

Background: Microglia respond to cell death by extending their processes toward ATP released at the site of damage.
Results: Norepinephrine induces process retraction in resting and activated microglia through β2 and α2A receptors, respectively.
Conclusion: Norepinephrine alters the ability of microglia to respond to ATP.
Significance: Adrenergic receptor signaling might alter the ability of microglia to detect and respond to tissue damage in vivo.
Keywords: Adrenergic Receptor, Imaging, Microglia, Migration, Purinergic Receptor, Brain Slice
Abstract
Microglia, the resident immune cells of the central nervous system (CNS), monitor the brain for disturbances of tissue homeostasis by constantly moving their fine processes. Microglia respond to tissue damage through activation of ATP/ADP receptors followed by directional process extension to the damaged area. A common feature of several neurodegenerative diseases is the loss of norepinephrine, which might contribute to the associated neuroinflammation. We carried out a high resolution analysis of the effects of norepinephrine (NE) on microglial process dynamics in acute brain slices from mice that exhibit microglia-specific enhanced green fluorescent protein expression. Bath application of NE to the slices resulted in significant process retraction in microglia. Analysis of adrenergic receptor expression with quantitative PCR indicated that resting microglia primarily express β2 receptors but switch expression to α2A receptors under proinflammatory conditions modeled by LPS treatment. Despite the differential receptor expression, NE caused process retraction in both resting and LPS-activated microglia cultured in the gelatinous substrate Matrigel in vitro. The use of subtype-selective receptor agonists and antagonists confirmed the involvement of β2 receptors in mediating microglial process dynamics in resting cells and α2A receptors in activated cells. Co-application of NE with ATP to resting microglia blocked the ATP-induced process extension and migration in isolated microglia, and β2 receptor antagonists prolonged ATP effects in brain slice tissues, suggesting the presence of cross-talk between adrenergic and purinergic signaling in microglia. These data show that the neurotransmitter NE can modulate microglial motility, which could affect microglial functions in pathogenic situations of either elevated or reduced NE levels.
Introduction
The locus coeruleus (LC)2 is the primary site of norepinephrine (NE) neurons in the brain with projections extending to the hippocampus, most cortical regions, and certain subcortical structures. The loss of LC neurons is an early hallmark of several neurodegenerative diseases, including Alzheimer disease, Parkinson disease, and dementia with Lewy bodies (1–5). The degeneration of the LC results in a decrease in NE levels in its projection areas (1, 4). In contrast, NE is released in large amounts during activation of the “fight-or-flight” response by stressful stimuli. Certain types of stress, especially acute stressors, can increase the LC firing rate and NE concentrations throughout the brain (6, 7). Both decreased and increased NE levels might lead to dysregulation of NE-dependent functions such as learning and memory, attention, and arousal (8) and could contribute to a pathological state. Experimental depletion of NE by either toxins or genetic manipulations exacerbated pathological processes in animal models of Alzheimer disease and Parkinson disease (9–11). In contrast, elevation of synaptic NE levels by preventing its uptake protects dopaminergic neurons from 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine toxicity (12).
In addition to its role as a neurotransmitter, NE can be released extrasynaptically and can act as a neuromodulator to influence the functions of glial cells and capillaries (13, 14). NE seems to possess anti-inflammatory actions as it has been shown to reduce proinflammatory gene expression in microglia, brain-resident immune cells (15–18). The beneficial role of NE is especially evident in animal models of Alzheimer disease where it might help resolve pathology in several ways. First, NE might enhance microglial chemoattraction to the amyloid β peptide (Aβ)-containing plaques associated with the disease (19). Second, it promotes the clearance of Aβ plaques by increasing microglial phagocytosis of Aβ and elevating the expression of Aβ-degrading enzymes (20). Third, NE treatment prevents the Aβ-induced increase in proinflammatory cytokine expression (11, 19), and lack of NE exacerbates microgliosis in a mouse model of Alzheimer disease (21).
The ability of microglia to constantly move their processes is thought to be essential for both their role in tissue surveillance and their response to tissue damage (22–24). NE is known to increase microglial motility in response to Aβ in Boyden chamber migration assays and in vivo (19), but the mechanism by which NE regulates motility has not been well described. We developed a new brain slice assay to study microglial motility in tissues and used an established in vitro assay to determine the receptor involvement and signaling pathways by which NE modulates microglial motility and the dynamics of individual processes. We examined both resting and activated microglia because of our previous findings showing that different G protein-coupled receptors control microglial motility in a manner dependent on microglial activation status (25, 26). We found that the expression of the α2A- and β2-adrenergic receptors markedly changes as a function of microglial activation and that both receptors can regulate microglial process dynamics. The α2A/β2 receptor pair is the second pair of Gi/Gs-coupled receptors that we have found to control microglial motility and that is altered by microglial phenotypic state (the other being purinergic P2Y12/adenosine A2A receptors) (26). These data suggest that both NE and ATP can control how microglia sense and respond to tissue damage, either independently or synergistically, which could hold therapeutic implications for the role of microglia in neurodegeneration.
EXPERIMENTAL PROCEDURES
Animals and Primary Microglial Culture
All procedures involving the use of animals were reviewed and approved by the Emory University Institutional Animal Care and Use Committee. Primary microglia were obtained by triturating the cortices of P0–P5 postnatal pups as described previously (26). Aspirating the media from the resulting astrocyte-microglia co-cultures following >14 days of incubation allowed us to obtain a microglial cell suspension that was 98.0 ± 1.2% pure (assessed by isolectin B4 staining). Cultured microglia for confocal imaging experiments were prepared from actin-enhanced green fluorescent protein (eGFP) mice that express eGFP in all cells under control of the actin promoter (provided by M. Okabe, Osaka University, Osaka, Japan). Microglia used for Ca2+ imaging experiments were isolated from wild type C57Bl/6 mice (Charles River). CX3CR1-eGFP mice that have microglia-specific eGFP expression driven by the CX3C-type chemokine receptor 1 promoter (27) were purchased from The Jackson Laboratory.
Preparation of Acute Brain Slices, Slice Imaging, and Data Analysis
Acute brain slices were prepared from 1–4-month-old CX3CR1-eGFP transgenic mice, which have microglia-specific eGFP expression (see Fig. 1A) (27). Coronal slices were cut at a thickness of 200 μm using a Leica VT1000S vibratome in ice-cold oxygenated cutting solution that contained 130 mm NaCl, 2.5 mm KCl, 1.25 mm NaH2PO4, 10 mm glucose, 24 mm NaHCO3, 3 mm MgSO4, and 1 mm CaCl2 equilibrated with 95% O2 and 5% CO2. The slices were then maintained in oxygenated artificial cerebrospinal fluid (aCSF) for at least 1 h at room temperature (to allow the tissue to recover from slicing) and up to 5 h before imaging. The composition of the aCSF was 130 mm NaCl, 2.5 mm KCl, 1.25 mm NaH2PO4, 10 mm glucose, 24 mm NaHCO3, 1.5 mm MgSO4, and 1.5 mm CaCl2. The cutting procedure and maintenance of slices in aCSF do not affect cytokine synthesis in this time frame (see Fig. 1B). For imaging, slices were placed on the stage of an Olympus IX51 inverted confocal microscope equipped with a disc spinning unit and constantly perfused with oxygenated aCSF at 32 °C. To prevent slice movement during solution flow, a platinum ring with nylon threads was placed above the slices. Imaging was carried out at 60× magnification using IPLab image acquisition software. The experimental protocol consisted of a baseline reading for 5 min followed by application of aCSF alone or different treatments in oxygenated aCSF. To better represent the complicated microglial morphology, ∼30–50 optical sections along the z axis of the slices were collected every 60 s; each section was 1 μm above the previous one. For image analysis, the optical sections at a given time point were projected onto the xy plane using the maximum intensity at each pixel position in the stack to obtain a two-dimensional representation of the brain slice. The two-dimensional maximum intensity projections were then used to calculate total process length with NIH ImageJ software. Each cell process was manually traced and saved as a region of interest. The length of the region of interest was measured at the beginning of imaging, and its length was tracked at 5-min intervals. The sum of all processes was normalized to the total process length (sum of all regions of interest) during the baseline recording to allow comparison between cells with different degrees of starting ramification. For illustration of the process traces in Figs. 2 and 3, the two-dimensional projections were imported into Adobe Photoshop, and the processes were manually traced out. For better visualization of the thin processes in movies and still images, the brightness and contrast for the whole movie were adjusted.
FIGURE 1.

Microglia in acute brain slices. A, 200-μm coronal cortical slices were prepared from CX3CR1-eGFP mice that have microglia-specific eGFP expression and imaged with a confocal microscope. B, IL-1β cytokine expression was assessed by RT-PCR using RNA isolated from acute brain slices. To compare cytokine expression in slices with normal brain, cortical lysates from PBS- (control) or LPS-injected (2 mg/kg intraperitoneally 2 days before slicing) animals were used as a control. C, maximum intensity projections of optical sections spanning 45 μm through representative cells before (left; t = 0 min) and following treatment with 30 μm ATP (right; t = 7 min). Arrowheads point to select processes that change over time. Scale bars, 5 μm. D, microglial processes for still images shown in C were manually traced out for visualization and quantification.
FIGURE 2.

Microglial motility in slices. Coronal slices were prepared from CX3CR1-eGFP mice and imaged as described. A, maximum intensity projection of a 31-μm section from a slice before (left) and following application of 30 μm NE (right). Arrowheads point to select processes that change over time. Scale bars, 5 μm. B, traced out processes for the cell shown in A. C, the total process length for each cell was calculated following ATP or NE treatment and normalized to the total process length during a 5-min baseline recording. D, process length of ATP- and NE-treated slices normalized to the average length of aCSF-treated slices at each time point. The numbers of cells analyzed for each treatment are shown in parentheses. Error bars, S.E. Statistical analysis was performed using two-way repeated measures ANOVA and Tukey's post hoc test. #, p < 0.05 compared with baseline response; *, p < 0.05 compared with aCSF at the corresponding time point.
FIGURE 3.
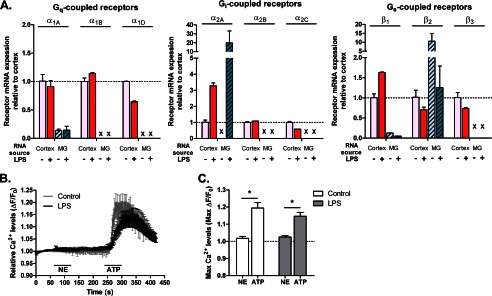
Expression of adrenergic receptors in microglia. A, quantitative real time PCR results for mouse adrenergic receptors in cortical lysates from PBS- (control) or LPS-injected (2 mg/kg) mice or purified primary microglia (MG) treated with 100 ng/ml LPS or HBSS (control) for 24 h. For each receptor, expression was calculated relative to cortical lysates of PBS-injected animals in three independent experiments, each performed in duplicate. Dotted line, relative expression = 1 compared with cortex. X, not detected. B, measurement of Ca2+ levels as a readout of Gq-linked receptor activation in primary microglia treated with HBSS (n = 12 cells) or 100 ng/ml LPS for 24 h (n = 15 cells). NE (30 μm) and ATP (30 μm; positive control) were each applied for 1 min. The average traces from all cells are shown. C, quantification of the maximum Fura-2 response following NE or ATP treatment. Error bars, S.E. Statistical analysis was performed using two-way repeated measures ANOVA and Tukey's post hoc test. *, p < 0.05.
Reverse Transcription-PCR (RT-PCR)
Total cellular RNA (isolated with a PureLink mini kit, Ambion) was treated with 2 units/reaction DNase I (Invitrogen) to remove any contaminating DNA. Semiquantitative reverse transcription-PCR was carried out with 50 ng of RNA as template using the SuperScript III One-Step RT-PCR System with Platinum Taq DNA polymerase (Invitrogen). The protocol included incubation at 60 °C for 30 min for cDNA synthesis before amplification, which consisted of heating to 94 °C for 2 min and 35 cycles of 94 °C for 15 s, 60 °C for 30 s, and 70 °C for 1 min with a final extension at 68 °C for 5 min. Increasing or decreasing the number of amplification cycles resulted in an altered level of product, suggesting that this protocol could detect changes in starting template levels. Primer sequences for mouse interleukin-1β (IL-1β) and β-actin have been described previously (28, 29).
Real Time Quantitative PCR
Total cellular RNA was isolated as described above. cDNA was synthesized from 1 μg of RNA using random primers and the High Capacity cDNA Reverse Transcription kit (Applied Biosystems), and 250 ng of the resulting cDNA was used as starting material for real time PCR using TaqMan Fast Universal PCR Master Mix (Applied Biosystems). Primers for the different adrenergic receptors and GAPDH are available from the Applied Bioscience TaqMan Gene Expression Assays: probes Mm00442668_m1 (α1A), Mm00431685_m1 (α1B), Mm01328600_m1 (α1D), Mm00845383_s1 (α2A), Mm00477390_s1 (α2B), Mm00431686_s1 (α2C), Mm00431701_s1 (β1), Mm02524224_s1 (β2), Mm00442669_m1 (β3), and Mm99999915_g1 (GAPDH). Amplification was carried out with the Applied Biosystems 7500 Fast Real-Time PCR System. Running conditions for all probes were 50 °C for 2 min and 95 °C for 10 min followed by 40 cycles of 95 °C for 15 s and 60 °C for 1 min. Data were analyzed using 7500 Software Suite (v2.0). RNA isolated from cortical lysates was used as a positive control for receptor expression. The relative expression of each receptor normalized to its expression in the cortex of PBS-injected mice was calculated using the 2−ΔΔCt method that compares the amplification cycle numbers at which a threshold fluorescence was reached between a sample of interest and a control sample (30). To activate primary microglia in vitro, the cells were treated with 100 ng/ml LPS (Sigma, catalog number L2654) for 24 h. In vivo activation was achieved by injecting mice intraperitoneally with 2 mg/kg LPS (Sigma, catalog number L2143) or vehicle (PBS) 2 days prior to tissue isolation.
Live Cell Ca2+ Imaging
Calcium imaging of primary wild type microglia loaded with 5 μm Fura-2 AM (Invitrogen) was performed in a manner similar to that described for primary astrocytes (31). Microglia were treated with Hank's balanced salt solution (HBSS; control) to preserve their resting phenotype or activated with 100 ng/ml LPS for 24 h before imaging. Imaging was performed at room temperature (23 °C) with dual excitation at 340- and 380-nm wavelengths and emission from both at 510 nm. Images were captured with a MicroMax camera (Princeton Scientific Instruments). The emission signals from each excitation (Ex) wavelength were used for ratio calculations (Ex 340/Ex 380) using Imaging Workbench software (Axon Instruments). The imaging protocol consisted of baseline reading for 60 s, application of 30 μm NE for 60 s, a wash for 120 s, application of 30 μm ATP for 60 s (positive control), and a final wash for 120 s. To facilitate comparisons between cells with different background levels of free calcium, all calculated ratios were normalized to the baseline reading according to the following formula.
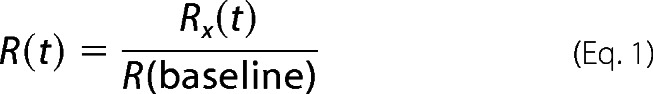 |
where R(t) is the normalized ratio, Rx(t) is the raw ratio at each time point, and R(baseline) is the average of the raw ratios in the first 60 s before cells were stimulated with NE.
Confocal Imaging and Three-dimensional Reconstructions
Microglial process dynamics were studied in vitro in the gelatinous three-dimensional substrate Matrigel (BD Biosciences) as described previously (26). When necessary, microglia were activated with 100 ng/ml LPS for 24 h. The cells were imaged using an Olympus IX51 inverted confocal microscope equipped with a disc spinning unit under 60× magnification with the IPLab image acquisition software. The imaging protocol included an initial period to establish baseline motility for 5 min, application of specified treatments for 5 min, and a 5-min washout. Optical sections were obtained along the z axis of the cells every 30 s; each section was 1 μm above the previous one for 25–35 sections per cell. The resulting time sequences were analyzed with Imaris software (Bitplane AG). The background was subtracted from all recordings using a 10-μm filter. Three-dimensional reconstructions of the cells were then generated from the z stacks. To improve the quality of the representations, they were subjected to a Gaussian smoothing algorithm with a 0.25-μm filter. The software then calculates various parameters of the reconstructed three-dimensional surface such as surface area and volume by breaking the surface into small triangles (for surface area) or voxels (for volume) and summing them up. We used the surface area-to-volume ratio at each time point to determine changes in cell ramification in response to various treatments. Surface area-to-volume ratio values show a strong correlation with measurements of individual process velocity (26) and the total process length from maximum intensity projections (data not shown). To quantitatively compare different treatments, we used the area under the ramification versus time curves starting at application of treatment (t = 5 min).
Iontophoresis and Microglial Migration
To study microglial chemotaxis, primary resting microglia from actin-eGFP mice were plated on Matrigel. On the next day, the cells were imaged over time. Only a single optical plane through the middle of the cell was recorded. An ATP gradient was generated by ejecting ATP from a micropipette as described previously (26). A micropipette with ∼3-megaohm resistance was filled with 0.5 mm ATP. To prevent ATP leak, a backing current of +700 nA was applied with a current generator (Dagan, model ION-100T). Ejection of ATP was achieved with a current of −1500 nA. Upon ejection, the ATP diffuses away from the pipette, generating a gradient. Cells were constantly perfused with imaging buffer with a flow rate of ∼1 ml/min; for some experiments, 30 μm NE was included in the perfusion solution. The time lapse recordings (5-min baseline and 25-min ATP application) were analyzed with Bitplane Imaris software. Cells were detected using the Spots function and tracked by the software over time using the Autoregressive algorithm. The spots had a minimum diameter of 5 μm to restrict tracking only to the cell body rather than processes. The migratory paths of each cell were visualized as tracks, and the magnitude of the vector displacement was calculated for quantification.
RESULTS
NE Modulates Microglial Process Motility in Tissues
Both in vitro and in vivo observations suggest that the presence of NE influences microglial motility in response to Aβ (19). To better understand the effects of NE on microglial motility in their native environment in real time, we developed a brain slice imaging system. We prepared 200-μm-thick slices from CX3CR1-eGFP mice that exhibit microglia-specific eGFP expression (Fig. 1A) (27). The cutting procedure itself, which likely affects microglia on a transient basis, does not lead to classical microglial activation over the course of several hours as evident by the absence of changes in IL-1β cytokine mRNA expression (Fig. 1A). In contrast, IL-1β induction can be observed in brain lysates following microglial activation in vivo achieved by an injection of the bacterial endotoxin LPS (2 mg/kg intraperitoneally 2 days before slicing; Fig. 1B) (26).
Microglia are highly motile under homeostatic conditions in vivo, constantly extending and retracting the tips of their fine processes with no net change in overall process length and ramification (23, 24). However, in the presence of tissue damage, microglia detect ATP released at the damaged site and extend their processes in response to ATP in vitro and in vivo (23, 26, 32, 33). To examine the effects of ATP on microglia in slices, we optically sectioned the slices over a 30-min time period, which allowed us to capture the complex three-dimensional morphology of microglial processes and their movement over time. Collapsing the 30–50-μm z stack of 1-μm optical sections from each time point into a two-dimensional image produced a time series of images with exceptional resolution of individual microglial processes. The morphology was reminiscent of the cells seen in their native environment in situ (Fig. 1C) (23). At baseline conditions, cells contained long processes with an average length of 10.5 μm. Bath application of 30 μm ATP to acute brain slices induced process extension by cortical microglia (Fig. 1, C and D, and supplemental Movie 1), suggesting that acute slices faithfully reproduce microglial behavior as seen in vivo and in vitro. Surprisingly, perfusion with 30 μm NE slowly induced process retraction as opposed to the extension seen following ATP treatment (Fig. 2, A and B, and supplemental Movie 2).
We quantified changes in microglial ramification in slices by measuring the total process length for each cell over the course of imaging (Fig. 2C). Slices perfused with aCSF maintained most of their processes over the course of the imaging; the process length often appeared to start decreasing after 15 min of imaging, but did not significantly decrease from baseline until the 30-min time point (Fig. 2C). ATP induced an 18% increase in total process length (n = 8 cells), which was significantly different from aCSF treatment after 15 min of imaging (Fig. 2C). In contrast, NE treatment led to a significant decrease in process length by 23% (n = 9 cells; Fig. 2C). It should be noted that changes in process length are likely underestimated because collapsing the stacks to a two-dimensional projection does not allow us to fully capture movement in the vertical direction (data not shown); errors will be the same for both control and experimental conditions and only modestly alter the ratio given that individual processes do not shift angular displacement with treatment. To adjust for the observed decrease in total process length over time (34) and more clearly compare the effects of NE and ATP, we normalized the process length of ATP- and NE-treated slices to aCSF-treated slices at each time point (Fig. 2D). Finally, addition of 0.5 μm tetrodotoxin, a voltage-gated sodium channel blocker, did not prevent the effects of NE (n = 9; data not shown). The inability of tetrodotoxin to block the NE-induced process retraction indicates that NE likely acts directly on microglia rather than through modulation of synaptic transmission and neurotransmitter release.
Noradrenergic Receptor Expression in Microglia Is Dependent on Their Activation Status
Although Heneka et al. (19) showed that β receptor agonists such as isoproterenol mimic the effects of NE on microglial motility in a Boyden chamber migration assay, no data were presented regarding which NE receptor subtypes were expressed on microglia. Thus, we assessed the expression of adrenergic receptors to determine which subtypes might be involved in regulating microglial motility. Because persistent inflammation occurs in neurodegenerative disease and microglial activation is known to affect the expression of genes involved in microglial motility (26), we examined adrenergic receptor expression in LPS-activated microglia. We assessed the expression of noradrenergic receptors at the mRNA level using real time quantitative PCR with RNA isolated from both resting and LPS-activated purified primary microglia. We used whole cortical lysates from control or LPS-injected mice (2 mg/kg intraperitoneally 2 days before slicing) as a positive control for receptor expression, and we were able to detect all receptors. In purified microglia, we detected mRNA for α1A, β1, and β2 receptors in resting microglia (Fig. 3A). However, the signal amplitudes for α1A and β1 receptors were only about 10% of the signals observed in whole cortical lysates. In contrast, the mRNA for β2 receptors was 10 times higher in microglia than in the mixed cell types present in cortical lysates. The expression of all three receptors decreased following LPS activation: α1A and β1 receptors decreased to almost undetectable levels, and β2 receptors decreased to the background expression levels in the cortex. Interestingly, LPS activation strongly induced the expression of α2A receptors in cultured microglia as well as in the cortical lysates (Fig. 3A). Thus, it seems likely that at least part of the increase in cortical expression of α2A receptors induced by LPS can be accounted for by an increase in microglial expression. In summary, resting microglia primarily express the Gαs-coupled β2 receptors, whereas activated microglia primarily express the Gαi-coupled α2A receptors.
To determine whether the low levels of α1 receptor mRNA detected by quantitative PCR lead to expression of functional receptors, we performed live cell calcium imaging of resting and LPS-activated (100 ng/ml for 24 h) primary microglia. Activation of Gαq-coupled receptors leads to an increase in intracellular calcium that can be detected with calcium-sensitive fluorescent dyes such as Fura-2. Application of 30 μm NE elicited a small Fura-2 response in one (of 12) control and two (of 15) LPS-treated microglia (Fig. 3, B and C). As a positive control, treatment with 30 μm ATP resulted in a strong increase of the Fura-2 response over baseline levels in all cells arising from activation of Gαq-coupled purinergic receptors (Fig. 3, B and C). These results show that α1 receptors are not expressed at an appreciable degree by microglia.
Norepinephrine Modulates Microglial Process Dynamics in Vitro
We recently described an assay that allows us to evaluate the motility of isolated primary microglia in vitro (26). Microglial cells from actin-eGFP mice plated on top of the gelatinous substrate Matrigel can enter the gel and assume a three-dimensional morphology; subsequent time lapse confocal imaging allows us to evaluate the behavior of individual cells at high resolution and to use pharmacological interventions to dissect the effects of different signaling pathways on microglial process dynamics. Using this assay, we showed that ATP causes process extension in resting microglia but process retraction in microglia activated with 100 ng/ml LPS for 24 h (26). Considering the unexpected finding that NE caused microglial process retraction in native tissues (Fig. 2), we imaged isolated primary cortical microglia grown in Matrigel in vitro to generate three-dimensional reconstructions of the cells with the assistance of image analysis software (Fig. 4 and supplemental Movie 3). As seen in acute brain slices, bath application of 30 μm NE to resting microglia in Matrigel induced process retraction (Figs. 4A and 5A). This was quantified by calculating the ramification of the cells as surface area-to-volume ratios from the three-dimensional reconstructions (Figs. 4A and 5C). The ability of NE to affect process dynamics in isolated microglia further suggests that the effects seen on microglia in slices are through direct modulation of microglia rather than the release of neuroactive substances.
FIGURE 4.

Imaging of primary microglia in Matrigel in vitro. Three-dimensional reconstructions of primary actin-eGFP microglia plated in Matrigel capture the complex, process-bearing morphology of primary microglia. The figure shows an example of a resting, HBSS-treated microglia (control; A) or a microglia activated with 100 ng/ml LPS (B). Treatment with 30 μm NE induced process retraction in both cases. Changes in the surface area-to-volume ratios (SA/V) correlate to changes in ramification. Scale bars, 10 μm.
FIGURE 5.

Control of microglial process dynamics by adrenergic receptors in vitro. Three-dimensional representations of primary actin-eGFP microglia plated in Matrigel were used to calculate cell ramification as surface area-to-volume ratios at each time point. HBSS-treated (control (Con); A) or LPS-activated microglia (100 ng/ml for 24 h; B) were treated with either 20 μm ATP or 30 μm NE. C, a comparison of the effects of NE and ATP by calculating the area under the ramification curves. The number of cells for each treatment is shown in parentheses. Error bars, S.E.
Despite the change in adrenergic receptor expression following LPS activation (Fig. 3A), NE produced a similar process retraction in activated microglia (Figs. 4B and 5B). The extent of process retraction can be quantitatively assessed by calculating the area under the time-response curves. This analysis showed that there is a similar degree of process retraction (∼20% decrease in ramification) for resting and activated microglia following NE treatment. Moreover, the extent of retraction was the same for NE and ATP in activated microglia (Fig. 5C).
Mechanisms Underlying NE Control of Microglial Process Motility
We next utilized subtype-selective adrenergic receptor agonists to determine whether NE mediates its effects on process dynamics through different receptors under resting and activating conditions. Bath application of the β receptor agonist isoproterenol (10 μm) induced on average a 16.7% decrease in ramification in resting microglia but had no effect on cell ramification in activated microglia (Fig. 6, A and C). This is consistent with the strong expression of β2 receptors in resting microglia and the down-regulation of β2 receptors in LPS-activated microglia (Fig. 3A). In contrast, the α2 receptor-selective agonist UK-14,304 (10 μm) had no effect on the ramification of resting microglia, consistent with the minimal expression of α2 receptors. UK-14,304 decreased ramification on average by 18.7% in activated microglia (Fig. 6, B and C), a result that is consistent with the increased expression of α2-adrenergic receptors seen with quantitative PCR (Fig. 3A).
FIGURE 6.

Mechanisms underlying the NE-regulated changes in microglial process dynamics. Primary actin-eGFP microglia were imaged in Matrigel to calculate cell ramification as surface area-to-volume ratios as described. A–C, effects of adrenergic agonists. Treatment of HBSS- (control (Con)) or LPS-treated (100 ng/ml for 24 h) microglia with the β2 receptor agonist isoproterenol (Iso; 10 μm; A) or the α2A receptor agonist UK-14,304 (UK; 10 μm; B). D–F, effects of adrenergic antagonists. D, treatment of HBSS-treated (control) microglia with 30 μm NE alone or 30 μm NE and the β receptor antagonist propranolol (Pro; 10 μm). E, treatment of LPS-activated (100 ng/ml for 24 h) microglia with 3 μm NE alone or 3 μm NE and the α receptor antagonist phentolamine (Phe; 10 μm). The effects of NE receptor agonists (C) and antagonists (F) were compared by calculating the area under the ramification curves with the number of cells for each treatment shown in parentheses. Error bars, S.E. Statistical analysis was performed using two-way ANOVA and Bonferroni's post hoc test compared with control (C) or NE-treated (F) cells. *, p < 0.05.
Additional support for the involvement of specific adrenergic receptor subtypes in the response to NE for resting and activated microglia was obtained through the use of subtype-selective antagonists. Addition of the β receptor antagonist propranolol (10 μm) prevented the NE-induced process retraction in resting microglia (Fig. 6, D and F), consistent with mediation by β2 receptors, which are highly expressed in resting microglia. Likewise, the α receptor antagonist phentolamine (10 μm) blocked the actions of 3 μm NE in activated microglia (Fig. 6, E and F) as predicted given the strong down-regulation of β2 receptors and up-regulation of α2A receptors by LPS. The lower concentration of NE used with phentolamine (3 versus 30 μm) was necessary to achieve an efficient blockade of NE signaling by the competitive antagonist phentolamine and was selected based on the relative potencies of both agonist and antagonist at the receptor (35, 36).
Adrenergic Receptor Activation Interferes with ATP Responses in Resting Microglia
Because both NE and ATP can affect microglial process dynamics, we next evaluated whether the two signaling pathways interact. Specifically, we wanted to determine whether NE signaling will interfere with the ability of microglia to respond to ATP, which is thought to be an important mediator of the ability of microglia to respond to cell death (23). We examined resting and LPS-activated (100 ng/ml for 24 h) microglia plated on Matrigel. The cells were treated either with 20 μm ATP or with 20 μm ATP in the presence of 30 μm NE. The inclusion of NE abolished the ATP-induced process extension in resting microglia (Fig. 7, A and C). Both ATP and ATP + NE induced process retraction in activated microglia (Fig. 7, B and C). Quantitative analysis of the average areas under the ramification curves showed that the degrees of retraction induced by ATP or ATP + NE in LPS-activated microglia were similar, and there are no additive or synergistic effects (Fig. 7C).
FIGURE 7.

Interaction between purinergic and adrenergic signaling in regulating microglial motility in vitro. Primary actin-eGFP microglia were imaged in Matrigel to calculate cell ramification as surface area-to-volume ratios as described. A–C, HBSS-treated (control; A) or LPS-activated microglia (100 ng/ml for 24 h; B) were treated with either 20 μm ATP or 20 μm ATP + 30 μm NE. C, quantification of cell ramification with the number of cells for each treatment shown in parentheses. Statistical analysis was performed using two-way ANOVA and Bonferroni's post hoc test compared with ATP-treated cells. *, p < 0.05. D–F, migration of primary actin-eGFP microglia to ATP released from a micropipette. The cells were perfused with either imaging buffer (D) or 30 μm NE (E). Paths of cell bodies were tracked with Bitplane Imaris and are shown in light gray; overall displacements are represented by blue (movement to pipette) or red (movement away) arrows. Scale bars, 5 μm. F, movement was quantified by measuring the magnitude of the vector displacement for each cell with positive displacement being movement toward the pipette. The number of cells for each treatment is shown in parentheses. Error bars, S.E. Statistical analysis was performed using Student's t test. *, p < 0.05.
To further confirm the interaction between NE and ATP in resting microglia, we determined whether NE affects microglial chemotaxis to ATP. We examined the motility of primary microglia in Matrigel in response to an ATP gradient. To generate the gradient, we locally applied ATP from a micropipette using iontophoresis; this allowed us to retain the ATP in the pipette by applying a positive current to the negatively charged ATP and release ATP at a specific time by applying negative current. Resting microglia migrated toward the ATP released from the pipette (Fig. 7D). However, if microglia were perfused with 30 μm NE (both during the baseline recording and the ATP application), they displayed reduced or no migration (Fig. 7E), which is reflected in significantly reduced cell displacement from an average of 12.9 to 2.57 μm (Fig. 7F). Together with the ability of NE to prevent ATP-induced process extension in resting microglia, these findings further suggest an interaction between adrenergic and purinergic signaling in microglia.
Finally, we wanted to determine whether the interaction between NE and ATP can be seen for microglia in the native tissues of acute brain slices that have endogenous NE. We hypothesized that the endogenous NE might be interfering with ATP-induced process extension and that blocking NE signaling will enhance the effects of ATP in tissues. Slices were perfused with either aCSF or aCSF containing the β-adrenergic receptor antagonist propranolol (10 μm). After a 5-min baseline recording, we applied 30 μm ATP in aCSF or aCSF + propranolol. As expected, ATP induced an increase in total process length that reached a peak early and then started decreasing (Fig. 8). Application of ATP in the presence of propranolol changed the kinetics of the ATP response such that ATP produced a sustained process extension, and we did not observe a time-associated decrease in ramification for the duration of imaging (Fig. 8). Thus, our results show that NE signaling and ATP signaling interact to modulate microglial motility both in vitro and in tissues.
FIGURE 8.

Interaction between purinergic and adrenergic signaling in regulating microglial motility in tissues. Coronal slices were prepared from CX3CR1-eGFP mice and imaged as described. The total process length of microglial processes in two-dimensional projections following ATP treatment was normalized to the total process length during baseline with the numbers of cells for each treatment shown in parentheses. Error bars, S.E. Statistical analysis was performed using two-way repeated measures ANOVA and Tukey's post hoc test. #, p < 0.05 compared with baseline response; *, p < 0.05 compared with ATP at the corresponding time point. Pro, propranolol.
DISCUSSION
Adrenergic neurons originating in the LC innervate almost all brain regions, and NE released at their terminals regulates both basic (sleep/wake) and higher level (memory/cognition) functions (8). In recent years, the ability of NE to act as a neuromodulator in the context of disease has gained attention. NE possesses anti-inflammatory properties (15–18), and thus loss of NE-expressing neurons may influence Alzheimer and Parkinson disease progression (1, 2, 4, 37).
In this study, we describe a novel neuromodulatory role of NE in microglial motility both in tissue and in vitro. Our data allow us to draw three main conclusions. First, we used confocal microscopy to study microglial motion in living brain tissue and found that NE caused microglial process retraction in the intact tissues of acute brain slices from CX3CR1-eGFP mice that have microglia-specific eGFP expression (Figs. 1 and 2). This result was supported by detailed three-dimensional time lapse imaging of isolated microglia obtained in Matrigel (Fig. 5). Second, through the use of receptor subtype-selective agonists and antagonists, we determined that NE exerted its effects through β2 receptors in resting microglia and through α2A receptors in LPS-activated microglia (Fig. 6). These functional data match the adrenergic receptor expression we observed with real time PCR (Fig. 3). Third, NE co-application to resting microglia blocked ATP-induced process extension and migration in vitro (Fig. 7), and the β2 receptor antagonist propranolol prolonged ATP-induced process extension in tissues (Fig. 8). These findings suggest that adrenergic signaling might modulate or control the ability of microglia to respond to tissue damage and subsequent ATP release in vivo.
Adrenergic Receptor Expression in Microglia
Although there are several reports that describe the functional effects of either α- or β-adrenergic receptor activation in cultured primary microglia (15, 19, 38–40), no data were provided regarding receptor expression. Here we conducted the first comprehensive evaluation of adrenergic receptor expression in primary mouse cortical microglia, examining both resting and LPS-activated microglia. Our findings that mouse cortical microglia primarily express β2 receptors in the resting state and α2A receptors in the activated state (Fig. 3) are consistent with most previous reports of expression of adrenergic receptors in microglia (16, 41, 42).
Despite the ∼10-fold down-regulation, there is detectable mRNA signal for the β2 receptor in activated microglia (Fig. 3A). However, treatment of activated microglia with the β2 agonist isoproterenol did not affect cell ramification in isolated primary cells (Fig. 6, A and C). It is known that β2 receptor expression is controlled at multiple levels, including mRNA translation and protein trafficking (43, 44). Thus, one possible explanation for the lack of functional effects of β2 agonists in activated microglia is the existence of post-translational mechanisms of expression regulation, but their elucidation is beyond the scope of this study.
Microglial Motility in Tissues
In the present study, we used a confocal imaging method to study the motility of eGFP-expressing microglia at high spatial and temporal resolution in tissues of acute brain slices. Although it is possible to image cortical microglia in the brains of alive, anesthetized animals using two-photon imaging (23, 24, 32), this approach requires specialized expertise and equipment and can only be applied to cells near the pial surface. In contrast, slices can be prepared from any region of interest, including deep brain loci such as nuclei in the midbrain that are affected in some neurodegenerative diseases. Although the slicing procedure might transiently affect microglia, our data suggest that the cells still exhibit primarily a “resting” phenotype, which was confirmed by their ability to respond to ATP with process extension (Figs. 1C and 2C) (32, 45). Furthermore, the expression of proinflammatory cytokines such as IL-1β was not induced by the process of slicing as opposed to clear induction following a peripheral LPS injection (2 mg/kg intraperitoneally) 2 days before slicing (Fig. 1B).
Differential Modulation of Microglial Process Dynamics by NE and ATP
Using high resolution, time lapse imaging, we showed that NE is capable of causing process retraction in both resting and LPS-activated primary microglia. The finding that the Gs-coupled β2- and the Gi-coupled α2A-adrenergic receptors modulate microglial motility represents the second pair of Gs/Gi receptors for neurotransmitters (the other being P2Y12/A2A receptors (25, 26)) that control microglial process dynamics (Fig. 9). Microglia express several neurotransmitter receptors (46) that might help them monitor normal brain function as abnormal increases or decreases in neurotransmitter levels might indicate a pathological event that warrants microglial intervention.
FIGURE 9.

A summary of the regulation of microglial motility by adrenergic and purinergic receptors. A, resting microglia express the purinergic P2Y12 and the β2-adrenergic (AR) receptors. Activation of the Gi-coupled P2Y12 receptors (P2Y12-R) by ATP results in process extension and migration to ATP possibly through PKC, β-arrestin-2 (β-arr-2)-mediated activation of ERK1/2, or Rac signaling. In contrast, β2 receptor activation by NE leads to process retraction and block of ATP-induced process extension and migration. Either Gi, Gs, or both types of G proteins might be involved in β2 receptor signaling. B, LPS-activated microglia express the purinergic A2A and the α2A-adrenergic receptors. Adenosine, a breakdown product of ATP, activates A2A receptors (A2A-R) to induce process retraction in a PKA-dependent manner. NE activation of α2A receptors also leads to process retraction. See text for references.
There are both similarities and differences in the manner by which ATP and NE affect microglial motility in resting and activated microglia. Whereas both neurotransmitters activate G protein-coupled receptors and influence cAMP signaling, microglial activation produces a switch from the Gi-coupled P2Y12 receptor to the Gs-coupled adenosine A2A receptor for which the ATP breakdown product adenosine is an agonist (26). By contrast, the adrenergic receptor switches during microglial activation from the Gs-coupled β2 receptors to the Gi-coupled α2A receptors, both of which are activated by the same neurotransmitter norepinephrine (Fig. 9). Furthermore, ATP/adenosine signaling through purinergic receptors in resting and activated microglia produces opposing effects on process dynamics and motility, whereas NE produces the same effect despite different G-protein coupled receptors.
β2 receptors are thought to couple to Gs proteins, and α2A receptors are thought to couple to Gi proteins. It is now clear that the signaling pathways initiated by these two adrenergic receptors are substantially more complex. β2 receptors can switch their coupling from Gs to Gi in a protein kinase A (PKA)-dependent manner (47) or might exist in a Gi-coupled state in the brain (48). Similarly, α2A receptors can activate or inhibit cAMP synthesis depending on agonist concentration and the adenylate cyclase isoform expressed in the particular cell type (49, 50). Activation of one of these non-canonical pathways in microglia might be responsible for the same downstream effect on process dynamics by β2 and α2A receptors. However, although the overall effect of adrenergic receptor activation in microglia appears the same (process retraction), NE is several orders of magnitude more potent at α2A receptors than at β2 receptors (36), suggesting that activated microglia will be more sensitive to any NE present in the extracellular milieu. On the contrary, β2 receptors on resting microglia might be activated only in pathological conditions (e.g. prolonged stress) that lead to release of large amounts of NE.
Considering the divergent effects of ATP and NE on microglia, it is likely that ATP- and NE-induced changes in microglial motility serve different functions. For example, increased extracellular ATP levels are thought to be a sign of cell damage and potentially a signal for assistance to microglia (22); microglia extend their processes to the site of tissue damage or local ATP release without any obvious cell body movement (23). Similarly, normal NE levels might suppress microglial reactivity (in terms of cytokine secretion) and motility; loss of NE as it occurs during neurodegeneration (4) will disinhibit microglia, allowing them to further extend their processes and monitor the cells around them.
It is also worth noting that the three phenotypes that result from process retraction following adenosine A2A or α2A-adrenergic receptor activation in LPS-treated microglia and β2-adrenergic receptor activation in resting microglia are all reminiscent of amoeboid microglia. However, it is likely that the three morphological phenotypes are three different functional states, which is consistent with the idea of microglial morphological and functional diversity (51, 52). We suggest that microglial exposure to various neurotransmitters (ATP and NE) might predispose them to a given functional phenotype that will determine the ultimate response that can be achieved following additional stimuli.
Interaction between ATP and NE Signaling in Microglia
The ability of NE to interfere with ATP signaling in resting microglia (Figs. 7 and 8) might represent an example of heterologous desensitization, a phenomenon in which activation of one receptor prevents or attenuates the signaling through another receptor (53–55). Cross-talk between adrenergic receptors and P2Y12 receptors has not been reported before, and the mechanisms by which it might occur remain unclear. In general, heterologous desensitization typically occurs after activation of protein kinases by one receptor that then phosphorylate and change the function of other receptors (53–55). The kinases most often implicated in this process are PKA, PKC, and to a lesser extent G-protein receptor kinases (53–56). In addition, both β2-adrenergic receptor and P2Y12 receptor can interact with β-arrestins (57, 58), and β-arrestin-2 is known to be involved in microglial chemotaxis to ATP (59). This suggests that the interaction between adrenergic and purinergic signaling might occur at the level of adaptor proteins and possibly affect the cellular localization of the receptors (Fig. 9). The ability of β receptor antagonists to prolong ATP responses in tissues might be a manifestation of their ability to block changes in P2Y12 receptor trafficking. Lastly, although the responses of activated microglia to ATP alone or NE + ATP were not different under the present conditions (Fig. 7B), it is possible that NE alters the subsequent response to ATP but that these effects cannot be detected by the analyses used here. Thus, the mechanisms of heterologous desensitization between adrenergic and purinergic receptors especially in the context of tissue damage and ATP release warrant further investigation.
Our finding that NE prevents ATP-induced process extension and migration (Figs. 7 and 8) suggests that the presence of NE might prevent microglia from efficiently responding to tissue damage and cell death. This could have implications in situations of stress that involve elevated NE levels (6, 7). In such situations, any normal disturbance of tissue integrity in the healthy brain might result in exacerbated tissue damage. Over time, prolonged exposure to stress could lead to accumulation of unresolved or poorly resolved damaging events that could later impair brain function.
Finally, process motility and response of microglia to tissue damage are only two of many functions that microglia perform in the CNS. Microglia are also well known phagocytes and sources of cytokines and other proinflammatory mediators (22, 60–63). Both ATP and NE have been reported to separately modulate these functions (11, 19, 20, 46, 64). Interestingly, NE pretreatment prevents the ATP-induced p38 phosphorylation and tumor necrosis factor-α (TNF-α) release by mouse spinal cord microglia (41), suggesting that adrenergic signaling and purinergic signaling might interact at multiple levels. In vivo, microglia are exposed to a variety of signals at the same time and express various neurotransmitter receptors (46). Thus, it is important to understand how these cells will respond to this complex milieu by examining the interactions among NE, ATP, and other signaling systems.
Supplementary Material
Acknowledgments
We thank Dr. Randy Hall, Dr. John Hepler, Dr. T. J. Murphy, and Dr. David Weinshenker for helpful discussion in preparing the manuscript. We are also thankful to Dr. Weinshenker for providing us with adrenergic primers for real time PCR and Dr. Allan Levey and Zoe White for sharing equipment and instructions for use.
This work was supported, in whole or in part, by National Institutes of Health Grants 1F31NS076215, a NINDS National Research Service Award (to S. G.); 5T32ES12870, an NIEHS toxicology institutional training grant (to S. G.); T32GM008602, a pharmacological sciences institutional training grant (to S. G.); and P50-NS071669, a NINDS internal pilot grant from the Udall Parkinson's Disease Research Center at Emory University (to S. F .T).

This article contains supplemental Movies 1–3.
- LC
- locus coeruleus
- Aβ
- amyloid β peptide
- aCSF
- artificial cerebrospinal fluid
- CX3CR1
- CX3C-type chemokine receptor 1
- eGFP
- enhanced green fluorescent protein
- HBSS
- Hanks' balanced salt solution
- NE
- norepinephrine
- ANOVA
- analysis of variance.
REFERENCES
- 1. Iversen L. L., Rossor M. N., Reynolds G. P., Hills R., Roth M., Mountjoy C. Q., Foote S. L., Morrison J. H., Bloom F. E. (1983) Loss of pigmented dopamine-β-hydroxylase positive cells from locus coeruleus in senile dementia of Alzheimer's type. Neurosci. Lett. 39, 95–100 [DOI] [PubMed] [Google Scholar]
- 2. German D. C., Manaye K. F., White C. L., 3rd, Woodward D. J., McIntire D. D., Smith W. K., Kalaria R. N., Mann D. M. (1992) Disease-specific patterns of locus coeruleus cell loss. Ann. Neurol. 32, 667–676 [DOI] [PubMed] [Google Scholar]
- 3. Leverenz J. B., Miller M. A., Dobie D. J., Peskind E. R., Raskind M. A. (2001) Increased α2-adrenergic receptor binding in locus coeruleus projection areas in dementia with Lewy bodies. Neurobiol. Aging 22, 555–561 [DOI] [PubMed] [Google Scholar]
- 4. Mann D. M., Lincoln J., Yates P. O., Stamp J. E., Toper S. (1980) Changes in the monoamine containing neurones of the human CNS in senile dementia. Br. J. Psychiatry 136, 533–541 [DOI] [PubMed] [Google Scholar]
- 5. Chan-Palay V., Asan E. (1989) Alterations in catecholamine neurons of the locus coeruleus in senile dementia of the Alzheimer type and in Parkinson's disease with and without dementia and depression. J. Comp. Neurol. 287, 373–392 [DOI] [PubMed] [Google Scholar]
- 6. Kvetnansky R., Sabban E. L., Palkovits M. (2009) Catecholaminergic systems in stress: structural and molecular genetic approaches. Physiol. Rev. 89, 535–606 [DOI] [PubMed] [Google Scholar]
- 7. Stanford S. C. (1995) Central noradrenergic neurones and stress. Pharmacol. Ther. 68, 297–342 [DOI] [PubMed] [Google Scholar]
- 8. Berridge C. W., Waterhouse B. D. (2003) The locus coeruleus-noradrenergic system: modulation of behavioral state and state-dependent cognitive processes. Brain Res. Rev. 42, 33–84 [DOI] [PubMed] [Google Scholar]
- 9. Rommelfanger K. S., Edwards G. L., Freeman K. G., Liles L. C., Miller G. W., Weinshenker D. (2007) Norepinephrine loss produces more profound motor deficits than MPTP treatment in mice. Proc. Natl. Acad. Sci. U.S.A. 104, 13804–13809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Fornai F., Alessandrì M. G., Torracca M. T., Bassi L., Corsini G. U. (1997) Effects of noradrenergic lesions on MPTP/MPP+ kinetics and MPTP-induced nigrostriatal dopamine depletions. J. Pharmacol. Exp. Ther. 283, 100–107 [PubMed] [Google Scholar]
- 11. Heneka M. T., Galea E., Gavriluyk V., Dumitrescu-Ozimek L., Daeschner J., O'Banion M. K., Weinberg G., Klockgether T., Feinstein D. L. (2002) Noradrenergic depletion potentiates β-amyloid-induced cortical inflammation: implications for Alzheimer's disease. J. Neurosci. 22, 2434–2442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Rommelfanger K. S., Weinshenker D., Miller G. W. (2004) Reduced MPTP toxicity in noradrenergic transporter knockout mice. J. Neurochem. 91, 1116–1124 [DOI] [PubMed] [Google Scholar]
- 13. Paspalas C. D., Papadopoulos G. C. (1998) Ultrastructural evidence for combined action of noradrenaline and vasoactive intestinal polypeptide upon neurons, astrocytes, and blood vessels of the rat cerebral cortex. Brain Res. Bull. 45, 247–259 [DOI] [PubMed] [Google Scholar]
- 14. Kalaria R. N., Stockmeier C. A., Harik S. I. (1989) Brain microvessels are innervated by locus ceruleus noradrenergic neurons. Neurosci. Lett. 97, 203–208 [DOI] [PubMed] [Google Scholar]
- 15. Färber K., Pannasch U., Kettenmann H. (2005) Dopamine and noradrenaline control distinct functions in rodent microglial cells. Mol. Cell. Neurosci. 29, 128–138 [DOI] [PubMed] [Google Scholar]
- 16. Mori K., Ozaki E., Zhang B., Yang L., Yokoyama A., Takeda I., Maeda N., Sakanaka M., Tanaka J. (2002) Effects of norepinephrine on rat cultured microglial cells that express α1, α2, β1 and β2 adrenergic receptors. Neuropharmacology 43, 1026–1034 [DOI] [PubMed] [Google Scholar]
- 17. Feinstein D. L., Heneka M. T., Gavrilyuk V., Dello Russo C., Weinberg G., Galea E. (2002) Noradrenergic regulation of inflammatory gene expression in brain. Neurochem. Int. 41, 357–365 [DOI] [PubMed] [Google Scholar]
- 18. Loughlin A. J., Woodroofe M. N., Cuzner M. L. (1993) Modulation of interferon-γ-induced major histocompatibility complex class II and Fc receptor expression on isolated microglia by transforming growth factor-β1, interleukin-4, noradrenaline and glucocorticoids. Immunology 79, 125–130 [PMC free article] [PubMed] [Google Scholar]
- 19. Heneka M. T., Nadrigny F., Regen T., Martinez-Hernandez A., Dumitrescu-Ozimek L., Terwel D., Jardanhazi-Kurutz D., Walter J., Kirchhoff F., Hanisch U.-K., Kummer M. P. (2010) Locus ceruleus controls Alzheimer's disease pathology by modulating microglial functions through norepinephrine. Proc. Natl. Acad. Sci. U.S.A. 107, 6058–6063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kong Y., Ruan L., Qian L., Liu X., Le Y. (2010) Norepinephrine promotes microglia to uptake and degrade amyloid β peptide through upregulation of mouse formyl peptide receptor 2 and induction of insulin-degrading enzyme. J. Neurosci. 30, 11848–11857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Jardanhazi-Kurutz D., Kummer M. P., Terwel D., Vogel K., Thiele A., Heneka M. T. (2011) Distinct adrenergic system changes and neuroinflammation in response to induced locus ceruleus degeneration in APP/PS1 transgenic mice. Neuroscience 176, 396–407 [DOI] [PubMed] [Google Scholar]
- 22. Hanisch U.-K., Kettenmann H. (2007) Microglia: active sensor and versatile effector cells in the normal and pathologic brain. Nat. Neurosci. 10, 1387–1394 [DOI] [PubMed] [Google Scholar]
- 23. Davalos D., Grutzendler J., Yang G., Kim J. V., Zuo Y., Jung S., Littman D. R., Dustin M. L., Gan W.-B. (2005) ATP mediates rapid microglial response to local brain injury in vivo. Nat. Neurosci. 8, 752–758 [DOI] [PubMed] [Google Scholar]
- 24. Nimmerjahn A., Kirchhoff F., Helmchen F. (2005) Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 308, 1314–1318 [DOI] [PubMed] [Google Scholar]
- 25. Gyoneva S., Orr A. G., Traynelis S. F. (2009) Differential regulation of microglial motility by ATP/ADP and adenosine. Parkinsonism Relat. Disord. 15, Suppl. S3, S195–S199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Orr A. G., Orr A. L., Li X.-J., Gross R. E., Traynelis S. F. (2009) Adenosine A2A receptor mediates microglial process retraction. Nat. Neurosci. 12, 872–878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Jung S., Aliberti J., Graemmel P., Sunshine M. J., Kreutzberg G. W., Sher A., Littman D. R. (2000) Analysis of fractalkine receptor CX3CR1 function by targeted deletion and green fluorescent protein reporter gene insertion. Mol. Cell. Biol. 20, 4106–4114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bianco F., Fumagalli M., Pravettoni E., D'Ambrosi N., Volonte C., Matteoli M., Abbracchio M. P., Verderio C. (2005) Pathophysiological roles of extracellular nucleotides in glial cells: differential expression of purinergic receptors in resting and activated microglia. Brain Res. Rev. 48, 144–156 [DOI] [PubMed] [Google Scholar]
- 29. Tha K. K., Okuma Y., Miyazaki H., Murayama T., Uehara T., Hatakeyama R., Hayashi Y., Nomura Y. (2000) Changes in expressions of proinflammatory cytokines IL-1β, TNF-α and IL-6 in the brain of senescence accelerated mouse (SAM) P8. Brain Res. 885, 25–31 [DOI] [PubMed] [Google Scholar]
- 30. Livak K. J., Schmittgen T. D. (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 25, 402–408 [DOI] [PubMed] [Google Scholar]
- 31. Lee C. J., Mannaioni G., Yuan H., Woo D. H., Gingrich M. B., Traynelis S. F. (2007) Astrocytic control of synaptic NMDA receptors. J. Physiol. 581, 1057–1081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Haynes S. E., Hollopeter G., Yang G., Kurpius D., Dailey M. E., Gan W.-B., Julius D. (2006) The P2Y12 receptor regulates microglial activation by extracellular nucleotides. Nat. Neurosci. 9, 1512–1519 [DOI] [PubMed] [Google Scholar]
- 33. Honda S., Sasaki Y., Ohsawa K., Imai Y., Nakamura Y., Inoue K., Kohsaka S. (2001) Extracellular ATP or ADP induce chemotaxis of cultured microglia through Gi/o-coupled P2Y receptors. J. Neurosci. 21, 1975–1982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Stence N., Waite M., Dailey M. E. (2001) Dynamics of microglial activation: a confocal time-lapse analysis in hippocampal slices. Glia 33, 256–266 [PubMed] [Google Scholar]
- 35. Atkinson B. N., Minneman K. P. (1991) Multiple adrenergic receptor subtypes controlling cyclic AMP formation: comparison of brain slices and primary neuronal and glial cultures. J. Neurochem. 56, 587–595 [DOI] [PubMed] [Google Scholar]
- 36. Zhang W.-P., Ouyang M., Thomas S. A. (2004) Potency of catecholamines and other l-tyrosine derivatives at the cloned mouse adrenergic receptors. Neuropharmacology 47, 438–449 [DOI] [PubMed] [Google Scholar]
- 37. Rommelfanger K. S., Weinshenker D. (2007) Norepinephrine: the redheaded stepchild of Parkinson's disease. Biochem. Pharmacol. 74, 177–190 [DOI] [PubMed] [Google Scholar]
- 38. Colton C. A., Chernyshev O. N. (1996) Inhibition of microglial superoxide anion production by isoproterenol and dexamethasone. Neurochem. Int. 29, 43–53 [DOI] [PubMed] [Google Scholar]
- 39. Fujita H., Tanaka J., Maeda N., Sakanaka M. (1998) Adrenergic agonists suppress the proliferation of microglia through β2-agrenergic receptor. Neurosci. Lett. 242, 37–40 [DOI] [PubMed] [Google Scholar]
- 40. Prinz M., Häusler K. G., Kettenmann H., Hanisch U. (2001) β-Adrenergic receptor stimulation selectively inhibits IL-12p40 release in microglia. Brain Res. 899, 264–270 [DOI] [PubMed] [Google Scholar]
- 41. Morioka N., Tanabe H., Inoue A., Dohi T., Nakata Y. (2009) Noradrenaline reduces the ATP-stimulated phosphorylation of p38 MAP kinase via β-adrenergic receptors-cAMP-protein kinase A-dependent mechanisms in cultured rat spinal microglia. Neurochem. Int. 55, 226–234 [DOI] [PubMed] [Google Scholar]
- 42. Tanaka K. F., Kashima H., Suzuki H., Ono K., Sawada M. (2002) Existence of functional β1- and β2-adrenergic receptors on microglia. J. Neurosci. Res. 70, 232–237 [DOI] [PubMed] [Google Scholar]
- 43. Kandasamy K., Joseph K., Subramaniam K., Raymond J. R., Tholanikunnel B. G. (2005) Translational control of β2-adrenergic receptor mRNA by T-cell-restricted intracellular antigen-related protein. J. Biol. Chem. 280, 1931–1943 [DOI] [PubMed] [Google Scholar]
- 44. Tholanikunnel B. G., Joseph K., Kandasamy K., Baldys A., Raymond J. R., Luttrell L. M., McDermott P. J., Fernandes D. J. (2010) Novel mechanisms in the regulation of G protein-coupled receptor trafficking to the plasma membrane. J. Biol. Chem. 285, 33816–33825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Avignone E., Ulmann L., Levavasseur F., Rassendren F., Audinat E. (2008) Status epilepticus induces a particular microglial activation state characterized by enhanced purinergic signaling. J. Neurosci. 28, 9133–9144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Pocock J. M., Kettenmann H. (2007) Neurotransmitter receptors on microglia. Trends Neurosci. 30, 527–535 [DOI] [PubMed] [Google Scholar]
- 47. Daaka Y., Luttrell L. M., Lefkowitz R. J. (1997) Switching of the coupling of the β2-adrenergic receptor to different G proteins by protein kinase A. Nature 390, 88–91 [DOI] [PubMed] [Google Scholar]
- 48. Schutsky K., Ouyang M., Castelino C. B., Zhang L., Thomas S. A. (2011) Stress and glucocorticoids impair memory retrieval via β2-agrenergic, Gi/o-coupled suppression of cAMP signaling. J. Neurosci. 31, 14172–14181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Federman A. D., Conklin B. R., Schrader K. A., Reed R. R., Bourne H. R. (1992) Hormonal stimulation of adenylyl cyclase through Gi-protein βγ subunits. Nature 356, 159–161 [DOI] [PubMed] [Google Scholar]
- 50. Eason M. G., Kurose H., Holt B. D., Raymond J. R., Liggett S. B. (1992) Simultaneous coupling of α2-adrenergic receptors to two G-proteins with opposing effects. J. Biol. Chem. 267, 15795–15801 [PubMed] [Google Scholar]
- 51. Colton C., Wilcock D. M. (2010) Assessing activation states in microglia. CNS Neurol. Disord. Drug Targets 9, 174–191 [DOI] [PubMed] [Google Scholar]
- 52. Scheffel J., Regen T., Van Rossum D., Seifert S., Ribes S., Nau R., Parsa R., Harris R. A., Boddeke H. W., Chuang H.-N., Pukrop T., Wessels J. T., Jürgens T., Merkler D., Brück W., Schnaars M., Simons M., Kettenmann H., Hanisch U.-K. (2012) Toll-like receptor activation reveals developmental reorganization and unmasks responder subsets of microglia. Glia 60, 1930–1943 [DOI] [PubMed] [Google Scholar]
- 53. Vázquez-Prado J., Casas-González P., García-Sáinz J. A. (2003) G protein-coupled receptor cross-talk: pivotal roles of protein phosphorylation and protein-protein interactions. Cell. Signal. 15, 549–557 [DOI] [PubMed] [Google Scholar]
- 54. Pierce K. L., Premont R. T., Lefkowitz R. J. (2002) Seven-transmembrane receptors. Nat. Rev. Mol. Cell Biol. 3, 639–650 [DOI] [PubMed] [Google Scholar]
- 55. Lohse M. J. (1993) Molecular mechanisms of membrane receptor desensitization. Biochim. Biophys. Acta 1179, 171–188 [DOI] [PubMed] [Google Scholar]
- 56. Böhm S. K., Grady E. F., Bunnett N. W. (1997) Regulatory mechanisms that modulate signalling by G-protein-coupled receptors. Biochem. J. 322, 1–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Goodman O. B., Jr., Krupnick J. G., Santini F., Gurevich V. V., Penn R. B., Gagnon A. W., Keen J. H., Benovic J. L. (1996) β-Arrestin acts as a clathrin adaptor in endocytosis of the β2-adrenergic receptor. Nature 383, 447–450 [DOI] [PubMed] [Google Scholar]
- 58. Li D., D'Angelo L., Chavez M., Woulfe D. S. (2011) Arrestin-2 differentially regulates PAR4 and ADP receptor signaling in platelets. J. Biol. Chem. 286, 3805–3814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Lee S.-H., Hollingsworth R., Kwon H.-Y., Lee N., Chung C. Y. (2012) β-Arrestin 2-dependent activation of ERK1/2 is required for ADP-induced paxillin phosphorylation at Ser83 and microglia chemotaxis. Glia 60, 1366–1377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Hanisch U.-K. (2002) Microglia as a source and target of cytokines. Glia 40, 140–155 [DOI] [PubMed] [Google Scholar]
- 61. Kim S. U., de Vellis J. (2005) Microglia in health and disease. J. Neurosci. Res. 81, 302–313 [DOI] [PubMed] [Google Scholar]
- 62. Smith J. A., Das A., Ray S. K., Banik N. L. (2012) Role of pro-inflammatory cytokines released from microglia in neurodegenerative diseases. Brain Res. Bull. 87, 10–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Napoli I., Neumann H. (2009) Microglial clearance functions in health and disease. Neuroscience 158, 1030–1038 [DOI] [PubMed] [Google Scholar]
- 64. Sperlágh B., Illes P. (2007) Purinergic modulation of microglial cell activation. Purinergic Signal. 3, 117–127 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.