV absorbance and confirmed after thawing. Solutions were protected from light throughout subsequent experiments to limit photolysis of the S-NO bond (26).
Steady-state Fluorescence
Steady-state fluorescence measurements were performed on a Jasco FP-6300 fluorimeter. Excitation was at 280 nm and emission light was collected at 342 nm. Equilibrium titrations were performed by measuring the fluorescence intensity of 2 μm S-nitrosated protein prepared as described above but with varying concentrations of GSNO or CysNO. The fluorescence intensities showed a hyperbolic dependence on reagent concentration, with the exception of the reaction between GSNO and C47S GSTP1-1, which fit better to a Hill equation with n = −0.5.
Kinetics Experiments
Kinetics of GSTP1-1 nitrosation or denitrosation were measured by following the change in intrinsic protein fluorescence using an SX-18MV stopped-flow instrument (Applied Photophysics). Excitation was at 280 nm and emission light was collected using a 320-nm long-pass filter. Final protein concentrations were 1 μm and all experiments were performed in 20 mm phosphate buffer with 150 mm NaCl, 2 mm EDTA, and 0.02% NaN3. At least three traces were recorded and averaged at each ligand concentration. α2 unfolding rates were measured under the same conditions by following the initial increase in protein fluorescence following mixing with 7.5 m urea (27).
Kinetic Data Analysis
Fluorescence transients for C101S or C47S GSTP1-1 nitrosation were fit to a double-exponential function using SigmaPlot version 11.0 or the stopped-flow instrument software. The second exponent was fixed to the kobs value from a single-exponential fit of the fluorescence transient for the reaction of C47S/C101S GSTP1-1 with GSNO. In this way, the kinetics data of the single mutants were corrected for the slow, substoichiometric S-nitrosation at the third site. The first exponent from the fits is subsequently reported here as the kobs for Cys47 nitrosation (C101S mutant) or Cys101 nitrosation (C47S mutant). The quality of the fits was judged by inspection of the residuals.
The data for Cys101 nitrosation were fit to Equation 1, for a single-step reaction.
For S-nitrosation at Cys47 at all temperatures, the data were fit to Equation 2, for a two-step reaction following a conformational selection mechanism.
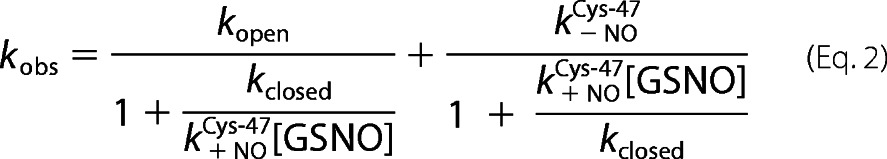 |
kopen and kclosed are the forward and reverse rate constants for the pre-nitrosation conformational equilibrium, and k+NOCys-47 and k−NOCys-47 are the rate constants for Cys47 nitrosation and denitrosation, respectively.
The data for GSH-mediated denitrosation at either Cys47-NO or Cys101-NO were fit to Equation 3, describing a single-step denitrosation dependent on GSH concentration.
The temperature dependence of Cys47 nitrosation or α2 unfolding was analyzed using an Eyring plot generated from Equation 4 (28).
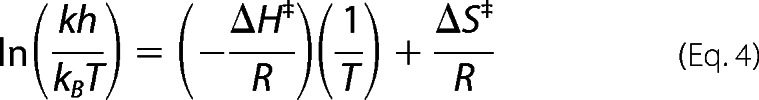 |
Where k is the nitrosation rate constant, h is Planck's constant, kB is the Boltzmann constant, T is absolute temperature, and R is the universal gas constant. ΔH‡ was calculated from the slope of a plot of the left-hand side of Equation 4 against 1/T. ΔS‡ was determined from Equations 5 and 6.
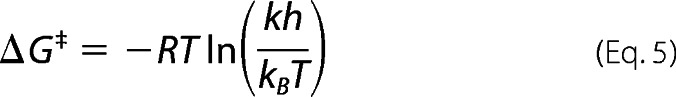 |
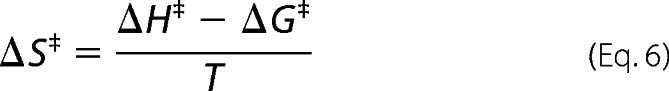 |
Isothermal Titration Calorimetry
ITC experiments were performed using a MicroCal VP-ITC instrument. 9.4 mm GSNO or 2 mm GSO3− in the syringe was injected into the sample cell containing 61.4 μm (C47S/C101S) or 114 μm (C101S) protein in 20 mm phosphate buffer with 150 mm NaCl, 2 mm EDTA, and 0.02% NaN3. The temperature was kept constant at 37 °C to match the kinetics experiments. Data were analyzed using Origin 7.0 (MicroCal).
RESULTS
GSTP1-1 S-Nitrosation Probed by Tryptophan Fluorescence
GSTP1-1 S-nitrosated at cysteines 47 and 101 has previously been discovered in vivo (19–21) and produced in vitro (22, 23) by reaction with GSNO. Here, we generated a series of cysteine mutants with the aim of examining the kinetics of nitrosation of GSTP1-1 in detail. Reaction of wild-type GSTP1-1 with GSNO or CysNO resulted in S-nitrosated protein with a stoichiometry of 2 ± 0.1 S-nitrosothiols per subunit, consistent with previous reports. Furthermore, mutation of either Cys47 or Cys101 to serine reduced the number of S-nitrosocysteines to 1.2 ± 0.1 per subunit. Surprisingly, we also detected a substoichiometric (0.3 ± 0.1/subunit) S-nitrosation of C47S/C101S double mutant GSTP1-1. The identity of the third S-nitrosation site (or sites) is unclear, but may represent S-nitrosation at another cysteine, or even nitration of tryptophan or tyrosine residues (29). Electrospray ionization-mass spectrometry did not suggest the presence of any additional modifications such as S-glutathionylation (data not shown), consistent with the mass spectrometric analysis of Lo Bello et al. (22). This alternate nitrosation event is unlikely to be physiologically interesting because it has not been detected in vivo, affects only a minor subpopulation of protein and occurs with very slow kinetics (Figs. 1C and 2B). We therefore corrected all kinetics data for S-nitrosation at the alternate site, as described under “Experimental Procedures.” In this way, only S-nitrosation at Cys47 (C101S mutant) or Cys101 (C47S mutant) were studied.
FIGURE 1.

Fluorescence equilibrium titration of cysteine mutants of GSTP1-1 with GSNO. A, representation of dimeric human GSTP1-1 (Protein Data Bank code 6GSS). The sites of nitrosation, Cys47 and Cys101, are indicated (pink). Trp28 and Trp38 (cyan) are probes of the nitrosation events. B, quenching of fluorescence upon reacting 2 μm wild-type and mutant GSTP1-1 with 100 μm GSNO at 37 °C for 1 h. Reduced (cyan), wild-type (black), C101S (green), C47S (red), C47S/C101S (blue) are indicated (λex = 280 nm). C, 2 μm protein was titrated with increasing amounts of GSNO and equilibrated for 1 h at 37 °C with λex = 280 nm and λem = 342 nm. The solid lines are hyperbolic fits to the data, except for the C47S mutant, where a Hill equation with n = −0.52 gave a better fit. KD values from the fits are reported in Table 1.
FIGURE 2.

Kinetics of GSTP1-1 nitrosation monitored by intrinsic fluorescence. A, representative transients showing the decrease in fluorescence when 1 μm of different cysteine mutants of GSTP1-1 was mixed with 500 μm nitrosating agent (GSNO or CysNO) at 37 °C. Curves were normalized to have the same starting fluorescence. B, S-nitrosation of 1 μm wild-type GSTP1-1 with 500 μm GSNO. The data were fit to a triple-exponential function. The residuals show an excellent fit to the data when the three exponential terms are fixed to the observed rate constants for nitrosation at Cys47, Cys101, and the alternate site, respectively, determined from fitting the data in B.
Cys101, and Cys47 especially, are located close to the two tryptophan residues (Trp28 and Trp38) in domain 1 of GSTP1-1 (Fig. 1A). These residues provide a convenient probe of S-nitrosation, as the modification results in significant quenching of intrinsic protein fluorescence, probably due to FRET between the indole ring and S-NO moiety (30) (Fig. 1B). Equilibrium fluorescence titrations of wild-type and mutant GSTP1-1 reveal that both Cys47 and Cys101 are S-nitrosated with relatively low affinity (low micromolar KD values) by either GSNO or CysNO (Fig. 1C and Table 1). The equilibrium titration of Cys101 with GSNO shows some evidence of negative cooperativity, with the data better described by a Hill equation with n = −0.5. This is discussed below with reference to Fig. 6. Consistent with the kinetics data in Fig. 2B, the alternate nitrosation site (C47S/C101S mutant) has a KD 10-fold higher than either of the canonical sites.
TABLE 1.
Kinetic and equilibrium constants for S-nitrosation of GSTP1-1 according to Scheme 1
| Ligand | Nitrosation site | kopenα2 | k+NO | k−NO | k−NOGSH | KD |
|---|---|---|---|---|---|---|
| s−1 | m−1 s−1 | s−1 | m−1 s−1 | μm | ||
| GSNO | Cys47 | 0.022 ± 0.001 | NDa | 0.0008 ± 0.0001 | 46 ± 3 | 10 ± 0.5 |
| Cys101 | NA | 40 ± 3b | 0.001 ± 0.0002 | 1.4 ± 0.06 | 4 ± 4 | |
| CysNO | Cys47 | 0.07 ± 0.02 | ND | 0.0008 ± 0.0001 | NAc | 8 ± 0.6 |
| Cys101 | NA | 100 ± 5 | 0.001 ± 0.0002 | NA | 15 ± 2 |
a ND, not determined.
b Estimated from fit to linear portion of curve.
c NA, not applicable.
FIGURE 6.

Cys101 nitrosation kinetics. A, 1 μm C47S GSTP1-1 was mixed with varying concentrations of GSNO (closed circles) or CysNO (open circles) under pseudo-first order conditions at 37 °C. The primary data were fit to a double exponential function. One exponential term was fixed to the observed nitrosation rate of C47S/C101S GSTP1-1, and the second rate is reported here as kobs, representing the nitrosation of Cys101 alone. The data for the CysNO reaction correspond to a one-step mechanism and were fit to Equation 1. Rate constants are reported in Table 1. The rate of the GSNO reaction appears to saturate at high [GSNO], which is likely due to steric hindrance at the dimer interface, visualized in B. B, model of GSTP1-1 (Protein Data Bank code 6GSS) showing GSH (cyan) attached to Cys101 (pink) on one subunit in a hypothetical transnitrosation reaction. It is apparent that a second GSNO molecule is not readily accommodated at the dimer interface, resulting in negative cooperativity for S-nitrosation of Cys101 on the adjacent subunit. C, structures of GSNO and CysNO.
The quenching of tryptophan fluorescence upon S-nitrosation of GSTP1-1 was used to probe the kinetics of the modification at Cys47 and Cys101. Representative transients from mixing 500 μm nitrosating agent with 1 μm protein reveal distinct differences between the rates and amplitudes of nitrosation at Cys47 and Cys101 (Fig. 2B). S-Nitrosation at Cys47 occurs more rapidly and produces a larger fluorescence change than at Cys101, whereas changing the nitrosation agent to CysNO results in even faster reaction kinetics. To demonstrate that the kinetics of nitrosation of the Cys-mutant GSTP1-1 reliably reports on the reactions of the wild-type protein, the fluorescence transient for wild-type GSTP1-1 mixed with GSNO was fit to a triple-exponential function (Fig. 2C). By fixing the three observed rate constants to the kobs calculated from fits to the C101S, C47S, and C47S/C101S nitrosation transients, respectively, an excellent fit was obtained for the wild-type data. This demonstrates that GSTP1-1 S-nitrosation kinetics at different sites can be reliably partitioned by cysteine mutagenesis.
S-Nitrosation at Cys47 Is Preceded by the Isomerization of Helix α2
To characterize the kinetics of Cys47 nitrosation, the concentration dependence of the observed rates at 37 °C were evaluated for both GSNO and CysNO under pseudo-first order conditions (Fig. 3). For both nitrosating agents, the plots appeared hyperbolic, indicating that more than one event contributes to Cys47 S-nitrosation kinetics. Assuming the simplest case (a two-step mechanism), two common scenarios are consistent with these data: 1) low affinity S-nitrosation of Cys47 followed by a slow conformational change (induced-fit, rapid equilibrium assumption); or 2) a slow equilibrium between protein conformers, followed by the chemical S-nitrosation event (conformational selection) (31). If the induced fit model is correct, then the data should correspond to an increasing hyperbolic function, whatever the relative magnitudes of the rate constants for the first and second steps. A conformational selection mechanism, however, will produce an inverted hyperbolic dependence on ligand concentration if the forward rate constant for the first step becomes smaller than the reverse rate constant for the second step. We therefore sought to discriminate between these two scenarios by following the reaction between GSNO and Cys47 of GSTP1-1 at different temperatures, potentially altering the relative magnitudes of the conformational and chemical steps. Fig. 4A clearly demonstrates that at 15 °C the concentration dependence of Cys47 nitrosation inverts to give a decreasing hyperbolic function. This is strong evidence that the formation of S-nitrosated GSTP1-1 is limited by a pre-equilibrium between protein conformers. With reference to Scheme 1, lowering the temperature to 15 °C causes kopen to become smaller than k−NOCys-47, giving the plot in Fig. 4A. Above 20 °C, kopen is larger than k−NOCys-47, resulting in an increasing hyperbolic function. The data at all temperatures were therefore fit to Equation 2, derived from Scheme 1. Because the two steps are not independently resolved in the fluorescence transients, only kopen (the saturation value) and k−NOCys-47 (the y intercept) can be resolved from these data. Deriving k−NOCys-47 from the plot in Fig. 3 is error-prone because the y intercept is close to 0, so this value was instead determined in an independent experiment (Fig. 7). Kinetic constants for S-nitrosation of Cys47 at 37 °C are reported in Table 1.
FIGURE 3.

Concentration dependence of the rate of S-nitrosation of Cys47 reveals a multistep mechanism. 1 μm C101S GSTP1-1 was mixed with varying concentrations of GSNO (closed circles) or CysNO (open circles) under pseudo-first order conditions at 37 °C. The primary data were fit to a double exponential function. One exponential term was fixed to the observed nitrosation rate of C47S/C101S GSTP1-1, and the second rate is reported here as kobs, representing the nitrosation of Cys47 alone. The data in the figure were fit to Equation 2.
FIGURE 4.

Temperature dependence of Cys47S-nitrosation suggests that a conformational equilibrium limits the chemical step. A, the experiment described in the legend to Fig. 3 was repeated at different temperatures, with varying [GSNO] and 1 μm C101S GSTP1-1. The solid lines are fits to Equation 4. Values of kopen from the fits were analyzed with an Eyring plot (B), yielding ΔH = 114 ± 23 kJ mol−1 and TΔS = 26 ± 23 kJ mol−1. C, the energetics of α2 unfolding were determined independently in an Eyring plot of the temperature dependence of the rate of unfolding of α2 (kU) in 7.5 m urea, yielding ΔH = 118 ± 3 kJ mol−1 and TΔS = 50 ± 3 kJ mol−1. D, domain 1 of GSTP1-1. When α2 is closed, the thiol of Cys47 is buried in a hydrophobic pocket (blue surface) at the active site. S-Nitrosation at Cys47 requires α2 to be in its open conformation, and is therefore limited by the equilibrium between the open and closed states (kclosed/kopen).
SCHEME 1.
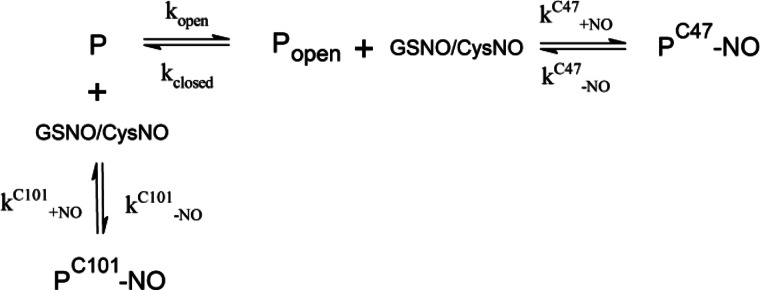
FIGURE 7.

Denitrosation of GSTP1-1-NO. A, S-nitrosated cysteine mutants of GSTP1-1 (μm) were prepared as described under “Experimental Procedures” and denitrosated using 1 mm GSH or DTT at 37 °C, causing an increase in intrinsic protein fluorescence. Observed rate constants for Cys47 (closed circles) or Cys101 (open circles) denitrosation with GSH were determined by fitting the primary data to a triple exponential function. Two of the exponents were fixed to the spontaneous denitrosation rate and the rate of denitrosation of C47S/C101S GSTP1-1, respectively, and the third rate is reported here as kobs, representing the rate of GSH-mediated denitrosation of Cys47 or Cys101 alone. The GSH concentration dependence of these rates is shown in B and the solid line is a fit to Equation 3.
Helix α2, covering the active site of GSTP1-1, is known to be highly dynamic in the apoenzyme (32) and has been linked to the reactivity of Cys47 (33). We therefore hypothesize that the conformational equilibrium that precedes chemical modification of Cys47 is between the open and closed states of α2 (Fig. 4D). When α2 is in its closed conformation, the Cys47 thiol is buried in a hydrophobic pocket in domain 1, inaccessible to transnitrosation by GSNO or CysNO. The rate of Cys47 nitrosation would therefore be limited by kopen, the rate constant for accessing the open state of α2. Eyring analysis of kopen gives ΔH‡ = 114 ± 23 kJ mol−1 and TΔS‡ = 26 ± 23 kJ mol−1 (Fig. 4B), suggesting a large, entropically favorable conformational change, such as the unfolding of α2. To test this with an independent approach, we examined the temperature dependence of the unfolding of α2 in 7.5 m urea (Fig. 4C). At high urea concentrations, α2 unfolding occurs rapidly, before the rest of the protein, and can be measured independently by stopped-flow fluorescence (27). Eyring analysis of the unfolding rate of α2 gives ΔH‡ = 118 ± 3 kJ mol−1 and TΔS‡ = 50 ± 3 kJ mol−1, in excellent agreement with the energetics of kopen. These values are also very close to estimates of α2 unfolding enthalpy from NMR (32) and ligand binding (34) experiments. Finally, high glycerol is known to impair GSTP1-1 activity by dampening α2 dynamics (33), and we found that 40% glycerol caused a 10-fold decrease in kopen (data not shown). Together, these data constitute strong evidence that S-nitrosation of GSTP1-1 at Cys47 is limited by an equilibrium between the open and closed conformations of α2.
GSNO binding has been proposed to precede S-nitrosation of GSTP1-1 (23). In support of this, GSNO was co-crystallized with GSTP1-1 and was found to bind the active site in essentially the same orientation as GSH (23, 35) (Fig. 5D). However, our finding that CysNO can readily nitrosate Cys47 of GSTP1-1 (Fig. 3) does not support the idea that GSNO binding necessarily precedes Cys47 S-nitrosation. To test this further, we examined the interaction of GSNO with GSTP1-1 at 37 °C using isothermal titration calorimetry. Titration of C101S GSTP1-1 with GSNO revealed at least one exothermic event (Fig. 5C), consistent with previous reports (23). However, we find no evidence from ITC for GSNO binding to C47S/C101S GSTP1-1 (Fig. 5A), with the heats essentially identical to those for GSNO titrated into buffer (Fig. 5B). This result does not appear to be due to an alteration of the protein structure by the C47S mutation, as the double mutant retains substantial activity (36) and binds glutathione sulfonate with similar affinity to the wild-type (data not shown), suggesting that the G-site of the protein is intact. The exothermic heats in Fig. 5A are too small to be attributed to any significant binding event, but may represent S-nitrosation at the third site. After correction for heats of dilution, the data for the interaction of GSNO with C101S GTP1-1 were fit to a model for a single set of binding sites. Accurate values for enthalpy and stoichiometry could not be derived from these data because the interaction was too weak to establish a thermogram pre-transition baseline. However, the KD, calculated from the slope of the transition, was 14.1 ± 0.8 μm, in good agreement with the KD determined from fluorescence titration for Cys47 nitrosation (Table 1).
FIGURE 5.

GSNO binding does not precede Cys47S-nitrosation. 9.4 mm GSNO in the ITC syringe was titrated into: A, 61.4 μm C47S/C101S GSTP1-1; B, buffer; or C, 114 μm C101S GSTP1-1. Injection volumes were 5 μl and the temperature was 37 °C for all experiments. The data in C was fit to a single-site binding model, giving a KD of 14 μm. D, single subunit of GSTP1-1, showing the binding mode of GSNO and the proximity of the NO moiety (black box) to the Cys47 thiol (Protein Data Bank code 2A2S).
S-Nitrosation at Cys101 Occurs in a Single Step, Limited by Steric Hindrance at the Dimer Interface
The rate of S-nitrosation of Cys101, at the dimer interface of GSTP1-1, was also investigated at 37 °C with increasing concentrations of GSNO or CysNO. Unlike S-nitrosation of Cys47, the concentration dependence of the observed rate constants for Cys101 nitrosation is qualitatively different for GSNO or CysNO (Fig. 6A). The rate of the reaction between Cys101 and CysNO shows a linear dependence on CysNO concentration, indicating that S-nitrosation occurs in a single step. In contrast, the same plot for Cys101 nitrosation by GSNO shows some curvature at high GSNO concentrations. Given that GSNO and CysNO function essentially identically as NO donors in transnitrosation reactions, it is unlikely that they would nitrosate Cys101 by substantially different mechanisms. In addition, there is no known GSNO (or GSH) binding site near Cys101, so it is also unlikely that a direct interaction between GSNO and the protein occurs before S-nitrosation of the cysteine thiol. Instead, we propose that the saturation effect in the plot in Fig. 6A is a result of negative cooperativity between Cys101 nitrosation sites on each subunit. This interpretation is consistent with the equilibrium titrations in Fig. 1C, where Cys101 nitrosation fits a Hill equation with n = −0.5. The solvent accessible cleft at the dimer interface of GSTP1-1 is relatively narrow, with only 6.5 Å separating the Cys101 thiols on each chain. Unless considerable structural rearrangement occurs, this space is insufficient to accommodate two GSNO molecules simultaneously without significant steric clashes. The situation can be visualized in Fig. 6B: GSH attached to Cys101 on one subunit (analogous to a hypothetical intermediate in the transnitrosation reaction with GSNO) occupies most of the solvent accessible region surrounding Cys101 on the adjacent subunit. At high concentrations of GSNO, steric hindrance could plausibly limit the rate of S-nitrosation of GSNO, giving the behavior in Fig. 6A. CysNO, in contrast, is much smaller than GSNO (Fig. 6C) and is therefore unlikely to experience competition for nitrosation sites at the dimer interface, resulting in linear dependence of the nitrosation rate on CysNO concentration. The rate of S-nitrosation of Cys101 by GSNO or CysNO was calculated from a fit of the data in Fig. 6A to Equation 1, for a single step reaction. For the GSNO reaction, k+NOCys-101 was estimated from the initial linear portion of the curve. All kinetic constants are reported in Table 1.
S-Nitrosated Cys101 Is More Resistant Than Cys47-NO to Reduction by GSH
To fully understand signaling by S-nitrosation, the mechanism and rates of denitrosation as well as nitrosation must be determined. We measured spontaneous denitrosation rates for GSTP1-1 as well as denitrosation mediated by GSH, the most abundant cellular thiol and therefore the most relevant cellular reducing agent. As with the nitrosation rates, denitrosation rates were corrected for denitrosation at the alternate site (C47S/C101S GSTP1-1). After S-nitrosating GSTP1-1 at either Cys47 or Cys101 by mixing with GSNO at 37 °C, excess GSNO was removed by buffer exchange and spontaneous denitrosation was measured by following the restoration of intrinsic protein fluorescence (Fig. 7A). The rate constant for spontaneous denitrosation was essentially the same for Cys101-NO and Cys47-NO, and is reported in Table 1. These rates were likely accelerated by the 280 nm excitation light (26) and are therefore not generally applicable beyond our experimental set-up. Of more interest are the rates of denitrosation in the presence of GSH. In this case, Cys101-NO and Cys47-NO were denitrosated at quite different rates. The linear dependence of the observed denitrosation rate on GSH concentration suggests that both Cys47-NO and Cys101-NO are denitrosated in a single step (Fig. 7B). Fitting the data to Equation 3 gives k−NOGSH 30-fold larger for Cys47-NO than Cys101-NO (Table 1), indicating that Cys101-NO is substantially more resistant than Cys47-NO to denitrosation by GSH. To test whether the mechanism of GSH-mediated denitrosation is related to GSH binding to the protein, we also performed denitrosation by DTT (Fig. 7A). Although DTT restored protein fluorescence at a much higher rate than GSH, the fluorescence transients were qualitatively similar to those for GSH, and also fit well to a triple exponential function (with two of the rates fixed to the spontaneous denitrosation rate and the rate of denitrosation of C47S/C101S GSTP1-1-NO, respectively).
DISCUSSION
Recently, protein S-nitrosation has emerged as an important cellular signaling mechanism, with a broad range of targets (37, 1) and significant implications for human health (6, 4). Unlike other physiologically important post-translational modifications (e.g. phosphorylation, glycosylation, and ubiquitination), S-nitrosation is largely not under enzymatic control. Instead, the dominant route of protein S-nitrosation appears to be transnitrosation by cellular GSNO, which exists in equilibrium with the pool of S-nitroso-proteins (12). It is therefore not obvious how S-nitrosation is controlled and how specific proteins and cysteines are targeted in vivo. Here, we examined in detail the mechanism of transnitrosation of the detoxification enzyme GSTP1-1, with the aim of providing insight into the molecular basis for the specificity and sensitivity of protein S-nitrosation. From transient kinetic analyses, we propose a minimal model for S-nitrosation of GSTP1-1 at both Cys47 and Cys101 (Scheme 1).
Cys101 is transnitrosated in a single step, whereas S-nitrosation at Cys47 is limited by a pre-equilibrium between the open and closed states of α2. The mechanism reported here suggests an intricate regulation of GSTP1-1 S-nitrosation. Because the rate of opening of helix α2 controls Cys47 nitrosation, ligands that bind the G-site and lock α2 into a closed conformation will prevent this residue from being modified. These include GSH, GSSG (oxidized glutathione disulfide), and the dinitrosyl-diglutathionyl-iron complex (38). The latter compound is an NO carrier in vivo, suggesting that Cys47 S-nitrosation can be either stimulated or inhibited depending on whether cellular NO is sequestered as GSNO or the dinitrosyl-diglutathionyl-iron complex. Cys101 nitrosation is less complex, occurring in a single step. However, modification of this cysteine is limited by negative cooperativity due to steric hindrance (Fig. 6), mitigating nitrosation at high GSNO. Cys101 is therefore nitrosated slowly, but is unusually stable; the rate constant for GSH-mediated denitrosation at Cys101-NO is 30 times smaller than at Cys47-NO (Table 1). This is consistent with a previous study that identified Cys101-NO of GSTP1-1 as an unusually long-lived nitrosation site in vivo (19). A possible explanation for this is the limited accessibility of Cys101-NO to GSH due to steric hindrance at the dimer interface (Fig. 6B). Consistent with this interpretation, the rate of spontaneous denitrosation was essentially identical for Cys47-NO and Cys101-NO. DTT-mediated denitrosation was qualitatively similar to denitrosation by GSH (Fig. 7A), suggesting that GSH binding does not influence the process of denitrosation at either site. Instead, GSH seems to be acting simply as a reducing agent.
Several enzymes capable of catalyzing denitrosation reactions have been identified (39). However, given the high cellular concentration of GSH and the lability of the S-nitrosothiols, spontaneous GSH-mediated denitrosation is likely highly significant physiologically. Physiological GSH concentrations can reach 1 to 10 mm (10). Based on the rate constants we report here (Table 1), Cys47-NO of GSTP1-1 would be denitrosated at a rate of 0.05–0.5 s−1, whereas Cys101-NO would be denitrosated at 0.001–0.01 s−1. This compares to nitrosation rates at physiological GSNO concentrations (1–5 μm (11)) of 0.0002 s−1 at Cys101 and 0.02 s−1 at Cys47 (kopen), indicating that nitrosation of GSTP1-1 is disfavored at high GSH concentrations. The rates reported here are consistent with those determined previously for l-cysteine (14), bovine serum albumin, and hemoglobin (15) and show that spontaneous protein transnitrosation by GSNO is generally not a rapid signaling mechanism. It is, however, clearly relevant to the function of many proteins, and can be regulated by a combination of ligand binding and the relative concentrations of GSNO and GSH.
S-Nitrosation of Cys47 significantly perturbs the detoxification activity of GSTP1-1 (22). Because Cys47 does not directly participate in catalysis (40), this is likely due to a disruption of the structure or dynamics of the active site. Nitrosation of Cys101 does not substantially affect the enzymatic function of the protein (22). Cys101 nitrosation may, however, affect the ability of GSTP1-1 to bind JNK (18) or peroxiredoxin (41). The unusual persistence of Cys101-NO in vivo also raises the possibility that this residue acts as a NO storage site or a shuttle for NO in protein-protein transnitrosation reactions.
The mechanism of S-nitrosation of GSTP1-1 provides some insight into S-nitrosation specificity. Although there is no clear consensus as to what constitutes an “S-nitrosation motif,” two recent structure-based analyses suggest a distant (within 8 Å) acid-base motif with exposed charged groups (42, 43). Cys101 of GSTP1-1 is not proximal to any obvious acid-base motif, but instead is distinguished from the three other cysteines on each chain of the protein by its high solvent accessibility. This factor is also important for Cys47 nitrosation: Cys47 is modified only when α2 is open and the thiol is solvent accessible. Lys44 has been proposed to enhance the reactivity of Cys47 by stabilizing the cysteine thiolate (44), and has also been identified as a relevant nitrosation motif for GSTP1-1 in dbSNO, a database of nitrosation sites (1). However, because the α2 equilibrium limits nitrosation at Cys47, its low pKa does not ultimately determine its reactivity. Eyring analysis in Fig. 4 reveals that activation enthalpy of kopen is essentially identical to the activation enthalpy for α2 unfolding in high urea. This suggests that α2 is unfolded or disordered in the open state, consistent with NMR data (32) and the lack of clear electron density for α2 in crystal structures of apo-GSTP1-1 (45). This further implies that, whereas the tertiary protein structure may be important for regulating the solvent accessibility of S-nitrosated cysteines, significant tertiary or even secondary structures are not necessary for S-nitrosation to occur. A similar association between intrinsic disorder and susceptibility to post-translational modifications has been observed for several proteins, including p53 (46) and histone tails (47). The ability of GSNO to dock to a site near the target cysteine residue has also been suggested as an important criterion for S-nitrosation specificity (43), and this has been demonstrated for several proteins (4, 48). GSNO has been co-crystallized with GSTP1-1, prompting the proposal that GSNO binding precedes Cys47 nitrosation (23). However, close analysis of the binding mode of GSNO does not support this proposal. Unsurprisingly, GSNO binds GSTP1-1 identically to GSH, which positions the NO group of GSNO distant from the Cys47 thiol (Fig. 5D). To transfer the NO group to Cys47, GSNO would have to undergo an energetically costly reorientation that would locate the GS− moiety at an unprecedented binding site. We therefore propose that, even if GSNO does bind GSTP1-1, this is not a precursor for S-nitrosation at Cys47. Consistent with this, CysNO (which has no known binding site on GSTP1-1) is able to S-nitrosate Cys47 with similar kinetics to GSNO (Fig. 3). Furthermore, our ITC experiments indicate that GSNO does not bind to GSTP1-1 with significant affinity (Fig. 5). Titrating C101S GSTP1-1 with GSNO resulted in larger exothermic events (Fig. 5C), which were attributed to GSNO binding by Téllez-Sanz et al. (23). In light of our results, these heats are more plausibly attributed to the S-nitrosation of Cys47 as well as any associated protein conformational changes. Consistent with this interpretation, the KD determined from ITC was very similar to the KD from the fluorescence titration (Table 1), which is not contributed to by GSNO binding (GSNO was removed by dialysis after S-nitrosation in the fluorescence experiments). Notably, the rate constant for the opening of α2, kopen, is three times lower when GSNO rather than CysNO is used as the nitrosating agent (Table 1). A possible explanation for this is that GSH, produced when GSNO donates NO to the protein, binds to the active site and reduces the apparent kopen by biasing the closed state of α2.
In this study, we present the mechanism of transnitrosation of GSTP1-1, a detoxification enzyme and key regulator of cell proliferation through the JNK pathway. The mechanism we report here is significant to understanding how this protein is regulated by spontaneous S-nitrosation, and also provides more general insights into the S-nitrosation of other proteins. Detailed mechanistic studies of S-nitrosation such as this one are vital to understanding how protein transnitrosation by small molecules is controlled and targeted.
This work was supported by the University of the Witwatersrand, South African National Research Foundation Grants 60810, 65510, and 68898 and South African Research Chairs Initiative of the Department of Science and Technology and National Research Foundation Grant 64788.
- GSNO
- S-nitrosoglutathione
- CysNO
- S-nitrosocysteine
- α2
- helix 2 of glutathione transferase P1-1
- ITC
- isothermal titration calorimetry.
REFERENCES
- 1. Lee T.-Y., Chen Y.-J., Lu C.-T., Ching W.-C., Teng Y.-C., Huang H.-D., Chen Y.-J. (2012) dbSNO. A database of cysteine S-nitrosylation. Bioinformatics 28, 2293–2295 [DOI] [PubMed] [Google Scholar]
- 2. Liu L., Yan Y., Zeng M., Zhang J., Hanes M. A., Ahearn G., McMahon T. J., Dickfeld T., Marshall H. E., Que L. G., Stamler J. S. (2004) Essential roles of S-nitrosothiols in vascular homeostasis and endotoxic shock. Cell 116, 617–628 [DOI] [PubMed] [Google Scholar]
- 3. Sarkar S., Korolchuk V. I., Renna M., Imarisio S., Fleming A., Williams A., Garcia-Arencibia M., Rose C., Luo S., Underwood B. R., Kroemer G., O'Kane C. J., Rubinsztein D. C. (2011) Complex inhibitory effects of nitric oxide on autophagy. Mol. Cell 43, 19–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Savidge T. C., Urvil P., Oezguen N., Ali K., Choudhury A., Acharya V., Pinchuk I., Torres A. G., English R. D., Wiktorowicz J. E., Loeffelholz M., Kumar R., Shi L., Nie W., Braun W., Herman B., Hausladen A., Feng H., Stamler J. S., Pothoulakis C. (2011) Host S-nitrosylation inhibits clostridial small molecule-activated glucosylating toxins. Nat. Med. 17, 1136–1141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hess D. T., Stamler J. S. (2012) Regulation by S-nitrosylation of protein posttranslational modification. J. Biol. Chem. 287, 4411–4418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Foster M. W., Hess D. T., Stamler J. S. (2009) Protein S-nitrosylation in health and disease. A current perspective. Trends Mol. Med. 15, 391–404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Madej E., Folkes L. K., Wardman P., Czapski G., Goldstein S. (2008) Thiyl radicals react with nitric oxide to form S-nitrosothiols with rate constants near the diffusion-controlled limit. Free Radic. Biol. Med. 44, 2013–2018 [DOI] [PubMed] [Google Scholar]
- 8. Goldstein S., Czapski G. (1996) Mechanism of the nitrosation of thiols and amines by oxygenated NO solutions. The nature of the nitrosating intermediates. J. Am. Chem. Soc. 118, 3419–3425 [Google Scholar]
- 9. Anand P., Stamler J. S. (2012) Enzymatic mechanisms regulating protein S-nitrosylation. Implications in health and disease. J. Mol. Med. 90, 233–244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Yap L.-P., Sancheti H., Ybanez M. D., Garcia J., Cadenas E., Han D. (2010) Determination of GSH, GSSG, and GSNO using HPLC with electrochemical detection. Methods Enzymol. 473, 137–147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Gaston B. (1993) Endogenous nitrogen oxides and bronchodilator S-nitrosothiols in human airways. Proc. Natl. Acad. Sci. U.S.A. 90, 10957–10961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Liu L., Hausladen A., Zeng M., Que L., Heitman J., Stamler J. S. (2001) A metabolic enzyme for S-nitrosothiol conserved from bacteria to humans. Nature 410, 490–494 [DOI] [PubMed] [Google Scholar]
- 13. Foster M. W., Forrester M. T., Stamler J. S. (2009) A protein microarray-based analysis of S-nitrosylation. Proc. Natl. Acad. Sci. U.S.A. 106, 18948–18953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hogg N. (1999) The kinetics of S-transnitrosation. A reversible second-order reaction. Anal. Biochem. 272, 257–262 [DOI] [PubMed] [Google Scholar]
- 15. Rossi R., Lusini L., Giannerini F., Giustarini D., Lungarella G., Di Simplicio P. (1997) A method to study kinetics of transnitrosation with nitrosoglutathione. Reactions with hemoglobin and other thiols. Anal. Biochem. 254, 215–220 [DOI] [PubMed] [Google Scholar]
- 16. Patel R. P., Hogg N., Spencer N. Y., Kalyanaraman B., Matalon S., Darley-Usmar V. M. (1999) Biochemical characterization of human S-nitrosohemoglobin. J. Biol. Chem. 274, 15487–15492 [DOI] [PubMed] [Google Scholar]
- 17. Barglow K. T., Knutson C. G., Wishnok J. S., Tannenbaum S. R., Marletta M. A. (2011) Site-specific and redox-controlled S-nitrosation of thioredoxin. Proc. Natl. Acad. Sci. U.S.A. 108, E600–E606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Adler V., Yin Z., Fuchs S. Y., Benezra M., Rosario L., Tew K. D., Pincus M. R., Sardana M., Henderson C. J., Wolf C. R., Davis R. J., Ronai Z. (1999) Regulation of JNK signaling by GSTp. EMBO J. 18, 1321–1334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Paige J. S., Xu G., Stancevic B., Jaffrey S. R. (2008) Nitrosothiol reactivity profiling identifies S-nitrosylated proteins with unexpected stability. Chem. Biol. 15, 1307–1316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Beltrán B., Orsi A., Clementi E., Moncada S. (2000) Oxidative stress and S-nitrosylation of proteins in cells. Br. J. Pharmacol. 129, 953–960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sinha V., Wijewickrama G. T., Chandrasena R. E., Xu H., Edirisinghe P. D., Schiefer I. T., Thatcher G. R. (2010) Proteomic and mass spectroscopic quantitation of protein S-nitrosation differentiates NO-donors. ACS Chem. Biol. 5, 667–680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lo Bello M., Nuccetelli M., Caccuri A. M., Stella L., Parker M. W., Rossjohn J., McKinstry W. J., Mozzi A. F., Federici G., Polizio F., Pedersen J. Z., Ricci G. (2001) Human glutathione transferase P1-1 and nitric oxide carriers. A new role for an old enzyme. J. Biol. Chem. 276, 42138–42145 [DOI] [PubMed] [Google Scholar]
- 23. Téllez-Sanz R., Cesareo E., Nuccetelli M., Aguilera A. M., Barón C., Parker L. J., Adams J. J., Morton C. J., Lo Bello M., Parker M. W., García-Fuentes L. (2006) Calorimetric and structural studies of the nitric oxide carrier S-nitrosoglutathione bound to human glutathione transferase P1-1. Protein Sci. 15, 1093–1105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chang M., Bolton J. L., Blond S. Y. (1999) Expression and purification of hexahistidine-tagged human glutathione S-transferase P1-1 in Escherichia coli. Protein Expr. Purif. 17, 443–448 [DOI] [PubMed] [Google Scholar]
- 25. Hart T. W. (1985) Some observations concerning the S-nitroso and S-phenylsulphonyl derivatives of l-cysteine and glutathione. Tetrahedron Lett. 26, 2013–2016 [Google Scholar]
- 26. Sexton D. J., Muruganandam A., McKenney D. J., Mutus B. (1994) Visible light photochemical release of nitric oxide from S-nitrosoglutathione. Photochem. Photobiol. 59, 463–467 [DOI] [PubMed] [Google Scholar]
- 27. Gildenhuys S., Wallace L. A., Burke J. P., Balchin D., Sayed Y., Dirr H. W. (2010) Class Pi glutathione transferase unfolds via a dimeric and not monomeric intermediate. Functional implications for an unstable monomer. Biochemistry 49, 5074–5081 [DOI] [PubMed] [Google Scholar]
- 28. Werbeck N. D., Kellner J. N., Barends T. R., Reinstein J. (2009) Nucleotide binding and allosteric modulation of the second AAA+ domain of ClpB probed by transient kinetic studies. Biochemistry 48, 7240–7250 [DOI] [PubMed] [Google Scholar]
- 29. Nuriel T., Hansler A., Gross S. S. (2011) Protein nitrotryptophan. Formation, significance and identification. J. Proteomics 74, 2300–2312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Chen X., Wen Z., Xian M., Wang K., Ramachandran N., Tang X., Schlegel H. B., Mutus B., Wang P. G. (2001) Fluorophore-labeled S-nitrosothiols. J. Org. Chem. 66, 6064–6073 [DOI] [PubMed] [Google Scholar]
- 31. Vogt A. D., Di Cera E. (2012) Conformational selection or induced-fit? A critical appraisal of the kinetic mechanism. Biochemistry 51, 5894–5902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hitchens T. K., Mannervik B., Rule G. S. (2001) Disorder-to-order transition of the active site of human class Pi glutathione transferase, GST P1-1. Biochemistry 40, 11660–11669 [DOI] [PubMed] [Google Scholar]
- 33. Ricci G., Caccuri A. M., Lo Bello M., Rosato N., Mei G., Nicotra M., Chiessi E., Mazzetti A. P., Federici G. (1996) Structural flexibility modulates the activity of human glutathione transferase P1-1. J. Biol. Chem. 271, 16187–16192 [DOI] [PubMed] [Google Scholar]
- 34. Caccuri A. M., Antonini G., Ascenzi P., Nicotra M., Nuccetelli M., Mazzetti A. P., Federici G., Lo Bello M., Ricci G. (1999) Temperature adaptation of glutathione S-transferase P1-1. J. Biol. Chem. 274, 19276–19280 [DOI] [PubMed] [Google Scholar]
- 35. Oakley A. J., Lo Bello M., Battistoni A., Ricci G., Rossjohn J., Villar H. O., Parker M. W. (1997) The structures of human glutathione transferase P1-1 in complex with glutathione and various inhibitors at high resolution. J. Mol. Biol. 274, 84–100 [DOI] [PubMed] [Google Scholar]
- 36. Ricci G., Lo Bello M., Caccurri A. M., Pastore A., Nuccetelli M., Parker M. W., Federici G. (1995) Site-directed mutagenesis of human glutathione transferase P1-1. J. Biol. Chem. 270, 1243–1248 [DOI] [PubMed] [Google Scholar]
- 37. Seth D., Stamler J. S. (2011) The SNO-proteome. Causation and classifications. Curr. Opin. Chem. Biol. 15, 129–136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. De Maria F., Pedersen J. Z., Caccuri A. M., Antonini G., Turella P., Stella L., Lo Bello M., Federici G., Ricci G. (2003) The specific interaction of dinitrosyl-diglutathionyl-iron complex, a natural NO carrier, with the glutathione transferase superfamily. Suggestion for an evolutionary pressure in the direction of the storage of nitric oxide. J. Biol. Chem. 278, 42283–42293 [DOI] [PubMed] [Google Scholar]
- 39. Benhar M., Forrester M. T., Stamler J. S. (2009) Protein denitrosylation. Enzymatic mechanisms and cellular functions. Nat. Rev. Mol. Cell. Biol. 10, 721–732 [DOI] [PubMed] [Google Scholar]
- 40. Kong K.-H., Inoue H., Takahashi K. (1991) Non-essentiality of cysteine and histidine residues for the activity of human class Pi glutathione S-transferase. Biochem. Biophys. Res. Commun. 181, 748–755 [DOI] [PubMed] [Google Scholar]
- 41. Ralat L. A., Misquitta S. A., Manevich Y., Fisher A. B., Colman R. F. (2008) Characterization of the complex of glutathione S-transferase π and 1-cysteine peroxiredoxin. Arch. Biochem. Biophys. 474, 109–118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Doulias P. T., Greene J. L., Greco T. M., Tenopoulou M., Seeholzer S. H., Dunbrack R. L., Ischiropoulos H. (2010) Structural profiling of endogenous S-nitrosocysteine residues reveals unique features that accommodate diverse mechanisms for protein S-nitrosylation. Proc. Natl. Acad. Sci. U.S.A. 107, 16958–16963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Marino S. M., Gladyshev V. N. (2010) Structural analysis of cysteine S-nitrosylation. A modified acid-based motif and the emerging role of trans-nitrosylation. J. Mol. Biol. 395, 844–859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Lo Bello M., Parker M. W., Desideri A., Polticelli F., Falconi M., Del Boccio G., Pennelli A., Federici G., Ricci G. (1993) Peculiar spectroscopic and kinetic properties of Cys-47 in human placental glutathione transferase. Evidence for an atypical thiolate ion pair near the active site. J. Biol. Chem. 268, 19033–19038 [PubMed] [Google Scholar]
- 45. Oakley A. J., Lo Bello M., Ricci G., Federici G., Parker M. W. (1998) Evidence for an induced-fit mechanism operating in π class glutathione transferases. Biochemistry 37, 9912–9917 [DOI] [PubMed] [Google Scholar]
- 46. Bell S., Klein C., Müller L., Hansen S., Buchner J. (2002) p53 contains large unstructured regions in its native state. J. Mol. Biol. 322, 917–927 [DOI] [PubMed] [Google Scholar]
- 47. Jenuwein T., Allis C. D. (2001) Translating the histone code. Science 293, 1074–1080 [DOI] [PubMed] [Google Scholar]
- 48. Kim S. O., Merchant K., Nudelman R., Beyer W. F., Jr., Keng T., DeAngelo J., Hausladen A., Stamler J. S. (2002) OxyR. A molecular code for redox-related signaling. Cell 109, 383–396 [DOI] [PubMed] [Google Scholar]