

Background: Lgr4/Gpr48 has been found to play an important role in development of different organs.
Results: Overactivated innate immune response was found in Lgr4-deficient mice.
Conclusion: Lgr4/Gpr48 could be a negative regulator in TLR2/4-associated immune responses.
Significance: Our novel findings provide new insights into the physiological functions of Lgr4/Gpr48 in immune diseases.
Keywords: CREB, G-protein-coupled Receptors (GPCRs), Pathogen-associated Molecular Pattern (PAMP), Sepsis, Toll-like Receptors (TLR), CD14, Lgr4
Abstract
The recognition of pathogen-associated molecular patterns by Toll-like receptors (TLRs) is pivotal in both innate and adaptive immune responses. Here we demonstrate that deletion of Lgr4/Gpr48 (G-protein-coupled receptor 48), a seven-transmembrane glycoprotein hormone receptor, potentiates TLR2/4-associated cytokine production and attenuates mouse resistance to septic shock. The expression of CD14, a co-receptor for TLR2/4-associated pathogen-associated molecular patterns, is increased significantly in Lgr4-deficient macrophages, which is consistent with the increased immune response, whereas the binding activity of cAMP-response element-binding protein is decreased significantly in Lgr4-deficient macrophages, which up-regulate the expression of CD14 at the transcriptional level. Together, our data demonstrate that Lgr4/Gpr48 plays a critical role in modulating the TLR2/4 signaling pathway and represents a useful therapeutic approach of targeting Lgr4/Gpr48 in TLR2/4-associated septic shock and autoimmune diseases.
Introduction
Immune cells, which are armed with pattern recognition receptors, can recognize numerous pathogen-associated molecular patterns (PAMPs),3 including lipopolysaccharide (LPS; Gram-negative bacteria), peptidoglycan (Gram-positive bacteria), and single- and double-stranded DNA or RNA (viruses) (1). Toll-like receptors (TLRs), which are widely distributed on antigen-presenting cells, function as primary sensors for invading pathogens in the innate immune system (2, 3). As of now, 10 human and 12 mouse TLRs have been identified, each of which has distinct recognition patterns. For example, TLR4 recognizes LPS from the outer membrane of Gram-negative bacteria, whereas TLR1, TLR2, and TLR6 recognize diacyl or triacyl lipoproteins from Gram-positive bacteria. TLR3 recognizes viral double-stranded RNA, whereas TLR7 and TLR8 recognize single-stranded RNA to protect the body from virus infection (4). The activation of TLRs by given agonists will stimulate transcription factor NF-κB and induce secretion of proinflammatory cytokines, chemokine, and type I interferons, such as TNF-α, IL-6, MCP-1, and IFN-β, to fight against invading pathogens and recruit additional cells of the immune response to the site of infection.
The recognition of PAMPs by TLRs is essential for activation of innate and adaptive immunity against pathogens and is involved in autoimmune, chronic inflammatory, and infectious diseases. For example, sepsis, experimental autoimmune encephalomyelitis (5), asthma (6), and atherosclerosis (7, 8) could be caused by overactivation of TLR signaling. Therefore, the activation of TLRs and their signaling pathways must be maintained under tight negative control. In the past few years, many negative regulators have been identified to limit toxic effects of excessive immune activation. For example, soluble decoy TLRs and intracellular negative regulators, including MyD88s (the short form of MyD88), IRAKM, SOCS1, NOD2, PI3K, TOLLIP, and A20, are all found to regulate the toxic effects of excessive TLR activation (9). However, identifying endogenous negative regulators of TLR signaling is still an enormous challenge and will have significant therapeutic value in our fight against inflammatory and autoimmune diseases.
CD14, a 55-kDa glycoprotein, was first described as a myeloid differentiation antigen on the surface of monocytes/macrophages in 1990 (10, 11). Similar to the conserved structure leucine-rich repeats in TLRs, CD14 is also composed of 11 leucine-rich repeats, which could be the key residues in pattern recognition (12). During TLR4-associated pattern recognition, CD14 plays a very important role in transferring LPS from LPS-binding protein to TLR4, which was regarded as the initiation of TLR4 signaling (13). Furthermore, CD14 also increases the TLR2-associated signaling through sensitizing the recognition of lipopeptides, LTA, lipoarabinomannans, and peptidoglycans (14). It has been shown that different regions of CD14 are required in the activation of TLR4 or TLR2 signaling (15). Therefore, regulation of CD14 is crucial in TLR2/4-mediated pattern recognition and immune responses.
Lgr4, also known as Gpr48 (G-protein-coupled receptor 48), belongs to the glycoprotein hormone receptor subfamily (16–18). Besides the seven-transmembrane regions, the N-terminal domain of Lgr4 contains a putative horseshoe-like structure composed of 18 leucine-rich repeats, which has the potential to be a ligand-binding site for this receptor (19). Although R-spondins have been found to interact with Lgr4 and mediate the potentiation of Wnt/β-catenin signaling, endogenous ligands of Lgr4 that activate classic signaling pathways of G-proteins have not been identified (20, 21). Lgr4/Gpr48 has been found widely expressed in multiple organs, including heart, brain, spleen, bone, lung, liver, muscle, kidney, and testis. Although Lgr4/Gpr48 has been found to play an important role in development of different organs (22–31), the function of Lgr4/Gpr48 in regulation of innate immunity has not been investigated.
In this study, we investigated the functions of Lgr4 in TLR-mediated immune responses using Lgr4-deficient mice. We demonstrate that deletion of Lgr4 makes the mice much more sensitive to TLR4-mediated septic shock and that Lgr4-deficient macrophages showed increased production of proinflammatory cytokines in response to TLR2/4 ligands. Furthermore, we show that inactivation of Lgr4 in macrophages substantially increased the expression level of CD14 by targeting the cAMP-PKA-CREB signaling pathway. Therefore, for the first time, we demonstrate that a G-protein coupled receptor (Lgr4) negatively regulates TLR2/4 signaling and innate immune responses through down-regulating the expression of CD14 protein.
EXPERIMENTAL PROCEDURES
Mice
Lgr4 knock-out mice were generated based on the secretory trap approach, and genotyping was performed by PCR as described previously (25). Lgr4+/− mice were back-crossed to the C57BL/6 strain for at least 7 generations to obtain a pure genetic background. Lgr4−/− and Lgr4+/+ littermates were used in all experiments. For the septic shock mouse model, 8–12-week-old female Lgr4−/− and Lgr4+/+ mice were challenged with LPS (20 mg/kg, administered intraperitoneally) and monitored for 48 h. Furthermore, a sample of blood was obtained, and plasma TNF-α, IL-6, and IL-1β were measured by ELISA after being challenged with LPS (20 mg/kg, administered intraperitoneally) for 2 h. All animal studies were according to National Institutes of Health guidelines for the use and care of live animals and were approved by the Animal Care and Use Committees of the Texas A&M University Health Science Center Institute of Biosciences and Technology.
Cells, Antibody, and Reagent
HEK293T and RAW 264.7 cells were from the American Type Culture Collection (ATCC). All cells were cultured as suggested by ATCC. Antibodies specific for CD14, CREB, and phospho-CREB were from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). Blocking antibody for CD14 was from BD Biosciences. Antibodies specific for IKKα, phospho-IKKα, IκBα, phospho-IκBα, ERK, phospho-ERK, JNK, phospho-JNK, and β-actin were from Cell Signaling Technology. The mouse TLR agonist kit (Pam3CSK4 for TLR1/2, HKLM for TLR2, poly(I:C) for TLR3, Escherichia coli K12 LPS for TLR4, Salmonella typhimurium flagellin for TLR5, FLS1 for TLR6/2, ssRNA40 for TLR8, and ODN18266 (type B) for TLR9) was from InvivoGen. Forskolin (Beyotime) was used to increase intracellular cAMP levels. Mouse R-spondin/recombinant protein was obtained from Sino Biological Inc.
Plasmids and Gene Transfer
Human pSM2 retroviral shRNAmir for Lgr4 (RHS1764-97192326) and non-silencing control (RHS1707) were prepared as recommended by the manufacturer (Open Biosystems). The plasmids encoding wild-type Lgr4, Lgr4T755I, PKI, and CREB were constructed in our laboratory as described (25). The DNA fragment of −996 to −186 bp of mouse CD14, which contained a putative CREB binding site (CTGACG, −859 to −854) was cloned into KpnI and XhoI sites of pGL3-basic with primers 5′-CGGGGTACCCTGCCAAATAGGTCAAAA (sense) and 5′-CCGCTCGACAATTTCCCACCGTTCA (antisense) to construct pGL3-CD14. Then the CREB binding site (−859 to −854) of pGL3-CD14 was deleted with the QuikChange® II site-directed mutagenesis kit, and the primers 5′-GGGAAGCAAGGTTTGCCGCATTGTCTTCATCATT-3′ (sense) and 5′-AATGATGAAGACAATGCGGCAAACCTTGCTTCCC-3′ (antisense) were used to get pGL3-CD14m. Indicated constructs were transfected into HEK293T cells with Lipofectamine 2000 (Invitrogen) as described by the manufacturer.
siRNAs against CD14
Shortly before transfection, Lgr4 knockdown RAW 264.7 cells were seeded into a 12-well plate at 5 × 105 cells/well in 200 μl of DMEM culture medium containing serum and antibiotics. 2 μl of diluted CD14 siRNA (sense, 5′-GGGCAGUUCACUGAUAUUATT-3′; antisense, 5′-UAAUAUCAGUGAACUGCCCTT-3′) and control siRNA molecules in 100 μl of culture medium without serum was added 9 μl of Lipofectamine 2000. siRNA was mixed by overtaxing. The samples were incubated for 5–10 min at room temperature to allow the formation of complexes. Adding the complexes dropwise onto cells ensures uniform distribution of the complexes. After incubation for 6 h, an additional 700 μl of DMEM containing 10% FBS and antibiotics were added to each well. At 48 h post-transfection, the cells were challenged with LPS for 6 h to detect TNF-α production by ELISA (52).
Preparation of Macrophages and Measurement of Cytokines
For peritoneal macrophages, 1 ml of 4% bacto brewer's thioglycolate (Sigma) was injected into the peritoneal cavity of Lgr4−/− and Lgr4+/+ mice. The animals were sacrificed on day 4 following the injection of thioglycolate, and the elicited macrophages were lavaged by intraperitoneal injection of Ca2+- and Mg2+-free PBS (5 ml, 4 °C). The cells were plated at density of 1 × 106/ml in a 6-well dish (Nunc), and after a 30-min rest, the non-adherent cells were removed by washing the Petri dishes with medium to enrich the cultures with macrophages. For bone marrow-derived macrophages, total BM cells were flushed out from the tibias and femurs of Lgr4−/− and Lgr4+/+ mice. Single-cell suspensions of total bone marrow (0.5 × 106/ml) were cultured in vitro in bone marrow-derived macrophage (BMM) medium (RPMI 1640 supplemented with 20% L929 conditioned medium (as a source of M-CSF), 20% equine FBS (Invitrogen), 2 mm l-glutamine, 100 units/ml gentamicin, and 50 μm β-mercaptoethanol). Medium was changed on day 4 of culture. After 6 days in culture, adherent macrophages were detached from the Petri dish, washed three times, and resuspended at a density of 0.5 × 106 cells/ml in 1% complete medium. The supernatants were collected at each time point to evaluate the levels of cytokines present by ELISA according to the manufacturer's protocol (BD Biosciences), the cell pellets were lysed, and the total cellular proteins were extracted for Western blot analysis.
Reporter Gene Assay
RAW 264.7 cells stably transfected with Lgr4 shRNA were transfected with NF-κB-luciferase reporter (pNF-κB-luc, Stratagene) with Lipofectamine 2000 (Invitrogen) as described by the manufacturer. Then, 48 h after transfection, cells were stimulated with different TLR agonists, and luciferase activity was measured with the Dual-Luciferase reporter assay system (Promega). Extracts were also normalized by β-galactosidase activity using the Galacto-Light Plus β-galactosidase reporter gene assay system (Promega).
Quantitative Real-time RT-PCR
First, macrophages were stimulated for 1 h with or without LPS, and then cDNA synthesis was performed using the Moloney murine leukemia virus reverse transcriptase kit (Promega), and the cDNA was analyzed in a quantitative real-time PCR system machine (Stratagene) using the RT real-time SYBR Green PCR Master Mix kit (SuperArray); results were normalized to Musculus acidic ribosomal phosphoprotein P0 (Arbp) expression. Primers for each cytokine and gene are described in supplemental Table 1.
Electrophoretic Mobility Shift Assays
Nuclear extracts were prepared as described previously (53). Briefly, nuclear extracts from LPS-treated cells were incubated with 32P-end-labeled 45-mer double-stranded NF-κB oligonucleotide (15 μg of protein with 16 fmol of DNA) from the HIV long terminal repeat, 5′-TTGTTACAAGGGACTTTCCGCTGGGGACTTTCCAGGGGGAGGCGTGG-3′ (boldface type indicates NF-κB-binding sites) for 30 min at 37 °C, and the DNA-protein complex formed was separated from free oligonucleotide on 6.6% native polyacrylamide gels. The dried gels were visualized with a Storm820 PhosphorImager, and radioactive bands were quantified using ImageQuant software (Amersham Biosciences).
ChIP Assay
ChIP assays were performed with the EZ-ChIPTM chromatin immunoprecipitation kit (Millipore). Briefly, RAW 264.7 cells were grown in 100-mm tissue culture plates, fixed with 1% formaldehyde for 10 min to cross-link the binding. Next, glycine was added to each dish to quench unreacted formaldehyde. Then DNA was sheared to fragments of 200–500 bp by seven 10-s sonications. The agarose beads were pelleted, and the precleared supernatant was incubated with antibodies to CREB overnight at 4 °C. The region between −889 and −635 of the CD14 promoter was amplified from the immunoprecipitated chromatin by using the following primers: 5′-AGGCTAATAGGAAATGACT-3′ (sense) and 5′-GACGACGACGATGACA-3′ (antisense).
Statistical Analysis
Student's t test (two-tailed) was used to analyze the significance of data comparison, except where indicated otherwise. For all tests, a p value of less than 0.05 was considered statistically significant.
RESULTS
Lgr4 Negatively Regulates the Production of TLR-mediated Proinflammatory Cytokines
TLR signaling is important in macrophage-associated pathogen recognition and in inducing the production of proinflammatory cytokines. To determine whether Lgr4 is involved in TLR-mediated macrophage activation, we challenged the peritoneal macrophages derived from Lgr4+/+ and Lgr4−/− mice with different TLR agonists (Pam3CSK4, HKLM, FSL1, LPS, poly(I:C), ST-FLA, and ssRNA40) and measured the induction of the proinflammatory cytokine TNF-α. As shown in Fig. 1A, ligands associated with TLR2/4 (such as Pam3CSK4, HKLM, FSL1, and LPS) significantly increased the production of TNF-α in Lgr4−/− macrophages, whereas the production of TNF-α induced by poly(I:C), ST-FLA, and ssRNA40 was almost the same in Lgr4+/+ and Lgr4−/− mice, suggesting that Lgr4 negatively regulates the production of TNF-α through the TLR2/4 signaling pathway.
FIGURE 1.
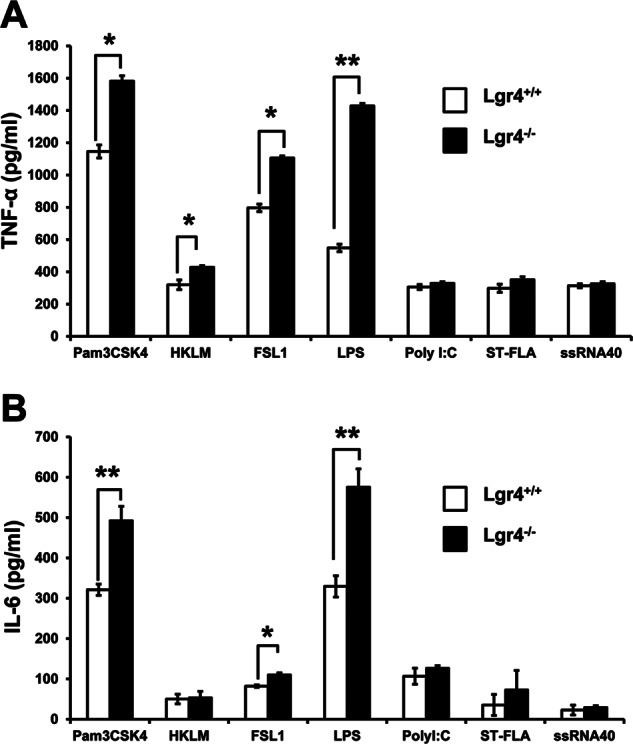
Increased cytokine production in Lgr4−/− macrophages upon TLR agonist stimulation. Peritoneal macrophages from wild-type and Lgr4−/− mice were stimulated with Pam3CSK4 (TLR1/2), HKLM (TLR2), poly(I:C) (TLR3), LPS (TLR4), ST-FLA (TLR5), FSL1 (TLR2/6), and ssRNA40 (TLR8) for 6 h. The concentrations of TNF-α (A) and IL-6 (B) in the supernatant were measured by ELISA, respectively. Data are representative of three independent experiments. *, p < 0.05; **, p < 0.01 (two-tailed Student's t test). Error bars, S.D.
To further confirm the role of Lgr4 in negative regulation of TLR signaling pathways, we measured the production of IL-6, another proinflammatory cytokine, in the macrophages from Lgr4-deficient mice. Consistent with the TNF-α result, the induction of IL-6 in Lgr4−/− macrophages was also enhanced when stimulated by ligands associated with TLR2/4 activation (such as Pam3CSK4, FSL1, and LPS), but no difference was found for the activation of other TLRs (Fig. 1B). These results indicate that Lgr4 negatively regulates only TLR2- and TLR4-associated signaling pathways and not others.
Lgr4 Attenuates LPS-induced Proinflammatory Cytokine Production
To examine how Lgr4 regulates LPS-activated TLR2/4 signaling and proinflammatory cytokine production, we measured LPS-induced proinflammatory cytokine production using ELISA and real-time quantitative PCR assays in wild-type and Lgr4-deficient peritoneal macrophages. As shown in Fig. 2, A and B, Lgr4 deficiency leads to increase of TNF-α production in a time- and dose-dependent manner (Fig. 2A). Furthermore, we analyzed the mRNA expression levels of different cytokines (IL-1α, IL-6, IFN-β, GM-CSF, CXCL10, and TNF-α) in wild-type and Lgr4-deficient macrophages when the cells were activated with LPS for about 1 h. As shown in Fig. 2B, the mRNA levels of cytokines, such as TNF-α, IL-6, IL-1α, IFN-β, CXCL10, and GM-CSF, were all increased significantly in Lgr4−/− macrophages. Meanwhile, we also treated RAW 264.7 cells with R-spondin to check the influence of Lgr4-mediated Wnt/β-catenin signaling in TLR4-associated innate immune response, but no obvious difference of TNF-α and IFN-β expression was observed by a quantitative PCR assay in R-spondin-treated cells (Fig. 2C).
FIGURE 2.
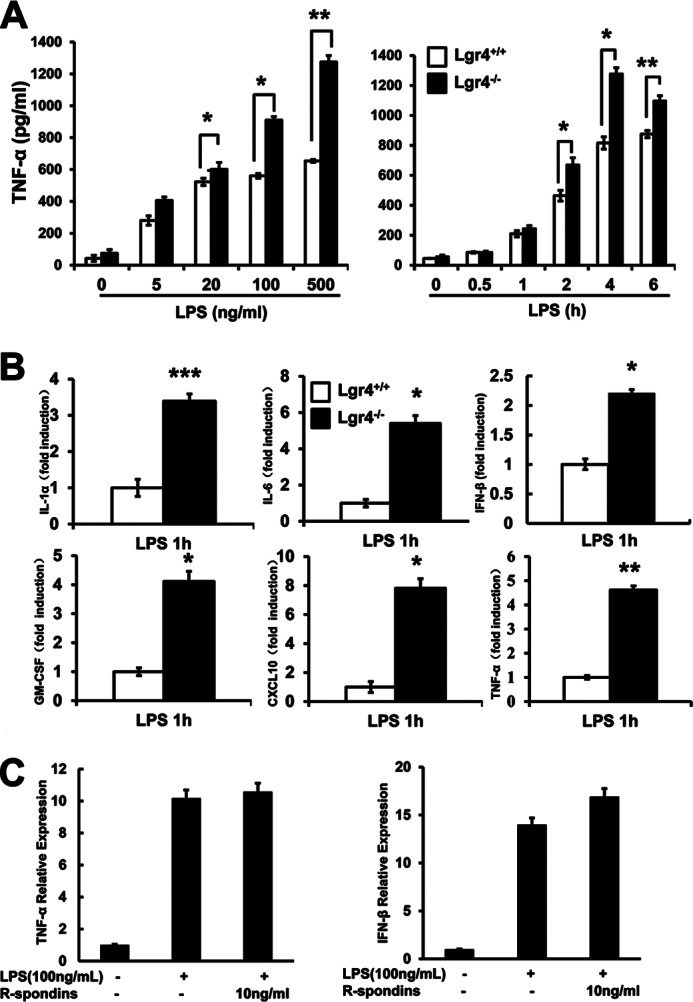
Lgr4 attenuates LPS-induced proinflammatory cytokine production. A, deletion of Lgr4 increased TNF-α concentration in Lgr4−/− peritoneal macrophages stimulated with LPS, as measured by ELISA in both a time-dependent (right) and dose-dependent (left) manner. *, p < 0.05; **, p < 0.01 (two-tailed Student's t test). Data are representative of three independent experiments. B, real-time PCR analysis of cytokine expression in Lgr4+/+- and Lgr4−/−-derived peritoneal macrophages stimulated with LPS for 1 h. Data are representative of three independent experiments. **, p < 0.01 (two-tailed Student's t test). C, real-time PCR analysis of cytokine expression in RAW 264.7 cells stimulated with LPS and R-spondins for 1 h. Data are representative of three independent experiments. Error bars, S.D.
To further confirm that the negative regulation of cytokines is mediated by Lgr4, not by other cellular factors, we examined the effects of Lgr4 knockdown using an in vitro cell model, where RAW 264.7 cells were stably transfected with Lgr4-specific shRNA to decrease endogenous Lgr4 expression up to 70% in mRNA level (supplemental Fig. 1A). As shown in Fig. 3, down-regulation of endogenous Lgr4 significantly increased LPS-induced TNF-α and IL-6 production in a dose- and time-dependent manner in cells transfected with Lgr4-specific shRNA (Fig. 3, A and B). On the other hand, overexpression of a constitutively active form of Lgr4 (Lgr4T755I) (supplemental Fig. 1B) in the RAW 264.7 cells significantly reduced the production of LPS-induced TNF-α and IL-6 in a time- and dose-dependent manner (Fig. 3, C and D). Together, these results suggested a critical role of Lgr4 in negatively regulating the production of TLR4-mediated proinflammatory cytokines.
FIGURE 3.
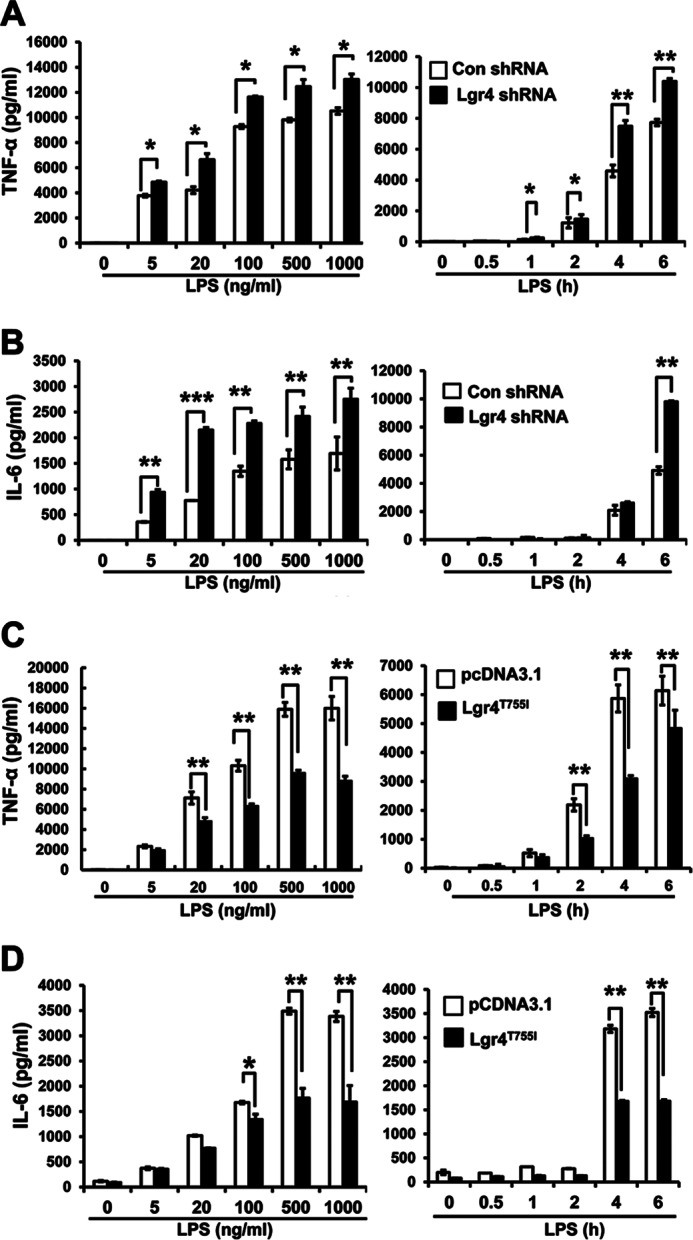
Lgr4 regulates LPS-induced cytokine production in RAW 264.7 cells. A and B, knockdown of Lgr4 increased the production of TNF-α (A) and IL-6 (B) induced by LPS. RAW 264.7 cells were stably transfected with Lgr4 shRNA to knock down the expression of Lgr4. Then the cells were treated with LPS in a time- and dose-dependent manner, and the concentrations of TNF-α (A) and IL-6 (B) were measured by ELISA. C and D, overexpression of active mutant Lgr4 (Lgr4T755I) inhibited the production of TNF-α (C) and IL-6 (D) in RAW 264.7 cells, respectively. Control or the active mutant receptor (Lgr4T755I) was transfected into RAW 264.7 cells. All the cells were challenged with LPS in a dose- and time-dependent manner. Then production of TNF-α (C) and IL-6 (D) in cell supernatant was measured by ELISA. Data are representative of three independent experiments. *, p < 0.05; **, p < 0.01 (two-tailed Student's t test). Error bars, S.D.
Lgr4-deficient Mice Are More Sensitive to LPS-induced Septic Shock
To investigate the function of Lgr4 in regulating LPS-mediated inflammation in vivo, we utilized a septic shock mouse model in Lgr4-deficient and wild-type mice through intraperitoneal injection of LPS, an agonist for TLR4. We monitored the survival of mice for 48 h and found that Lgr4-deficient mice were more susceptible to LPS-induced septic shock, with much higher mortality than wild-type mice (Fig. 4A). Because proinflammatory cytokines are the main reason for LPS-induced septic shock (32), we analyzed the circulating concentration of TNF-α, IL-6, and IL-1β at 2 h after LPS was injected into the mice. As shown in Fig. 4B, much higher TNF-α, IL-6, and IL-1β accumulation was observed in Lgr4-deficient mouse serum when compared with that in wild-type mouse serum, which is consistent with the higher mortality in Lgr4−/− mice. These results suggest that Lgr4 negatively regulates TLR4-mediated induction of proinflammatory cytokines and plays a crucial role in LPS-induced septic shock and mouse death (Fig. 4).
FIGURE 4.

Lgr4-deficient mice are more sensitive to LPS-induced septic shock. A, survival curve of wild-type and Lgr4 null mice after LPS-induced septic shock. Age- and sex-matched Lgr4+/+ (n = 10) and Lgr4−/− (n = 12) mice (8–12 weeks old) were injected intraperitoneally with 20 mg/kg LPS and monitored every 2 h for 48 h. Results are plotted as a percentage of total numbers. B, serum concentrations of TNF-α, IL-6, and IL-1β in wild-type and Lgr4 null mice at 2 h after injection of LPS. Mouse TNF-α, IL-6, and IL-1β in serum were obtained at 2 h after injection of LPS for ELISA. *, p < 0.05; **, p < 0.01 (two-tailed Student's t test). Data are representative of two (A) or three (B) independent experiments. Error bars, S.D.
Deletion of Lgr4 Increases the TLR2/4-mediated NF-κB Activation and Signaling Pathway
In most immune response, NF-κB-associated signaling is activated to induce the releasing of cytokines and chemokines (33). Thus, the up-regulation of NF-κB activity is the key step in host defense against invading pathogens. To investigate the role of Lgr4 in regulating NF-κB activity, we examined the effects of Lgr4 deletion on LPS-stimulated NF-κB activation in peritoneal macrophages and RAW cells. As shown in Fig. 5, LPS-stimulated NF-κB activation was significantly increased in Lgr4−/− macrophages in EMSAs (Fig. 5A). Meanwhile, knockdown of Lgr4 in RAW cells showed a partial enhancement of NF-κB activation upon stimulation by LPS (Fig. 5B). These results suggest that Lgr4 negatively regulates NF-κB activation in the LPS-induced signaling pathway. Furthermore, using an NF-κB-responsive luciferase reporter gene, we measured the NF-κB activation in Lgr4 WT and knockdown RAW cells with different ligands for the TLRs. As shown in Fig. 5C, agonists for TLR2/4 activated much more NF-κB-responsive luciferase reporter in Lgr4 knockdown cells (Fig. 5C, black bars) as compared with control cells (Fig. 5C, white bars). However, there was no difference for the activation of NF-κB luciferase reporter when other TLRs were stimulated, suggesting a specific function of Lgr4 in TLR2/4-mediated NF-κB activation (Fig. 5C).
FIGURE 5.

Increased signaling in Lgr4−/− macrophages upon LPS stimulation. A and B, deletion or knockdown of Lgr4 increases NF-κB activity in both peritoneal macrophages (A) and RAW 264.7 cells (B), respectively. EMSA was performed to measure the DNA binding activity of NF-κB in the nuclear extracts of Lgr4+/+- and Lgr4−/−-derived peritoneal macrophages (A) or Lgr4 knockdown RAW 264.7 cells (B), unstimulated (left) or stimulated (right) with LPS (100 ng/ml). C, knockdown of Lgr4 increased the NF-κB-luciferase activity in RAW cells transfected with an NF-κB-luciferase reporter and then stimulated for 1 h with agonists to different TLRs. *, p < 0.05; **, p < 0.01 (two-tailed Student's t test). D, immunoblot analysis of IKKα, IκBα, ERK, and JNK phosphorylation (p-) in wild-type and Lgr4-deficient BMDMs stimulated for 0–60 min with LPS (100 ng/ml). β-Actin serves as a loading control. Error bars, S.D.
The activation of TLRs usually accompany with two distinct signaling pathways involved by NF-κB and MAPKs. To investigate the influence of Lgr4 on those signaling pathways, we treated BMMs from Lgr4-deficient mice with TLR4-specific agonists (LPS). Our data show that Lgr4 deficiency enhanced LPS-induced phosphorylation of IKKα, IκBα, ERK, and JNK in BMMs (Fig. 5D). We also checked the function of Lgr4 in the TLR3-associated signaling pathway by poly(I:C) activation. No obvious difference was observed between wild-type and Lgr4-deficient BMMs (data not shown), which indicates that Lgr4 has little influence on the TLR3-dependent signaling pathway.
Deletion of Lgr4 Increases the Expression of CD14 in TLR Signaling Pathways
To examine the target genes regulated by Lgr4 in TLR signaling, we measured the expression levels of TLR-associated genes in wild-type and Lgr4-deficient peritoneal macrophages by quantitative PCR. As shown in Fig. 6A, a significant increase in CD14 expression was observed in Lgr4-deficient cells both in resting (LPS 0 h, Fig. 6A, top) and in activated status (LPS 1 h, Fig. 6A, bottom), suggesting that CD14 is a potential target gene regulated by Lgr4 in the macrophages. To further determine whether CD14 is up-regulated in Lgr4-deficient macrophages, we analyzed the protein expression level of CD14 in peritoneal macrophages by FACS using a specific antibody for CD14. As expected, Lgr4-deficient macrophages showed a much higher expression level of CD14 on the cell membrane, suggesting a negative regulation of CD14 by Lgr4 signaling (Fig. 6B). A similar observation was also obtained in Lgr4 knockdown RAW cells (Gpr48 shRNA), which showed increased CD14 expression level by immunoblot analysis using a specific antibody against CD14 (Fig. 6C). Together, our results suggest that Lgr4-mediated signaling inhibited the expression level of CD14 in macrophages, a possible key mechanism in negative regulation of the TLR2/4 signaling pathway by Lgr4 in macrophages.
FIGURE 6.

Regulation of TLR signaling and target genes by Lgr4. A, deletion of Lgr4 increased the expression of CD14 mRNA. Real-time PCR analysis of mRNA expression in Lgr4+/+- and Lgr4−/−-derived peritoneal macrophages. The cells were unstimulated (top, LPS 0 h) or stimulated with LPS for 1 h (bottom, LPS 1 h). Data were presented relative to the expression of genes in Lgr4+/+ samples. **, p < 0.01 (two-tailed Student's t test). B, flow cytometry analysis of the surface expression of CD14 protein on Lgr4+/+- and Lgr4−/−-derived peritoneal macrophages. C, immunoblot analysis of CD14 in RAW 264.7 cells transfected with Lgr4-specific shRNA and then stimulated with LPS (100 ng/ml) for different times. D, the production of TNF-α was analyzed by ELISA at different concentrations of CD14 blocking antibody in control and Lgr4 knockdown RAW 264.7 cells (Lgr4 shRNA) stimulated with LPS (100 ng/ml) for 6 h. Data are representative of three independent experiments. E, production of TNF-α was analyzed by ELISA at different concentrations of CD14 siRNA in Lgr4 knockdown RAW 264.7 cells stimulated with LPS (100 ng/ml) for 6 h. Data are representative of three independent experiments. Error bars, S.D.
CD14 Antibody and siRNA Rescue Lgr4 Deficiency-mediated Cytokine Production
To further investigate the function of CD14 in LPS-induced cytokine production, we treated Lgr4 knockdown RAW cells, which have increased CD14 expression, with CD14 antibody to block CD14-enhanced TLR4 signaling. As shown in Fig. 6D, the production of TNF-α was decreased significantly by CD14 antibody in a dose-dependent manner, and the increased TNF-α production enhanced by Lgr4 knockdown was also rescued by CD14-blocking antibody (Fig. 6D), suggesting a crucial role of CD14 in Lgr4-regulated TLR4 signaling. Furthermore, we also decreased the expression of CD14 by siRNA in Lgr4 knockdown and control cells to rescue Lgr4 deficiency-mediated TNF-α increase. Similar to the above observation, the production of TNF-α was decreased by CD14-specific siRNA in a dose-dependent manner and returned to a similar level prior to the knockdown of Lgr4 in the cells (Fig. 6E). Taken together, Lgr4 negatively regulated the expression of CD14, and knockdown of Lgr4 enhanced the expression of CD14 in the TLR2/4-mediated signaling pathway.
Lgr4 Regulates CD14 at the Transcriptional Level through the cAMP-PKA-CREB Signaling Pathway
To explore the mechanism of Lgr4 in regulating CD14 expression, we analyzed the promoter region of CD14 and identified a conserved cAMP-responsive element site (CREB binding site) at −859 to −854 in the murine CD14 promoter (Fig. 7A). We then generated the wild type (pGL3-CD14) and the CREB-binding site-deleted mutant promoter (pGL3-CD14m) of murine CD14 into a luciferase reporter construct. To determine whether Lgr4 regulates CD14 expression through this CRE site, we measured the luciferase activity of the reporter gene under different conditions. As shown in Fig. 7B (left), the CD14-luciferase activity was increased by knockdown of Lgr4 (Lgr4 shRNA) and by the specific PKA inhibitor (PKI). On the other hand, overexpression of Lgr4 or its active mutant receptor (Lgr4T755I) or CREB in HEK293T cells directly inhibited the luciferase activity of the CD14 luciferase promoter gene with the intact CRE site. To confirm that the CRE site is essential for Lgr4-mediated CD14 promoter activity, we measured the luciferase activity of the CD14 promoter with mutation in the CRE binding site (pGL3-CD14m). As shown in Fig. 7B (right), no obvious influence was observed with the mutant CD14 promoter (pGL3-CD14m) in all conditions tested, suggesting that the regulation of CD14 promoter by Lgr4 is mediated by the PKA-CREB pathway.
FIGURE 7.

CREB negatively regulates CD14 expression. A, the schematic diagram represents the mouse CD14 promoter with a conserved CREB binding site. Putative binding sites for CREB transcription factor and promoter mutation were revealed. B, regulation of CD14 promoter by Lgr4, PKA, and CREB. HEK-293T cells transfected with pGL3-CD14 or pGL3-CD14m, which contained the original (left) or mutated CREB binding site (right), were transiently co-transfected with Lgr4, Lgr4T755I, Lgr4 shRNA, PKI (a specific inhibitor of PKA), and CREB and then subjected to the luciferase assay as described under “Experimental Procedures.” *, p < 0.05; **, p < 0.01; ***, p < 0.001 (two-tailed Student's t test). Data are representative of two independent experiments. C, immunoblot analysis of CREB phosphorylation in RAW 264.7 cells transfected with control and Lgr4-specific shRNA and then stimulated with LPS (100 ng/ml) at different times. D, knockdown of Lgr4 decreased CREB binding to the CD14 promoter by ChIP analysis. Control and Lgr4 knockdown RAW cells were analyzed for CREB occupancy at the CD14 promoter using RT-PCR, presented relative to control RAW cells. IgG, immunoprecipitation with an irrelevant antibody (negative control). Data are representative of three independent experiments. E, production of TNF-α and IL-6 was analyzed by quantitative PCR at different concentrations of forskolin for 4 h in Lgr4 knockdown RAW 264.7 cells stimulated with LPS (100 ng/ml). Data are representative of three independent experiments. Error bars, S.D.
To examine whether deletion of Lgr4 affects CREB phosphorylation and activation, we investigated the phosphorylation levels of CREB (phosphorylated at serine 133) in Lgr4 knockdown macrophages using a specific anti-phospho-CREB antibody in Western blot analysis. Our data indicated that knockdown of Lgr4 leads to the decrease of CREB phosphorylation in macrophages (Fig. 7C). Furthermore, to confirm the inhibitory effect of Lgr4 on the binding activity of the CREB transcription factor, we examined the specific binding of CREB to the CD14 promoter region by a ChIP assay. As shown in Fig. 7D, the binding of CREB to the CD14 promoter was decreased significantly in Lgr4 knockdown RAW cells (Fig. 7D), suggesting a direct correlation of Lgr4 expression and CREB binding activity to the CD14 promoter. These data are consistent with those of the luciferase assays in which CREB is the negative regulator for CD14 transcription. To further confirm the function of the cAMP-PKA-CREB signaling pathway in the regulation of TLR4-associated immune response, we used forskolin to increase endogenous cAMP in Lgr4 knockdown cells. As shown in Fig. 7E, the expression of TNF-α and IL-6 was suppressed significantly by forskolin in a dose-dependent manner, which is consistent with the negative regulation of Lgr4 in TLR2/4-associated immune response. Together, our data demonstrate that Lgr4 negatively regulates CD14 transcriptional activation and CD14-mediated immune response through the cAMP-PKA-CREB signaling pathway.
DISCUSSION
G-protein coupled receptors have been found with a variety of physiological functions and have become the most popular drug targets in pharmaceutical markets. Although a novel orphan G-protein-coupled receptor, Lgr4 has been found to play a crucial role in the development of different organs and in the regulation of Wnt signaling (20, 34). The role of Lgr4 in innate immune response remains unknown. In this study, we have shown that Lgr4-deficient mice are much more susceptible to and have much higher mortality from LPS stimulation. Lgr4 negatively regulates TLR2/4-associated pattern recognition and proinflammatory cytokine production by decreasing the expression of CD14 through the cAMP-PKA-CREB signaling pathway. Increased expression of CD14 leads to the activation of the TLR2/4 signaling pathway and the production of proinflammatory cytokines (Fig. 8). Our results are the first to suggest that Lgr4 is a novel negative regulator in innate immune response and could be a potential therapeutic target for inflammatory disorders.
FIGURE 8.

Model for the regulation of TLR signaling by Lgr4. Model of Lgr4-mediated signaling cascades in TLR2/4-mediated immune response. Lgr4 can activate the cAMP-PKA-CREB pathway to negatively regulate CD14 expression through CREB binding to the CD14 promoter. Down-regulation of CD14 reduced the production of inflammatory cytokines in TLR2/4-mediated innate immune response.
The activation of TLR signaling is a double-edged sword (35). Although the production of cytokines can activate the immune system to fight infection, uncontrolled immune responses also have serious side effects. Thus, the immune system needs to keep a balance between activation and inhibition to avoid detrimental and inappropriate inflammatory responses. Sepsis is a serious body-wide response to infection and is considered to be a severe disease with high mortality. In the United States, the total number of deaths from septic shock continue to increase (36). Therefore, finding negative regulators in TLR-mediated immune responses will be important in understanding the mechanisms of immune disorders. In this paper, we investigated the function of Lgr4 in modulating pattern recognition and innate immunity using a septic shock mouse model by intraperitoneal injection of a high dose of LPS. We demonstrate that almost all Lgr4−/− mice were dead from LPS injection after 48 h, whereas more than 60% of Lgr4+/+ mice remained alive, suggesting that Lgr4 is a negative regulator for LPS signaling and response. Because Lgr4 and TLRs are transmembrane proteins with multiple leucine-rich repeat regions in the extracellular domain, it will be fascinating to explore the possible interaction of Lgr4 and TLR-mediated innate immune responses.
Macrophages play a key role in the innate immune system, which includes both acute and chronic inflammation (37). Macrophages can prevent invading pathogens through nonspecific uptake of particles at the first recognition and then present the antigen information to the acquired immune system as antigen-presenting cells. Therefore, macrophage has been regarded as the co-regulator between innate and acquired immune systems. An overly activated macrophage response, however, can lead to pathological symptoms that contribute significantly to septic shock, autoimmune diseases, and various granulomatous disorders (38). In this research, the bone marrow-derived and peritoneal macrophages from Lgr4-deficient mice were challenged with different TLR agonists to investigate the effects of Lgr4 in macrophage-mediated functions. We showed that Lgr4 negatively modulates PAMP-induced immune responses. The loss of Lgr4 in macrophages induces the activation of NF-κB and the overproduction of cytokines by LPS, Pam3CSK4, HKLM, and FSL1, which is the key reason for septic shock responses in the mouse models.
CD14 is a well known pattern recognition receptor in the innate immune system, which was strongly up-regulated during the differentiation of monocytic precursor cells into monocytes (39). CD14 exists in membrane-bound (mCD14) and soluble form (sCD14), and both contribute to host clearance of LPS. The interaction between CD14 and LPS is also responsible for the toxic effects of LPS in the liver and in systemic circulation after the release of inflammatory cytokines and mediators. Thus, inhibition of CD14 expression is an alternative way to rescue LPS-induced septic shock. Treatment with CD14 antibody, which blocks the binding of CD14 to LPS, attenuates the release of proinflammatory cytokines (40). In this study, deletion of Lgr4 leads to an obvious increase of CD14 both in mRNA and protein levels. Accordingly, the release of cytokines, such as IL-6 and TNF-α, in the serum and cell culture medium was enhanced in response to LPS and Pam3CSK4. Meanwhile, a series of LPS-induced cytokines, such as IL-1α, IFN-β, IL-10, GM-CSF, and CXCL10, were all enhanced in Lgr4-deficient macrophages, suggesting that both MyD88-dependent and MyD88-independent signaling pathways are regulated by the loss of Lgr4. When we treated the Lgr4 knockdown cells with CD14-blocking antibody or CD14-specific siRNA, the production of TNF-α was rescued to the normal level, suggesting an important role of CD14 in Lgr4-regulated immune responses. It has been found that the increased soluble circulating CD14 is associated with high mortality in Gram-negative septic shock and even in HIV infection (41, 42), suggesting a potential therapeutic role of Lgr4 in CD14-mediated immune diseases.
Most recently, it has been shown that R-spondin can interact with Lgr4 to activate Wnt/β-catenin signaling through noncanonical G-protein signaling pathway (20, 21). Although R-spondin1 has been found clinically useful in the therapeutic treatment of inflammatory bowel disease by stimulating crypt cell growth, accelerating mucosal regeneration, and restoring intestinal architecture (43), no obvious influence on TLR4-associated innate immune response was observed by R-spondin in macrophages (Fig. 2C), whereas we demonstrated that Lgr4 can regulate innate immune response in classical cAMP-PKA signaling pathway. Therefore, it will be interesting to explore the cross-talk between Wnt/β-catenin and Lgr4 classic signaling pathways in different immune systems.
It has been shown that the activation of Lgr4 by constitutively mutant receptor leads to an increase in intracellular cAMP and the activation of PKA, which phosphorylates and activates the transcription factor CREB (25, 30, 44, 45). Although CREB acts usually as a transactivator, it could also function as a repressor when phosphorylated at Ser-142 (46) or heterodimerized with the co-repressor forms of the cAMP-response element modulator (CREM and ICER) (47). The negative regulation of CREB in gene transcription has also been shown on CYR61 and necdin (48, 49). In this study, we demonstrate that CREB binds to the promoter region of CD14 to negatively regulate the expression of CD14. Deletion of the CREB-binding site in CD14 will terminate the inhibitory effects of Lgr4 and CREB to CD14, confirming a crucial role of cAMP and CREB in regulating CD14-associated signaling, which is consistent with previous studies (50, 51). In fact, the recognition of LPS by TLR4 not only activates the NF-κB signaling pathway but also increases the activity of CREB binding to CD14 (Fig. 7C). It is reasonable to hypothesize that CREB regulates the expression of CD14 through a feedback pathway to keep the response to PAMPs at an appropriate level. The mCD14 or sCD14 helps TLRs to recognize the invading pathogen and activates the downstream signaling pathway, including NF-κB and CREB activity. Then the increased cAMP/CREB activity reduces the expression level of CD14 and eventually restricts the immune response to an appropriate level (Fig. 7E). Our results showed that Lgr4 functions as a co-regulator through the cAMP-PKA-CREB pathway to keep the immune response in balance. The evidence we provided here shows the potential of Lgr4 in curing TLR-associated septic shock and autoimmune diseases. The finding of Lgr4 as a regulator in innate immunity will provide new insights into the physiological functions of this G-protein-coupled receptor in immune diseases.
Supplementary Material
This work was supported, in whole or in part, by National Institutes of Health Grant R01CA134731. This work was also supported by National Basic Research Program of China Grant 2012CB910400, National Natural Science Foundation of China Grants 31000398 and 81272369, and the Science and Technology Commission of Shanghai Municipality Grants 09JC1405200 and 11DZ2260300.

This article contains supplemental Table 1 and Fig. 1.
- PAMP
- pathogen-associated molecular pattern
- TLR
- Toll-like receptor
- CREB
- cAMP-response element-binding protein
- BMM
- bone marrow-derived macrophage.
REFERENCES
- 1. Akira S., Uematsu S., Takeuchi O. (2006) Pathogen recognition and innate immunity. Cell 124, 783–801 [DOI] [PubMed] [Google Scholar]
- 2. Takeda K., Kaisho T., Akira S. (2003) Toll-like receptors. Annu. Rev. Immunol. 21, 335–376 [DOI] [PubMed] [Google Scholar]
- 3. Janeway C. A., Jr., Medzhitov R. (2002) Innate immune recognition. Annu. Rev. Immunol. 20, 197–216 [DOI] [PubMed] [Google Scholar]
- 4. Kawai T., Akira S. (2010) The role of pattern-recognition receptors in innate immunity. Update on Toll-like receptors. Nat. Immunol. 11, 373–384 [DOI] [PubMed] [Google Scholar]
- 5. Kerfoot S. M., Long E. M., Hickey M. J., Andonegui G., Lapointe B. M., Zanardo R. C., Bonder C., James W. G., Robbins S. M., Kubes P. (2004) TLR4 contributes to disease-inducing mechanisms resulting in central nervous system autoimmune disease. J. Immunol. 173, 7070–7077 [DOI] [PubMed] [Google Scholar]
- 6. Eisenbarth S. C., Piggott D. A., Huleatt J. W., Visintin I., Herrick C. A., Bottomly K. (2002) Lipopolysaccharide-enhanced, Toll-like receptor 4-dependent T helper cell type 2 responses to inhaled antigen. J. Exp. Med. 196, 1645–1651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Michelsen K. S., Wong M. H., Shah P. K., Zhang W., Yano J., Doherty T. M., Akira S., Rajavashisth T. B., Arditi M. (2004) Lack of Toll-like receptor 4 or myeloid differentiation factor 88 reduces atherosclerosis and alters plaque phenotype in mice deficient in apolipoprotein E. Proc. Natl. Acad. Sci. U.S.A. 101, 10679–10684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Björkbacka H., Kunjathoor V. V., Moore K. J., Koehn S., Ordija C. M., Lee M. A., Means T., Halmen K., Luster A. D., Golenbock D. T., Freeman M. W. (2004) Reduced atherosclerosis in MyD88-null mice links elevated serum cholesterol levels to activation of innate immunity signaling pathways. Nat. Med. 10, 416–421 [DOI] [PubMed] [Google Scholar]
- 9. Liew F. Y., Xu D., Brint E. K., O'Neill L. A. (2005) Negative regulation of Toll-like receptor-mediated immune responses. Nat. Rev. Immunol. 5, 446–458 [DOI] [PubMed] [Google Scholar]
- 10. Wright S. D., Ramos R. A., Tobias P. S., Ulevitch R. J., Mathison J. C. (1990) CD14, a receptor for complexes of lipopolysaccharide (LPS) and LPS binding protein. Science 249, 1431–1433 [DOI] [PubMed] [Google Scholar]
- 11. Goyert S. M., Ferrero E., Rettig W. J., Yenamandra A. K., Obata F., Le Beau M. M. (1988) The CD14 monocyte differentiation antigen maps to a region encoding growth factors and receptors. Science 239, 497–500 [DOI] [PubMed] [Google Scholar]
- 12. Viriyakosol S., Kirkland T. N. (1996) The N-terminal half of membrane CD14 is a functional cellular lipopolysaccharide receptor. Infect. Immun. 64, 653–656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wurfel M. M., Hailman E., Wright S. D. (1995) Soluble CD14 acts as a shuttle in the neutralization of lipopolysaccharide (LPS) by LPS-binding protein and reconstituted high density lipoprotein. J. Exp. Med. 181, 1743–1754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Means T. K., Lien E., Yoshimura A., Wang S., Golenbock D. T., Fenton M. J. (1999) The CD14 ligands lipoarabinomannan and lipopolysaccharide differ in their requirement for Toll-like receptors. J. Immunol. 163, 6748–6755 [PubMed] [Google Scholar]
- 15. Muroi M., Ohnishi T., Tanamoto K. (2002) Regions of the mouse CD14 molecule required for Toll-like receptor 2- and 4-mediated activation of NF-κB. J. Biol. Chem. 277, 42372–42379 [DOI] [PubMed] [Google Scholar]
- 16. Mazerbourg S., Bouley D. M., Sudo S., Klein C. A., Zhang J. V., Kawamura K., Goodrich L. V., Rayburn H., Tessier-Lavigne M., Hsueh A. J. (2004) Leucine-rich repeat-containing, G protein-coupled receptor 4 null mice exhibit intrauterine growth retardation associated with embryonic and perinatal lethality. Mol. Endocrinol. 18, 2241–2254 [DOI] [PubMed] [Google Scholar]
- 17. Hsu S. Y., Liang S. G., Hsueh A. J. (1998) Characterization of two LGR genes homologous to gonadotropin and thyrotropin receptors with extracellular leucine-rich repeats and a G protein-coupled, seven-transmembrane region. Mol. Endocrinol. 12, 1830–1845 [DOI] [PubMed] [Google Scholar]
- 18. Loh E. D., Broussard S. R., Kolakowski L. F. (2001) Molecular characterization of a novel glycoprotein hormone G-protein-coupled receptor. Biochem. Biophys. Res. Commun. 282, 757–764 [DOI] [PubMed] [Google Scholar]
- 19. Kajava A. V. (1998) Structural diversity of leucine-rich repeat proteins. J. Mol. Biol. 277, 519–527 [DOI] [PubMed] [Google Scholar]
- 20. Carmon K. S., Gong X., Lin Q., Thomas A., Liu Q. (2011) R-spondins function as ligands of the orphan receptors LGR4 and LGR5 to regulate Wnt/β-catenin signaling. Proc. Natl. Acad. Sci. U.S.A. 108, 11452–11457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. de Lau W., Barker N., Low T. Y., Koo B. K., Li V. S., Teunissen H., Kujala P., Haegebarth A., Peters P. J., van de Wetering M., Stange D. E., van Es J. E., Guardavaccaro D., Schasfoort R. B., Mohri Y., Nishimori K., Mohammed S., Heck A. J., Clevers H. (2011) Lgr5 homologues associate with Wnt receptors and mediate R-spondin signalling. Nature 476, 293–297 [DOI] [PubMed] [Google Scholar]
- 22. Yamashita R., Takegawa Y., Sakumoto M., Nakahara M., Kawazu H., Hoshii T., Araki K., Yokouchi Y., Yamamura K. (2009) Defective development of the gall bladder and cystic duct in Lgr4− hypomorphic mice. Dev. Dyn. 238, 993–1000 [DOI] [PubMed] [Google Scholar]
- 23. Luo J., Zhou W., Zhou X., Li D., Weng J., Yi Z., Cho S. G., Li C., Yi T., Wu X., Li X. Y., de Crombrugghe B., Höök M., Liu M. (2009) Regulation of bone formation and remodeling by G-protein-coupled receptor 48. Development 136, 2747–2756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Gao Y., Shan Z. Y., Wang H., Zhang H. M., Teng W. P. (2009) Inhibitory effect of shRNA targeting GPR48 on invasion and metastasis of human cervical carcinoma cell line HeLa. Ai Zheng 28, 104–107 [PubMed] [Google Scholar]
- 25. Weng J., Luo J., Cheng X., Jin C., Zhou X., Qu J., Tu L., Ai D., Li D., Wang J., Martin J. F., Amendt B. A., Liu M. (2008) Deletion of G protein-coupled receptor 48 leads to ocular anterior segment dysgenesis (ASD) through down-regulation of Pitx2. Proc. Natl. Acad. Sci. U.S.A. 105, 6081–6086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Song H., Luo J., Luo W., Weng J., Wang Z., Li B., Li D., Liu M. (2008) Inactivation of G-protein-coupled receptor 48 (Gpr48/Lgr4) impairs definitive erythropoiesis at midgestation through down-regulation of the ATF4 signaling pathway. J. Biol. Chem. 283, 36687–36697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Mohri Y., Kato S., Umezawa A., Okuyama R., Nishimori K. (2008) Impaired hair placode formation with reduced expression of hair follicle-related genes in mice lacking Lgr4. Dev. Dyn. 237, 2235–2242 [DOI] [PubMed] [Google Scholar]
- 28. Jin C., Yin F., Lin M., Li H., Wang Z., Weng J., Liu M., Da Dong X., Qu J., Tu L. (2008) GPR48 regulates epithelial cell proliferation and migration by activating EGFR during eyelid development. Invest. Ophthalmol. Vis. Sci. 49, 4245–4253 [DOI] [PubMed] [Google Scholar]
- 29. Kato S., Mohri Y., Matsuo T., Ogawa E., Umezawa A., Okuyama R., Nishimori K. (2007) Eye-open at birth phenotype with reduced keratinocyte motility in LGR4 null mice. FEBS Lett. 581, 4685–4690 [DOI] [PubMed] [Google Scholar]
- 30. Li X. Y., Lu Y., Sun H. Y., Wang J. Q., Yang J., Zhang H. J., Fan N. G., Xu J., Jiang J. J., Liu R. Y., Li D. L., Liu M. Y., Ning G. (2010) G protein-coupled receptor 48 upregulates estrogen receptor α expression via cAMP/PKA signaling in the male reproductive tract. Development 137, 151–157 [DOI] [PubMed] [Google Scholar]
- 31. Wang Z., Jin C., Li H., Li C., Hou Q., Liu M., Dong Xda E., Tu L. (2010) GPR48-induced keratinocyte proliferation occurs through HB-EGF mediated EGFR transactivation. FEBS Lett. 584, 4057–4062 [DOI] [PubMed] [Google Scholar]
- 32. Beutler B., Milsark I. W., Cerami A. C. (1985) Passive immunization against cachectin/tumor necrosis factor protects mice from lethal effect of endotoxin. Science 229, 869–871 [DOI] [PubMed] [Google Scholar]
- 33. Kawai T., Akira S. (2007) TLR signaling. Semin. Immunol. 19, 24–32 [DOI] [PubMed] [Google Scholar]
- 34. Mustata R. C., Van Loy T., Lefort A., Libert F., Strollo S., Vassart G., Garcia M. I. (2011) Lgr4 is required for Paneth cell differentiation and maintenance of intestinal stem cells ex vivo. EMBO Rep. 12, 558–564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Cook D. N., Pisetsky D. S., Schwartz D. A. (2004) Toll-like receptors in the pathogenesis of human disease. Nat. Immunol. 5, 975–979 [DOI] [PubMed] [Google Scholar]
- 36. Martin G. S., Mannino D. M., Eaton S., Moss M. (2003) The epidemiology of sepsis in the United States from 1979 through 2000. N. Engl. J. Med. 348, 1546–1554 [DOI] [PubMed] [Google Scholar]
- 37. Hoffmann J. A., Kafatos F. C., Janeway C. A., Ezekowitz R. A. (1999) Phylogenetic perspectives in innate immunity. Science 284, 1313–1318 [DOI] [PubMed] [Google Scholar]
- 38. Dinarello C. A. (2000) Proinflammatory cytokines. Chest 118, 503–508 [DOI] [PubMed] [Google Scholar]
- 39. Griffin J. D., Ritz J., Nadler L. M., Schlossman S. F. (1981) Expression of myeloid differentiation antigens on normal and malignant myeloid cells. J. Clin. Invest. 68, 932–941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Spek C. A., Verbon A., Aberson H., Pribble J. P., McElgunn C. J., Turner T., Axtelle T., Schouten J., Van Der Poll T., Reitsma P. H. (2003) Treatment with an anti-CD14 monoclonal antibody delays and inhibits lipopolysaccharide-induced gene expression in humans in vivo. J. Clin. Immunol. 23, 132–140 [DOI] [PubMed] [Google Scholar]
- 41. Landmann R., Zimmerli W., Sansano S., Link S., Hahn A., Glauser M. P., Calandra T. (1995) Increased circulating soluble CD14 is associated with high mortality in Gram-negative septic shock. J. Infect. Dis. 171, 639–644 [DOI] [PubMed] [Google Scholar]
- 42. Lawn S. D., Labeta M. O., Arias M., Acheampong J. W., Griffin G. E. (2000) Elevated serum concentrations of soluble CD14 in HIV− and HIV+ patients with tuberculosis in Africa. Prolonged elevation during anti-tuberculosis treatment. Clin. Exp. Immunol. 120, 483–487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Zhao J., de Vera J., Narushima S., Beck E. X., Palencia S., Shinkawa P., Kim K. A., Liu Y., Levy M. D., Berg D. J., Abo A., Funk W. D. (2007) R-spondin1, a novel intestinotrophic mitogen, ameliorates experimental colitis in mice. Gastroenterology 132, 1331–1343 [DOI] [PubMed] [Google Scholar]
- 44. Gao Y., Kitagawa K., Shimada M., Uchida C., Hattori T., Oda T., Kitagawa M. (2006) Generation of a constitutively active mutant of human GPR48/LGR4, a G-protein-coupled receptor. Hokkaido Igaku Zasshi 81, 101–105, 107,, 109 [PubMed] [Google Scholar]
- 45. Wang J., Li X., Ke Y., Lu Y., Wang F., Fan N., Sun H., Zhang H., Liu R., Yang J., Ye L., Liu M., Ning G. (2012) GPR48 increases mineralocorticoid receptor gene expression. J. Am. Soc. Nephrol. 23, 281–293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Sun P., Enslen H., Myung P. S., Maurer R. A. (1994) Differential activation of CREB by Ca2+/calmodulin-dependent protein kinases type II and type IV involves phosphorylation of a site that negatively regulates activity. Genes Dev. 8, 2527–2539 [DOI] [PubMed] [Google Scholar]
- 47. Borlikova G., Endo S. (2009) Inducible cAMP early repressor (ICER) and brain functions. Mol. Neurobiol. 40, 73–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Dobroff A. S., Wang H., Melnikova V. O., Villares G. J., Zigler M., Huang L., Bar-Eli M. (2009) Silencing cAMP-response element-binding protein (CREB) identifies CYR61 as a tumor suppressor gene in melanoma. J. Biol. Chem. 284, 26194–26206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Cypess A. M., Zhang H., Schulz T. J., Huang T. L., Espinoza D. O., Kristiansen K., Unterman T. G., Tseng Y. H. (2011) Insulin/IGF-I regulation of necdin and brown adipocyte differentiation via CREB- and FoxO1-associated pathways. Endocrinology 152, 3680–3689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Katsuno G., Takahashi H. K., Iwagaki H., Sugita S., Mori S., Saito S., Yoshino T., Nishibori M., Tanaka N. (2006) The effect of ciprofloxacin on CD14 and Toll-like receptor-4 expression on human monocytes. Shock 25, 247–253 [DOI] [PubMed] [Google Scholar]
- 51. Zhu N., Cui J., Qiao C., Li Y., Ma Y., Zhang J., Shen B. (2008) cAMP modulates macrophage development by suppressing M-CSF-induced MAPKs activation. Cell Mol. Immunol. 5, 153–157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Lei M., Jiao H., Liu T., Du L., Cheng Y., Zhang D., Hao Y., Man C., Wang F. (2011) siRNA targeting mCD14 inhibits TNF-α, MIP-2, and IL-6 secretion and NO production from LPS-induced RAW264.7 cells. Appl. Microbiol. Biotechnol. 92, 115–124 [DOI] [PubMed] [Google Scholar]
- 53. Sung B., Pandey M. K., Aggarwal B. B. (2007) Fisetin, an inhibitor of cyclin-dependent kinase 6, down-regulates nuclear factor-κB-regulated cell proliferation, antiapoptotic and metastatic gene products through the suppression of TAK-1 and receptor-interacting protein-regulated IκBα kinase activation. Mol. Pharmacol. 71, 1703–1714 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}