

Background: Huntington disease (HD) is caused by an expanded polyglutamine (poly(Q)) domain in huntingtin (htt), leading to aggregation.
Results: Specific flanking sequences adjacent to the poly(Q) domain modulate htt aggregation on lipid bilayers.
Conclusion: Lipid-mediated htt aggregation may lead to membrane dysfunction in HD.
Significance: Flanking sequences may play a role in membrane dysfunction associated with Huntington disease.
Keywords: Atomic Force Microscopy, Huntingtons Disease, Lipids, Polyglutamine, Polyglutamine Disease, Protein Aggregation
Abstract
Huntington disease (HD) is caused by an expanded polyglutamine (poly(Q)) repeat near the N terminus of the huntingtin (htt) protein. Expanded poly(Q) facilitates formation of htt aggregates, eventually leading to deposition of cytoplasmic and intranuclear inclusion bodies containing htt. Flanking sequences directly adjacent to the poly(Q) domain, such as the first 17 amino acids on the N terminus (Nt17) and the polyproline (poly(P)) domain on the C-terminal side of the poly(Q) domain, heavily influence aggregation. Additionally, htt interacts with a variety of membraneous structures within the cell, and Nt17 is implicated in lipid binding. To investigate the interaction between htt exon1 and lipid membranes, a combination of in situ atomic force microscopy, Langmuir trough techniques, and vesicle permeability assays were used to directly monitor the interaction of a variety of synthetic poly(Q) peptides with different combinations of flanking sequences (KK-Q35-KK, KK-Q35-P10-KK, Nt17-Q35-KK, and Nt17-Q35-P10-KK) on model membranes and surfaces. Each peptide aggregated on mica, predominately forming extended, fibrillar aggregates. In contrast, poly(Q) peptides that lacked the Nt17 domain did not appreciably aggregate on or insert into lipid membranes. Nt17 facilitated the interaction of peptides with lipid surfaces, whereas the poly(P) region enhanced this interaction. The aggregation of Nt17-Q35-P10-KK on the lipid bilayer closely resembled that of a htt exon1 construct containing 35 repeat glutamines. Collectively, this data suggests that the Nt17 domain plays a critical role in htt binding and aggregation on lipid membranes, and this lipid/htt interaction can be further modulated by the presence of the poly(P) domain.
Introduction
An expanded polyglutamine (poly(Q))3 repeat near the N terminus of the protein huntingtin (htt) causes Huntington disease (HD), a fatal neurodegenerative disorder (1). This poly(Q) expansion leads directly to the formation of fibrils and other aggregates that deposit inclusion bodies in HD brain tissues (2). In HD, the age of onset and severity of disease are correlated with poly(Q) length. That is, poly(Q) domains shorter than ∼35 are not associated with disease; 35–39 repeats may cause disease; 40–60 repeats lead to adult onset; and greater than 60 repeats causes juvenile forms of HD (3). Although htt is over 3000 amino acids, htt undergoes proteolysis, resulting in a variety of truncation products (4, 5). In particular, the first exon of htt may play an important role in HD. The expression of htt exon1 with an expanded poly(Q) tract is sufficient to cause a progressive neurological phenotype in transgenic mice (2, 6), and N-terminal fragments similar to exon1 are detected in knock-in mouse models of HD that express full-length htt (7). Fragments slightly larger than exon1 have been detected in HD patients (5).
Flanking sequences directly adjacent to the poly(Q) domain within htt strongly influence aggregation (8, 9). The first 17 amino acids of htt exon1 (Nt17) has been shown to promote oligomer formation in synthetic poly(Q) peptides (10, 11), and targeting the Nt17 domain is an effective way of altering the aggregates formed and the kinetics of formation (10, 12). Studies with synthetic peptides indicated that the addition of a 10-residue polyproline (poly(P)) sequence to the C terminus of a poly(Q) peptide altered aggregation kinetics and conformational properties of the resulting poly(Q) tract (13). Flanking poly(P) sequences can also inhibit the formation of the β-sheet structure in poly(Q) peptides by inducing PPII-like helix structure (14).
Approximately 50% of endogenous htt distributes with membranes after subcellular fractionation of neuron-like clonal striatal cells (15), and htt aggregates have been shown to accumulate brain lipids in mouse models of HD (16, 17). Studies, based on structural analysis, demonstrated a potential role of Nt17 in htt binding to lipids (18, 19). Nt17 functions as a membrane targeting domain and in trafficking of htt to the endoplasmic reticulum, mitochondria, Golgi, and vesicles (20–22). Furthermore, N-terminal fragments of htt are associated with membranes isolated from brain (4), and there is an apparent correlation with poly(Q) length and insertion into lipid membranes (23). This interaction between htt and lipid membranes could result in altered membrane properties and dysfunction.
Although the importance of htt aggregation in HD has been appreciated for some time, the role of quasi-“solid/liquid” interfaces, such as membranous surfaces, on the aggregation process of htt is poorly understood. The two-dimensional liquid environments associated with lipid membranes can profoundly influence protein structure and dynamics via a variety of specific and nonspecific interactions. Importantly for HD, lipid bilayers may modulate protein conformation and exert influence on htt aggregation. Here, we characterize the role of flanking sequences associated with htt exon1 in the aggregation of poly(Q) peptides in the presence of surfaces, including lipid bilayers. The poly(Q) peptides sequences studied here were chosen based on several considerations: 1) studies demonstrating the 35 repeat Gln in synthetic peptides is sufficient for aggregation (11); 2) the poly(P) domain length (10 prolines) is similar to that directly adjacent to poly(Q) in htt exon1; and 3) the known lipid-binding properties of the first 17 amino acids of htt (20) (Fig. 1).
FIGURE 1.
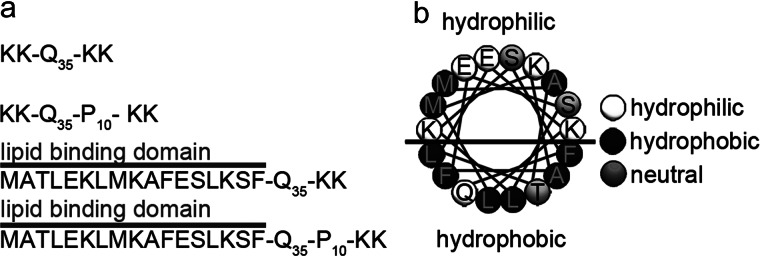
a, schematic representations of the poly(Q) peptides used in this study. The lipid binding domain is abbreviated as Nt17 in the article and (b) forms an amphipathic α-helix.
EXPERIMENTAL PROCEDURES
Peptide Preparation
KK-Q35-KK, KK-Q35-P10-KK, Nt17-Q35-KK, and Nt17-Q35-P10-KK were obtained via custom synthesis (Keck Biotechnology Resource Laboratory, New Haven, CT). Lysines were added to the N- and C-terminal sides to aid in solubility. Disaggregation and solubilization of poly(Q) peptides was achieved based on published protocols (24). In short, crude peptide was dissolved for 3 h in a 1:1 mixture of trifluoroacetic acid (Acros Organics) and hexafluoroisopropanol (Acros Organics) to a concentration of 0.5 mg/ml. After rigorous vortexing, the solvent was then evaporated off with a gentle stream of N2 and placed in a Vacufuge concentrator (Eppendorf, Hauppauge, NY), producing a thin peptide film. The remaining peptide residue was resuspended in ultrapure water adjusted to pH 3 with trifluoroacetic acid to 200 μm, snap frozen, and stored at −80 °C. This stock solution was thawed and immediately diluted with phosphate-buffered saline (PBS) (Fisher Scientific) to a final concentration of 20 μm at a pH of 7.3 prior to any AFM experiment.
Purification of GST-htt-Exon1 Fusion Proteins
Glutathione S-transferase (GST)-htt exon1 35Q fusion protein was purified as described elsewhere (25). Expression of GST-htt fusion proteins was induced in Escherichia coli by isopropyl β-d-thiogalactoside (4 h at 30 °C). The cells were lysed with the addition of 0.5 mg/ml of lysozyme. The fusion proteins were purified from lysate by liquid chromatography (Bio-Rad) with a GST affinity column. Gel electrophoresis was utilized to verify the pertinent fractions and determine purity. Freshly purified GST-htt exon1 35Q fusion proteins were used for each experiment. Prior to any experiment, solutions of the GST-htt exon1 35Q fusion protein were centrifuged at 20,000× g for 30 min at 4 °C to remove pre-existing aggregates. Cleavage of the GST moiety by Factor Xa (Promega, Madison, WI) initiated aggregation. Factor Xa and the GST-htt exon1 fusion protein were incubated for 1 h on ice to ensure efficient GST cleavage. All experiments were carried out in buffer A (50 mm Tris-HCl, pH 7.0, 150 mm NaCl, 1 mm DTT).
AFM and SPAM Imaging Conditions
In situ AFM experiments were performed with a Nanoscope V MultiMode scanning probe microscope (Veeco, Santa Barbara, CA) equipped with a closed-loop “vertical engage” J-scanner and a sealable tapping fluid cell. Images were acquired with V-shaped oxide-sharpened silicon nitride cantilevers (Veeco, Santa Barbara, CA) with nominal spring constants of ∼0.5 N/m. Scan rates were set at 1–2 Hz with cantilever drive frequencies ranging from ∼8 to 10 kHz. The free amplitude of the cantilever was ∼25 nm, and the tapping amplitude was set at 75% of free amplitude.
For SPAM experiments, 5 × 1.25-μm images were captured with 512 × 128 pixel resolution. Using a signal access module (Veeco, Santa Barbara, CA) and CompuScope 14100 data acquisition card (Gage, Lachine, Quebec) cantilever deflection trajectories were captured at 2.5 MS/s and 14-bit resolution with a vertical range of 4 V. These deflection trajectories were used to reconstruct the time-resolved tip/sample force as described (26). The Matlab image processing toolbox (Mathworks, Natick, MA) was used to analyze AFM images. Physical dimensions of aggregates were measured automatically as described elsewhere in literature (27, 28). The molecular weight of oligomers was estimated from AFM volume measurements. Images were imported into Matlab such that a flattening algorithm could be applied to correct for curvature due to the imaging process. A height threshold was then applied to the flattened images to generate binary maps of aggregate location. This was accomplished by assigning values of 0 or 1 to any pixel of the image that represented a height below or above the allocated threshold, respectively. By applying pattern recognition algorithms to the binary map, discrete aggregates were located. Once a discrete aggregate was located, physical properties such as height and position within the image were measured automatically.
Determining the Number of Peptides per Oligomer from AFM Images
Volume measurements were partially corrected using geometric models for error associated with the finite size of the AFM tip (28). The volume of an individual poly(Q) peptide was estimated based on the molecular weight of each peptide and the average density of globular proteins (29, 30). By dividing the measured, corrected volume of individual aggregates by the estimated volume of a single monomer, the number of molecules per each oligomer was calculated. This calculation assumes that individual monomers are efficiently packed into aggregates and that protein density is the same in the aggregated and unaggregated state.
Preparation of Supported Total Brain Lipid Extract (TBLE) Bilayers for AFM Experiments
Lyophilized TBLE (Avanti Polar Lipids, Alabaster, AL) was resuspended in phosphate-buffered saline to a concentration of 1 mg/ml. Using liquid nitrogen, bilayers and multilayer lipid sheets were formed by five cycles of freeze-thaw treatment. Prior to experimentation, these lipid suspensions were bath sonicated for 15 min to promote the formation of vesicles used to create bilayers for AFM experiments. All AFM experiments were performed with the same lot of lipids.
Langmuir Trough Set-up
Surface activity and monolayer insertion experiments were performed using a computer controlled Teflon Langmuir trough with Delrin-coated symmetric moving barriers (large inverted microscopy model, NIMA Technologies, Coventry, England). A stationary Wilhelmy balance (NIMA Technologies, Coventry, England) was used to measure surface pressure. All surface activity and monolayer insertion measurements were collected at 30 ± 0.5 °C and trough temperature was maintained by a circulating water bath (Isotemp 3016D water circulator, Thermo Fisher Scientific).
Measuring Peptide Surface Activity
To prepare peptide samples, pH 3 trifluoroacetic acid (Sigma) was added to solvate a lyophilized peptide film and subsequently vortexed. The stock solution was diluted with PBS (Invitrogen) to a final concentration of 100–300 μm at pH 7.3 and used immediately. To determine peptide affinity for the air/water interface, surface activity was measured. Trough barriers were compressed to form a 195-cm2 surface area and peptide was injected under the barriers using an L-shaped syringe (Hamilton, Reno, Nevada) to a final peptide concentration of 900 nm. After mixing, surface pressure was monitored; within 20 min, material equilibrated at the interface and longer wait times, up to 90 min, had no effect on the surface activity.
Measuring Peptide Insertion into Lipid Monolayers
Insertion experiments were carried out to determine the magnitude of the poly(Q) peptide insertion into a lipid monolayer. 1,2-Ditetradecanoyl-sn-glycero-3-phosphocholine (DMPC) and TBLE (Avanti Polar Lipids) were obtained in powder form and used without further purification. Spreading solutions of 0.4 mg/ml were created in chloroform (Sigma) and stored in glass vials at −20 °C. The lipid monolayer was formed by dropwise addition of spreading solution on the PBS surface, and the organic solvent was allowed to evaporate for 15 min. The lipid monolayer was compressed to a surface pressure of 30 mN/m to mimic the lipid packing density of a normal bilayer (31). When this surface pressure was attained, the barriers were switched from compression to feedback mode that maintained constant pressure by adjusting monolayer surface area. Freshly prepared peptide was then injected beneath the monolayer to a final concentration of 750 (DMPC monolayers) or 900 nm (TBLE monolayers). After 10 min, if no increase in average area per molecule to signify insertion was observed, the surface pressure was lowered by a 2 mN/m step and then the process repeated. If peptide insertion did occur, the pressure was held for additional 10-min time increments until insertion ceased. This was continued until the barriers expanded to the fully open position or the surface pressure could no longer be lowered. Percent insertion of each poly(Q) peptide was determined by summing the area increase percent ((Afinal − Ainitial)/(Ainitial)) × 100) for each surface pressure.
Vesicle Preparation for Leakage Assay
Large unilamellar egg phosphatidylcholine (Avanti Polar Lipids) vesicles were prepared using a mini extruder (Avanti Polar Lipids). Briefly, a thin lipid film of egg phosphatidylcholine, dried under nitrogen from a chloroform stock, was hydrated in 70 mm calcein (Sigma) and vortexed for 10 min. This solution was treated with 7 freeze-thaw cycles using ethanol/dry ice and warm water baths and then extruded 11 times through a 100-nm pore polycarbonate filter. Vesicles were separated from excess dye with a Sephadex G-50 (Sigma) size exclusion column. Dynamic light scattering measurements with a Zetasizer ZS90 (Malvern, Worcestershire, UK) were used to measure vesicle diameter of ∼120 nm and ensure sample homogeneity. Lipid concentration was determined by a phosphorus assay adapted from Anderson and Rouser (32, 33). An aliquot of vesicle solution (500 μl) and 8.9 n H2SO4 (225 μl) were combined and heated at 155 °C for 20 min. After cooling, 75 μl of 6% H2O2 was added and heated for 1 h at 155 °C. After cooling, 2 ml of deionized water, 225 μl of 5% molybdic acid (Sigma), and 225 μl of 10% ascorbic acid (Sigma) were added. The resulting solution was placed in a boiling water bath for 10 min and absorbance measured at 820 nm with a Jasco V-550 UV/visible spectrophotometer (Jasco, Easton, MD). Absorbance values were compared with a KH2PO4 reference plot to determine the phosphorous amount and calculate lipid concentration.
Vesicle Leakage Assays
The release of calcein after 5 min of peptide exposure to LUVs (34, 35) with peptide:lipid ratios ranging from 1:64 to 1:1 was observed with a RF-1501 spectrofluorometer (Shimadzu, Columbia, MD) at 515 nm (excitation 495 nm); maximum fluorescence was observed within the first 5 min. Vesicles were then fully lysed by addition of 1% Triton X-100 (Sigma) until no further increase in fluorescence was observed. Vesicle leakage caused by peptide was calculated by,
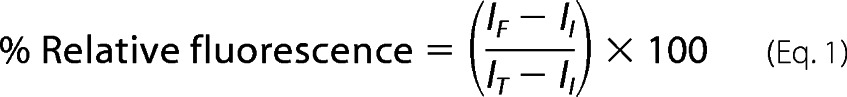 |
where IF is the measured fluorescence intensity, II is the initial fluorescence of vesicles, and IT is the total fluorescence intensity after LUV lysis. Vesicle leakage experiments were repeated 48 h later with the same prepared peptide samples to determine the effect of incubation with non-monomeric peptides.
RESULTS
Flanking Sequences Do Not Alter Aggregate Morphology on a Mica Surface
To determine how flanking sequences associated with htt exon1 modulate poly(Q) aggregation at solid/liquid interfaces, the aggregation of four synthetic poly(Q) peptides, KK-Q35-KK, KK-Q35-P10-KK, Nt17-Q35-KK, and Nt17-Q35-P10-KK (Fig. 1A), were monitored on mica via in situ AFM, which allows for the tracking of discrete aggregates as a function of time. For these studies, freshly cleaved mica was systematically exposed to newly prepared monomeric 20 μm solutions of each peptide, and the aggregation was observed by successive AFM images of the same area. To verify that the aggregation on the mica surface was not influenced by the continual scanning associated with AFM, the imaging frame was offset after each experiment to a new area of the surface that was not previously scanned. By comparison of the aggregate morphology and aggregate coverage observed on these areas, it was determined that the continual scanning was not affecting the aggregation process on the surface for any of the peptides.
On the mica surface, all four poly(Q) peptides predominantly formed extended, fibrillar aggregates that were highly branched and interconnected. These fibrillar aggregates grew to cover large areas of the mica surface, forming extensive meshes of fibril aggregates (Fig. 2). The fibrillar aggregates were morphologically indistinguishable between the four different peptides with fibril heights ranging from ∼4.5 to 7 nm (Fig. 3A). This observation suggests that the 35 glutamines common to each peptide formed a similar sized core of the fibril structure. Although these large meshes of fibrils were the predominant aggregate formed on the mica surface, a very small population (less than 2% of the total aggregated peptide) of discrete oligomers was also observed for all four peptides. This result is consistent with a previous report on poly(Q) peptide aggregation (no flanking sequences) of various length on mica forming a small population of oligomeric species (36). Although a small percentage of these oligomers were stable for extended periods of time (upwards of 30–50 min), most of the oligomers either directly nucleated fibril growth or were quickly assimilated into the larger fibril networks. The appearance of similar aggregate morphologies for all four peptides was surprising based on previous reports that indicated that poly(Q) peptides in bulk solution form fibrils directly from monomer, whereas, the addition of Nt17 to poly(Q) peptides resulted in an aggregation mechanism that favored the formation of an oligomeric intermediate between monomers and fibrils (10, 11). This indicates that the presence of the mica surface may promote the direct formation of fibrils from monomer despite any influence on aggregation associated with flanking sequences.
FIGURE 2.

Freshly prepared 20 μm solutions of KK-Q35-KK, KK-Q35-P10-KK, Nt17-Q35-KK, or Nt17-Q35-P10-KK were exposed to an anionic mica surface and imaged continuously in solution using atomic force microscopy. Time-lapse images of the same 2.5 × 2.5-μm region of mica are presented to demonstrate the aggregation of all four peptides. Although extensive meshes of fibrils are the predominant aggregate form, some oligomers were observed for all four peptides (blue arrows).
FIGURE 3.
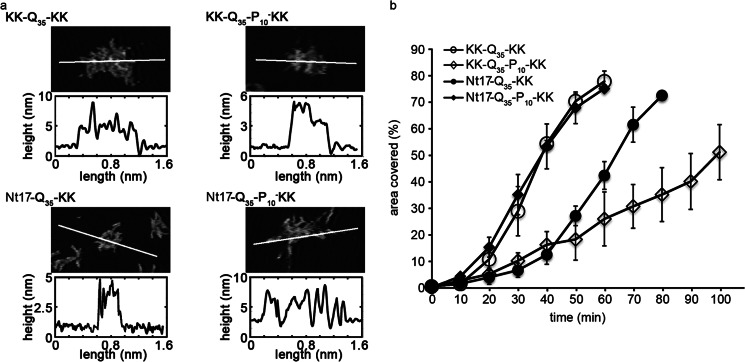
a, zoomed in AFM images of fibril meshes are presented for KK-Q35-KK, KK-Q35-P10-KK, Nt17-Q35-KK, or Nt17-Q35-P10-KK that had aggregated in the presence of mica. The white line in each image corresponds to the height profile directly below the image. b, the area of the surface covered as a function of time demonstrates the relative aggregation rate of KK-Q35-KK, KK-Q35-P10-KK, Nt17-Q35-KK, and Nt17-Q35-P10-KK on a mica surface. Error bars represent the S.D. over 3 separate experiments for each peptide.
To analyze the rate of aggregation on the mica surface, the percent of the mica surface covered by peptide aggregates was determined as a function of time (Fig. 3B). Contrary to previous studies in free solution (11) (without the presence of a surface), Nt17-Q35-KK aggregated at a slower rate than KK-Q35-KK as measured by the percentage of surface covered by peptide aggregates. The addition of just the poly(P) region (KK-Q35-P10-KK) appeared to retard aggregation on the surface as well; however, the addition of the Nt17 and poly(P) flanking sequences (Nt17-Q35-P10-KK) resulted in a similar aggregation rate as KK-Q35-KK.
The Interaction of Poly(Q) Peptides with Lipid Membranes Is Dependent on Flanking Sequences
To investigate the role of flanking sequences on the aggregation of poly(Q) peptides on lipid surfaces, supported lipid bilayers on mica were exposed to the various poly(Q) peptides and images using in situ AFM as a function of time. The supported bilayers were produced via the fusion of TBLE vesicles. The resulting supported lipid bilayers preserve many properties associated with free membranes, such as lateral fluidity (37). TBLE contains a physiologically relevant ratio of membrane components and a mixture of zwitterionic and negatively charged lipids, more specifically 9.6% phosphatidylcholine, 16.7% phosphatidylethanolamine, 1.6% phosphatidylinositol, 10.6% phosphatidylserine, 2.8% phosphatidic acid, and 58.7% unknown. Only defect-free TBLE bilayers, as assessed by AFM imaging, were exposed to the poly(Q) peptides, and observations were limited to regions of the surface that were verified to be defect-free. The fluid cell was washed with PBS three times to reduce the amount of lipid components not bound to the mica substrate. Once formed, control experiments verified that the supported TBLE bilayers were stable on the mica surface for at least 14–15 h, i.e. there were no visible morphological changes as assessed by AFM imaging in the absence of any peptide.
Similar to experiments performed on mica, the supported bilayers were exposed to freshly prepared aliquots of each poly(Q) peptide at a final concentration of 20 μm. Although fibrillar aggregates of KK-Q35-KK and KK-Q35-P10-KK were observed on mica within 10 min, these two peptides did not appreciably aggregate on the lipid surface during the 90-min time frame of the experiment, and no morphological changes of the bilayer were observed (Fig. 4). This indicates that poly(Q) alone does not interact directly with lipid membranes and that the addition of a poly(P) region was not sufficient to facilitate the interaction between poly(Q) peptides with a lipid bilayer.
FIGURE 4.

Freshly prepared TBLE bilayers were exposed to 20 μm solutions of KK-Q35-KK, KK-Q35-P10-KK, Nt17-Q35-KK, or Nt17-Q35-P10-KK and imaged continuously in solution using atomic force microscopy. Time-lapse images of the same 2.0 × 2.0-μm region of the bilayer are presented as three-dimensional surfaces with two-dimensional insets zooming into 500 × 500-nm regions of the surface to provide morphological detail of specific aggregates. In the two-dimensional representations, blue arrows represent discrete oligomers, and green arrows represent fibrillar aggregates.
When the TBLE bilayer was exposed to Nt17-Q35-KK, small, discrete oligomers were observed on the lipid bilayer within 15–30 min after the injection of peptide (Fig. 4). In contrast to the small population of oligomers observed on mica, these oligomers of Nt17-Q35-KK were numerous, stable, and did not transition to a fibrillar morphology. Whether these oligomers of Nt17-Q35-KK formed directly on the bilayer or in solution before binding to the bilayer cannot be determined directly from the experiment; however, it is clear that the preferred aggregate form of Nt17-Q35-KK on the lipid bilayer was oligomeric. The appearance of these oligomers of Nt17-Q35-KK on the lipid bilayer demonstrates that the Nt17 domain facilitates the interaction between htt and lipid membranes.
Upon exposure to the TBLE bilayer, Nt17-Q35-P10-KK formed a smaller number of discrete oligomers compared with Nt17-Q35-KK; however, the dominant aggregate form associated with Nt17-Q35-P10-KK was large amorphous accumulations of peptide that appeared to have some fibrillar character around their periphery (Fig. 4). It is also likely that some lipid components are directly incorporated into these large aggregates. In some experiments, these areas appeared as slightly rougher areas of the lipid surface; in others, these were much larger accumulations of protein and lipid (Fig. 5). These larger aggregates, whereas immobile on the surface, grew in size (both height and diameter) in successive AFM images (Fig. 4). Again, the Nt17 domain was necessary to facilitate the interaction between poly(Q) peptides and the lipid bilayer; however, in this case, the presence of the poly(P) flanking sequence on the C-terminal side of the poly(Q) domain modulated the interaction with the bilayer, resulting in aggregation beyond the formation of oligomers. For both Nt17-Q35-KK and Nt17-Q35-P10-KK, the morphology of the aggregates on the surface were distinct from those formed on mica, indicating that the presence of the lipid bilayer directly influenced aggregation.
FIGURE 5.

AFM images of separate TBLE bilayers exposed to Nt17-Q35-P10-KK demonstrate the morphological variability of the large regions of aggregation.
To determine the relative rate of aggregation of the various poly(Q) peptides in the presence of the TBLE bilayer, the number of discrete aggregates per square micron and the total volume of aggregates occupying the surface was determined as a function of time (Fig. 6, a and b). Although KK-Q35-KK and KK-Q35-P10-KK resulted in few aggregates, if any, on the bilayer, Nt17-Q35-KK and Nt17-Q35-P10-KK formed aggregates that increased in number with time. Nt17-Q35-KK formed discrete aggregates on the bilayer at a significantly faster rate compared with Nt17-Q35-P10-KK. However, Nt17-Q35-P10-KK was capable of forming larger aggregate structures on the bilayer, resulting in a larger total volume of protein deposited on the surface after 60 min. The average height of the oligomers formed by Nt17-Q35-KK remained constant as a function of time (∼0.6 ± 0.2 nm) with a tight distribution (Fig. 6c). The measured volume of oligomers of Nt17-Q35-KK was corrected for the contribution due to the finite size of the AFM tip and used to estimate the molecular weight of the aggregates (28). Oligomers of Nt17-Q35-KK were ∼78 kDa in size and contained ∼10 peptides (Fig. 6d). The height and volume distribution of Nt17-Q35-KK oligomers as a function of time demonstrates that the oligomers were highly stable on the bilayer surface. In contrast, the oligomers (excluding the large amorphous aggregates) formed from Nt17-Q35-P10-KK exposed to bilayer were more diverse, as their height ranged from 0.8 to 1.5 nm with a gradual increase in size as a function of time (Fig. 6, c and d). Oligomers of Nt17-Q35-P10-KK were comprised of ∼10 peptides at early time points but increased to ∼12–13 peptides (∼97 kDa) after 90 min. The regions of the bilayer displaying perturbed morphology and amorphous aggregates were more variable in size (Fig. 6e) as the distance across these regions ranged from 25 to 225 nm and occupied a region of the surface ranging from ∼0.05 to 0.1 μm2. Perturbed regions that did not contain large amorphous aggregates had a tallest feature on the range of ∼1.2–4 nm, and the amorphous features were typically larger than 5 nm (Fig. 6e).
FIGURE 6.

a, the number of discrete aggregates and b, the total volume of aggregates appearing on the TBLE bilayer surface as a function of time demonstrates the relative aggregation rate of KK-Q35-KK, KK-Q35-P10-KK, Nt17-Q35-KK, and Nt17-Q35-P10-KK on the TBLE bilayer. Error bars represent the S.D. over 3 separate experiments for each peptide. c, histogram heights of oligomers formed by Nt17-Q35-KK or Nt17-Q35-P10-KK as a function of time demonstrate the relative stability of these aggregates. d, based on corrected volume measurements and the molecular mass of each peptide, the number of molecules per aggregate and the molecular weight for oligomers formed by Nt17-Q35-KK or Nt17-Q35-P10-KK were estimated as a function of time. e, histograms of diameter, area occupied, and height of the large amorphous aggregates formed by Nt17-Q35-P10-KK.
Nt17 Facilitates the Interactions of Poly(Q) Peptides with Lipid Monolayers
When injected into the aqueous subphase of a Langmuir trough, peptide molecules partition between the bulk and the air/water interface until a certain surface pressure, the equilibrium spreading pressure, is attained. Surface pressure is defined as surface tension of the aqueous interface minus surface tension of the monolayer (Π = γ0 − γ). Therefore, a higher surface pressure measurement means the peptide is more surface active and this may correlate to increased insertion into or association with the amphipathic cell membrane. The addition of monomeric htt peptides containing the Nt17 region to the subphase at a final concentration of 900 nm gives rise to an equilibrium spreading pressure of 19.2 mN/m for Nt17-Q35-KK and 17.3 mN/m for Nt17-Q35-P10-KK (Table 1). The peptides without Nt17, KK-Q35-KK and KK-Q35-P10-KK, induced a negligible change in surface pressure (Table 1) indicating the Nt17 lipid binding domain (Fig. 1) is necessary for the poly(Q) peptide to be surface active. To directly assess the role of flanking sequences on poly(Q) peptide insertion into lipid surfaces, we performed experiments to measure the amount of peptide monomer insertion into a lipid monolayer, a model for the outer leaflet of the cell membrane. Relatively low peptide concentrations (750–900 nm) were used so as to determine the role of the flanking regions of individual peptides, as opposed to larger oligomeric/fibril structures, in the interaction of the peptide with a cell membrane. A lipid monolayer at the air/water interface was compressed to a physiologically relevant surface pressure of 30 mN/m (31, 38) and held at this pressure, whereas freshly prepared peptide was injected into the subphase beneath the monolayer. No immediate change in area per molecule for any of the peptides was observed at this pressure for a period of 10 min. The pressure was then stepped down by 2 mN/m increments with 10-min equilibration times until a measureable increase in area, indicating peptide insertion, occurred. For a zwitterionic DMPC monolayer, chosen as the simplest uncharged membrane model, 750 nm poly(Q) peptide containing both flanking sequences, Nt17-Q35-P10-KK, inserted the most at 14%, whereas removal of the poly(P) region resulted in ∼6% insertion (Table 1). Both poly(Q) peptides without the Nt17 terminus displayed negligible insertion and inserted at lower surface pressures (Table 1).
TABLE 1.
Surface activity and lipid monolayer insertion of polyQ peptides with different flanking sequences
| Peptide construct | Surface activity, surface pressure (mN/m) 900 nm peptide | Insertion |
|||
|---|---|---|---|---|---|
| DMPC |
TLBE |
||||
| Insertiona | Insertionb | Insertiona | Insertion | ||
| Nt17-Q35-KK | 19.2 ± 0.9c | 26 mN/m | 5.8 ± 0.5% | 24 mN/m | 26 ± 3% |
| Nt17-Q35-P10-KK | 17.3 ± 0.7 | 22 mN/m | 14 ± 2% | 22 mN/m | 38 ± 5% |
| KK-Q35-KK | 0.2 ± 0.1 | 14 mN/m | 2.0 ± 0.5% | 18 mN/m | 10 ± 2% |
| KK-Q35-P10-KK | 0.1 ± 0.1 | 14 mN/m | 1.1 ± 0.5% | 18 mN/m | 10 ± 3% |
a Surface pressure at which insertion into lipid monolayer is first detected.
b % insertion determined be summing percentage area increase for each surface pressure step.
c Reported error is S.D. from three independent measurements.
To gain insight about how the poly(Q) peptides associate with the more physiologically relevant lipid mixtures used for the AFM morphological studies, insertion into TBLE monolayers was also measured. In similar fashion, the poly(Q) peptide with both the Nt17 and poly(P) flanking regions caused the greatest increase in area due to insertion (38%), whereas those lacking the N-terminal region had the least insertion (10% for both KK-Q35-P10-KK and KK-Q35-KK) (Table 1). For both monolayer types, Nt17-Q35-KK began inserting at a higher surface pressure than Nt17-Q35-P10-KK even though the total magnitude of insertion was significantly lower; this is likely due to the slightly lower surface activity caused by the poly(P) region (Table 1). It should be noted that in all cases, poly(Q) peptides insert into the lipid monolayer at a surface pressure higher than the equilibrium spreading pressure indicating a driving force for the peptide to interact with the membrane and not merely to reside at the air/water interface. The results of the insertion experiments show the poly(Q) peptides with the Nt17 flanking sequence insert considerably more into a model membrane than the corresponding peptides without the N terminus. The poly(P) region mediates this insertion, but only in the case of the poly(Q) peptide with the Nt17 flanking region does the addition of the 10 proline repeat lead to increased insertion into the lipid monolayer; KK-Q35-P10-KK and KK-Q35-KK behave similarly.
Nt17 and Poly(P) Flanking Region of Poly(Q) Peptides Necessary to Permeabilize Membranes
The measured insertion of the poly(Q) peptides into lipid monolayers suggested the capability of these amphipathic peptides to permeabilize membranes. Permeability was followed by calcein dye release from LUVs where disruption of the membrane results in calcein release, dilution, and fluorescence increase from the unquenched dye. To determine the effects of poly(Q) peptides on vesicle membrane permeability, monomeric peptides were mixed with egg phosphatidylcholine vesicles in various lipid:peptide ratios ([peptide] = 7.34–470 nm) and fluorescence measured. To parallel the Langmuir trough measurements, nanomolar peptide concentrations were used; this ensured monomeric peptides with different flanking regions would interact with the model cell membrane. As shown in Fig. 7, only Nt17-Q35-P10-KK was able to induce a significant calcein release from LUVs compared with control volumes of trifluoroacetic acid/PBS buffer. The importance of these flanking sequences is echoed in work where the C-terminal proline-rich region of genetically engineered mouse brain htt exon1 fragments was required for disruption of brain lipid vesicles (16). At certain peptide:lipid ratios, Nt17-Q35-KK caused slightly greater vesicle permeability compared with the poly(Q) peptides without the Nt17 region, suggesting insertion of the peptide without membrane disruption. After mixing of the vesicles and Nt17-Q35-P10-KK peptide, the fluorescence emission increased sharply for 3 min and plateaued within 5 min indicating the membrane disruption was complete; the effect of the three other peptides was seen within 1 min of mixing and can primarily be attributed to solvent effects (kinetics data not shown). Similar vesicle leakage assays were repeated 48 h after the peptide solutions were initially prepared to compare the effect of monomers and more complex monomer/oligomeric/fibrillar mixtures. After a 48-h waiting period, the Nt17-Q35-P10-KK peptide solution was markedly less efficient at inducing calcein release; as an example, for a 0.125 peptide:lipid ratio, the % release went from ∼85 to ∼20%. For the Nt17-Q35-KK, KK-Q35-KK, and KK-Q35-P10-KK peptides, the leakage remained comparable with buffer exposure. These results, in conjunction with the monolayer insertion experiments, suggest that the Nt17 region is necessary for insertion of the monomeric poly(Q) peptides into the lipid membrane and addition of the C-terminal poly(P) segment induces a structural change, which promotes an increase in membrane disruption.
FIGURE 7.
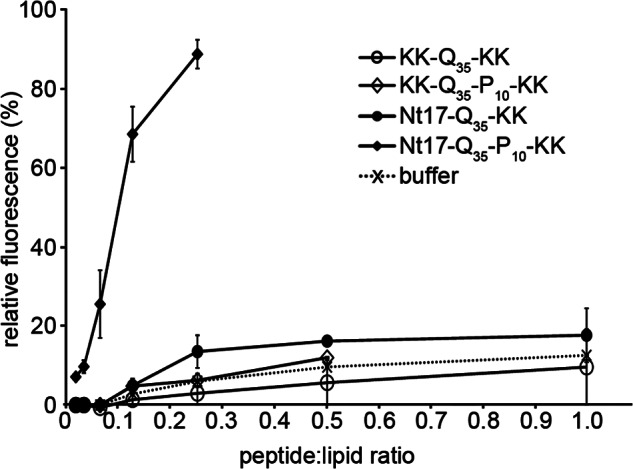
Calcein leakage, measured as relative fluorescence, from LUVs exposed to KK-Q35-KK, KK-Q35-P10-KK, Nt17-Q35-KK, Nt17-Q35-P10-KK, or buffer (acting as a control) plotted as a function of the peptide:lipid ratio. Error bars represent the S.D. over 3 separate experiments for each peptide.
Nt17-Q35-P10-KK Interacts with Lipid Membranes Similarly to htt exon1(35Q)
To determine whether the poly(Q) peptides interacted with bilayers in a similar fashion compared with purified htt exon1, supported TBLE bilayers were exposed to a htt exon1 fragment with Gln35 (the same length as the synthetic peptides). Although Nt17-Q35-KK and Nt17-Q35-P10-KK both formed aggregates on the lipid membrane, the aggregation of htt exon1(35Q) on the bilayer more closely resembled that of Nt17-Q35-P10-KK (Fig. 8). Although some discrete oligomers of htt exon1(35Q) were observed, the dominant morphological feature associated with the interaction of htt exon1(35Q) was amorphous regions of increased roughness, reminiscent of those observed for Nt17-Q35-P10-KK. These increased areas of roughness may represent an accumulation of htt exon1 proteins with lipid components, resulting in the observed patches of increased roughness. There were some differences between the morphologies of these larger aggregate types formed by Nt17-Q35-P10-KK and htt exon1(35Q). First, these roughened regions were typically shorter for the htt exon1 interaction with lipids compared with Nt17-Q35-P10-KK. Second, there was no obvious fibrillar structure around the periphery of these regions for htt exon1(35Q).
FIGURE 8.
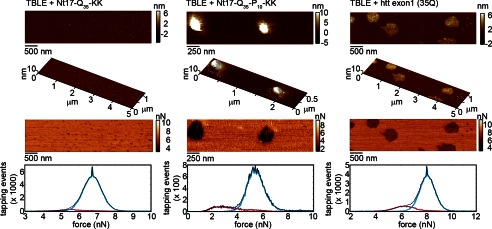
Exposing TBLE bilayers to (a) Nt17-Q35-KK, (b) Nt17-Q35-P10-KK, or (c) htt exon1(35Q) resulted in similar aggregate morphologies and local regions of reduced Young's modulus. The features in the topography image (top) correspond to regions associated with reduced values of maximum tapping force in the Fmax images (middle) and histograms (bottom) recovered from SPAM analysis. Values of Fmax are directly related to the relative rigidity (Young's modulus) of the surface.
Next, the mechanical impact of aggregates on the lipid surface were analyzed using scanning probe acceleration microscopy (SPAM) to spatially resolve the rigidity of lipid bilayers exposed to Nt17-Q35-KK, Nt17-Q35-P10-KK, or htt exon1(35Q). SPAM is suitable for this purpose because it is capable of reconstructing the time-resolved forces between the probe tip and sample associated with every tapping event during tapping mode AFM imaging in solution. Although the total force per oscillation cycle remains constant at any given set of operation parameters, features of the time-resolved tip/sample force changes in response to specific mechanical properties of the surfaces. Specifically, the maximum tapping force (Fmax), defined as the peak or largest positive force experienced between the tip and surface, increases with an increase in surface rigidity (measured as the Young's modulus) with a power law dependence (39). As a result, spatially resolved maps of Fmax can be used to correlate the information the relative rigidity to specific topographical features of surfaces. Spatially resolved Fmax images that correspond to the topographical images of lipid bilayers exposed to Nt17-Q35-KK, Nt17-Q35-P10-KK, or htt exon1(35Q) deposited on lipid bilayers were obtained. In the Fmax images of bilayers exposed to these peptides, there is a distinct contrast between the unperturbed regions of bilayer and regions containing either oligomers or disrupted regions of the bilayer. The stable oligomers of Nt17-Q35-KK were associated with a slight decrease in the rigidity of the bilayer (lower Fmax, Fig. 8a). In both cases, the domains where Nt17-Q35-P10-KK or htt exon1(35Q) associated with the lipid were associated with shifts to smaller magnitudes of Fmax, indicating that these domains were less rigid compared with unperturbed lipid bilayers (Fig. 8, b and c). This result demonstrates that not only does Nt17-Q35-P10-KK form similar aggregates on lipid bilayers compared with htt exon1(35Q), but also alters the local rigidity of the lipid bilayer in a similar fashion. The relative shift of the Fmax values associated with oligomers of Nt17-Q35-KK compared with unperturbed bilayer was smaller than the shift in Fmax associated with perturbed bilayer regions associated with Nt17-Q35-P10-KK or htt exon1(35Q), indicating that without the poly(P) domain the mechanical changes in the bilayer are less pronounced.
DISCUSSION
In this study, we investigated the role of flanking sequences on the aggregation of poly(Q) peptides on surfaces with an emphasis on lipid membranes. We report several findings. First, we demonstrate that, unlike aggregation in free solution (10, 11, 40), flanking sequences do not alter the aggregation pathway of poly(Q) on an anionic mica surfaces. Second, we provide direct evidence that the Nt17 domain is essential for the insertion of poly(Q) peptides into and interaction of poly(Q) aggregates with lipid membranes. Third, we show that lipid membranes can stabilize specific aggregate structures of poly(Q) peptides containing just the Nt17 domain. Fourth, we show that the addition of a C-terminal poly(P) domain, whereas not being sufficient alone to induce interaction of poly(Q) peptides with lipid surface, modulates the aggregation of poly(Q) peptides containing the Nt17 domain and increases the extent to which the peptides can permeabilize vesicles. Fifth, we demonstrate that the Nt17-Q35-P10-KK peptide aggregates in a similar fashion on brain lipid extract bilayers when compared with a htt exon1 construct containing Gln35. Finally, we provide evidence that the aggregation of Nt17-Q35-P10-KK and htt exon1(35Q) on lipid membranes is associated with a similar local reduction in the Young's modulus of the membrane. These results further support the hypothesis that flanking amino acids sequences modulate the conformation and aggregation of poly(Q) tracts and likely their biological activities.
The importance of Nt17 in htt aggregation and toxicity has received considerable attention. Our studies indicate a role of Nt17 in the interaction between htt and lipid membranes. Nt17 forms an amphipathic α-helical structure (20), which can influence the first few glutamines in the poly(Q) domain to adopt an α-helical conformation (41). The amphipathic properties of the Nt17 α-helix (Fig. 1B) can act as a lipid-binding domain (19), consistent with our observations that it facilitates interaction of poly(Q) peptides with lipid monolayers and bilayers. The importance of the α-helical Nt17 domain in facilitating the interaction between htt and lipid membranes is reminiscent of similar mechanisms of other amyloid-forming proteins. For example, the interaction of α-syn with lipid structures is directed by the first N-terminal 60 amino acids, containing an amphipathic α-helix structure (42), and IAPP also appears to interact with lipids via an α-helical binding domain (43). Several aggregation pathways have been proposed for the formation of fibrils of poly(Q)-containing proteins. Two of the more prominent aggregation schemes are: 1) re-arrangement of a monomer to a thermodynamically unfavorable conformation that directly nucleates fibril formation (44) and 2) the formation of soluble oligomeric intermediates that slowly undergo structural re-arrangement into a β-sheet-rich structure leading to fibrils (10, 11, 36). These aggregation pathways may not be mutually exclusive, but environmental factors, such as surfaces, may be able to promote a specific aggregation pathway (45). In our studies, the presence of lipid bilayers resulted in predominately either stabilized oligomers (Nt17-Q35-KK) or in large amorphous aggregates (Nt17-Q35-P10-KK), suggesting that the interaction of flanking sequences with membranes further modulates the aggregation pathway.
An important aspect of htt aggregation not addressed here is the role of poly(Q) expansion on the stability of flanking sequences and the consequences of poly(Q) expansion on the interaction with lipid membranes. Computational studies suggest that the Nt17 domain suppresses poly(Q) aggregation but expanding the poly(Q) domain destabilizes the Nt17 α-helix (40). Analysis of circular dichroism (CD) spectra of synthetic peptides containing Nt17 demonstrated an increased α-helical content associated with longer poly(Q) domain length, suggesting that poly(Q) expansion may act to stabilize the Nt17 domain (10). However, caution must be used in interpreting this result as CD is incapable of assigning a secondary structure to specific protein domains. Further clarification of the impact of poly(Q) expansion on the structure and stability of the Nt17 domain is warranted as this could further influence the interaction of htt with lipid membranes. In this regard, we have observed an increased rate of aggregation of htt exon1 proteins as a function of increased poly(Q) length in the presence of lipid membranes4; however, the aggregate morphology and mechanical impact on the bilayer is consistent with the observations reported here.
Despite the clear influence of the Nt17 and poly(P) domain on htt aggregation in bulk solution (11–13), poly(Q) peptides, with or without the Nt17 domain, aggregated via the same pathway on mica based on tracking and observing changes in morphology of individual aggregates over time. The four poly(Q) peptides studied here displayed the same aggregation pattern on mica, although the rate of aggregation formation was altered by flanking sequences; although, the effect of the different flanking sequences was not systematic. The similarities in the aggregation of the four peptides on mica demonstrate that the presence of the anionic surface dictated the aggregation pathway despite the presence of flanking sequences. As the only common motif between the four peptides was the 35-amino acid long poly(Q) sequence, one can reasonably conclude the specific interaction between the mica surface and the peptides was mediated by the poly(Q) domain.
Interestingly, the aggregation of poly(Q) peptides on a lipid bilayer was greatly influenced by the presence of flanking sequences. Although other studies suggested pure polyglutamine peptides are capable of forming discrete channels in planar lipid bilayers (46, 47), we did not observe any appreciable aggregation of pure poly(Q) on the TBLE bilayers. Additionally, the pure poly(Q) peptides did not insert into lipid monolayers or disrupt vesicles. As neither the poly(Q) domain nor poly(P) domain possess a net lipophilic hydropathy index, it is unsurprising that neither KK-Q35-KK nor KK-Q35-P10-KK appreciably aggregated on a lipid bilayer, suggesting that the poly(Q) and poly(P) domains are not essential for lipid association. The addition of only the Nt17 domain stabilized small, well defined oligomers of poly(Q) peptides on a bilayer. It is not clear if these oligomers form on the bilayer surface or in solution prior to binding to the bilayer. If these oligomers form in solution, they may represent similar oligomeric species observed in other studies (10). Such results indicate that surfaces influence aggregate formation and that surface effects in a physiological environment may influence disease pathology.
The different aggregate morphologies observed between Nt17-Q35-KK and Nt17-Q35-P10-KK on the lipid bilayer provides additional support for the importance of the poly(P) domains in htt aggregation. Although the addition of the poly(P) domain alone was not enough to induce an interaction with the bilayer, the poly(P) domain caused larger, amorphous aggregates to form on the bilayer when the Nt17 lipid binding domain was present. This notion is in agreement with our Langmuir trough studies, which demonstrate the Nt17 leads to membrane insertion, whereas the poly(P) domain enhances this interaction. The interaction and aggregation of Nt17-Q35-P10-KK on the lipid bilayer was similar to that of an actual htt exon1(35Q) construct. Beyond morphology, both the Nt17-Q35-P10-KK and htt exon1(35Q) softened the bilayer surface locally. It has been demonstrated that the addition of a poly(P) sequence to the C terminus of a poly(Q) peptide influences the conformation of the poly(Q) domain and alters the rate of aggregation (13). The poly(P) domain has also been shown to facilitate the ability of Nt17 to associate specifically with the ER and Golgi (22). Flanking poly(P) sequences induce a PPII-like helical structure on the adjacent poly(Q) domain, which can increase the length of the poly(Q) domain that is needed to induce fibril formation (14, 48). Our studies further support the importance of poly(P) in htt aggregation but in the context of lipid membranes.
The interaction of htt with membranous surfaces may mediate pathogenesis in a variety of ways. Interactions with lipids could stabilize or promote the formation of specific aggregate species that may prove toxic. Anchoring to membranes can lead to high local concentrations of peptide that facilitate aggregation. In such a scenario, post-translational modification of Nt17 that would reduce the affinity of htt for membranes would reduce toxicity, as is observed for phosphorylation of Nt17 (49). This is also consistent with observations that phosphomimetic and phosphoresistant mutations in the Nt17 domain differentially altered pathogenicity (50). Altered mechanical integrity of membranes due to htt binding and aggregation could lead directly to their dysfunction as was seen with the disruption of vesicles by Nt17-Q35-P10-KK. Lipid/htt interactions could also play a role in the trafficking of htt, which has been shown to be mediated by Nt17. Our results demonstrate that these interactions are directly mediated by the flanking sequences directly adjacent to the poly(Q) domain, suggesting therapeutic strategies designed to target these domains (such as intrabodies) may be appropriate to modulate membrane/htt interactions and neutralize toxic mutant htt assemblies (51, 52).
It has been documented that the poly(Q) repeat in htt adopts several conformations in monomers (41) and other aggregated forms (53). It is plausible that a subset of these conformers mediate pathogenesis in HD, and understanding environmental factors, such as membrane interactions that dictate the relative abundance of these different forms could provide insights into mechanisms associated with htt aggregation and toxicity. In light of these observed instances of htt aggregate heterogeneity, the ability of lipid bilayers to stabilize discrete, well defined oligomer species of the Nt17-Q35-KK and to promote different aggregate morphologies in Nt17-Q35-P10-KK was striking, especially considering the distinct fibrillar morphology observed on mica for aggregates of these two peptides. Recent EPR structural studies of htt aggregates suggested that the Nt17 domain form a rigid, interior core of the fibril (54). The stabilization of oligomers by the lipid bilayer may partially be caused by a strong interaction between the bilayer and the amphipathic Nt17 domain, as suggested by the insertion of the Nt17 containing peptides into monolayers, creating an energetic barrier to rearrangement of the Nt17 domain to form the core of the fibril. No matter the mechanism, the stabilization of oligomeric aggregates may play a role in a variety of toxic mechanisms.
It has long been hypothesized that membrane elasticity directly influences membrane stability and that alterations in this mechanical property can lead to dysfunction and disease (55, 56). Our results demonstrate Nt17-Q35-P10-KK shares a similar aggregate morphology and mechanical impact on a lipid bilayer compared with htt exon1(35Q). Both Nt17-Q35-P10-KK and htt exon1(35Q) produce roughened domains that are more compressible than unperturbed bilayers. This observed mechanical change is consistent with a decreased efficiency of lipid component packing as they stretch themselves to compensate for protein insertion/binding, thus resulting in a rougher bilayer that is more easily compressed by the AFM tip. By binding to lipid membranes, inserting, and aggregating, mutant huntingtin could disorder the lipid bilayer and disturb subcellular membrane function, consistent with our observation that the compression modulus decrease in regions of the bilayer displaying increased roughness or containing protein aggregates. These observations are important in light of studies comparing control and HD brains indicate that membranes exposed to htt with expanded poly(Q) have altered physical properties (57).
In the interaction of htt with lipid membranes, the flanking sequences appear to play a significant role. The initial interaction of htt with a variety of model membranes was dependent on the addition of the Nt17 flanking region. Interestingly, the ability of Nt17-Q35-P10-KK to cause significant leakage of calcein dye from lipid vesicles is consistent with the large extent of morphological and mechanical changes caused by Nt17-Q35-P10-KK compared with the other peptides, as observed by AFM. Collectively, these observations suggest that although Nt17 is necessary for lipid binding, poly(P) may facilitate membrane damage associated with htt aggregation.
This work was supported by the Brodie Discovery and Innovation fund, National Science Foundation Grant 1054211, and Gettysburg College (to K. J. K. and S. L. F.).
K. A. Burke, C. S. Umbaugh, K. M. Hensal, M. Chaibva, and J. Legleiter, unpublished data.
- poly(Q)
- polyglutamine
- HD
- Huntington disease
- AFM
- atomic force microscopy
- htt
- huntingtin
- TBLE
- total brain lipid extract
- poly(P)
- polyproline
- Nt17
- the first 17 amino acids of htt
- SPAM
- scanning probe acceleration microcopy, Fmax, maximum tapping force
- DMPC
- 1,2-ditetradecanoyl-sn-glycero-3-phosphocholine
- mN
- millinewton
- LUV
- large unilamellar vesicles.
REFERENCES
- 1. The Huntington Disease Collaborative Research Group (1993) A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes. Cell 72, 971–983 [DOI] [PubMed] [Google Scholar]
- 2. DiFiglia M., Sapp E., Chase K. O., Davies S. W., Bates G. P., Vonsattel J. P., Aronin N. (1997) Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science 277, 1990–1993 [DOI] [PubMed] [Google Scholar]
- 3. Ravina B., Romer M., Constantinescu R., Biglan K., Brocht A., Kieburtz K., Shoulson I., McDermott M. P. (2008) The relationship between CAG repeat length and clinical progression in Huntington's disease. Movement Disord. 23, 1223–1227 [DOI] [PubMed] [Google Scholar]
- 4. Kim Y. J., Yi Y., Sapp E., Wang Y., Cuiffo B., Kegel K. B., Qin Z. H., Aronin N., DiFiglia M. (2001) Caspase 3-cleaved N-terminal fragments of wild-type and mutant huntingtin are present in normal and Huntington's disease brains, associate with membranes, and undergo calpain-dependent proteolysis. Proc. Natl. Acad. Sci. U.S.A. 98, 12784–12789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ratovitski T., Gucek M., Jiang H., Chighladze E., Waldron E., D'Ambola J., Hou Z., Liang Y., Poirier M. A., Hirschhorn R. R., Graham R., Hayden M. R., Cole R. N., Ross C. A. (2009) Mutant Huntingtin N-terminal fragments of specific size mediate aggregation and toxicity in neuronal cells. J. Biol. Chem. 284, 10855–10867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Davies S. W., Turmaine M., Cozens B. A., DiFiglia M., Sharp A. H., Ross C. A., Scherzinger E., Wanker E. E., Mangiarini L., Bates G. P. (1997) Formation of neuronal intranuclear inclusions underlies the neurological dysfunction in mice transgenic for the HD mutation. Cell 90, 537–548 [DOI] [PubMed] [Google Scholar]
- 7. Landles C., Sathasivam K., Weiss A., Woodman B., Moffitt H., Finkbeiner S., Sun B., Gafni J., Ellerby L. M., Trottier Y., Richards W. G., Osmand A., Paganetti P., Bates G. P. (2010) Proteolysis of mutant Huntingtin produces an exon 1 fragment that accumulates as an aggregated protein in neuronal nuclei in Huntington disease. J. Biol. Chem. 285, 8808–8823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bulone D., Masino L., Thomas D. J., San Biagio P. L., Pastore A. (2006) The interplay between polyQ and protein context delays aggregation by forming a reservoir of protofibrils. PLoS One 1, e111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ignatova Z., Gierasch L. M. (2006) Extended polyglutamine tracts cause aggregation and structural perturbation of an adjacent β barrel protein. J. Biol. Chem. 281, 12959–12967 [DOI] [PubMed] [Google Scholar]
- 10. Jayaraman M., Kodali R., Sahoo B., Thakur A. K., Mayasundari A., Mishra R., Peterson C. B., Wetzel R. (2012) Slow amyloid nucleation via α-helix-rich oligomeric intermediates in short polyglutamine-containing Huntingtin fragments. J. Mol. Biol. 415, 881–899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Thakur A. K., Jayaraman M., Mishra R., Thakur M., Chellgren V. M., Byeon I.-J., Anjum D. H., Kodali R., Creamer T. P., Conway J. F., Gronenborn A. M., Wetzel R. (2009) Polyglutamine disruption of the huntingtin exon 1 N terminus triggers a complex aggregation mechanism. Nat. Struct. Mol. Biol. 16, 380–389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Mishra R., Jayaraman M., Roland B. P., Landrum E., Fullam T., Kodali R., Thakur A. K., Arduini I., Wetzel R. (2012) Inhibiting the nucleation of amyloid structure in a Huntingtin fragment by targeting α-helix-rich oligomeric intermediates. J. Mol. Biol. 415, 900–917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bhattacharyya A., Thakur A. K., Chellgren V. M., Thiagarajan G., Williams A. D., Chellgren B. W., Creamer T. P., Wetzel R. (2006) Oligoproline effects on polyglutamine conformation and aggregation. J. Mol. Biol. 355, 524–535 [DOI] [PubMed] [Google Scholar]
- 14. Darnell G., Orgel J. P., Pahl R., Meredith S. C. (2007) Flanking polyproline sequences inhibit β-sheet structure in polyglutamine segments by inducing PPII-like helix structure. J. Mol. Biol. 374, 688–704 [DOI] [PubMed] [Google Scholar]
- 15. Kegel K. B., Kim M., Sapp E., McIntyre C., Castaño J. G., Aronin N., DiFiglia M. (2000) Huntingtin expression stimulates endosomal-lysosomal activity, endosome tubulation, and autophagy. J. Neurosci. 20, 7268–7278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Suopanki J., Götz C., Lutsch G., Schiller J., Harjes P., Herrmann A., Wanker E. E. (2006) Interaction of huntingtin fragments with brain membranes. Clues to early dysfunction in Huntington's disease. J. Neurochem. 96, 870–884 [DOI] [PubMed] [Google Scholar]
- 17. Valencia A., Reeves P. B., Sapp E., Li X., Alexander J., Kegel K. B., Chase K., Aronin N., DiFiglia M. (2010) Mutant Huntingtin and glycogen synthase kinase 3-β accumulate in neuronal lipid rafts of a presymptomatic knock-in mouse model of Huntington's disease. J. Neurosci. Res. 88, 179–190 [DOI] [PubMed] [Google Scholar]
- 18. Kegel K. B., Sapp E., Yoder J., Cuiffo B., Sobin L., Kim Y. J., Qin Z. H., Hayden M. R., Aronin N., Scott D. L., Isenberg G., Goldmann W. H., DiFiglia M. (2005) Huntingtin associates with acidic phospholipids at the plasma membrane. J. Biol. Chem. 280, 36464–36473 [DOI] [PubMed] [Google Scholar]
- 19. Michalek M., Salnikov E. S., Werten S., Bechinger B. (2013) Membrane interactions of the amphipathic amino terminus of Huntingtin. Biochemistry 52, 847–858 [DOI] [PubMed] [Google Scholar]
- 20. Atwal R. S., Xia J., Pinchev D., Taylor J., Epand R. M., Truant R. (2007) Huntingtin has a membrane association signal that can modulate huntingtin aggregation, nuclear entry and toxicity. Hum. Mol. Genet. 16, 2600–2615 [DOI] [PubMed] [Google Scholar]
- 21. Trettel F., Rigamonti D., Hilditch-Maguire P., Wheeler V. C., Sharp A. H., Persichetti F., Cattaneo E., MacDonald M. E. (2000) Dominant phenotypes produced by the HD mutation in STHdh(Q111) striatal cells. Hum. Mol. Genet. 9, 2799–2809 [DOI] [PubMed] [Google Scholar]
- 22. Rockabrand E., Slepko N., Pantalone A., Nukala V. N., Kazantsev A., Marsh J. L., Sullivan P. G., Steffan J. S., Sensi S. L., Thompson L. M. (2007) The first 17 amino acids of Huntingtin modulate its subcellular localization, aggregation and effects on calcium homeostasis. Hum. Mol. Genet. 16, 61–77 [DOI] [PubMed] [Google Scholar]
- 23. Kegel K. B., Sapp E., Alexander J., Valencia A., Reeves P., Li X., Masso N., Sobin L., Aronin N., DiFiglia M. (2009) Polyglutamine expansion in huntingtin alters its interaction with phospholipids. J. Neurochem. 110, 1585–1597 [DOI] [PubMed] [Google Scholar]
- 24. Chen S., Wetzel R. (2001) Solubilization and disaggregation of polyglutamine peptides. Protein Sci. 10, 887–891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Muchowski P. J., Schaffar G., Sittler A., Wanker E. E., Hayer-Hartl M. K., Hartl F. U. (2000) Hsp70 and Hsp40 chaperones can inhibit self-assembly of polyglutamine proteins into amyloid-like fibrils. Proc. Natl. Acad. Sci. U.S.A. 97, 7841–7846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Legleiter J., Park M., Cusick B., Kowalewski T. (2006) Scanning probe acceleration microscopy (SPAM) in fluids. Mapping mechanical properties of surfaces at the nanoscale. Proc. Natl. Acad. Sci. U.S.A. 103, 4813–4818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Burke K. A., Godbey J., Legleiter J. (2011) Assessing mutant huntingtin fragment and polyglutamine aggregation by atomic force microscopy. Methods 53, 275–284 [DOI] [PubMed] [Google Scholar]
- 28. Legleiter J., DeMattos R. B., Holtzman D. M., Kowalewski T. (2004) In situ AFM studies of astrocyte-secreted apolipoprotein E- and J-containing lipoproteins. J. Colloid. Interface Sci. 278, 96–106 [DOI] [PubMed] [Google Scholar]
- 29. Gekko K., Noguchi H. (1979) Compressibility of globular proteins in water at 25 °C. J. Phys. Chem. 83, 2706–2714 [Google Scholar]
- 30. Squire P. G., Himmel M. E. (1979) Hydrodynamics and protein hydration. Arch. Biochem. Biophys. 196, 165–177 [DOI] [PubMed] [Google Scholar]
- 31. Seelig A. (1987) Local anesthetics and pressure: a comparison of dibucaine binding to lipid monolayers and bilayers. Biochim. Biophys. Acta 899, 196–204 [DOI] [PubMed] [Google Scholar]
- 32. Anderson R. L., Davis S. (1982) An organic phosphorus assay which avoids the use of hazardous perchloric acid. Clin. Chim. Acta 121, 111–116 [DOI] [PubMed] [Google Scholar]
- 33. Rouser G., Fkeischer S., Yamamoto A. (1970) Two-dimensional then layer chromatographic separation of polar lipids and determination of phospholipids by phosphorus analysis of spots. Lipids 5, 494–496 [DOI] [PubMed] [Google Scholar]
- 34. Chou H. T., Wen H. W., Kuo T. Y., Lin C. C., Chen W. J. (2010) Interaction of cationic antimicrobial peptides with phospholipid vesicles and their antibacterial activity. Peptides 31, 1811–1820 [DOI] [PubMed] [Google Scholar]
- 35. Lamazière A., Burlina F., Wolf C., Chassaing G., Trugnan G., Ayala-Sanmartin J. (2007) Non-metabolic membrane tubulation and permeability induced by bioactive peptides. PLoS One 2, e201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Legleiter J., Mitchell E., Lotz G. P., Sapp E., Ng C., DiFiglia M., Thompson L. M., Muchowski P. J. (2010) Mutant Huntingtin fragments form oligomers in a polyglutamine length-dependent manner in vitro and in vivo. J. Biol. Chem. 285, 14777–14790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Groves J. T., Ulman N., Boxer S. G. (1997) Micropatterning fluid lipid bilayers on solid supports. Science 275, 651–653 [DOI] [PubMed] [Google Scholar]
- 38. Seelig A., Blatter X. L., Frentzel A., Isenberg G. (2000) Phospholipid binding of synthetic talin peptides provides evidence for an intrinsic membrane anchor of talin. J. Biol. Chem. 275, 17954–17961 [DOI] [PubMed] [Google Scholar]
- 39. Shamitko-Klingensmith N., Molchanoff K. M., Burke K. A., Magnone G. J., Legleiter J. (2012) Mapping the mechanical properties of cholesterol-containing supported lipid bilayers with nanoscale spatial resolution. Langmuir 28, 13411–13422 [DOI] [PubMed] [Google Scholar]
- 40. Williamson T. E., Vitalis A., Crick S. L., Pappu R. V. (2010) Modulation of polyglutamine conformations and dimer formation by the N-terminus of Huntingtin. J. Mol. Biol. 396, 1295–1309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kim M. W., Chelliah Y., Kim S. W., Otwinowski Z., Bezprozvanny I. (2009) Secondary structure of Huntingtin amino-terminal region. Structure 17, 1205–1212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Clayton D. F., George J. M. (1998) The synucleins. A family of proteins involved in synaptic function, plasticity, neurodegeneration and disease. Trends Neurosci. 21, 249–254 [DOI] [PubMed] [Google Scholar]
- 43. Knight J. D., Miranker A. D. (2004) Phospholipid catalysis of diabetic amyloid assembly. J. Mol. Biol. 341, 1175–1187 [DOI] [PubMed] [Google Scholar]
- 44. Chen S., Ferrone F. A., Wetzel R. (2002) Huntington's disease age-of-onset linked to polyglutamine aggregation nucleation. Proc. Natl. Acad. Sci. U.S.A. 99, 11884–11889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Burke K. A., Yates E. A., Legleiter J. (2013) Biophysical insights into how surfaces, including lipid membranes, modulate protein aggregation related to neurodegeneration. Front. Neurol. 4, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Hirakura Y., Azimov R., Azimova R., Kagan B. L. (2000) Polyglutamine-induced ion channels. A possible mechanism for the neurotoxicity of Huntington and other CAG repeat diseases. J. Neurosci. Res. 60, 490–494 [DOI] [PubMed] [Google Scholar]
- 47. Monoi H., Futaki S., Kugimiya S., Minakata H., Yoshihara K. (2000) Poly-l-glutamine forms cation channels. Relevance to the pathogenesis of the polyglutamine diseases. Biophys. J. 78, 2892–2899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Darnell G. D., Derryberry J., Kurutz J. W., Meredith S. C. (2009) Mechanism of cis-inhibition of polyQ fibrillation by polyP. PPII oligomers and the hydrophobic effect. Biophys. J. 97, 2295–2305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Atwal R. S., Desmond C. R., Caron N., Maiuri T., Xia J., Sipione S., Truant R. (2011) Kinase inhibitors modulate Huntingtin cell localization and toxicity. Nat. Chem. Biol. 7, 453–460 [DOI] [PubMed] [Google Scholar]
- 50. Gu X., Greiner E. R., Mishra R., Kodali R., Osmand A., Finkbeiner S., Steffan J. S., Thompson L. M., Wetzel R., Yang X. W. (2009) Serines 13 and 16 are critical determinants of full-length human mutant Huntingtin induced disease pathogenesis in HD mice. Neuron 64, 828–840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Southwell A. L., Ko J., Patterson P. H. (2009) Intrabody gene therapy ameliorates motor, cognitive, and neuropathological symptoms in multiple mouse models of Huntington's disease. J. Neurosci. 29, 13589–13602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Colby D. W., Garg P., Holden T., Chao G., Webster J. M., Messer A., Ingram V. M., Wittrup K. D. (2004) Development of a human light chain variable domain (V-L) intracellular antibody specific for the amino terminus of Huntingtin via yeast surface display. J. Mol. Biol. 342, 901–912 [DOI] [PubMed] [Google Scholar]
- 53. Nekooki-Machida Y., Kurosawa M., Nukina N., Ito K., Oda T., Tanaka M. (2009) Distinct conformations of in vitro and in vivo amyloids of huntingtin-exon1 show different cytotoxicity. Proc. Natl. Acad. Sci. U.S.A. 106, 9679–9684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Bugg C. W., Isas J. M., Fischer T., Patterson P. H., Langen R. (2012) Structural features and domain organization of huntingtin fibrils. J. Biol. Chem. 287, 31739–31746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Janmey P. A., Weitz D. A. (2004) Dealing with mechanics. Mechanisms of force transduction in cells. Trends Biochem. Sci. 29, 364–370 [DOI] [PubMed] [Google Scholar]
- 56. Vogel V., Sheetz M. (2006) Local force and geometry sensing regulate cell functions. Nat. Rev. Mol. Cell Biol. 7, 265–275 [DOI] [PubMed] [Google Scholar]
- 57. Lloyd K. G., Davidson L. (1979) H-3 GABA binding in brains from Huntington chorea patients, Altered regulation by phospholipids. Science 205, 1147–1149 [DOI] [PubMed] [Google Scholar]