

Background: FVIIIa binds FIXa through interactions involving A2 and A3C1C2 subunits.
Results: A2 subunit shows a high affinity interaction for FIXa with contribution of residues 707-714 to this interaction.
Conclusion: Sequences in A2 subunit make significant contributions to binding FIXa.
Significance: This study shows an important contribution of A2 subunit in forming FXase and identifies residues participating in this interaction.
Keywords: Coagulation Factors, Factor VIII, Mutagenesis, Protein Domains, Surface Plasmon Resonance (SPR)
Abstract
Factor (F) VIIIa forms a number of contacts with FIXa in assembling the FXase enzyme complex. Surface plasmon resonance was used to examine the interaction between immobilized biotinylated active site-modified FIXa, and FVIII and FVIIIa subunits. The FVIIIa A2 subunit bound FIXa with high affinity (Kd = 3.9 ± 1.6 nm) that was similar to the A3C1C2 subunit (Kd = 3.6 ± 0.6 nm). This approach was used to evaluate a series of baculovirus-expressed, isolated A2 domain (bA2) variants where alanine substitutions were made for individual residues within the sequence 707-714, the C-terminal region of A2 thought to be FIXa interactive. Three of six bA2 variants examined displayed 2- to 4-fold decreased affinity for FIXa as compared with WT bA2. The variant bA2 proteins were also tested in two reconstitution systems to determine activity and affinity parameters in forming FXase and FVIIIa. Vmax values for all variants were similar to the WT values, indicating that these residues do not affect cofactor function. All variants showed substantially greater increases in apparent Kd relative to WT in reconstituting the FXase complex (8- to 26-fold) compared with reconstituting FVIIIa (1.3- to 6-fold) suggesting that the mutations altered interaction with FIXa. bA2 domain variants with Ala replacing Lys707, Asp712, and Lys713 demonstrated the greatest increases in apparent Kd (17- to 26-fold). These results indicate a high affinity interaction between the FVIIIa A2 subunit and FIXa and show a contribution of several residues within the 707-714 sequence to this binding.
Introduction
FVIIIa2 serves as a cofactor for the serine protease FIXa in the conversion of FX to FXa during the propagation phase of coagulation, increasing the kcat for this reaction by several orders of magnitude (1, 2). FVIII is synthesized as a multidomain, single chain precursor with domain structure A1-A2-B-A3-C1-C2. Segments rich in acidic residues border the A domains of FVIII and are designated a1 (residues 337–372) and a2 (residues 711–740), which directly follow the A1 and A2 domains, respectively, and a3 (residues 1649–1689) preceding the A3 domain. FVIII circulates primarily as a noncovalent heterodimer comprised of a heavy chain (A1a1A2a2B domains) and light chain (LC) (a3A3C1C2 domains) resulting from proteolysis at the B-A3 junction (see Ref. 3 for review). FVIII is converted to the active cofactor, FVIIIa, following limited proteolysis catalyzed by thrombin or FXa at the a1-A2, a2-B, and a3-A3 junctions. The resulting FVIIIa heterotrimer (4, 5) is comprised of the heavy chain-derived A1 subunit in a stable association with the light chain-derived A3C1C2 subunit, whereas the A2 subunit is weakly associated with the A1/A3C1C2 dimer through electrostatic interactions (6, 7). FVIIIa can be reconstituted from the isolated A2 subunit and A1/A3C1C2 dimer to generate high levels of cofactor activity (5, 7).
FIXa has been shown to stabilize FVIIIa in the presence of Ca2+ and phospholipid (8) by promoting association of A2 subunit with A1/A3C1C2 dimer (9, 10). This stabilization appears to result from multiple interactions with FIXa involving residues within the A3 domain of the A3C1C2 subunit and residues within the A2 subunit. The former interaction is of relatively high affinity (Kd ∼15–50 nm) as determined by solid phase, nonequilibrium (11) and steady state fluorescence binding assays (12). Studies on assessing the affinity of A2 subunit for FIXa have been limited to activity-based assays and suggested a relatively weaker affinity interaction (Kd ∼300 nm) (13). A number of studies assessing inhibition by peptides and antibodies as well as analyses of point mutations in the hemophilia A database have identified several sequences as FIXa interactive (see Ref. 14 for a review). One FIXa-interactive region in the A2 subunit, the 558-loop (15) appears to modulate the active site of the enzyme (16) as judged by reduced kcat values following site-specific mutations in this sequence. More recently, Jagannathan et al. (17) developed a baculovirus-expression system for the isolated bA2 domain and showed by FVIIIa reconstitution that this domain effectively recapitulated FVIIIa activity when combined with the A1/A3C1C2 dimer. Furthermore, that study showed FXase could be reconstituted using the isolated bA2 domain, A1/A3C1C2 dimer, FIXa, and a phospholipid membrane. Comparison of reconstitution systems showed an ∼6-fold increase in affinity of A2 for FXase compared with FVIIIa reconstitution, consistent with the stabilizing activity of FIXa in A2 subunit retention. In addition, that study also examined a number of point mutations where residues in and around the 558-loop were replaced with Ala and demonstrated that the A2 subunit affinity relative to the WT bA2, as determined by FXase reconstitution, was significantly more affected than affinity determined by FVIIIa reconstitution, and identified a number of residues that contribute to both the FIXa binding interaction and cofactor function.
In the current study, we examine protein-protein interactions for several FVIII and FVIIIa-derived substrates with FIXa by surface plasmon resonance (SPR) and demonstrate a previously unidentified high affinity interaction of the isolated FVIIIa A2 subunit for the enzyme. We also extend this analysis to a putative FIXa-interactive region, residues 707-714, localized to the C-terminal region of the A2 subunit and show by both SPR and functional FVIIIa and FXase reconstitution assays that a number of variants possessing Ala mutations at individual residues within the 707-714 sequence possess reduced affinity for FIXa. These results suggest that the 707-714 region, which is in close proximity to the 558-loop, contributes to an extended interactive surface for FIXa.
EXPERIMENTAL PROCEDURES
Reagents
SPR chips and cross-linking reagents were purchased from GE Healthcare. FIXa and FX were acquired from Enzyme Research Laboratory (South Bend, IN) and Pefa-5523 chromogenic substrate from Centerchem (Norwalk, CT). Recombinant FVIII (Kogenate) was a generous gift from Dr. Lisa Regan of Bayer Corp. (Berkeley, CA). Phospholipids were obtained from Avanti Polar Lipids and PS/PC/PE vesicles containing 20% phosphatidylserine (PS), 40% phosphatidylcholine (PC), and 40% phosphatidylethanolamine (PE) were prepared as previously described (18). Biotinylated-FPR chloromethylketone and human α-thrombin were purchased from Hematologic Technologies Inc. (Essex Junction, VT).
Preparation of FVIII Subunits
FVIII LC and the FVIIIa subunits A2, A3C1C2, and A1/A3C1C2 dimer were prepared from recombinant FVIII using previously published methods. Briefly, LC was isolated by treating FVIII (overnight) in buffer containing 50 mm EDTA to separate the heavy and light chains. FVIII LC (a3A3C1C2) was isolated by SP-Sepharose column chromatography (19). To remove the a3 acidic region preceding the A3 domain, LC was treated with thrombin and the product re-purified using SP-Sepharose chromatography. FVIII heavy chain was further purified by Q-Sepharose column chromatography and was used as a source of A2 subunit following digestion with thrombin and isolation by Hi-Trap Heparin HPLC (20). The A1/A3C1C2 dimer was prepared by thrombin digestion of FVIII and removal of A2 by use of a anti-A2 subunit monoclonal antibody coupled to Affi-Gel 10 (21). The purity of all subunits was >95%, as determined by SDS-PAGE, and the concentrations were determined by the method of Bradford (22).
Mutagenesis, Expression, and Purification of Wild Type and Variant FVIII
The QuikChange site-directed mutagenesis kit was used to introduce single alanine mutations into the plasmid pFastBac-wtA2 (17), containing a His6 tag followed by TEV cleavage site. The Cys711 residue was not changed to alanine as it forms a disulfide bond with Cys630 that is essential for function (23). The A2 wild type (WT) and variants were prepared from Sf9 cells using the Bac-to-Bac Expression System, and expressed in High Five cells (Invitrogen). These A2 subunits were designated as bA2 to indicate the source of the proteins. WT and variant proteins were purified as previously described (17). Briefly, supernatant from baculovirus-infected Hi5 cells expressing bA2 protein was concentrated and partially purified on a heparin-Sepharose column. A TalonTM immobilized metal affinity chromatography column was used to isolate the protein through use of the His6 tag, followed by cleavage of the tag by AcTEV protease. Protease was removed and protein was concentrated by SP-Sepharose column chromatography and final concentrations were determined by the Bradford assay. The bA2 proteins obtained were >90% pure and samples were quick-frozen and stored at −80 °C. The V708A variant protein was refractory to purification, and was not used in the study.
Modification of FIXa with bFPRck
FIXa was modified with a biotin-labeled active site-directed inhibitor, bFPRck following incubation of 10 μm FIXa with 50 μm bFPRck at 23 °C for 2 h, then 18 h at 4 °C in a buffer of 0.2 m Tris, pH 7.4, 0.15 m NaCl, and 20% ethylene glycol. The resulting bFPR-FIXa was assayed in a FXa generation system to ensure reaction completion, filtered on a PD10 column to remove unreacted peptide, and protein concentration was determined by Bradford assay.
FVIIIa Subunit Interaction with FIXa by SPR
The kinetics of FIXa interaction with various FVIII(a) subunits were monitored at 37 °C by SPR using a Biacore T100 instrument (GE Healthcare). bFPR-FIXa was immobilized on a streptavidin chip. Binding (association) of the FVIIIa subunit at various concentrations (0.78 to 50 nm) in HEPES-buffered saline buffer (20 mm HEPES, pH 7.2, 100 mm NaCl, 0.01% Tween 20) with 5 mm CaCl2 was monitored for 3 min, followed by dissociation of bound subunit for 3–4 min by switching to buffer alone. Nonspecific binding was accounted for by running parallel samples on an uncoated chip, and the resulting signal subtracted from the sample signal. Following each analysis, chip surfaces were regenerated with 500 mm NaCl, 25 mm NaOH for 30 s. Using the BIAevalution software, kinetics parameters were determined by globally fitting the data to a 2-state binding model described by Equation 1.
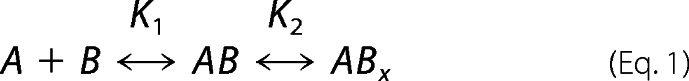 |
In which analyte (A) binds ligand (B) to form an initial complex AB, then undergoes further change to yield the final ABx complex. The individual equilibrium constants are defined by K1 = ka1/kd1 and K2 = ka2/kd2 and overall Kd = 1/(K1 (1 + K2)). Using this binding model, fits were found to be superior to those of a 1:1 Langmuir model, by comparison of χ2 values. Experiments were repeated a minimum of 3 times.
FXa Generation Assays
The rate of conversion of FX to FXa was monitored in a purified system at 23 °C. FXase was reconstituted by combining WT or variant bA2 (3–1600 nm) with purified A1/A3C1C2 dimer (6.8 nm) in the presence of PS/PC/PE vesicles (10 μm) and limiting FIXa (1 nm) were incubated for 10 min, and reactions initiated by addition of 300 nm FX and run for 1 min. Alternatively, FVIIIa was reconstituted by adding WT or variant bA2 (12.5–1600 nm) to limiting A1/A3C1C2 dimer (1 nm) for 10 min at 23 °C followed by addition of FIXa (40 nm) and PS/PC/PE vesicles (10 μm). Following incubation for 1 min, reactions were initiated by addition of FX (300 nm) and run for 1 min. All reactions were stopped by the addition of EDTA (final concentration 50 mm), and rates of FXa generation determined by the addition of chromogenic substrate Pefa-5523 (0.46 mm final concentration) and read for 6 min at 385 nm using a Vmax microtiter plate reader (Molecular Devices).
Data Analysis
FXa generation data were fit to a single-site ligand binding model using nonlinear least-squares regression analysis,
where A is the activity of FXase (nm min−1(nm FIX)−1) or FVIIIa (nm min−1(nm A1/A3C1C2)−1). [A2] is the bA2 subunit concentration, Vmax is the maximum velocity, and Kd is the dissociation constant for A2 in FXase or FVIIIa.
RESULTS
Interaction between FVIIIa Subunits and FIXa by SPR
Selected subunits derived from FVIII (a3A3C1C2) and FVIIIa (A2, A3C1C2, A1/A3C1C2) were purified as described under “Experimental Procedures.” Subunits were >95% pure as judged by SDS-PAGE (results not shown). SPR was used to determine the affinity between FIXa and each FVIII or FVIIIa subunit. FIXa was tethered in an orientation-specific manner to a streptavidin chip by use of a biotin-labeled peptide inhibitor, bFPRck, that binds covalently near the active site of the enzyme. FIXa modified in its active site with FPRck reagents has been shown to fully block the interaction of FVIIIa with native FIXa as judged by a FXa generation assay (12), indicating that blocking the active site in the proteinase does not impact FVIIIa binding. Various concentrations of the FVIII and/or subunits (0.78–50 nm) were then used in the mobile phase, and binding was monitored as described under “Experimental Procedures.” Data from SPR were globally fitted to a 2-state model as described under “Experimental Procedures.” This model is consistent with a multiphase process because more than one FIXa-interactive site has been identified in isolated FVIIIa subunits. For example, A2 subunit residues contained within the 558-loop as well as the C-terminal region of the A2 domain have been implicated in interacting with FIXa (see Refs. 3 and 14). Furthermore, superior fits of the data were obtained with the two-state model compared with a single-site binding model as shown by comparison of the fits in Fig. 1 and χ2 values in Table 1. Values for Kd derived from the single-site model are also presented in Table 1 for comparison.
FIGURE 1.
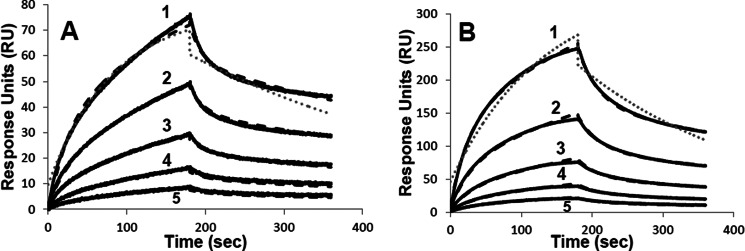
Binding of A2 and a3A3C1C2 to FIXa by SPR. Biotinylated FPR-FIXa was immobilized on a streptavidin chip and the binding kinetics with the FVIII A2 subunit (A) and a3A3C1C2 (B) were monitored at a flow rate of 10 μl/min for a 3-min association with subunit followed by a 3-min dissociation after return to buffer flow. Representative curves correspond to subunit concentrations of (1) 12.5 nm, (2) 6.25 nm, (3) 3.13 nm, (4) 1.56 nm, and (5) 0.78 nm with data represented by the bold black line, and the fitted curve using the two-state binding model shown by the black dashed line. A representative fit using the single-site binding model is shown by the gray dotted line for the 12.5 nm subunit concentrations.
TABLE 1.
Kinetic parameters for binding of FVIIIa subunits and FIXa
Kinetic parameters were determined by SPR as described under “Experimental Procedures” using FVIII or FVIII(a)-derived subunits in the mobile phase and bFPR-FIXa bound to a SA chip. Samples were run at least three times, and mean values with standard deviations shown. Values for Kd and χ2 are shown for both the two-state and single-site models for comparison.
| SA analyte | ka1 | kd1 | ka2 | kd2 | Two-state |
Single-site |
||
|---|---|---|---|---|---|---|---|---|
| Kd | χ2 | Kd | χ2 | |||||
| m−1 s−1 | s−1 | s−1 | s−1 | nm | nm | |||
| A2 | (2.3 ± 0.7) × 107 | (0.52 ± 0.2) | (1.3 ± 0.2) × 10−2 | (2.6 ± 0.8) × 10−3 | 3.9 ± 1.6 | 0.87 | 3.9 ± 0.4 | 2.54 |
| A3C1C2 | (4.1 ± 0.8) × 105 | (0.9 ± 0.4) × 10−2 | (3.5 ± 0.6) × 10−3 | (0.7 ± 0.1) × 10−3 | 3.6 ± 0.6 | 135 | 11.3 ± 0.1 | 101 |
| a3A3C1C2 | (6.6 ± 0.7) × 105 | (3.0 ± 0.2) × 10−2 | (8.2 ± 1.4) × 10−3 | (2.4 ± 0.6) × 10−3 | 10.5 ± 0.9 | 6.6 | 16.7 ± 0.2 | 115 |
| A1/A3C1C2 | (6.6 ± 0.4) × 105 | (1.7 ± 0.2) × 10−2 | (7.0 ± 1.6) × 10−3 | (3.1 ± 1.1) × 10−3 | 7.5 ± 0.9 | 15 | 12.7 ± 0.3 | 90.0 |
| FVIII | (8.0 ± 1.0) × 105 | (3.1 ± 0.2) × 10−2 | (8.8 ± 0.3) × 10−3 | (2.1 ± 0.1) × 10−3 | 7.3 ± 0.4 | 6.7 | 11.1 ± 0.8 | 238 |
Results obtained for several concentrations of A2 and a3A3C1C2 subunits are shown in Fig. 1 with parameter values obtained from binding experiments for all subunits presented in Table 1. The A2 subunit was observed to bind with high affinity to FIXa (Fig. 1A), with a Kd of 3.9 nm. This value was markedly reduced compared with earlier results assessing an affinity value from a functional assay based upon the capacity of the isolated A2 subunit to stimulate FIXa-catalyzed activation of FX (Kd ∼300 nm, (13)), and suggests a large disparity between static and kinetic affinity values for this interaction. Although the A2 subunit appears to have ka1 and kd1 values that were 2 orders of magnitude larger than those for other FVIII-derived subunits, these approach instrument limitations (Table 1). Refitting the A2 data with local (versus global) Rmax resulted in values of the same order of magnitude as the other FVIII-derived subunits, with no significant effect on the overall Kd value (data not shown). In contrast, the light chain of FVIII (a3A3C1C2) showed a Kd value (10.5 nm) that was ∼3-fold weaker than that for A2 (Fig. 1B) and was similar to a value obtained for the FVIII light chain (Kd = 14 nm) from an earlier study employing a solid phase binding assay (11). Values for Kd estimated from the analysis of RU versus either the A2 subunit concentration or FVIII light chain concentration yielded ∼11 and ∼18 nm, respectively (data not shown), and represented modest increases (∼3- and <2-fold, respectively) compared with values calculated from the fitted kinetic constants.
Both intact FVIII and the A1/A3C1C2 dimer had Kd values that were intermediate between the A2 subunit and FVIII light chain. The A3C1C2 subunit of FVIIIa had a Kd value essentially identical to that of the A2 subunit, indicating that removal of the a3 acidic segment enhanced the interaction of A3C1C2 with FIXa. For reasons not understood, the sensorgram describing this interaction could not be fit with a high degree of confidence compared with the other subunits (averaged χ2 value of 135 using 2-state model). Attempts to run reciprocal binding experiments using a CM5 chip to immobilize FVIII or FVIIIa subunits while using FIXa in the mobile phase were not successful, yielding data that in many cases were highly irreproducible. The reason(s) for this is not clear but may reflect masking of interactive sites due to the random orientation of the immobilized FVIII subunit substrates.
Interactions between WT and Variant bA2 Subunits and FIXa by SPR
The C-terminal region of the A2 subunit, residues 707-714 (KVSSCDKN), has been proposed to be interactive with FIXa (12), although experimental evidence supporting this interaction is limited. To gain insights into this potential interaction, a baculovirus expression system for the isolated A2 domain of FVIII was utilized to obtain WT and point mutants where the original residue was replaced with Ala. This expression system was employed previously to obtain isolated bA2 domain variants to assess contributions of the 558-loop in the affinity of the FVIIIa subunit for FIXa (17). For this analysis, SPR was performed as above using bFPR-FIXa immobilized to a streptavidin chip with WT or variant bA2 subunits in the mobile phase. Results in Fig. 2 show representative binding curves for a single concentration (25 nm) of bA2.
FIGURE 2.
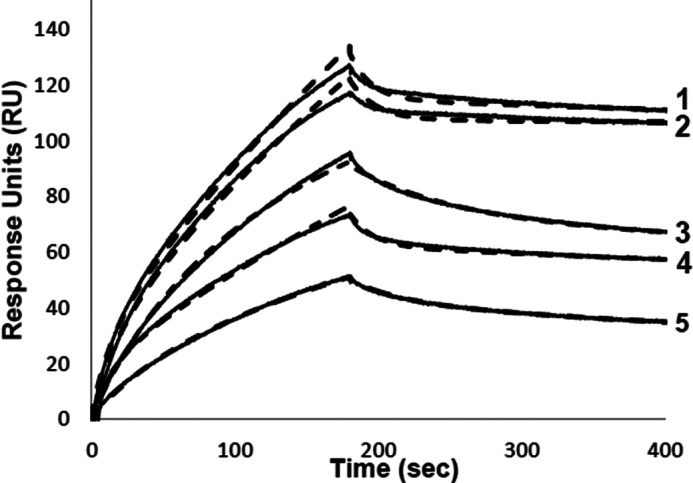
Binding of bA2 WT and variants with FIXa. Biotinylated FPR-FIXa was immobilized on a streptavidin chip and the kinetics with baculovirus-expressed A2 domain (25 nm) were monitored at a flow rate of 20 μl/min for a 3-min association with subunit followed by a 4-min dissociation after return to buffer flow. Representative curves for bA2 forms correspond to (1) WT, (2) K707A, (3) S709A, (4) S710A, and (5) K713A with data represented by the bold line, and fitted curve shown by the dashed line.
All bA2 forms reversibly bound the immobilized bFPR-FIXa. Results with WT bA2 yielded a similar Kd value as observed for the A2 subunit derived from FVIIIa. Although both ka and kd rates were reduced by ∼2 orders of magnitude compared with the WT A2 (Table 2), these values were more in line with the parameter values for other FVIII-derived substrates (see Table 1, and above comment). Results for the Ala variants showed little if any effect of the Lys709 substitution, whereas Kd values for bA2 variants with Ala substitutions at Asp712 and Asn714 were slightly increased, and Ala substitutions at Ser710 and Lys713 yielded ∼3–4-fold increases in Kd values. Surprisingly the K707A variant showed an ∼4-fold lower Kd value than WT. Overall, these results suggest this sequence may contribute to interactions of the A2 subunit with FIXa with residues Ser710, Asp712, Lys713, and Asn714 making a positive contribution to binding, whereas Lys707 appears to discourage this interaction, perhaps by some repulsive interaction.
TABLE 2.
Kinetic parameters for binding of bA2 variants and FIXa
Kinetic parameters were determined by SPR as described under “Experimental Procedures” using A2 in the mobile phase and bFPR-FIXa bound to a SA chip. Samples were run at least three times, and mean ± S.D. shown.
| A2 analyte | ka1 | kd1 | ka2 | kd2 | Kd | χ2 |
|---|---|---|---|---|---|---|
| m−1 s−1 | s−1 | s−1 | s−1 | nm | ||
| WT | (3.4 ± 0.3) × 105 | (1.5 ± 0.1) × 10−2 | (1.3 ± 0.1) × 10−2 | (6.5 ± 2.4) × 10−4 | 2.1 ± 0.02 | 4.2 |
| K707A | (2.6 ± 0.2) × 105 | (3.5 ± 0.05) × 10−2 | (2.5 ± 0.5) × 10−2 | (6.6 ± 0.4) × 10−5 | 0.41 ± 0.04 | 0.71 |
| S709A | (2.9 ± 0.3) × 105 | (0.8 ± 0.2) × 10−2 | (6.9 ± 0.2) × 10−3 | (5.7 ± 1.3) × 10−4 | 2.1 ± 0.1 | 1.5 |
| S710A | (1.6 ± 0.4) × 105 | (4.3 ± 0.2) × 10−2 | (1.9 ± 0.02) × 10−3 | (0.54 ± 0.04) × 10−3 | 8.0 ± 2.5 | 0.20 |
| D712A | (3.2 ± 0.5) × 105 | (1.8 ± 0.4) × 10−2 | (1.3 ± 0.1) × 10−2 | (8.3 ± 0.5) × 10−4 | 3.4 ± 0.1 | 0.20 |
| K713A | (2.7 ± 1.0) × 105 | (1.8 ± 0.3) × 10−2 | (1.2 ± 0.05) × 10−2 | (1.5 ± 0.1) × 10−3 | 7.3 ± 0.1 | 0.15 |
| N714A | (1.1 ± 0.2) × 105 | (3.3 ± 0.1) × 10−2 | (1.9 ± 0.1) × 10−2 | (2.9 ± 0.7) × 10−3 | 4.7 ± 0.2 | 1.0 |
Reconstitution of FXase and FVIIIa with bA2 WT and Variants
Activity-based assays employing reconstitutions of the FXase enzyme complex or FVIIIa with bA2 WT and variants were performed to further evaluate the 707-714 region as FIXa interactive. In the former assay, reconstitution of FXase employs a limiting concentration of FIXa (1 nm) plus an excess of A1/A3C1C2 dimer (6.8 nm) and PS/PC/PE vesicles (10 μm). Phospholipid vesicles were included during the initial stages of FXase reconstitution to help orient FIXa on the membrane for more efficient interaction with the isolated bA2 subunit. Reactions were then titrated with variable concentrations of bA2 subunit (3.1–1600 nm) and initiated with a Vmax concentration of factor X (300 nm). Under these conditions, rates of FXa generated are an indicator of the amount of functional FXase that is formed. Thus this assay monitors the affinity of bA2 subunit for the membrane-bound FIXa-A1/A3C1C2 complex. FVIIIa is reconstituted from limiting A1/A3C1C2 dimer (1 nm) that was titrated with a variable concentration of bA2 subunit (12.5–1600 nm) prior to addition of PS/PC/PE vesicles (10 μm), FIXa (40 nm), and initiation of the reaction with a Vmax concentration of factor X (300 nm). Rates of FXa generation from this titration are used to assess the affinity of the bA2 subunit for the A1/A3C1C2 dimer in forming functional FVIIIa.
Results from the FXase and FVIIIa reconstitutions are shown in Figs. 3 and 4, respectively, with Kd and Vmax values presented in Table 3. Comparison of the values obtained for WT bA2 in the two assays shows an ∼6-fold reduction in the Kd value in FXase reconstitution relative to FVIIIa reconstitution. This increase in affinity reflects the contribution of FIXa to partially stabilize the association of bA2 subunit in the inter-FVIIIa subunit interaction. These affinity values were equivalent to those obtained for WT bA2 and A2 subunit purified from recombinant FVIII using similar reconstitution assays (17). Although the Vmax value for the reconstituted FXase complex with WT bA2 (63 nm min−1(nm FIXa)−1) was equivalent to that obtained in the previous study, the Vmax value for the FVIIIa reconstituted with WT bA2 (21 nm min−1(nm FIXa)−1) was reduced by ∼3-fold and the reason(s) for this is not clear.
FIGURE 3.

Effects of alanine mutation of 707-714 residues on FXase reconstitution as measured by FXa generation assays. FXase was reconstituted with the indicated concentrations of bA2, 6.8 nm A1/A3C1C2 dimer, 1 nm FIXa, and 10 μm PS/PC/PE vesicles and reactions were initiated with the addition of 300 nm FX. Initial rates were plotted as a function of A2 subunit concentration and fitted to a single-site ligand binding equation by nonlinear least squares regression. The assays were performed at least three times and mean ± S.D. are shown. Curves represent WT (●), K707A (■), S709A (▴), S710A, (♦), D712A (○), K713A (▵), and N714A (□).
FIGURE 4.
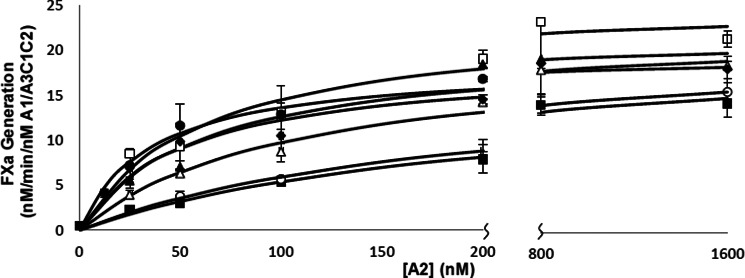
Effects of alanine mutation of 707-714 residues on FVIIIa reconstitution as measured by FXa generation assays. bA2 subunit was titrated in the presence of 1 nm A1/A3C1C2 dimer to reconstitute FVIIIa, followed by addition of 40 nm FIXa and 10 μm PS/PC/PE vesicles. Reactions were initiated with the addition of 300 nm FX. Initial rates were plotted as a function of A2 subunit concentration and fitted to a single-site ligand binding equation by nonlinear least squares regression. Assays were performed at least three times and mean ± S.D. are shown. Curves represent WT, (●), K707A (■), S709A (▴), S710A (♦), D712A (○), K713A (▵), and N714A (□).
TABLE 3.
Kinetic and binding parameters for FXase and FVIIIa reconstituted with WT and variant bA2
| A2 variant | FXase reconstitutiona |
FVIIIa reconstitutionb |
||
|---|---|---|---|---|
| Kd | Vmax | Kd | Vmax | |
| nm | nm FXa min−1 (nm IXa)−1 | nm | nm FXa min−1 (nm A1/A3C1C2)−1 | |
| WT | 5.5 ± 1.3 | 63.4 ± 1.5 | 36.3 ± 3.8 | 21.2 ± 2.8 |
| K707A | 125.0 ± 23.2 | 51.9 ± 1.6 | 217.1 ± 25.0 | 19.2 ± 2.0 |
| S709A | 45.1 ± 6.4 | 64.7 ± 2.6 | 62.5 ± 7.9 | 23.4 ± 1.3 |
| S710A | 44.1 ± 6.5 | 63.8 ± 4.4 | 48.6 ± 0.3 | 21.3 ± 1.2 |
| D712A | 145.2 ± 14.0 | 59.1 ± 1.8 | 189.0 ± 19.4 | 19.4 ± 2.0 |
| K713A | 91.1 ± 0.6 | 79.9 ± 8.3 | 106.8 ± 14.8 | 23.0 ± 0.03 |
| N714A | 43.7 ± 3.9 | 90.5 ± 5.7 | 62.9 ± 8.3 | 26.9 ± 0.03 |
a Reconstitution of FXase was performed using limiting FIXa (1 nm).
b Reconstitution of FVIIIa was performed using limiting A1/A3C1C2 dimer (1 nm).
Examination of the bA2 variants showed that Vmax values for all variants were similar to the WT value and dependent upon the type of reconstitution assay performed. This result indicated that these residues do not affect cofactor function. However, marked differences were observed in the Kd parameter values. The bA2 variants S709A, S710A, and N714A yielded modest (<2-fold) increases in Kd using the FVIIIa reconstitution assay, whereas these variants yielded ∼8-fold increases in Kd assessed by FXase reconstitution. An approximate 3-fold increase in Kd was observed for the K713A bA2 variant in FVIIIa reconstitution, whereas this value was increased ∼17-fold in FXase reconstitution. Finally, K707A and D712A bA2 variants showed 5–6-fold increases in Kd by FVIIIa reconstitution and >20-fold increases in the FXase reconstitution assay. Thus the observation that all the bA2 Ala variants showed markedly greater decreases in affinity in the FXase reconstitution compared with the FVIIIa reconstitution assay was consistent with mutations within this sequence altering interaction of the A2 subunit with FIXa.
DISCUSSION
Multiple protein-protein contacts have been documented in the interactions of FVIIIa and FIXa to form the FXase complex (see Ref. 3 for review). FIXa-interactive sites are localized to the FVIII LC-derived A3C1C2 subunit and the A2 subunit. Earlier work employing a nonequilibrium solid phase binding assay showed an apparent Kd = 14.8 nm for the binding of the FVIII LC to FIXa (11). This result is similar to results presented in the current report employing equilibrium binding by SPR (Kd = 10.5 nm for the a3A3C1C2). Furthermore, removal of the a3 acidic segment yielded a higher affinity interaction (Kd = 3.6 nm) for the A3C1C2 subunit. These results further confirm that neither the highly acidic a3 region that precedes the A3 domain and is removed by thrombin cleavage nor the A1 subunit when bound to A3C1C2 (Kd = 7.5 nm for the A1/A3C1C2 dimer) contributes to interactions with FIXa. These data are consistent with the accepted notion that the A3C1C2 subunit of FVIIIa contributes the majority of the binding energy to complex assembly.
However, whereas functional studies based upon the capacity for the isolated A2 subunit to stimulate FXa generation catalyzed by FIXa (13, 20) yielded an apparent Kd ∼300 nm (13), the results from the SPR analysis indicate a much higher affinity for the bA2-FIXa interaction (Kd = 2.1–3.9 nm) that is similarly observed for the A3C1C2 subunit. The reason for this large disparity in the interactions of bA2 subunit with FIXa is not clear, but may reflect differences in the static affinity of the bA2 subunit for the protease assessed by SPR compared with kinetic affinity wherein the membrane-bound bA2-FIXa complex is actively catalyzing conversion of substrate to product (see below). Furthermore, we note that the affinity measured by SPR for A2 subunit is greater than that measured for FVIII (Kd = 7.3 nm). This observation is consistent with previous work showing that FIXa-interactive site(s) in the A2 domain are cryptic or masked in FVIII and required cleavage of heavy chain during the activation of FVIII to FVIIIa for these sites to be exposed (19). Thus results from the present study support a prominent role for the A2 subunit in contributing to the binding energy for the interaction of FVIIIa and FIXa in assembling FXase. Although both A2 and A3C1C2 subunits show nanomolar affinity for FIXa, these values are not additive in FVIIIa due to the weak affinity interaction of the A2 subunit for the A1/A3C1C2 dimer (7).
Specific sites in the A2 subunit have been identified as contributing to binding FIXa. One previously identified site, the 558-loop (15), contains a scissile bond cleaved by activated protein C (24) and FXa (25) that inactivates cofactor function. In a recent study Jagannathan et al. (17) employed FVIIIa and FXase reconstitution assays as used in the current report to identify residues within the 558-loop region that contribute to binding and catalysis following Ala scanning mutagenesis and purification of the isolated A2 domain variants from the baculovirus expression system. Results from that study showed an ∼6-fold reduced Kd value for interaction of the WT bA2 subunit within the reconstituted FXase compared with reconstituted FVIIIa, consistent with the capacity for FIXa to partially stabilize the interaction with A2 subunit. Furthermore, Ala scanning mutagenesis revealed that several residues (Ser558, Val559, Asp560, Gly563, and Ile566) showed 9–16-fold increases in Kd values for FXase reconstitution, whereas only marginal increases in FVIIIa reconstitution were observed. Interestingly, an overlapping set of residues (Tyr555, Asp560, Gly563, Ile566, and Asp569) also showed marked (∼20–40-fold) reductions in the Vmax parameter. These results indicated that residues within the 558-loop region make significant contributions to both binding FIXa and cofactor function by modulating the FIXa active site.
In the present study we evaluate residues localized near the C-terminal end of the A2 domain (Fig. 5A) using SPR binding and functional reconstitution assays. The basis for examining this region is prior modeling studies of Bajaj et al. (26), suggesting that residues Asp712 and Lys713 of A2 subunit reside at the interface with the protease domain of FIXa, and with Lys713 potentially interacting with Glu240 in FIXa. In addition, previous work from our laboratory showed that a peptide to residues 708–717 inhibited FVIIIa activity in a FXa generation assay and that a FVIII possessing an D712A mutation in the A2 domain showed an ∼8-fold reduction in affinity of the variant FVIIIa for FIXa as compared with WT FVIIIa (12). In the present study, SPR analysis of bA2 variants S710A and K713A showed modest (3–4-fold) increases in the Kd for FIXa. Thus the result with the Lys713 bA2 variant was consistent with potential participation in FIXa binding. Interestingly, whereas the bA2 D712A variant showed essentially no effect on Kd in the SPR analysis, this and other variants showed more prominent effects in the functional reconstitution assays.
FIGURE 5.

Orientation of residues 707-714 within the A2 subunit. Schematic representation of the (A) A2 subunit of FVIII (PDB code 2R7E) with residues 555–571 in red and residues 707-714 in orange stick representation. B, residues 707-714 in stick representation with oxygen in red, nitrogen in blue, and sulfur in yellow. Residues on the 558-loop found to be important for FIXa binding and function are highlighted in green, and Cys630, which forms an essential disulfide bond with Cys711 is highlighted in yellow.
Comparison of FXase and FVIIIa reconstitution studies with these variants showed significantly larger decreases in the affinity of the bA2 subunit in the former assay consistent with a defect in the interaction with FIXa. Residues most effected were Lys707, Asp712, and Lys713, which showed ∼17–27-fold increases in the Kd parameter compared with WT in reconstituting FXase. The residues showing the greatest reductions in affinity are charged residues compared with the more modest (∼8-fold) increases in Kd observed for Ala replacement of the polar residues Ser709, Ser710, and Asn714. This observation may suggest a role for the side chains in salt bridging in the association of the A2 subunit with FIXa. However, none of the Ala mutations within this sequence resulted in significant changes in the Vmax parameter values, suggesting that unlike the 558-loop, this region of the A2 subunit does not impact the active site of the enzyme.
To reconcile the modest differences in affinity values of the bA2 Ala variants for FIXa observed by SPR compared with the more pronounced affinity differences observed for these variants when assessed as functional FXase complexes, we speculate a static affinity in the absence of catalysis. This interaction would be of high affinity and may in fact be little influenced by the 707-714 sequence. In contrast, we further speculate a kinetic affinity where residues within the 707-714 sequences exert greater contribution when the A2 subunit is bound within FXase and facilitating catalysis. For example, there may be a microscopic step(s) in the catalytic mechanism where the A2 subunit of the cofactor, which likely directly modulates the active site of FIXa, undergoes a change in configuration that may be less stable than the static binding configuration to the enzyme. This could explain a greater static affinity compared with a kinetic affinity value for the variant A2 forms.
The intermediate resolution x-ray structures of FVIII (27, 28) show a close proximity of the 558-loop and residues 707-714 (Fig. 5B) that are localized to the same face of the A2 subunit. Thus these sequences may form an extended interactive surface for binding FIXa. Based upon several lines of information including experimental evidence that the A2 558-loop interacts with the 330-helix in the FIXa protease domain (26) and the localization of a FVIIIa-interactive site to FIXa residues 301–303 (29), Ngo et al. (28) constructed a model of the membrane-bound FVIIIa-FIXa complex that suggests residues within the 707-714 region of the A2 subunit interact with FIXa residues in or around 301–303 (Lys-Phe-Gly). However, this proposed interaction is not well defined.
The A2 subunit of FVIIIa serves an essential role in modulating the active site of FIXa for efficient activation of substrate FX. However, limited structural information on this critical interaction is available. Given the apparent high affinity for this interaction as described in this report, it may be possible to prepare co-crystals of the A2-FIXa complex for detailed structural study.
Acknowledgments
We thank Lisa Regan of Bayer Corp. for the FVIII concentrate (Kogenate) and Indu Jagannathan for helpful discussions and experimental contributions to the early stages of this study. The Biacore T100 was obtained from National Institutes of Health shared instrument Grant NCRR 1S10 RR027241.
This work was supported, in whole or in part, by National Institutes of Health Grants HL76213 and HL38199.
- FVIII
- factor VIII
- FVIIIa
- factor VIIIa
- FIXa
- factor IXa
- FX
- factor X
- FXa
- factor X
- SPR
- surface plasmon resonance
- bFPRck
- biotin-labeled Phe-Pro-Arg-chloromethyl ketone
- LC
- light chain
- bA2
- A2 subunit expressed in insect cells using a baculovirus construct
- PS
- phosphatidylserine
- PC
- phosphatidylcholine
- PE
- phosphatidylethanolamine.
REFERENCES
- 1. van Dieijen G., Tans G., Rosing J., Hemker H. C. (1981) The role of phospholipid and factor VIIIa in the activation of bovine factor X. J. Biol. Chem. 256, 3433–3442 [PubMed] [Google Scholar]
- 2. Duffy E. J., Lollar P. (1992) Intrinsic pathway activation of factor X and its activation peptide-deficient derivative, factor Xdes-143–191. J. Biol. Chem. 267, 7821–7827 [PubMed] [Google Scholar]
- 3. Fay P. J. (2004) Activation of factor VIII and mechanisms of cofactor action. Blood Rev. 18, 1–15 [DOI] [PubMed] [Google Scholar]
- 4. Lollar P., Parker C. G. (1989) Subunit structure of thrombin-activated porcine factor VIII. Biochemistry 28, 666–674 [DOI] [PubMed] [Google Scholar]
- 5. Fay P. J., Haidaris P. J., Smudzin T. M. (1991) Human factor VIIIa subunit structure. Reconstitution of factor VIIIa from the isolated A1/A3-C1-C2 dimer and A2 subunit. J. Biol. Chem. 266, 8957–8962 [PubMed] [Google Scholar]
- 6. Lollar P., Parker C. G. (1990) pH-dependent denaturation of thrombin-activated porcine factor VIII. J. Biol. Chem. 265, 1688–1692 [PubMed] [Google Scholar]
- 7. Fay P. J., Smudzin T. M. (1992) Characterization of the interaction between the A2 subunit and A1/A3-C1-C2 dimer in human factor VIIIa. J. Biol. Chem. 267, 13246–13250 [PubMed] [Google Scholar]
- 8. Lollar P., Knutson G. J., Fass D. N. (1984) Stabilization of thrombin-activated porcine factor VIII:C by factor IXa phospholipid. Blood 63, 1303–1308 [PubMed] [Google Scholar]
- 9. Lamphear B. J., Fay P. J. (1992) Factor IXa enhances reconstitution of factor VIIIa from isolated A2 subunit and A1/A3-C1-C2 dimer. J. Biol. Chem. 267, 3725–3730 [PubMed] [Google Scholar]
- 10. Fay P. J., Beattie T. L., Regan L. M., O'Brien L. M., Kaufman R. J. (1996) Model for the factor VIIIa-dependent decay of the intrinsic factor Xase. Role of subunit dissociation and factor IXa-catalyzed proteolysis. J. Biol. Chem. 271, 6027–6032 [DOI] [PubMed] [Google Scholar]
- 11. Lenting P. J., Donath M. J., van Mourik J. A., Mertens K. (1994) Identification of a binding site for blood coagulation factor IXa on the light chain of human factor VIII. J. Biol. Chem. 269, 7150–7155 [PubMed] [Google Scholar]
- 12. Jenkins P. V., Dill J. L., Zhou Q., Fay P. J. (2004) Contribution of factor VIIIa A2 and A3-C1-C2 subunits to the affinity for factor IXa in factor Xase. Biochemistry 43, 5094–5101 [DOI] [PubMed] [Google Scholar]
- 13. Fay P. J., Koshibu K. (1998) The A2 subunit of factor VIIIa modulates the active site of factor IXa. J. Biol. Chem. 273, 19049–19054 [DOI] [PubMed] [Google Scholar]
- 14. Fay P. J., Jenkins P. V. (2005) Mutating factor VIII: lessons from structure to function. Blood Rev. 19, 15–27 [DOI] [PubMed] [Google Scholar]
- 15. Fay P. J., Beattie T., Huggins C. F., Regan L. M. (1994) Factor VIIIa A2 subunit residues 558–565 represent a factor IXa interactive site. J. Biol. Chem. 269, 20522–20527 [PubMed] [Google Scholar]
- 16. Jenkins P. V., Freas J., Schmidt K. M., Zhou Q., Fay P. J. (2002) Mutations associated with hemophilia A in the 558–565 loop of the factor VIIIa A2 subunit alter the catalytic activity of the factor Xase complex. Blood 100, 501–508 [DOI] [PubMed] [Google Scholar]
- 17. Jagannathan I., Ichikawa H. T., Kruger T., Fay P. J. (2009) Identification of residues in the 558-loop of factor VIIIa A2 subunit that interact with factor IXa. J. Biol. Chem. 284, 32248–32255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Mimms L. T., Zampighi G., Nozaki Y., Tanford C., Reynolds J. A. (1981) Phospholipid vesicle formation and transmembrane protein incorporation using octyl glucoside. Biochemistry 20, 833–840 [DOI] [PubMed] [Google Scholar]
- 19. Fay P. J., Mastri M., Koszelak M. E., Wakabayashi H. (2001) Cleavage of factor VIII heavy chain is required for the functional interaction of A2 subunit with factor IXa. J. Biol. Chem. 276, 12434–12439 [DOI] [PubMed] [Google Scholar]
- 20. Koszelak Rosenblum M. E., Schmidt K., Freas J., Mastri M., Fay P. J. (2002) Cofactor activities of factor VIIIa and A2 subunit following cleavage of A1 subunit at Arg-336. J. Biol. Chem. 277, 11664–11669 [DOI] [PubMed] [Google Scholar]
- 21. Lapan K. A., Fay P. J. (1997) Localization of a factor X interactive site in the A1 subunit of factor VIIIa. J. Biol. Chem. 272, 2082–2088 [DOI] [PubMed] [Google Scholar]
- 22. Bradford M. M. (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72, 248–254 [DOI] [PubMed] [Google Scholar]
- 23. Selvaraj S. R., Scheller A. N., Miao H., Kaufman R. J., Pipe S. W. (2012) Bioengineering of coagulation factor VIII for efficient expression through elimination of a dispensable disulfide loop. J. Thromb. Haemostasis 10, 107–115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Fay P. J., Smudzin T. M., Walker F. J. (1991) Activated protein C-catalyzed inactivation of human factor VIII and factor VIIIa. Identification of cleavage sites and correlation of proteolysis with cofactor activity. J. Biol. Chem. 266, 20139–20145 [PubMed] [Google Scholar]
- 25. Plantier J. L., Rolli V., Ducasse C., Dargaud Y., Enjolras N., Boukerche H., Négrier C. (2010) Activated factor X cleaves factor VIII at arginine 562, limiting its cofactor efficiency. J. Thromb. Haemost. 8, 286–293 [DOI] [PubMed] [Google Scholar]
- 26. Bajaj S. P., Schmidt A. E., Mathur A., Padmanabhan K., Zhong D., Mastri M., Fay P. J. (2001) Factor IXa:factor VIIIa interaction. Helix 330–338 of factor IXa interacts with residues 558–565 and spatially adjacent regions of the A2 subunit of factor VIIIa. J. Biol. Chem. 276, 16302–16309 [DOI] [PubMed] [Google Scholar]
- 27. Shen B. W., Spiegel P. C., Chang C. H., Huh J. W., Lee J. S., Kim J., Kim Y. H., Stoddard B. L. (2008) The tertiary structure and domain organization of coagulation factor VIII. Blood 111, 1240–1247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ngo J. C., Huang M., Roth D. A., Furie B. C., Furie B. (2008) Crystal structure of human factor VIII. Implications for the formation of the factor IXa-factor VIIIa complex. Structure 16, 597–606 [DOI] [PubMed] [Google Scholar]
- 29. Kolkman J. A., Lenting P. J., Mertens K. (1999) Regions 301–303 and 333–339 in the catalytic domain of blood coagulation factor IX are factor VIII-interactive sites involved in stimulation of enzyme activity. Biochem. J. 339, 217–221 [PMC free article] [PubMed] [Google Scholar]