

Background: The pyruvate dehydrogenase complex produces acetyl-CoA and NADH, utilizing three protein components whose structural interactions need elucidation.
Results: The E3 structure and interaction loci between E2 and E3 were determined.
Conclusion: The peripheral subunit-binding domain of E2 establishes key interactions with E1 and E3.
Significance: The multifaceted approach used could help delineate interaction surfaces in other 2-oxoacid dehydrogenase complexes.
Keywords: Crystallography, Enzyme Structure, NMR, Protein-Protein Interactions, Pyruvate Dehydrogenase Complex, H/D Exchange MS, Lipoamide Dehydrogenase
Abstract
Multifaceted structural approaches were undertaken to investigate interaction of the E2 component with E3 and E1 components from the Escherichia coli pyruvate dehydrogenase multienzyme complex (PDHc), as a representative of the PDHc from Gram-negative bacteria. The crystal structure of E3 at 2.5 Å resolution reveals similarity to other E3 structures and was an important starting point for understanding interaction surfaces between E3 and E2. Biochemical studies revealed that R129E-E2 and R150E-E2 substitutions in the peripheral subunit-binding domain (PSBD) of E2 greatly diminished PDHc activity, affected interactions with E3 and E1 components, and affected reductive acetylation of E2. Because crystal structures are unavailable for any complete E2-containing complexes, peptide-specific hydrogen/deuterium exchange mass spectrometry was used to identify loci of interactions between 3-lipoyl E2 and E3. Two peptides from the PSBD, including Arg-129, and three peptides from E3 displayed statistically significant reductions in deuterium uptake resulting from interaction between E3 and E2. Of the peptides identified on E3, two were from the catalytic site, and the third was from the interface domain, which for all known E3 structures is believed to interact with the PSBD. NMR clearly demonstrates that there is no change in the lipoyl domain structure on complexation with E3. This is the first instance where the entire wild-type E2 component was employed to understand interactions with E3. A model for PSBD-E3 binding was independently constructed and found to be consistent with the importance of Arg-129, as well as revealing other electrostatic interactions likely stabilizing this complex.
Introduction
Multienzyme complexes catalyze successive steps of a multistep chemical reaction (1). One of the best characterized of the 2-oxoacid dehydrogenase multienzyme complexes (2) is the pyruvate dehydrogenase complex (PDHc)4 from Escherichia coli, with multiple copies of each of the following three enzymatic components: the thiamin diphosphate-dependent pyruvate dehydrogenase (E1, EC 1.2.4.1, mass of 99,474 Da); dihydrolipoamide acetyltransferase (E2, EC 2.3.1.12, mass of 65,959 Da) containing 1–3 covalently bonded lipoyl groups (lipoamides); and dihydrolipoamide dehydrogenase (E3, EC 1.8.1.4, mass of 50,554 Da) binding FAD and NAD+. E2 components form the core of the complex, to which E1 and E3 components bind noncovalently. The key metabolic product acetyl coenzyme A is generated on the E2 components. The E. coli E2 has a multidomain structure; proceeding from the N terminus, it consists of three lipoyl domains (3-lip E2, ∼80 amino acids each), a peripheral subunit-binding domain (PSBD, ∼45 amino acids), and a large core-forming acetyltransferase domain (∼250 amino acids), all linked together by flexible 25–30-residue segments rich in alanine, proline, and charged amino acids (3, 4). To simplify some of our studies with the E2 component, a 1-lip E2, bearing a single hybrid domain, was constructed, which behaves similarly to 3-lip E2 in every respect (5).
The E3 components of the complex catalyze re-oxidation of dihydrolipoyl groups (created during the overall reaction) attached to lysine residues on the E2 lipoyl domains, after the acetyl group is transferred to coenzyme A. This restores the initial redox states of the lipoamides allowing the multienzyme complex to cycle, and it reduces NAD+ to NADH in the process. Unlike in higher eukaryotes, where there is an E1 recognition site on E2 and an E3 recognition site via an E3-binding protein (6–8), in E. coli the PSBD of E2 binds both E1 and E3 (9). Structural, spectroscopic, biochemical, and kinetic studies on the E. coli PDHc have formed a core project in our laboratories for years. X-ray structural work on the E1 component as well as its variants, a complex of E1 with the inhibitor thiamin thiazolone diphosphate, a complex with phosphonolactylthiamin diphosphate (an analog of the pre-decarboxylation intermediate), and the E1 apoenzyme free of thiamin diphosphate (10–15) have yielded valuable information on the basic folding, conformation, and function of homodimeric (α2) E1. They also provided information regarding the role of water in enzyme inhibition and catalysis, large scale disorder-to-order transitions, disorders in specific amino acids in the active site area, differences in the extent of hydrogen bonding networks, and insight into a possible proton wire hypothesis facilitating communication between remote active sites. In this work, directed mutagenesis on the E. coli E2 followed by size-exclusion chromatography identifies some residues that are involved in the interaction with E1. Furthermore, a possible E3-PSBD binding mode is suggested based on available structure and sequence information. It is noteworthy that the identical E3 protein examined in these studies on PDHc is also utilized in the α-ketoglutarate dehydrogenase complex and the glycine cleavage system in E. coli (16–19); thus, the crystal structure reported here will be important in the understanding of those complex functions as well, once structural information becomes available for all their components.
E3 has also been found in organisms that do not have 2-oxoacid dehydrogenases, leading to suggestions that the enzyme may also function outside the framework of the multienzyme complexes (20, 21). The tertiary structures of several E3s and glutathione reductase, a related oxidoreductase, have been elucidated through x-ray studies. A list of these enzymes chosen for comparison with E. coli E3 is given in Table 1, along with their PDB codes and literature references (9, 22–34). In subsequent discussion, these enzymes are identified in the text by their PDB codes for brevity. All E3 enzymes function as dimers and contain in their active center a reactive disulfide bridge directly involved in catalysis. They share a common prosthetic group (FAD) and secondary substrate (NAD+ or NADP+), but the sequence identity between E3 proteins from various sources is moderate, between 28 and 47% (Table 1) (35, 36), except for E3 from Neisseria meningitidis (64%); their chain lengths also differ by up to about 30 amino acid residues.
TABLE 1.
Crystal structures of glutathione reductase and lipoamide dehydrogenases selected for comparison with E. coli lipoamide dehydrogenase E3
| Identification | SIDa | PDB code | Resolution | Citation |
|---|---|---|---|---|
| % | Å | |||
| E. coli glutathione reductase | 28 | 1GER | 1.86 | Mittl and Schulz (22) |
| Azobacter vinelandii | 40 | 3LAD | 2.20 | Mattevi et al. (23) |
| Pseudomonas fluorescens | 43 | 1LPF | 2.80 | Mattevi et al. (24) |
| N. meningitides | 64 | 1OJT | 2.75 | de la Sierra et al. (25) |
| Yeast | 41 | 1JEH | 2.40 | Toyoda et al. (9) |
| Pisum sativum | 49 | 1DXL | 3.15 | Faure et al. (26) |
| Mycobacterium tuberculosis | 38 | 2A8X | 2.40 | Rajashankar et al. (27) |
| P. putida; complexed with NAD+ | 38 | 1LVL | 2.45 | Mattevi et al. (28) |
| Human; complexed with NAD+ | 43 | 1ZMC | 2.53 | Brautigam et al. (29) |
| Yeast; complexed with NAD+ | 41 | 1V59 | 2.20 | –b |
| B. stearothermophilus; complexed with acetyltransferase-binding domain | 47 | 1EBD | 2.60 | Mande et al. (30) |
| Human; complexed with E3BP | 44 | 1ZY8 | 2.59 | Ciszak et al. (31) |
| Human; complexed with E3BP | 43 | 2F5Z | 2.18 | Brautigam et al. (32) |
| T. thermophilus; complexed with PSBD | 44 | 2EQ8 | 1.94 | Nakai et al. (33) |
| Human; complexed with subunit binding domain | 44 | 3RNM | 2.40 | Brautigam et al. (34) |
a SID is Sequence Identity with E. coli E3, determined by protein BLAST sequence alignment (37).
b Adachi, W., Suzuki, K., Tsunoda, M., Sekiguchi, T., Reed, L. J., and Takenaka, A. (2003) RCSB Protein Data Bank.
The goal of this study was to identify the loci of interaction of the E2 component with E3 on the basis of the x-ray structure of E3, H/D exchange MS experiments, NMR protein-protein interaction studies, and the kinetics of E2 variants created by substitutions on the PSBD at amino acid residues hypothesized to be involved in inter-component interactions. Indeed, peptides from both the E2 PSBD and E3 were identified that support the stabilization of the E2-E3 subcomplex, consistent with a model independently constructed. Our results clearly demonstrate the need for such a multifaceted approach to understand these important issues in the absence of high resolution structural data on the intact subcomplexes.
EXPERIMENTAL PROCEDURES
Construction of Singly Substituted Variants of E2p
The pET-22b(+)-1-lip E2 plasmid encoding E. coli E2p and two synthetic oligonucleotide primers complementary to opposite strands of DNA were used for site-directed mutagenesis according to the protocol supplied with the QuikChange site-directed mutagenesis kit from Stratagene (La Jolla, CA) for 16 cycles. The presence of substitutions was confirmed by DNA sequencing at the University of Medicine and Dentistry of New Jersey, Newark.
Expression and Purification of E1p
E1p was expressed and purified as described previously (38).
Expression and Purification of 1-lip E2p
On the basis of our past observations that the kinetic behavior of 1-lip PDHc was very similar to that of 3-lip PDHc (39), the 1-lip E2p and its truncated version of the E2p didomain were created as reported earlier (5). The lipoyl domain in 1-lip E2p is a hybrid wherein the 85-amino acid residues starting from the N terminus include residues 1–33 from the N-terminal end of the first lipoyl domain and residues 238–289 from the C-terminal end of the third lipoyl domain of the wild-type 3-lip E2p component (40). The expression and purification of 1-lip E2p and 1-lip E2p variants followed the general procedure reported earlier from our laboratory (5) with modifications (41, 42).
Construction of Plasmid, Expression and Purification of 1-lip E2p Didomain
The 1-lip E2p didomain comprising residues 1–190 from 1-lip E2p corresponding to the hybrid lipoyl domain and the peripheral subunit-binding domain (PSBD) with linkers connecting them was constructed as reported earlier (41).
Preparation of 15N- and 13C-Labeled 1-lip E2p Didomain for NMR Studies
For the production of the doubly (15N and 13C) labeled E. coli PDHc E2p didomain with and without the N-terminal His6 tag (removed through a thrombin-cleavable site), minimal medium supplemented with 15NH4Cl (1.0 g/liter) as the sole source of nitrogen, [U-13C]glucose (4.0 g/liter) as the sole source of carbon, and 0.3 mm dl-lipoic acid was used. The cells were grown at 37 °C, and expression was induced with 1 mm IPTG at A600 = 0.8. Cells were grown for 6 h at 37 °C. Purification of the His6 tag E2p didomain was carried out by nickel affinity column method. Thrombin (10 units/mg protein) cleavage was carried out in 20 mm KH2PO4 (pH 7.0) with 0.15 m NaCl at 22 °C for 16–20 h, and the protein was then purified by anion exchange and size-exclusion chromatography.
Expression and Purification of 3-lip E2p
Wild-type 3-lip E2p was expressed and purified in a manner similar to 1-lip E2p. The pCA24N plasmid encoding 3-lip E2p was obtained from the National BioResource Project, National Institute of Genetics, Japan. The AG1 cells were grown at 37 °C in LB medium supplemented with 30 μg/ml chloramphenicol and 0.30 mm lipoic acid. The expression of 3-lip E2p was induced by IPTG (0.5 mm) for 4 h at 37 °C. The purification protocol for 3-lip E2p was similar to that presented above for 1-lip E2p.
Expression and Purification of E3
E. coli BL21(DE3) cells (Novagene, Madison, WI) transformed with the pET-22b(+)-E3 plasmid encoding E3 were grown on an LB plate supplemented with 50 μg/ml ampicillin for 16 h (42). A single colony was used to inoculate 500 ml of LB medium supplemented with 50 μg/ml ampicillin. Cells were grown at 37 °C to A650 = 0.4–0.6, and E3 expression was induced by 1 mm IPTG for 3–4 h. Cells were collected, washed with 20 mm KH2PO4 (pH 8.0) containing 1 mm EDTA, and stored at −20 °C. The E3 was purified using a phenyl-Sepharose high performance hydrophobic column (GE Healthcare) pre-equilibrated with 50 mm KH2PO4 (pH 8.0) containing 1 mm EDTA and 40% ammonium sulfate. The E3 was eluted at 15% saturation of ammonium sulfate when a decreasing gradient of ammonium sulfate was applied. Fractions containing E3 were analyzed by SDS-PAGE and were dialyzed against 50 mm KH2PO4 (pH 8.0) containing 1 mm EDTA. The E3 was stored at −20 °C.
Activity Measurements
The activities of 1-lip E2p and its variants were measured after reconstitution of the PDH complex from its individual E1p, E2p, and E3 components using a E1/E2/E3 mass ratio of 1:1:0.5 (mg/mg/mg). Pyruvate-dependent reduction of NAD+ was monitored at 340 nm as reported earlier (43).
Size-exclusion Chromatography Experiments to Study Interaction between Binary Component Subcomplexes
A TSK G4000-SW size-exclusion column equilibrated with 50 mm KH2PO4 (pH 7.0) containing 0.5 m NaCl was used. The column was calibrated with the following standards (Mr values listed): thyroglobulin (669,000), ferritin (440,000), catalase (232,000), aldolase (158,000), bovine serum albumin (67,000), ovalbumin (43,000), chymotrypsinogen (25,000), and ribonuclease A (13,700). E2p (1 mg) or its variants (1 mg) were incubated with E1p (1 mg) or E3 (1 mg) in 50 mm KH2PO4 (pH 7.0) containing 0.5 m NaCl in a total volume of 0.20 ml at 25 °C.
Reductive Acetylation of E2p and Its Variants by E1p and Pyruvate as Monitored by MALDI-TOF Mass Spectrometry
E2p or its variants (2 mg/ml) were incubated with E1p (0.1 mm) and pyruvate (2.0 mm) in Tris-HCl (pH 8.0) containing 0.3 m NaCl, 2 mm MgCl2, and 0.2 mm thiamin diphosphate in a total volume of 0.1 ml at 25 °C. After 1 min of incubation, trypsin was added to digest E2p or its variants (mass ratio of trypsin/E2p = 200:1). After 30 min of incubation with trypsin, the reaction was stopped by the addition of benzamidine hydrochloride (0.15 mg/ml), and aliquots were diluted 10-fold with 0.1% trifluoroacetic acid. Samples (1 ml) were withdrawn and mixed with 1 ml of sinapinic acid on the target plate.
X-ray Structure Determination of the E. coli E3
The E. coli E3 enzyme was concentrated to 13.5 mg/ml in 10 mm HEPES (pH 7.0), containing 0.5 mm EDTA, 0.2% sodium azide, and 1 μm leupeptin. The protein was crystallized at 18 °C by the sitting drop vapor diffusion method by mixing equal volumes of protein and precipitant solutions. The best crystals were obtained with a precipitant solution containing 2.4 m ammonium sulfate and 0.1 m MES buffer at pH 6.4. Crystal growth took 4–6 weeks, and the crystals were typically 0.6 × 0.5 × 0.1 mm.
Data were collected at −180 °C at the Advanced Photon Source (SE Regional Collaborative Access Team, sector 22-BM). Although diffraction initially extended to 2.2 Å resolution, the data were truncated to 2.5 Å due to rapid decay that resulted in rather high merging residuals and very low I/σ values at the high resolution limits. The crystals are tetragonal with a = 180.0 and c = 196.9 Å in space group I422. The data were processed with the d*TREK package (44). The Matthews coefficient (45) Vm is 3.94 Å3/Da with one E3 dimer per asymmetric unit, yielding a solvent content of 68.8%.
For initial structure determination, data to 3 Å were used to derive phases by molecular replacement using the program PHASER (46). The search model was derived from the structure of the E3 domain in the protein p64k of a surface antigen from Neisseria meningitidis (PDB code 1OJT (25)). The model (polyalanine) contained 482 residues that originally had the highest sequence homology with the E. coli E3 protein (64% identity). Residues in the region 135–594 of the N. meningitidis E3 domain generally matched the sequence of E. coli E3 residues in the region 19–467, and the results clearly indicated two E3 monomers per asymmetric unit.
After sequence adjustment where needed, refinement was performed with PHENIX (47), and no noncrystallographic symmetry restraints were applied during the initial stages. The polypeptide chain was subsequently extended to residue 2 in both monomers, and all-atom rigid body refinement and simulated annealing followed. A simulated annealing composite omit map calculated at this stage clearly revealed the two FAD molecules (one associated with each protein subunit), which were then included. The entire structure was analyzed with the graphics program COOT (48), and several residues were rebuilt. After further refinement with adjustments of atomic positions and individual B values, solvent molecules were located using a (Fo − Fc) difference map and a minimum electron density level of 2.8σ. In addition to 417 water molecules, several large blobs of density were located that could be identified as MES buffer molecules or SO42− ions. For subsequent refinement, the two independent protein subunits A and B, along with their associated FAD cofactors, were defined as separate translation/libration/screw groups; the rest of the atoms were treated isotropically, and noncrystallographic symmetry restraints between molecules were included. On least squares superposition of the Cα atoms from protein subunits A and B, an r.m.s. difference of 0.06 Å was observed. The average B value for the structure was fairly high, 64.9 Å2, perhaps indicating a general static disorder. The structure was checked for possible side chain flips and unfavorable contacts with the program MOLPROBITY (49). Residues that had to be fine-tuned were then regularized to ensure proper geometry. Crystallographic data and final refinement statistics are detailed in Table 2. Graphical representations of protein molecules were prepared with the program RIBBONS (51).
TABLE 2.
Crystallographic data and refinement statistics for E. coli lipoamide dehydrogenase (E3)
Values in parentheses are for the last shell.
| Data statistics | |
| Space group | I422 |
| Unit-cell parameters | a, b = 180.0 Å and c = 196.9 Å |
| Diffraction limit | 2.50 Å |
| Completeness | 94.4% (90.3%) |
| Total no. of reflections | 407,934 |
| No. of unique reflections | 52,714 |
| Rmerge (on I) | 0.115 (0.561) |
| X-ray source | Synchrotron (APS, sector 22BM) |
| Wavelength | 1.00 Å |
| Refinement statistics | |
| Resolution range | 35.3 to 2.50 Å |
| No. of reflections | 52,714 |
| No. of reflections for test set | 2687 |
| R factor | 0.214 (0.411) |
| Rfree | 0.256 (0.416) |
| No. of residues | 942 |
| No. of protein atoms | 7076 |
| No. of cofactor (FAD) atoms | 106 |
| No. of solvent atoms | 417 |
| Average B value | |
| All atoms | 64.9 Å2 |
| Protein atoms | 64.5 Å2 |
| Cofactor (FAD) atoms | 53.9 Å2 |
| Solvent, MES, and SO4 atoms | 72.8 Å2 |
| Estimated coordinate errora | 0.29 Å |
| r.m.s deviations | |
| Bond lengths | 0.010 Å |
| Bond angles | 1.281° |
| Molprobity results | |
| Clashscore | 16.15 (83rd percentile) |
| Molprobity score | 2.01 (95th percentile) |
a Data are from a Luzzati plot (50).
NMR Spectroscopy
NMR experiments were carried out using a 0.7 mm solution of the uniformly 13C,15N-labeled E2p didomain in 20 mm phosphate (pH 7.0) with 150 mm NaCl. All NMR experiments for backbone sequence-specific resonance assignments were run at 25 °C on a Varian INOVA 600 MHz spectrometer equipped with 5-mm HCN probes. Chemical shift assignments for the E2p didomain were determined using conventional triple resonance NMR methods from the Varian standard Biopack pulse sequence library. Spectra were processed with NMR DRAW 5.96 (52) and transferred to CARA 1.9.0b3 (53) for further analysis. HNCA, HN(CO)CACB, HNCACB, and CBCA(CO)NH experiments were used to link sequential amide groups via matching pairs of Cα/Cβ peaks (54). To study E2 didomain-E3 interactions by chemical shift mapping methodology, 0.3 mm 15N-labeled E2-didomain was mixed with 0.3 mm unlabeled E3 at pH 7.0, and two-dimensional 1H-15N-TROSY HSQC spectra were acquired.
H/D Exchange by Mass Spectrometry
For the H/D exchange studies, the wild-type 3-lipoyl E2 (3-lip E2) component was used. The H/D exchange analysis was conducted on an HPLC system (1200 series HPLC, Agilent) interfaced to a Fourier transform-mass spectrometer (Apex-ultra 70 hybrid Fourier transform-mass spectrometer, Bruker Daltonics), equipped with an in-house built refrigeration system, constructed to be similar to the one described by Keppel et al. (55).
Prior to H/D exchange, E2p and E3 proteins were exchanged into 10 mm potassium phosphate (pH 7.0) with 50 mm KCl. Three samples were prepared as follows: the first sample was 80 μm E2p by itself, the second was 80 μm E3 by itself, and the third contained 80 μm each of E2p and E3. These samples were incubated at 20 °C for at least 1 h. The H/D exchange reactions were initiated by dilution of 15 μl of the previous samples into 285 μl of 10 mm potassium phosphate, 50 mm potassium chloride in 99.9% D2O (pH 7.0) at 20 °C. After incubation at 20 °C for various lengths of time (20 s and 1, 3, 10, 30, 100, 300, and 1000 min), 30-μl aliquots of the exchange reaction were rapidly quenched by 30 μl of ice-cold quench buffer (0.2 m potassium phosphate (pH 2.6)). The samples were immediately frozen in liquid nitrogen and stored at −80 °C before analysis. Un-deuterated samples were generated following the same procedure except that protein samples were diluted into aqueous buffer and incubated for 1 min followed by the quench process. All experiments were run in triplicates.
The frozen deuterated sample was quickly thawed and loaded with an ice-cold syringe into a 20-μl sample loop inside the refrigeration system. The protein sample (∼40 pmol) was carried by a 0.1 ml/min digestion flow (0.1% formic acid) into an immobilized pepsin column (Poroszyme Immobilized Pepsin Cartridge, 2.1 × 30 mm, Applied Biosystems) and digested at 15 °C for 1 min. The resultant peptides were immediately cooled down to 0 °C through a heat exchanger and were concentrated and desalted on a peptide trap (Michrom Peptide MacroTrap, 3 × 8 mm). The peptides were eluted and separated in 15 min through a reversed-phase C18 HPLC column (Agilent Poroshell 300SB-C18, 2.1 × 75 mm) at a flow rate of 0.2 ml/min with a 0 °C 2–40% acetonitrile gradient containing 0.1% formic acid. ESI-Fourier transform-mass spectrometry measurements began 5 min after the initiation of the elution process and lasted 10 min. The time from initiation of digestion to elution of the last peptide was less than 20 min.
Bruker Daltonics DataAnalysis 4.0 was used for spectrum analysis and data treatment. Peptides were identified from undeuterated samples by a customized program DXgest, which matches experimental peptide mass with theoretically generated peptic peptide mass by using statistical data (56) for the pepsin cleavage pattern under H/D exchange conditions. Mass tolerance was set at <2.0 ppm. H/D exchange data for each individual peptide at various time points were processed using the program HX-Express (57) to determine deuterium uptake and peak width. No correction was applied for back exchange. The number of backbone amides, peptide coverage, and H/D heat map were generated by using MSTools (58). Origin (OriginLab, Northampton, MA) and Microsoft Excel were used to produce time-dependent deuterium uptake plot, butterfly plot, and difference plot (59).
RESULTS
Crystal Structure of the E. coli E3
Each monomer of E3 is composed of four domains. In E. coli, these are as follows: the FAD-binding domain (residues 1–149), the NAD-binding domain (150–282), the central domain (283–350), and the interface domain (351–474). The functional dimer of E3 is shown as a ribbon diagram in Fig. 1A. The tertiary structures of glutathione reductase and all 14 E3s listed in Table 1 are generally similar to that of E. coli E3, and the dimer in each structure could be boxed in a rectangle of approximate dimensions 110 × 65 × 65 Å. Comparison of an E. coli E3 subunit to a subunit from each of the other E3 structures yields an r.m.s. deviation (Cα) between 0.9 and 1.58 Å. The primary differences in the conformations of the E3 structures elucidated to date are in the regions 36–42, 126–136, and 251–264 (the numbers referring to the residue ranges in the current E. coli E3 structure) and include addition or deletion of residues or small differences in folding. These regions are all parts of surface loops and generally result in no additional or fewer interactions of the protein with the FAD cofactor, except as indicated in the footnote to Table 3. They also do not seem to participate in interactions with the NAD substrate as can be seen from crystal structures of E3 complexed with NAD (1LVL, 1ZMC, and 1V59). These regions may, however, be important in specific assembly interactions with other components in multienzyme complexes from their respective species.
FIGURE 1.

A, functional dimer of E. coli E3. One of the E3 monomers is shown in cyan. In the other, the FAD-binding domain is shown in red, NAD-binding domain in green, central domain in yellow, and the interface domain in purple. The FAD molecules are shown as CPK models in gray and orange. B, superposition of the E. coli (green) and Pseudomonas putida (1LVL) (orange) E3 structures illustrating hydrogen bond formation (shown by dashed line) in the former between OE2 of Glu-319 and N of Met-317 arising from the cis-conformation of Pro-318.
TABLE 3.
Interactions (d, in Å) between FAD atoms and protein/solvent atoms (averaged between the two subunits)
The ′ indicates residues of the other subunit in the functional dimer.
| FAD atom | Protein/solvent atom | d |
|---|---|---|
| Å | ||
| N6A | Gly-117(O) | 3.04 |
| N6A | Solvent 333 (HOH-O) | 2.50a |
| N1A | Gly-117(N) | 2.97 |
| N3A | Arg-37(N) | 3.21b |
| N7A | Solvent 417, solvent 25 (HOH-O) | 2.80 |
| N7A | Solvent 333 (HOH-O) | 3.20a |
| O2B | Glu-36(OE2) | 2.60 |
| O2B | Arg-37(NE) | 3.10c |
| O2B | Solvent 227, solvent 327 (HOH-O) | 3.33 |
| O3B | Glu-36(OE1) | 2.58 |
| O3B | Glu-36(OE2) | 3.17 |
| O3B | Solvent 90, solvent 73 (HOH-O) | 3.01 |
| O1A | Val-44(N) | 3.02 |
| O2A | Solvent 34, solvent 240 (HOH-O) | 2.65 |
| O2A | Solvent 92, solvent 244 (HOH -O) | 2.65 |
| O1P | Ala-17(N) | 3.15 |
| O1P | Solvent 1, solvent 6 (HOH-O) | 2.61 |
| O2P | Asp-313(N) | 3.01 |
| O2P | Solvent 91, solvent 93 (HOH-O) | 2.80 |
| O4′ | Cys-45(N) | 3.36 |
| O3′ | Asp-313(OD1) | 2.91 |
| O3′ | Asp-313(OD2) | 3.23 |
| O3′ | Met-319(O) | 3.09 |
| O3′ | Leu-320(N) | 3.51 |
| O3′ | Ala-321(N) | 3.45 |
| O2 | Ala-321(N) | 2.74 |
| O2 | His-445′(O) | 3.25 |
| O2 | His-322(ND1) | 3.38d |
| N3 | His-445′(O) | 2.81 |
| N3 | Solvent 253, solvent 210 (HOH-O) | 2.90 |
| O4 | Lys-54(NZ) | 3.11 |
| O4 | Solvent 75, solvent 55 (HOH-O) | 2.73 |
| O4 | Solvent 339, solvent 127(HOH-O) | 3.57 |
| N5 | Solvent 339, solvent 127(HOH-O) | 2.81 |
a Water molecule and interaction are seen in subunit B only.
b This is absent in 2A8X.
c This is absent in 1LPF, 1LVL, 3LAD, 1V59, 1GER, and 1DXL.
d This is absent in 1GER.
cis-Conformation of Peptide Bonds Formed by Three Prolines
In E. coli E3, peptides of three prolines, Pro-318, Pro-355, and Pro-446, are in cis-conformations. Residue 318 is conserved as proline except in 1LPF, 2A8X, 3LAD, and 1GER, but only E. coli E3, 1OJT, and 2EQ8 contain a cis-proline at that position. In E. coli E3 and 1OJT, the cis-peptide bond results in an orientation that allows OE1 of the preceding glutamine residue to form a hydrogen bond with the main chain nitrogen of the succeeding methionine residue (Fig. 1B); in 2EQ8, the preceding residue is also a proline. In 1LVL, where the corresponding proline (residue 310) is trans, OE1 or OE2 of glutamate 309 are involved in no hydrogen bonds. In all the other structures, the residue corresponding to Gln-317 in E. coli E3 is Gly, Ala, or Leu, possessing no side chains capable of hydrogen bonding interactions. The residue at the position equivalent to 355 in E. coli is always a proline, and the peptide bond is in cis-conformation in all except 1DXL and 1EBD. This residue falls in a tight β-turn on the anti-parallel β-sheet of the interface domain featuring a characteristic O(i)—N(i + 3) type hydrogen bond between the carbonyl oxygen of Thr-353 and the amide nitrogen of Glu-356. This hydrogen bond is absent in 1GER alone where the residue following the cis-proline is another proline, but the hairpin bend itself is observed in all the E3 structures. The orientation of His-445 in E. coli E3 is dictated by the hydrogen bonds its ND1 atom makes with the OE1 and OE2 atoms of Glu-450 in the same subunit. Position 446 is conserved as proline and occurs in a cis-form in all the E3 structures discussed; this orientation results in a hydrogen bond between the carbonyl oxygen of His-445′ and N3 of the flavin ring in E. coli E3, similar to what has been discussed for 3LAD (23); this hydrogen bond is found in all the other E3 structures as well.
FAD Cofactor and Its Interactions with E3
The FAD molecules assume elongated conformations in all E3 structures. The two FAD molecules in the E3 dimer are separated by 31.2 ± 1.0 Å in the various structures at their closest points (the O4 atoms). As in 1GER, a B value gradient is observed along the FAD molecules in E. coli E3. The average isotropic B values, with numbers for the second subunit in the dimer given in parentheses, are 64.8 (65.5), 60.8 (55.8), 51.2 (53.7), 49.6 (51.1), and 45.5 (50.5) Å2 for the adenine, adenine ribose, pyrophosphate, ribitol, and isoalloxazine moieties, respectively, indicating that the isoalloxazine end is the most tightly bound. The characteristic sequence GXGXXG responsible for the binding of FAD and NAD starts at residue 13 (GAGPAG) in the FAD-binding domain and at residue 182 (GGGIIG) in the NAD-binding domain. These characteristic regions comprising residues 13–18 and 182–187 act as hinges between the β-strand and the α-helix in each binding domain, as in 1OJT. The βαβ unit corresponding to the FAD-binding domain is β(8–12), α(16–27), and β(32–36).
The interactions FAD makes with the two E3 subunits in E. coli are listed in Table 3. Fig. 2 also illustrates these interactions together with the relatively short contacts (∼3.5 Å) observed between His-445 of one subunit of E3 and the reactive cysteines Cys-45 and Cys-50 of the other subunit. Both subunits contribute residues to each active site and to interactions with each FAD molecule. The inter-subunit interactions in E. coli E3 are listed in Table 4. The active disulfide bridge Cys-45 to Cys-50 in each catalytic site is also located at the dimer interface involving residues from both the FAD-binding and central domains of the same subunit, as well as residues His-455 and Glu-450 from the other subunit, possibly explaining the necessity of the dimeric form for enzymatic activity.
FIGURE 2.

Interactions of the FAD molecule with E3 subunits. The hydrogen bonds formed between His-445′ and Glu-450′ are also shown, as well as the short (<3.5 Å) contacts between His-445′ and the sulfur atoms of Cys-45 and Cys-50 involved in the disulfide bond. ′ denotes residues from the other subunit.
TABLE 4.
Intersubunit interactions (d, in Å) in E. coli E3 (averaged between the two subunits)
The ′ indicates residues of the other subunit in the functional dimer.
| Residues/atoms | d |
|---|---|
| Å | |
| Asp-27(OD2)–Asn-468′(ND2) | 2.87 |
| His-58(NE2)–Ser-390′(O) | 3.41 |
| His-72(O)–Ile-86′(N) | 2.87 |
| Gly-73(O)–Asp-82′(N) | 2.75 |
| Val-75(N)–Lys-80′(O) | 2.90 |
| Val-75(O)–Lys-80′(N) | 3.01 |
| Arg-108(NH1)–Leu-466′(O) | 3.32 |
| Arg-108(NH2)–Ile-463′(O) | 3.30 |
| Arg-108(NH2)–Leu-466′(O) | 2.77 |
| His-322(ND1)–His-445′(N) | 3.19 |
| His-329(NE2)– Thr-464′(OG1) | 2.84 |
| Tyr-352(OH)–Pro-446′(O) | 3.57 |
| Gly- 419(O)–His-449′(ND1) | 3.54 |
| Glu-420(OE1)–Leu-448′(N) | 3.41 |
| Glu-420(OE1)–His-449′(N) | 2.93 |
| Glu-420(OE1)–His-449′(ND1) | 2.81 |
| Glu-420(OE2)–Leu-448′(N) | 2.80 |
| Gly-423(N)–His-449′(NE2) | 3.41 |
| His-445′(O)–FAD-600(O2) | 3.24 |
| His-445′(O)–FAD-600(N3) | 2.81 |
In 1LVL, Mattevi et al. (28) have indicated that the side chain of Tyr-181 points toward the flavin ring as it does in 1GER, in what they suggest is an energetically unfavorable conformation. We note, however, that the conformation adopted is one of the commonly allowed rotamers for tyrosine; furthermore, the same conformation is observed in 1EBD and is one of two equally populated disordered conformations reported in 1DXL, the only two other E3 structures available where the corresponding residue is a tyrosine. In all the other structures listed in Table 1, the corresponding residue (185 in E. coli E3) is isoleucine, valine, or alanine.
Catalytically Important Disulfide Bridge between Cys-45 and Cys-50
Approximately 70% of the residues in E. coli E3 are distributed as α-helices and β-sheets in roughly equal amounts. The longest of these helices (Gly-43 to Ala-70) includes cysteines 45 and 50 that form the catalytically important disulfide bridge. This helix is distorted between residues 51 and 53 because of the presence of a proline at position 52, which naturally breaks the conventional N(i + 4)—O(i) hydrogen bonding pattern observed in a regular α-helix. The disulfide bonds between residues 45 and 50 are 1.96 and 2.04 Å in the two monomers with χSS dihedral angles (Cβ-Sγ-Sγ-Cβ) of 29 and 46°, respectively (Fig. 3A). In the other E3 structures, these angles range between −130 and −82°. With molecular orbital calculations, Boyd (60) postulated that the disulfide bond is strongest at the orthogonal dihedral angles of ±90°. In the light of this information, the Sγ atoms of residues Cys-45 and Cys-50 in E. coli E3 were readjusted several times to yield different χSS dihedral angles in these ranges, but subsequent refinement always returned the original values of 29 and 46°, which were therefore accepted as correct.
FIGURE 3.

A, disulfide bond between Cys-45 and Cys-50, shown in yellow for subunit A. The electron density from a composite omit map is contoured at 1.5σ. B, distorted rectangular cluster of four sulfur atoms featuring Met-431 and Cys-433 in both subunits, with distances shown in Å.
Other Miscellaneous Features
In the E. coli E3 dimer, a distorted rectangular cluster of four sulfur atoms 3.5 × 6.8 Å in size forms due to the orientation of Cys-433 and Met-431, as shown in Fig. 3B. In only one other structure, 1OJT, does a similar arrangement occur, with the four sulfur atoms forming a cluster of 3.4 × 6.2 Å. In all the other E3 structures, the corresponding residues are not both sulfur-containing amino acids. In most of the E3 structures elucidated to date, the hydroxyl group of the tyrosine residue immediately following the characteristic GXGXXG sequence responsible for FAD binding is linked by a hydrogen bond to the side chain of the C-terminal residue closest to the catalytic center, always a histidine, asparagine, or aspartate. The only exceptions are the current E. coli E3 structure where the side chains of the corresponding residues Tyr-19 and Asp-465′ are involved in no direct hydrogen bonds at all, and 1EBD, 1V59 and 1GER, which are shorter and have no residue equivalent to the C-terminal residue involved. It has been reported that asparagine 473 is important to the efficient catalytic function of human E3 (61) based on the fact that mutation of this residue to leucine reduced the E3 activity about 37-fold. However, the role of the C-terminal residues that form an arm in some of the E3 structures or the lack of this arm itself in others and the significance of the hydrogen bond between tyrosine 19 and asparagine 473 are not clearly understood.
Kinetic Experiments on E2p and Its Variants with Substitutions in PSBD and the Catalytic Domain
The role of the N-terminal region of E1p (residues 1–55) important for interaction with E2p had been reported earlier (62). The N-terminal region of E1p contains acidic amino acid residues, whereas the PSBD of E2p has a high content of basic amino acid residues. This led to the hypothesis that the N-terminal region of E1p interacts with the PSBD of E2p through electrostatic contacts. To identify the loci of interactions of E1p and E3 on E2p, several amino acid substitutions were carried out on the PSBD (residues 129–161) as well as at Lys-191 and Arg-202 from the catalytic domain (Table 5). As shown in Table 5, the overall PDHc activities of R129E, R150E, R150A, R150K, and R202E E2 variants reconstituted with independently expressed E1p and E3 components were less than 10% that of the parental E2p. A multisite substituted E2 variant with G144A, G146A, and G149A substitutions displayed only 3% activity. The effect of substitutions on the overall PDHc activity was not significant in other E2p variants with activities in the 34–100% range. It is evident that the R129E and R150E substitutions in PSBD and the R202E substitution in the catalytic domain, with a charge-reversed glutamate in place of arginine greatly diminished the overall PDHc activity.
TABLE 5.
Kinetic, size-exclusion HPLC, and MALDI-TOF experiments probing the interaction of E1p and E3p with the E2p component and its variants
| Substitutions in E2 | Overall activity | Interaction between E1p and E2pa | Interaction between E3 and E2pa | Reductive acetylation of the E2b |
|
|---|---|---|---|---|---|
| Reductively acetylated lipoyl domainc | Unacetylated lipoyl domainc | ||||
| % | |||||
| None | 100 | + | + | + | − |
| R129A | 34 | + | − | + | + |
| R129K | 77 | + | + | + | + |
| R129E | 3.0 | − | − | − | + |
| R130A | 100 | NA | NA | NA | NA |
| R130E | 57 | NA | NA | NA | NA |
| R150A | 8.0 | − | + | NA | NA |
| R150K | 5.0 | − | + | + | + |
| R150E | 2.4 | − | − | − | + |
| R153K | 83 | + | + | + | + |
| R153E | 60 | + | + | + | + |
| K161A | 65 | NA | NA | + | + |
| K161E | 84 | NA | NA | NA | NA |
| K191R | 79 | NA | NA | NA | NA |
| K191E | 50 | NA | NA | NA | NA |
| R202K | 86 | + | + | + | + |
| R202E | 10 | + | + | NA | NA |
a + indicates E2 variants converted quantitatively to E1p-E2p or E3-E2p subcomplexes according to size-exclusion HPLC experiment; − indicates E2 variants that did not form the subcomplexes with E1p or E3 according to size-exclusion HPLC experiment. NA means data were not available.
b The reductive acetylation of the E2p lipoyl domain was monitored by MALDI-TOF MS as presented under “Experimental Procedures.”
c The molecular mass of the acetylated lipoyl domain = 10,154 Da and that of the unacetylated lipoyl domain = 10,110 Da.
Size-exclusion HPLC Experiments Probing the Interaction of E1p and E3 with E2p and Its Variants
Experiments by size-exclusion chromatography demonstrated that the R129E substitution strongly affects the interaction of E2p with both E1p and E3. The R129A E2p interacts with E1p but not with E3, whereas R129K E2p forms the subcomplex with both E1p and E3 (Table 5). These results suggest moderately different binding modes for E1p and E3 to the PSBD of E2p. Similarly to Arg-129, the charge-reversed substitution at Arg-150 strongly affects interaction between components. Although lysine should be the most conservative substitution for arginine, R150K E2p does not display binding to E1p, suggesting a multipoint interaction between E1p and E2p. According to size-exclusion experiments, the Arg-150 variants (R150A, R150K, and R150E) do not interact with E1p; yet R150A and R150K variants interact with E3 but R150E does not. These studies revealed the following: 1) the interaction between E2p PSBD and E1p or E3 is strongly affected by charge-reversed substitutions at Arg-129 and Arg-150; 2) substitution at Arg-129 more strongly affects the binding of E2p to E3, whereas that at Arg-150 more strongly affects the binding of E2p to E1p, and 3) the E2p variants with K191R, K191E, and R202K substitutions in the N-terminal region of the catalytic domain exhibited high activities and could still form the subcomplex with E1p or E3, suggesting that the residues Lys-191 and Arg-202 are not critically involved in the binding of E2p to E1p or E3.
Reductive Acetylation of E2p and Its Variants by E1p and Pyruvate Monitored by MALDI-TOF MS
To test the effect of the E2 substitutions on functional interaction between E1p and 1-lip E2p, the reductive acetylation of the E2 lipoyl domain was analyzed by MALDI-TOF MS. The molecular mass of the lipoyl domain derived from 1-lip E2p by trypsin digestion is 10,110 Da. After acetylation by E1p and pyruvate, the mass of the lipoyl domain increased to 10,154 Da indicating reductive acetylation of the lipoic acid covalently attached to the lipoyl domain. It was demonstrated that within 1 min of incubation of 1-lip E2p with E1p and pyruvate, the lipoyl domain derived from E2p was fully acetylated (Table 5). However, only the unacetylated form of the lipoyl domain was detected for R129E and R150E variants, although for all other E2p variants, both acetylated and unacetylated forms were in evidence (Table 5).
H/D Exchange Mass Spectrometry
H/D exchange by MS (HDX-MS) was performed as described under “Experimental Procedures” for wild-type 3-lip E2p and E3 individually, as well as for the 3-lip E2p-E3 subcomplex. A total of 25 peptides from 3-lip E2p with 61% sequence coverage (supplemental Table S1 and Fig. 4), and 26 peptides from E3 with 84% sequence coverage (supplemental Table S2 and Fig. 4) were chosen for data analysis. The time-dependent deuterium uptake patterns of each peptide in the single protein (3-lip E2p or E3) and in the binary protein complex (3-lip E2p + E3) were compared. Heat map (supplemental Fig. S1 and Fig. S2), butterfly plot, and difference plot (Fig. 5) showed a snapshot of deuterium uptake pattern change of either 3-lip E2p or E3 in the presence and absence of the other component. Overall, two peptides from 3-lip E2p (Fig. 5, A and B) and three peptides from E3 (Fig. 5, C and D) exhibited statistically significant changes in deuterium uptake pattern resulting from complexation as explained below; the other peptides exhibited almost identical H/D exchange patterns in the individual or complexed component.
FIGURE 4.
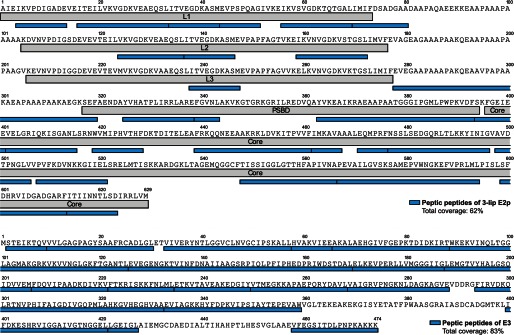
Sequence coverage of the selected peptides from 3-lip E2 (top) and E3 (bottom) digested by pepsin. See “Experimental Procedures” for details of digestion.
FIGURE 5.

HDX-MS analysis of 3-lip E2p and E3 in the absence and presence of the other component. A, butterfly plot representing average relative deuterium incorporation percentage (y axis) (deuterons exchanged/maximum exchangeable amides × 100%) of peptic fragments from 3-lip E2p (x axis, listed from N to C terminus) in the absence of E3 (top) versus in the presence of E3 (bottom) based on three independent experiments. B, difference plot showing deuterium incorporation changes of peptic fragments of 3-lip E2p in the absence and presence of E3 (deuterons exchanged in the absence of E3 minus deuterons exchanged in the presence of E3). C, butterfly plot representing average relative deuterium incorporation percentage (y axis) (deuterons exchanged/maximum exchangeable amides × 100%) of peptic fragments from E3 (x axis, listed from N to C terminus) in the absence of 3-lip E2p (top) versus in the presence of 3-lip E2p (bottom) based on three independent replicates. D, difference plot showing deuterium incorporation changes of peptic fragments of E3 in absence and presence of 3-lip E2p (deuterons exchanged in the absence of 3-lip E2p minus deuterons exchanged in presence of 3-lip E2p).
The two identified peptides in 3-lip E2p were both from the PSBD domain, 325YVHATPLIRRLARE338 (corresponding to residues 121–134 in the 1-lip E2p didomain) and 363YVKEAIKRAEAAPAATGGGIPGMLPWPKVDF393 (corresponding to residues 159–189 in the 1-lip E2 didomain). Deuterium uptake of 325YVHATPLIRRLARE338 in PSBD of 3-lip E2p was reduced by the binding to E3 (Fig. 6A). The maximum deuterium uptake difference of 1.2 Da was detected after 3 min of exchange. Peak width analysis of 325YVHATPLIRRLARE338 (Fig. 6B) displays EX1 exchange mechanism (partial unfolding, H/D exchange, followed by refolding; see Refs. 63, 64) with a partial unfolding half-life of about 10 min for E2p alone and about 30 min for E2p-E3 subcomplex, indicating that E3 alters the dynamics of 325YVHATPLIRRLARE338 in E2p by tripling the partial unfolding half-life.
FIGURE 6.

HDX-MS analysis of peptide YVHATPLIRRLARE (325–338) from 3-lip E2p alone (black squares) versus 3-lip E2p-E3 subcomplex (red circles). A, relative deuterium incorporation as a function of exchange time of 20 s to 1000 min (time in log scale). B, relative peak width analysis by determining peptide isotopic mass envelope width at 20% of the maximal peak height. The error bars for A and B were plotted as the standard deviation of three independent experiments. C, mass spectra of 2+ charged form of peptic fragment YVHATPLIRRLARE (3-lip E2p 325–338). Deuterium uptake of the peptide from 3-lip E2p alone (left) versus 3-lip E2p-E3 subcomplex (right) at 0 s (nondeuterated reference) and 20 s and 1, 3, 10, 30, 100, and 1000 min is shown.
The Butterfly plot also revealed several regions that were highly protected in the E2p core domain even in the absence of E3 as follows: 419WVMIPHVTHF428, 462IMKAVA467, 567PEVAIL572, and 614ITIINNTLSD623, which had almost no deuterium uptake in 30 min.
The three lipoyl domains were too rigid to be digested by pepsin, resulting in low peptide coverage. Also, because of their sequence similarity, it was difficult to determine from which lipoyl domain the peptide was cleaved.
In E. coli E3, two peptides 33VIVERYNTLGGVCLNVGCIPSKALL57 and 89WKEKVINQLTGGLAGMAKGRKVKVVNGLGKFTGANTL125 from the FAD-binding domain (the first with two reactive cysteines Cys-45 and Cys-50) and a third peptide 458VFEGSITDLPNPKAKKK474 from the C terminus located at the subunit interface (in the interface domain) were identified to undergo reduced H/D exchange indicating protection by the presence of 3-lip E2.
Although interactions of E3 with lipoyl domains do not lend themselves readily to delineation by the H/D exchange method (because of highly structured domains and near identity of the three domains), NMR complements the procedure.
NMR Spectroscopy
A representative 1H-15N TROSY HSQC spectrum and a segment of the three-dimensional HNCACB strip of the E2 didomain construct are shown in Fig. 7. Partial backbone assignments of the E2p didomain were made, and backbone chemical shifts of the assigned residues are summarized in supplemental Table S3. The E2p didomain consists of 194 amino acid residues, including 16 prolines. “Peak picking” identified 135 peaks in the 1H-15N TROSY HSQC spectrum, ∼75% of the total number of residues. At present, we have sequence-specific assignments mainly for the hybrid lipoyl domain region. The linker between the lipoyl domain and PSBD is proline-rich and flexible, and most of the resonances from these regions remain unassigned. To probe possible interactions of E2p didomain with E3, two-dimensional TROSY-HSQC spectra of 0.3 mm 15N-E2p didomain were acquired in the absence of and in complex with 0.3 mm unlabeled E3 at 25 °C in 20 mm KH2PO4 (pH 7.0 with 150 mm NaCl) and 10% (v/v) 2H2O. The two-dimensional 15N-TROSY HSQC NMR spectra of E2p didomain in the presence of E3 at a 1:1 molar ratio revealed negligible changes in chemical shift indicating that NMR detected little or no interaction between E3 and the lipoyl domain.
FIGURE 7.
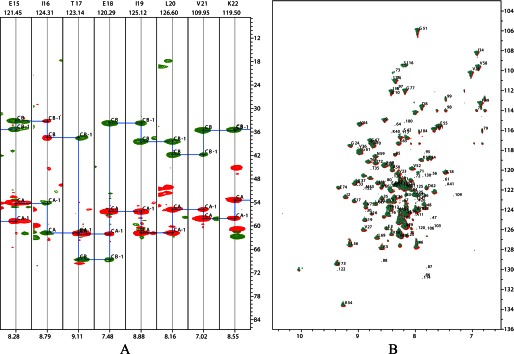
Representative NMR spectra of E2 didomain in the absence and presence of E3. A, plots displaying the strips from three-dimensional HNCACB for E2 didomain. B, overlaid two-dimensional 1H-15N TROSY HSQC spectra of E2 didomain (0.3 mm in red) with that of the E2 didomain-E3 subcomplex (0.3 mm of each protein in green) recorded at 25 °C in 20 mm sodium phosphate (pH 7.0) with 0.15 m NaCl.
The NMR method here used is complementary to the x-ray structure determination and especially the MS-detected H/D exchange; the former clearly identifies the lipoyl domain but the latter two cannot, and the H/D exchange clearly identifies peptides from the PSBD, which participate in interactions with E3.
DISCUSSION
It must be emphasized that no crystal structure of an intact E2 component from any source is currently available, making studies such as the present one important for a more complete understanding of the interactions of the E1 and E3 components with the E2 component.
The biochemical studies summarized in Table 5 gave clear support to the notion that E1p and E3 bind to nonidentical but strongly overlapping epitopes of E2p, mostly localized to the PSBD. As was shown recently (41), this does not require competition of E1p and E3 for unique E2p-binding sites, because E2p components are always organized in an oligomeric structure as trimers at the corners of a core (2, 65–67). The fact that the hot spots identified also interfered with reductive acetylation (according the MALDI-TOF MS) confirms that these hot spots have important functional significance in the E1p-E2p interaction as well. The NMR results indicated weak interaction between the lipoyl domain of the 1-lip E2p didomain and E3.
Several crystal structures of E3 complexed to its binding domain (30–34) as well as a solution NMR study of the interaction of a 15N-labeled PSBD with the dimer of the interface domain of B. stearothermophilus E3 (68) have been published. The novelty in the current research compared with those above is as follows. 1) For the first time an attempt was undertaken to analyze the binding of the wild-type 3-lip E2p to E3, rather than any truncated version of either one. 2) The first H/D MS experiment applied to such complexes demonstrates that notwithstanding the accepted aggregation of intact E2p components, our high resolution method is capable of elucidating important information about protein-protein interactions, often unobtainable by either x-ray or NMR techniques. 3) The peptide-specific H/D exchange results are consistent with all previous structural reports.
From Fourier transform-mass spectrometer H/D exchange experiments for E2p, the following is evident. 1) A significantly reduced level of deuteration was detected for the peptide 325YVHATPLIRRLARE338 (corresponding to residues 121–134 in 1-lip E2p) from the PSBD region of 3-lip E2 on complexation with E3. Starting with the lone Pro in this sequence, all amino acids are part of the α-helix (H1), which is the only one of the three helixes (H1, H2, and H3) that interacts with the E3 dimer in all cases with the exception of the E2 component of the 2-oxoglutarate dehydrogenase complex (33), and it provides electrostatic stabilization of the complex. The residue Arg-333 (corresponding to Arg-129 in 1-lip E2p) is here identified by both biochemical and MS methods as a “hot spot” for interactions with both E1p and E3. This residue is highly conserved in known PSBDs (30). 2) The second peptide from 3-lip E2p identified with reduced deuterium uptake on complexation with E3 is 363YVKEAIKRAEAAPAATGGGIPGMLPWPKVDF393 (corresponding to residues 159–189 in the 1-lip E2 didomain) and contains the first four residues (YVKE) from H2 of PSBD, the remainder coming from the hinge region and suggests that H2 is also protected by E3, as envisioned for the 2-oxoglutarate dehydrogenase complex (33).
Regarding Fourier transform-mass spectrometer H/D exchange experiments for E3, the following is evident. 1) The peptide 458VFEGSITDLPNPKAKKK474 displays low deuterium uptake when complexed with 3-lip E2p and contains residues from the E3 subunit interface domain involved in interaction with E2p, an interaction that is conserved across all E3-PSBD structures. 2) Two peptides (the first one including the two reactive cysteines Cys-45 and Cys-50), 33VIVERYNTLGGVCLNVGCIPSKALL57 and 89WKEKVINQLTGGLAGMAKGRKVKVVNGLGKFTGANTL125 from the FAD-binding domain, also display low deuterium uptake, not evident from structures without the intact E2p component, suggesting a conformational change in the E3 active center on complexation.
To interpret in stereochemical terms the results presented above concerning the influence of reverse charge substitutions R129E, R150E, R153E, and K161E on the binding of PSBD to E3, a model of the E3-PSBD interface in E. coli was postulated based on an analogy with the known crystal structure of the corresponding interface in the E3b-PSBDp complex from Thermus thermophilus (PDB code 2EQ8 (33)). This approach was chosen because of the large number of common amino acid residues in the PSBDs of E. coli and T. thermophilus, as shown in Tables 6 and 7. A comparison by PROTEIN BLAST software indicated a sequence homology plus identity of 65% between the E3 components of E. coli and T. thermophilus. To further illustrate the relationship between sequence and structural similarity between them, each molecule of E3 from the two sources above was divided into regions where such sequence similarity was high and into those where it was low. Least square comparisons of main chain atoms were made between the two structures in each region. The average r.m.s. deviation was 0.47Å in regions of high sequence similarity and 1.43 Å in the rest of the regions. Furthermore, for a particular PSBD binding region on E3 of T. thermophilus and for the corresponding region on the uncomplexed E3 of E. coli, a least squares superposition of Cα atoms showed that the two molecules matched each other with an r.m.s. deviation between 0.13 and 0.50 Å in that specific region; simultaneous inclusion of all the three interface binding regions on the E3 increased the variability to 0.77Å.
TABLE 6.
Sequences of the E3-binding domains
Sequences of known crystal structures of E3-binding domains from different sources are compared with that of the E3-binding domain from the dihydrolipoamide succinyltransferase core from the pyruvate dehydrogenase multienzyme complex of E. coli. Residues at equivalent locations identical to those in E. coli are shown in green, homologous residues in blue, and other residues in red. The last two columns indicate the percentage of residues identical with (Iden) or homologous to (Hom) those in E. coli.

TABLE 7.
Sequences of the appropriate binding regions of the E3s
Residues at equivalent locations identical to those in E. coli E3 are shown in green, homologous residues in blue, and other residues in red. The last two columns indicate the percentage of residues identical with (Iden) or homologous to (Hom) those in E. coli.

Accordingly, it was assumed that the main chains in the E. coli and T. thermophilus E3-PSBD interfaces were folded virtually identically and also that the side chains of residues identical in both interfaces possess virtually the same torsion angles. Hence, the coordinates of T. thermophilus main chain atoms were assigned to the main chain atoms of E. coli in the appropriate region, as were the side chain atoms when the residues were identical. The nonidentical side chains were generally allowed to follow the conformations of the corresponding residues in T. thermophilus as far as was reasonable stereochemically. Finally, a few minor adjustments in side chains were made to avoid stereochemically unacceptable contacts. Ultimately, among residues identical in E. coli and T. thermophilus, only one residue in the E. coli PSBD, Arg-130, had to be assigned a significantly different side chain rotamer conformation than its counterpart Arg-137 in T. thermophilus to result in excellent salt bridge contacts, charge distribution, and packing considerations. Fortuitously, this new rotamer choice turned out to be one even more commonly observed in crystal structures than the original assignment; it was also noted that neither in the T. thermophilus crystal structure nor in the E. coli model could any allowed rotamer, other than that selected in each case, have been chosen without causing severe short contacts. Although the residue is an arginine in the PSBD of both E. coli and T. thermophilus, the difference in its conformation results from a difference in sequence of the E3 residues in its vicinity. Interactions observed in the resulting model of the E. coli E3-PSBD interface are shown in Fig. 8A, and those in the T. thermophilus crystal structure itself (PDB code 2EQ8 (33)) are shown in Fig. 8B, to illustrate the salient differences between the two. The coordinates of the model of the E. coli E3-PSBD interface are shown in supplemental Table S4.
FIGURE 8.

A, diagram illustrating electrostatic binding interactions (≤3.5 Å) in the model of the E3-PSBD interface from E. coli. Bonds in residues from E3 are shown in green and those in residues from the PSBD in orange. Hydrogen bond and salt bridge contacts are indicated by dashed lines. This diagram is a model based on the crystal structure of the E3b-PSBDp complex from T. thermophilus described by Nakai et al. (33), with coordinates from the Protein Data Bank (PDB code 2EQ8). Please see text for details. B, diagram illustrating observed electrostatic binding interactions (≤3.5 Å) between the E3 and PSBD in the crystal structure of the E3b-PSBDp complex from T. thermophilus described by Nakai et al. (33), with coordinates from the Protein Data Bank (PDB code 2EQ8). Bonds in residues from E3 are shown in green and those in residues from the PSBD in orange. Hydrogen bond and salt bridge contacts are indicated by dashed lines.
Based on the E. coli E3-PSBD model, in E. coli variant R129E the conserved electrostatic binding interactions of Arg-129 with Asp-343 and Glu-430 are replaced by electrostatic repulsions between negatively charged oxygens on Glu-129, Asp-343, and Glu-430. An analogous situation occurs with the variant R150E. In this case, the favorable electrostatic interaction between Arg-150 and the main chain nucleophilic carbonyl oxygen of Met-431 of E3 is replaced by electrostatic repulsion between Glu-150 and the Met-431 carbonyl oxygen. In the model, Arg-129 assumes an extended conformation stretching deeply into the E3 surface where its guanidinium nitrogen atoms form hydrogen bonds with E3 carboxylate or carbonyl oxygen atoms. Additionally, its main chain carbonyl oxygen atom makes a hydrogen bond with the hydroxyl group of Tyr-341 on E3. This network of hydrogen bonds implies the existence of very strong electrostatic bonding in the E3-PSBD interface at Arg-129. The situation at Arg-130, however, is quite different. Here part of the side chain assumes a folded conformation and constitutes a part of the PSBD interface. The Arg-130 guanidinium group protrudes a short distance into the interface to make hydrogen bonding contacts with a carboxylate oxygen on Asp-343, which also is hydrogen bonded to the Arg-129 guanidinium group. The much greater involvement of Asp-343 in Arg-129 hydrogen bonds may limit the favorable possibilities for interaction with Arg-130. Nevertheless, the guanidinium nitrogens of Arg-130 are hydrogen bonded to only two oxygens. Accordingly, Arg-130 is bound far more weakly to E3 than Arg-129. Replacement of Arg-130 with Glu-130 transforms a weak binding interaction into a weakly dissociative one. These stereochemical features of the model are consistent with the biochemical results cited in Table 5 concerning variants of Arg-129 and Arg-130. The embedment of Arg-129 into the E3 surface apparently insulates this residue from external influences as also indicated by the H/D exchange experiments, whereas Arg-130 is not shielded so thoroughly because part of its side chain is contained in the PSBD surface.
The E. coli E3-PSBD model also indicates that Arg-153 does not bind electrostatically to an E3 residue but that its main chain carbonyl oxygen forms hydrogen bonds to the amide groups of the PSBD main chain residues Val-156 and Gln-157. In the crystal structure of the T. thermophilus enzyme, the counterpart of Arg-153 in E. coli is a glutamate forming hydrogen bonds to main chain nitrogen atoms of residues that are counterparts of Val-156 and Gln-157 in E. coli but again not to E3. In other words, the same intra-PSBD main chain hydrogen-bonding motif occurs in both the native and variant forms Arg-153 and R153E. In fact, this intra-PSBD main chain hydrogen bonding feature is observed in all known crystal structures of E3-PSBD complexes from various sources, perhaps contributing to a general similarity in the folding and conformation of the subunit-binding domains. Interestingly, in T. thermophilus, the side chain atoms of Glu-160 and Arg-164 form an intra-PSBD salt bridge, whereas in the E. coli model the side chain atoms of the corresponding equivalent residues Arg-153 and Gln-157 form hydrogen bonds, likely indicating a weaker interaction in the E. coli complex.
The E. coli E3-PSBD model and the presently known crystal structures also give the answer straightaway as to what structural basis underlies the absence of an effect of K161E on interface binding in E. coli as was observed in the biochemical experiments. Significantly, this particular residue, located at the end of the last helix in the PSBD, is too far from E3 for it to participate in interfacial binding interactions. A variety of amino acids, such as Lys, Gly, Ala, Gln, and Glu, are known to occupy this site in wild-type PSBDs. In T. thermophilus, for example, the residue corresponding to Lys-161 is a glycine. In the crystal structure, this glycine projects into a channel between molecules in regions distant from the E3-PSBD interface. In the other crystal structures referred to in Tables 6 and 7, the residues corresponding to Lys-161 also protrude out into similar channels. Accordingly, Lys-161 and K161E in the E. coli model also exhibit this structural feature. Clearly amino acids that occupy this site can hardly be expected to influence the binding of E3 to PSBD appreciably.
Supplementary Material
This work was supported, in whole or in part, by National Institutes of Health Grants GM061791 (to W. F.) and GM050380 (to F. J.). This work was also supported by Veterans Affairs Merit Review (to W. F.).

This article contains supplemental Tables S1–S4 and Figs. S1 and S2.
The atomic coordinates and structure factors (code 4JDR) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- PDHc
- pyruvate dehydrogenase complex
- E1p
- first pyruvate dehydrogenase component of PDHc
- E2p
- second dihydrolipoamide acetyltransferase component of PDHc
- 3-lip E2p
- the wild-type three-lipoyl E2p component
- 1-lip E2p
- single-lipoyl construct of E2p
- E3
- third dihydrolipoamide dehydrogenase component of PDHc
- PSBD
- peripheral subunit-binding domain
- 1-lip E2p didomain
- E2p construct comprising residues 1–190 from 1-lip E2p corresponding to the hybrid lipoyl domain and PSBD
- E3b-PSBDp
- one of only three specific E3-E2 complexes that can form for T. thermophilus depending on substrate specificity, with b representing branched-chain 2-oxo acid dehydrogenase and p representing pyruvate dehydrogenase
- PDB
- RCSB Protein Data Bank
- r.m.s.
- root mean square
- IPTG
- isopropyl 1-thio-β-d-galactopyranoside
- HSQC
- heteronuclear single quantum coherence
- H/D
- hydrogen/deuterium.
REFERENCES
- 1. Perham R. (1991) Domains, motifs, and linkers in 2-oxo acid dehydrogenase multienzyme complexes: a paradigm in the design of a multifunctional protein. Biochemistry 30, 8501–8512 [DOI] [PubMed] [Google Scholar]
- 2. Reed L. J. (1974) Multienzyme complexes. Acc. Chem. Res. 7, 40–46 [Google Scholar]
- 3. Bleile D. M., Munk P., Oliver R. M., Reed L. J. (1979) Subunit structure of dihydrolipoyl transacetylase component of pyruvate dehydrogenase complex from Escherichia coli. Proc. Natl. Acad. Sci. U.S.A. 76, 4385–4389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Stephens P. E., Darlison M. G., Lewis H. M., Guest J. R. (1983) The pyruvate dehydrogenase complex of Escherichia coli K12. Nucleotide sequence encoding the dihydrolipoamide acetyltransferase component. Eur. J. Biochem. 133, 481–489 [DOI] [PubMed] [Google Scholar]
- 5. Wei W., Li H., Nemeria N., Jordan F. (2003) Expression and purification of the dihydrolipoamide acetyltransferase and dihydrolipoamide dehydrogenase subunits of the Escherichia coli pyruvate dehydrogenase multienzyme complex: a mass spectrometric assay for reductive acetylation of dihydrolipoamide acetyltransferase. Protein Expr. Purif. 28, 140–150 [DOI] [PubMed] [Google Scholar]
- 6. Patel M. S., Korotchkina L. G., Sidhu S. (2009) Interaction of E1 and E3 components with the core proteins of the human pyruvate dehydrogenase complex. J. Mol. Catal. B Enzym. 61, 2–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Patel M. S., Roche T. E. (1990) Molecular biology and biochemistry of pyruvate dehydrogenase complexes. FASEB J. 4, 3224–3233 [DOI] [PubMed] [Google Scholar]
- 8. Patel M. S., Korotchkina L. G. (2003) The biochemistry of the pyruvate dehydrogenase complex. Biochem. Mol. Biol. Educ. 31, 5–15 [Google Scholar]
- 9. Toyoda T., Suzuki K., Sekiguchi T., Reed L. J., Takenaka A. (1998) Crystal structure of eukaryotic E3, lipoamide dehydrogenase from yeast. J. Biochem. 123, 668–674 [DOI] [PubMed] [Google Scholar]
- 10. Arjunan P., Nemeria N., Brunskill A., Chandrasekhar K., Sax M., Yan Y., Jordan F., Guest J. R., Furey W. (2002) Structure of the pyruvate dehydrogenase multienzyme complex E1 component from Escherichia coli at 1.85 Å resolution. Biochemistry 41, 5213–5221 [DOI] [PubMed] [Google Scholar]
- 11. Arjunan P., Chandrasekhar K., Sax M., Brunskill A., Nemeria N., Jordan F., Furey W. (2004) Structural determinants of enzyme binding affinity: the E1 component of pyruvate dehydrogenase from Escherichia coli in complex with the inhibitor thiamin thiazolone diphosphate. Biochemistry 43, 2405–2411 [DOI] [PubMed] [Google Scholar]
- 12. Arjunan P., Sax M., Brunskill A., Chandrasekhar K., Nemeria N., Zhang S., Jordan F., Furey W. (2006) A thiamin-bound, pre-decarboxylation reaction intermediate analogue in the pyruvate dehydrogenase E1 subunit induces large scale disorder-to-order transformations in the enzyme and reveals novel structural features in the covalently bound adduct. J. Biol. Chem. 281, 15296–15303 [DOI] [PubMed] [Google Scholar]
- 13. Chandrasekhar K., Arjunan P., Sax M., Nemeria N., Jordan F., Furey W. (2006) Active-site changes in the pyruvate dehydrogenase multienzyme complex E1 apoenzyme component from Escherichia coli observed at 2.32 Å resolution. Acta Crystallogr. D Biol. Crystallogr. 62, 1382–1386 [DOI] [PubMed] [Google Scholar]
- 14. Nemeria N. S., Arjunan P., Chandrasekhar K., Mossad M., Tittmann K., Furey W., Jordan F. (2010) Communication between thiamin cofactors in the Escherichia coli pyruvate dehydrogenase complex E1 component active centers: evidence for a “direct pathway” between the 4′-aminopyrimidine N1′ atoms. J. Biol. Chem. 285, 11197–11209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kale S., Arjunan P., Furey W., Jordan F. (2007) A dynamic loop at the active center of the Escherichia coli pyruvate dehydrogenase complex E1 component modulates substrate utilization and chemical communication with the E2 component. J. Biol. Chem. 282, 28106–28116 [DOI] [PubMed] [Google Scholar]
- 16. Pettit F. H., Reed L. J. (1967) α-Keto acid dehydrogenase complexes. 8. Comparison of dihydrolipoyl dehydrogenases from pyruvate and α-ketoglutarate dehydrogenase complexes of Escherichia coli. Proc. Natl. Acad. Sci. U.S.A. 58, 1126–1130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Perham R. N., Harrison R. A., Brown J. P. (1978) The lipoamide dehydrogenase component of the 2-oxo acid dehydrogenase multienzyme complexes of Escherichia coli. Biochem. Soc. Trans. 6, 47–50 [DOI] [PubMed] [Google Scholar]
- 18. Guest J. R., Darlison M. G., Spencer M. E., Stephens P. E. (1984) Cloning and sequence analysis of the pyruvate and 2-oxoglutarate dehydrogenase complex genes of Escherichia coli. Biochem. Soc. Trans. 12, 220–223 [DOI] [PubMed] [Google Scholar]
- 19. Steiert P. S., Stauffer L. T., Stauffer G. V. (1990) The lpd gene product functions as the L protein in the Escherichia coli glycine cleavage enzyme system. J. Bacteriol. 172, 6142–6144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Danson M. J., Eisenthal R., Hall S., Kessell S. R., Williams D. L. (1984) Dihydrolipoamide dehydrogenase from halophilic archaebacteria. Biochem. J. 218, 811–818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Danson M. J., Conroy K., McQuattie A., Stevenson K. J. (1987) Dihydrolipoamide dehydrogenase from Trypanosoma brucei. Characterization and cellular location. Biochem. J. 243, 661–665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Mittl P. R., Schulz G. E. (1994) Structure of glutathione reductase from Escherichia coli at 1.86 Å resolution: comparison with the enzyme from human erythrocytes. Protein Sci. 3, 799–809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Mattevi A., Schierbeek A. J., Hol W. G. (1991) Refined crystal structure of lipoamide dehydrogenase from Azotobacter vinelandii at 2.2 Å resolution. A comparison with the structure of glutathione reductase. J. Mol. Biol. 220, 975–994 [DOI] [PubMed] [Google Scholar]
- 24. Mattevi A., Obmolova G., Kalk K. H., van Berkel W. J., Hol W. G. (1993) Three-dimensional structure of lipoamide dehydrogenase from Pseudomonas fluorescens at 2.8 Å resolution. Analysis of redox and thermostability properties. J. Mol. Biol. 230, 1200–1215 [DOI] [PubMed] [Google Scholar]
- 25. Li de la Sierra I., Pernot L., Prangé T., Saludjian P., Schiltz M., Fourme R., Padrón G. (1997) Molecular structure of the lipoamide dehydrogenase domain of a surface antigen from Neisseria meningitidis. J. Mol. Biol. 269, 129–141 [DOI] [PubMed] [Google Scholar]
- 26. Faure M., Bourguignon J., Neuburger M., MacHerel D., Sieker L., Ober R., Kahn R., Cohen-Addad C., Douce R. (2000) Interaction between the lipoamide-containing H-protein and the lipoamide dehydrogenase (L-protein) of the glycine decarboxylase multienzyme system 2. Crystal structures of H- and L-proteins. Eur. J. Biochem. 267, 2890–2898 [DOI] [PubMed] [Google Scholar]
- 27. Rajashankar K. R., Bryk R., Kniewel R., Buglino J. A., Nathan C. F., Lima C. D. (2005) Crystal structure and functional analysis of lipoamide dehydrogenase from Mycobacterium tuberculosis. J. Biol. Chem. 280, 33977–33983 [DOI] [PubMed] [Google Scholar]
- 28. Mattevi A., Obmolova G., Sokatch J. R., Betzel C., Hol W. G. (1992) The refined crystal structure of Pseudomonas putida lipoamide dehydrogenase complexed with NAD+ at 2.45 Å resolution. Proteins Struct. Funct. Genet. 13, 336–351 [DOI] [PubMed] [Google Scholar]
- 29. Brautigam C. A., Chuang J. L., Tomchick D. R., Machius M., Chuang D. T. (2005) Crystal structure of human dihydrolipoamide dehydrogenase: NAD+/NADH binding and the structural basis of disease-causing mutations. J. Mol. Biol. 350, 543–552 [DOI] [PubMed] [Google Scholar]
- 30. Mande S. S., Sarfaty S., Allen M. D., Perham R. N., Hol W. G. (1996) Protein-protein interactions in the pyruvate dehydrogenase multienzyme complex: dihydrolipoamide dehydrogenase complexed with the binding domain of dihydrolipoamide acetyltransferase. Structure 4, 277–286 [DOI] [PubMed] [Google Scholar]
- 31. Ciszak E. M., Makal A., Hong Y. S., Vettaikkorumakankauv A. K., Korotchkina L. G., Patel M. S. (2006) How dihydrolipoamide dehydrogenase-binding protein binds dihydrolipoamide dehydrogenase in the human pyruvate dehydrogenase complex. J. Biol. Chem. 281, 648–655 [DOI] [PubMed] [Google Scholar]
- 32. Brautigam C. A., Wynn R. M., Chuang J. L., Machius M., Tomchick D. R., Chuang D. T. (2006) Structural insight into interactions between dihydrolipoamide dehydrogenase (E3) and E3-binding protein of human pyruvate dehydrogenase complex. Structure 14, 611–621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Nakai T., Kuramitsu S., Kamiya N. (2008) Structural bases for the specific interactions between the E2 and E3 components of the Thermus thermophilus 2-oxo acid dehydrogenase complexes. J. Biochem. 143, 747–758 [DOI] [PubMed] [Google Scholar]
- 34. Brautigam C. A., Wynn R. M., Chuang J. L., Naik M. T., Young B. B., Huang T.-H., Chuang D. T. (2011) Structural and thermodynamic basis for weak interactions between dihydrolipoamide dehydrogenase and subunit-binding domain of the branched-chain α-ketoacid dehydrogenase complex. J. Biol. Chem. 286, 23476–23488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Rice D. W., Schulz G. E., Guest J. R. (1984) Structural relationship between glutathione reductase and lipoamide dehydrogenase. J. Mol. Biol. 174, 483–496 [DOI] [PubMed] [Google Scholar]
- 36. Schierbeek A. J., Swarte M. B., Dijkstra B. W., Vriend G., Read R. J., Hol W. G., Drenth J., Betzel C. (1989) X-ray structure of lipoamide dehydrogenase from Azotobacter vinelandii determined by a combination of molecular and isomorphous replacement techniques. J. Mol. Biol. 206, 365–379 [DOI] [PubMed] [Google Scholar]
- 37. Altschul S. F., Gish W., Miller W., Myers E. W., Lipman D. J. (1990) Basic local alignment search tool. J. Mol. Biol. 215, 403–410 [DOI] [PubMed] [Google Scholar]
- 38. Nemeria N., Yan Y., Zhang Z., Brown A. M., Arjunan P., Furey W., Guest J. R., Jordan F. (2001) Inhibition of the Escherichia coli pyruvate dehydrogenase complex E1 subunit and its tyrosine 177 variants by thiamin 2-thiazolone and thiamin 2-thiothiazolone diphosphates. Evidence for reversible tight-binding inhibition. J. Biol. Chem. 276, 45969–45978 [DOI] [PubMed] [Google Scholar]
- 39. Yi J., Nemeria N., McNally A., Jordan F., Machado R. S., Guest J. (1996) Effect of substitutions in the thiamin diphosphate-magnesium fold on the activation of the pyruvate dehydrogenase complex from Escherichia coli by cofactors and substrate. J. Biol. Chem. 271, 33192–33200 [DOI] [PubMed] [Google Scholar]
- 40. Ali S. T., Guest J. R. (1990) Isolation and characterization of lipoylated and unlipoylated domains of the E2p subunit of the pyruvate dehydrogenase complex of Escherichia coli. Biochem. J. 271, 139–145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Song J., Jordan F. (2012) Interchain acetyl transfer in the E2 component of bacterial pyruvate dehydrogenase suggests a model with different roles for each chain in a trimer of the homooligomeric component. Biochemistry 51, 2795–2803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Park Y.-H. (2008) Protein-Protein Interactions in the Pyruvate Dehydrogenase Complex from Escherichia coli. Ph.D. thesis, Rutgers University [Google Scholar]
- 43. Nemeria N., Volkov A., Brown A., Yi J., Zipper L., Guest J. R., Jordan F. (1998) Systematic study of the six cysteines of the E1 subunit of the pyruvate dehydrogenase multienzyme complex from Escherichia coli: none is essential for activity. Biochemistry 37, 911–922 [DOI] [PubMed] [Google Scholar]
- 44. Pflugrath J. W. (1999) The finer things in x-ray diffraction data collection. Acta Crystallogr. D Biol. Crystallogr. 55, 1718–1725 [DOI] [PubMed] [Google Scholar]
- 45. Matthews B. W. (1968) Solvent content of protein crystals. J. Mol. Biol. 33, 491–497 [DOI] [PubMed] [Google Scholar]
- 46. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L.-W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., Richardson J. S., Terwilliger T. C., Zwart P. H. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Emsley P., Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 49. Davis I. W., Murray L. W., Richardson J. S., Richardson D. C. (2004) MOLPROBITY: Structure validation and all-atom contact analysis for nucleic acids and their complexes. Nucleic Acids Res. 32, W615–W619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Luzzati V. (1952) Traitement statistique des erreurs dans la determination des structures cristallines. Acta Crystallogr. 5, 802–810 [Google Scholar]
- 51. Carson M. (1991) RIBBONS 2.0. J. Appl. Crystallogr. 24, 958–961 [Google Scholar]
- 52. Delaglio F., Grzesiek S., Vuister G. W., Zhu G., Pfeifer J., Bax A. (1995) NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 6, 277–293 [DOI] [PubMed] [Google Scholar]
- 53. Keller R. L. (2004) The Computer Aided Resonance Assignment, Cantina Verlag, Goldau, Switzerland [Google Scholar]
- 54. Yamazaki T., Lee W., Arrowsmith C. H., Muhandiram D. R., Kay L. E. (1994) A suite of triple resonance NMR experiments for the backbone assignment of 15N-, 13C-, 2H labeled proteins with high sensitivity. J. Am. Chem. Soc. 116, 11655–11666 [Google Scholar]
- 55. Keppel T. R., Jacques M. E., Young R. W., Ratzlaff K. L., Weis D. D. (2011) An efficient and inexpensive refrigerated LC system for H/D exchange mass spectrometry. J. Am. Soc. Mass Spectrom. 22, 1472–1476 [DOI] [PubMed] [Google Scholar]
- 56. Hamuro Y., Coales S. J., Molnar K. S., Tuske S. J., Morrow J. A. (2008) Specificity of immobilized porcine pepsin in H/D exchange compatible conditions. Rapid Commun. Mass Spectrom. 22, 1041–1046 [DOI] [PubMed] [Google Scholar]
- 57. Weis D. D., Engen J. R., Kass I. J. (2006) Semi-automated data processing of hydrogen exchange mass spectra using HX-Express. J. Am. Soc. Mass Spectrom. 17, 1700–1703 [DOI] [PubMed] [Google Scholar]
- 58. Kavan D., Man P. (2011) MSTools—Web based application for visualization and presentation of HXMS data. Int. J. Mass Spectrom. 302, 53–58 [Google Scholar]
- 59. Nambi S., Badireddy S., Visweswariah S. S., Anand G. S. (2012) Cyclic AMP-induced conformational changes in mycobacterial protein acetyltransferases. J. Biol. Chem. 287, 18115–18129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Boyd D. B. (1972) Conformational dependence of the electronic energy levels in disulfides. J. Am. Chem. Soc. 94, 8799–8804 [Google Scholar]
- 61. Kim H. (2005) Asparagine 473 residue is important to the efficient function of human dihydrolipoamide dehydrogenase. J. Biochem. Mol. Biol. 38, 248–252 [DOI] [PubMed] [Google Scholar]
- 62. Park Y.-H., Wei W., Zhou L., Nemeria N., Jordan F. (2004) Amino-terminal residues 1–45 of the Escherichia coli pyruvate dehydrogenase complex E1 subunit interact with the E2 subunit and are required for activity of the complex but not for reductive acetylation of the E2 subunit. Biochemistry 43, 14037–14046 [DOI] [PubMed] [Google Scholar]
- 63. Zhang Z., Smith D. L. (1993) Determination of amide hydrogen exchange by mass spectrometry: a new tool for protein structure elucidation. Protein Sci. 2, 522–531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Weis D. D., Wales T. E., Engen J. R., Hotchko M., Ten Eyck L. F. (2006) Identification and characterization of EX1 kinetics in H/D exchange mass spectrometry by peak width analysis. J. Am. Soc. Mass Spectrom. 17, 1498–1509 [DOI] [PubMed] [Google Scholar]
- 65. Oliver R. M., Reed L. J. (1982) in Electron Microscopy of Proteins (Harris J. R., ed) Vol. 2, pp. 1–48, Academic Press, London [Google Scholar]
- 66. Knapp J. E., Mitchell D. T., Yazdi M. A., Ernst S. R., Reed L. J., Hackert M. L. (1998) Crystal structure of the truncated cubic core component of the Escherichia coli 2-oxoglutarate dehydrogenase multienzyme complex. J. Mol. Biol. 280, 655–668 [DOI] [PubMed] [Google Scholar]
- 67. Mattevi A., Obmolova G., Schulze E., Kalk K. H., Westphal A. H., de Kok A., Hol W. G. (1992) Atomic structure of the cubic core of the pyruvate dehydrogenase multienzyme complex. Science 255, 1544–1550 [DOI] [PubMed] [Google Scholar]
- 68. Allen M. D., Broadhurst R. W., Solomon R. G., Perham R. N. (2005) Interaction of the E2 and E3 components of the pyruvate dehydrogenase multienzyme complex of Bacillus stearothermophilus. Use of a truncated protein domain in NMR spectroscopy. FEBS J. 272, 259–268 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.