

Abstract
In these studies, we analyzed the effects of ozone on bronchiolar epithelium. Exposure of rats to ozone (2 ppm, 3h) resulted in rapid (within 3h) and persistent (up to 72h) histological changes in the bronchiolar epithelium, including hypercellularity, loss of cilia, and necrotizing bronchiolitis. Perivascular edema and vascular congestion were also evident, along with a decrease in Clara cell secretory protein in bronchoalveolar lavage, which was maximal 24h post-exposure. Ozone also induced the appearance of 8-hydroxy-2′-deoxyguanosine, Ym1, and heme oxygenase-1 in the bronchiolar epithelium. This was associated with increased expression of cleaved caspase-9 and beclin-1, indicating initiation of apoptosis and autophagy. A rapid and persistent increase in galectin-3, a regulator of epithelial cell apoptosis, was also observed. Following ozone exposure (3–24h), increased expression of cyclooxygenase-2, inducible nitric oxide synthase, and arginase-1 was noted in bronchiolar epithelium. Ozone-induced injury and oxidative stress in bronchiolar epithelium were linked to methacholine-induced alterations in pulmonary mechanics. Thus, significant increases in lung resistance and elastance, along with decreases in lung compliance and end tidal volume, were observed at higher doses of methacholine. This indicates that ozone causes an increase in effective stiffness of the lung as a consequence of changes in the conducting airways. Collectively, these studies demonstrate that bronchiolar epithelium is highly susceptible to injury and oxidative stress induced by acute exposure to ozone; moreover, this is accompanied by altered lung functioning.
Key Words: ozone, bronchiole, epithelium, oxidative stress, pulmonary mechanics
Ozone is a highly reactive oxidant and a common urban air pollutant. The toxicological effects of ozone are attributed to its ability to cause oxidation and peroxidation of membrane lipids and proteins, either directly or indirectly, through the generation of reactive intermediates (Pryor and Church, 1991). Distal structures of the lung including the terminal bronchioles, the bronchiole-alveolar duct junction, and proximal alveolar regions are considered primary targets of ozone. Thus, following exposure to ozone, necrosis of ciliated cells, deciliation and degeneration of secretory cells, and necrosis of Type I epithelial cells are observed. This is accompanied by altered vascular permeability and an accumulation of inflammatory cells in the lung, which are thought to contribute to the pathogenic response by releasing cytotoxic oxidants and proinflammatory cytokines (Laskin et al., 2011). Although ozone-induced inflammation and oxidative stress in alveolar epithelial regions of the lung have been well characterized, the response of epithelial cells in the terminal bronchioles is poorly described. As airway function is known to be altered by low-level ozone exposure, it seems likely that injury to the bronchiolar epithelium is important in the pathogenic response to this pulmonary irritant.
Epithelial cells in the terminal bronchioles (bronchiolar epithelial cells) are multifunctional cells that play a key role in inflammatory responses to lung injury (Proud and Leigh, 2011). Whereas initially they release bioactive lipids, cytokines, and chemokines that promote inflammation, subsequently they generate mediators involved in antioxidant defense and tissue repair. Airway epithelial cell wound repair responses also involve migration to cover damaged areas, followed by proliferation and differentiation to replace injured cells (Tesfaigzi, 2003). These findings suggest that persistent injury to the bronchiolar epithelium and impaired functioning may have significant consequences for restoring lung homeostasis. This is supported by reports linking dysregulated tissue repair processes in the bronchiolar epithelium with diseases such as asthma, chronic obstructive pulmonary disease, and pulmonary fibrosis and to alterations in lung functioning (Hackett and Knight, 2007; Puchelle et al., 2006).
In this study, we analyzed the effects of acute exposure of rats to ozone on the persistence of injury and inflammatory/antioxidant responses in the bronchiolar epithelium. The functional consequences of these alterations were also analyzed. Our findings that the bronchiolar epithelium is highly sensitive to ozone may be important in the development of new therapeutics for irritant-induced lung injury.
MATERIALS AND METHODS
Animals and exposures.
Female specific pathogen-free Wistar rats (200–225g) were obtained from Harlan Laboratories (Indianapolis, IN). Animals were housed in filter-top microisolation cages and maintained on food and water ad libitum. Animals received humane care in compliance with the institution’s guidelines, as outlined in the Guide for the Care and Use of Laboratory Animals, published by the National Institutes of Health. Animals were exposed to air or 2 ppm ozone for 3h in a whole-body Plexiglas chamber. Ozone was generated from oxygen gas via an ultraviolet-light ozone generator and mixed with air. Ozone concentrations inside the chamber were monitored using a Photometric analyzer (model 400E, Teledyne Instruments, City of Industry, CA). Animals were euthanized 3–72h after exposure by ip injection of Nembutal (250mg/kg). The lung was perfused in situ via the portal vein with 50ml of warm (37°C) perfusion medium (25mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid [HEPES], 0.5mM ethylene glycol tetraacetic acid, and 4.4mM NaHCO3 in Hank’s balanced salt solution [HBSS], pH 7.3), followed by perfusion with Ca2+/Mg2+-free HBSS (22mM HEPES and 4.2mM NaHCO3, pH 7.3) at a rate of 22ml/min.
Histology.
Following lung perfusion, the left bronchus was clamped and the largest lobe of the lung inflated in situ via the trachea with PBS containing 3% paraformaldehyde. After 4h on ice, the tissue was transferred to 50% ethanol. Histological sections (4 μm) were prepared, stained with hematoxylin and eosin (HE), and assessed blindly by a board-certified veterinary pathologist (Sherritta Ridgely, DVM, PhD). Images were acquired at high resolution using an Olympus VS120 Virtual Microscopy System, scanned using VS-ASW version 2.4 software and viewed using OlyVIA version 2.4 software (Center Valley, PA). Lung sections from four rats per treatment group were evaluated.
Collection of bronchoalveolar lavage.
Ice-cold PBS (10ml) was slowly instilled and withdrawn into the right bronchus. Bronchoalveolar lavage (BAL) fluid was centrifuged (300 × g, 10min, 4°C), and cell-free supernatants were aliquoted and stored at −80°C until analysis. BAL protein content was measured using a BCA Protein Assay kit (Pierce Biotechnologies Inc., Rockford, IL) with bovine serum albumin as the standard.
Western blotting.
BAL protein (0.5 μg) was fractionated on 20% SDS-polyacrylamide gel electrophoresis and transferred to polyvinylidene fluoride membranes (0.45 μm pore size; Millipore, Billerica, MA). Nonspecific binding was blocked by incubation of the blots for 2h at room temperature with 5% nonfat dry milk in 0.1% TBS-T buffer (0.02M Tris base, 0.137M sodium chloride, and 0.1% Tween-20). Blots were incubated overnight at 4°C with rabbit polyclonal anti-Clara cell secretory protein (CCSP, 1:10,000, Millipore). After five washes in 0.1% TBS-T buffer, blots were incubated with anti-rabbit (1:20,000) horseradish peroxidase–conjugated secondary antibody (Cell Signaling, Danvers, MA) for 1h at room temperature. Bands were visualized using an ECL detection system (GE, Healthcare Bio-Science Corp., Piscataway, NJ).
Immunohistochemistry.
Tissue sections were deparaffinized and subjected to antigen retrieval using citrate buffer (10.2mM sodium citrate and 0.05% Tween-20, pH 6.0). Sections were then incubated with 3% H2O2 for 15min to quench endogenous peroxidase and then with goat serum (2–10%, room temperature, 1h) to block nonspecific binding. This was followed by overnight incubation at 4°C with rabbit polyclonal antibody to iNOS (1:50; Santa Cruz Biotechnology Inc., Santa Cruz, CA), cyclooxygenase (COX)-2 (1:400; Abcam, Cambridge, MA), heme oxygenase (HO)-1 (1:200; Enzo Life Sciences, Plymouth Meeting, PA), cleaved caspase-9 (1:100; Cell Signaling), beclin-1 (1:200; Abcam), Ym1 (1:200; Stem Cell Technologies, Vancouver, Canada), or mouse monoclonal antibody to 8-hydroxy-2′-deoxyguanosine (8-OHdG, 1:500; Abcam), arginase-1 (1:50; BD Biosciences, Rockville, MD), galectin-3 (1:400; R&D Systems, Minneapolis, MN), or appropriate IgG controls. Sections were then incubated with biotinylated secondary antibody (Vector Labs, Burlingame, CA) for 30min at room temperature. Binding was visualized using a Peroxidase substrate kit DAB (Vector Labs). Sections from three rats per treatment group were analyzed.
Measurement of pulmonary mechanics.
Animals were anesthetized with ketamine (80mg/kg) and xylazine (10mg/kg). After 5min, tracheotomy was performed using a 15-G cannula; the animals were then attached to a SCIREQ flexiVent (Montreal, Canada), and baseline pulmonary mechanics assessed using forced oscillation and PV loop manipulations at a positive end-expiratory pressure of 3cm H2O. Animals were subsequently treated intratracheally with increasing doses of methacholine (0–96mg/ml) and measurements of pulmonary mechanics repeated. Input impedance data (ZL) were generated using an 8-s broadband flow perturbation. Resistance (RRS) and elastance (ERS) spectra were generated from ZL. Data were analyzed using flexiVent software version 5.2. Since the forced oscillation data failed to fit to the constant-phase model (Hirai et al., 1999), they were analyzed using an empirical model that takes into account greater regional variability in lung function (Groves et al., 2012). Matlab software was used for this analysis with the following equations:
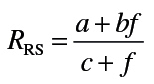 |
In these equations, f is the frequency, a the measure of low-frequency resistance, b the measure of high-frequency resistance, E0 the theoretical elastance at 0 Hz and thus a measure of inherent tissue stiffness, ΔE the magnitude of the change in frequency-dependent elastance, and β the measure of the rate of elastance change as a function of frequency.
Statistical analysis.
All experiments were repeated at least three times. For comparison of lung mechanics parameters, data from three to six rats per treatment group were analyzed by a nonpaired two-tailed Student’s t-test, and a p value of ≤ 0.05 was considered statistically significant. For each resistance and elastance spectrum, a best fit line was generated using nonlinear regression. For hypothesis testing using the mechanical impedance data, the residual error from curve fitting with nonlinear regression was used as a measure of goodness of fit. Comparison between air- and ozone-treated groups was made by evaluating the reduction in model error, when two independent curves were fit to the data (alternative hypotheses fit), versus the residual error when only one curve was used to fit the data (null hypotheses fit). This reduction in error allowed for the calculation of an F-statistic, which was used to determine statistical significance.
RESULTS
Ozone-Induced Injury and Oxidative Stress in the Terminal Bronchioles
Exposure of animals to ozone resulted in significant histological alterations in bronchiolar regions of the lung (Fig. 1). This included focal desquamation and hypercellularity of bronchiolar epithelium, loss of cilia, necrotizing bronchi- olitis, characterized by necrotic debris in the lumen, and interstitial inflammation. Desquamation and hypercellularity of pleural epithelium, mild pleural and perivascular edema and inflammation, and vascular congestion were also observed. These effects were evident within 3h of ozone exposure and persisted for at least 72h.
FIG. 1.

Effects of ozone on terminal airway histology. Tissue sections, prepared 3–72h after exposure of rats to air or ozone, were stained with H & E. Images were acquired using a VS120 Virtual Microscopy system. One representative lung section from four rats per treatment group is shown. Left panels: Original magnification, ×10. Right panels: Original magnification, ×40. ndl, necrotic debris in lumen; pve, perivascular edema; vc, vascular congestion; am, alveolar macrophage; hc, hypercellularity of bronchiolar epithelium; boxes show areas of higher magnification.
CCSP is secreted by Clara cells and is important in airway homeostasis, repair, and regeneration (Wong et al., 2009). CCSP was detected in bronchiolar epithelium and in BAL from control animals (Fig. 2; data not shown). Exposure of rats to ozone resulted in a time-related decrease in BAL CCSP levels. This was most notable 24h post-exposure; reduced levels persisted for 48h, returning to control by 72h postexposure. In contrast, no major changes were noted in bronchiolar epithelium (data not shown).
FIG. 2.
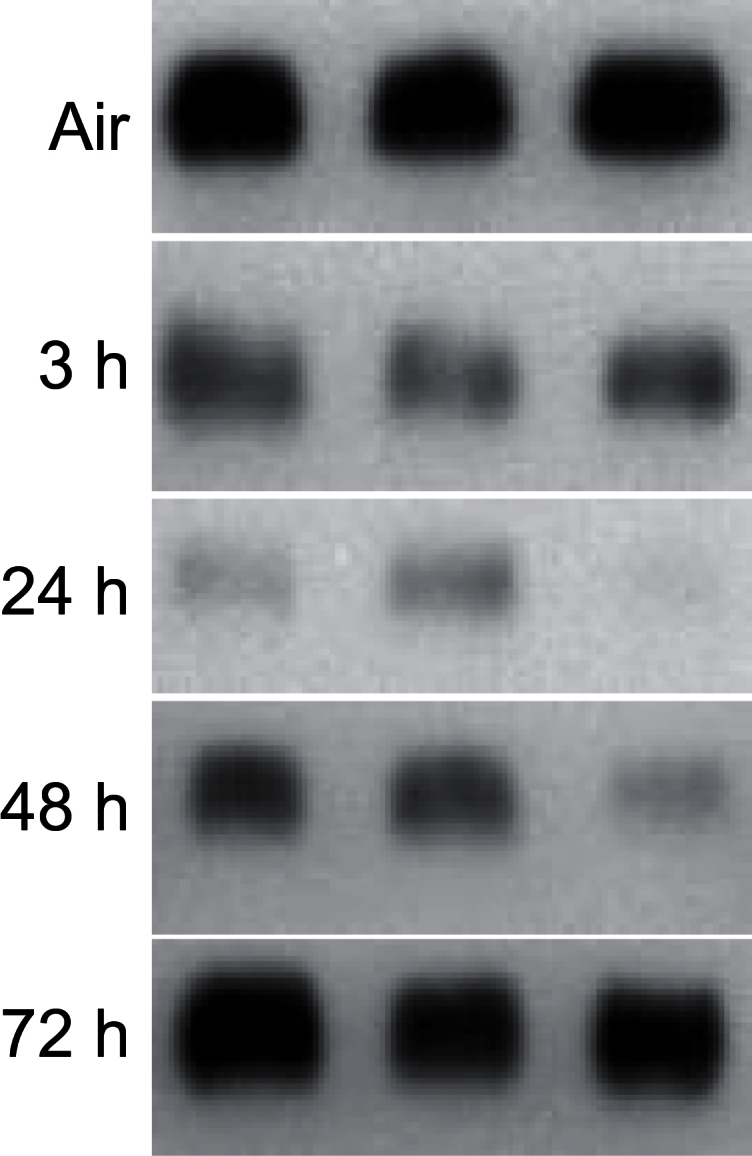
Effects of ozone on CCSP. BAL, collected 3–72h after exposure of rats to air or ozone, was analyzed for CCSP levels by Western blotting. Each lane represents BAL from one rat.
Ozone-induced structural alterations in the terminal bronchioles and decreases in CCSP levels were associated with the appearance of markers of oxidative stress including 8-OHdG and Ym1 (Fig. 3). Whereas increases in Ym1 were rapid, occurring within 3h, 8-OHdG was delayed for 24h. Expression of the antioxidant, HO-1, also increased in bronchiolar epithelium after ozone exposure; this response was biphasic, increasing initially after 3–6h, and then again at 48h (Fig. 3).
FIG. 3.

Effects of ozone on markers of oxidative stress. Lung sections, prepared 3–72h after exposure of rats to air or ozone, were stained with antibody to 8-OHdG, Ym1, or HO-1. Binding was visualized using a peroxidase DAB substrate kit. One representative section from three rats per treatment group is shown (original magnification, ×20).
Oxidative stress is known to result in activation of pathways associated with apoptosis and autophagy (Ryter and Choi, 2010). Consistent with ozone-induced oxidative stress, we observed a rapid (within 3h) and persistent (up to 48h) increase in bronchiolar epithelial cell expression of cleaved caspase-9, a marker of apoptosis (Fig. 4). This response was correlated with increases in expression of galectin-3, a β-galactoside-binding lectin reported to stimulate epithelial cell apoptosis (Pilette et al., 2007), 3–72h post-ozone exposure. We also found that expression of beclin-1, a marker of autophagy, increased in bronchiolar epithelium following ozone inhalation; however, this was delayed for 24h (Fig. 4).
FIG. 4.

Effects of ozone on markers of apoptosis and autophagy. Lung sections, prepared 3–72h after exposure of rats to air or ozone, were stained with antibody to cleaved caspase-9, galectin-3, or beclin-1. Binding was visualized using a peroxidase DAB substrate kit. One representative section from three rats per treatment group is shown (original magnification, ×20).
Effects of Ozone on Expression of Inflammatory Proteins in Terminal Bronchioles
We next analyzed expression of the proinflammatory proteins, COX-2 and iNOS, which have been implicated in ozone-induced lung injury (Laskin et al., 2011). Constitutive expression of COX-2 protein was noted in bronchiolar epithelium from control animals (Fig. 5). Exposure of rats to ozone resulted in increased expression of COX-2, as well as iNOS. While COX-2 increased rapidly (within 3h) and persisted for at least 72h, iNOS expression was transient, becoming most prominent at 24h and returning to control levels by 72h (Fig. 5). We also found that exposure of rats to ozone resulted in an increase in arginase-1 expression, which peaked after 6h (Fig. 5).
FIG. 5.

Effects of ozone on iNOS, COX-2, and arginase-1 expression. Lung sections, prepared 3–72h after exposure of rats to air or ozone, were stained with antibody to iNOS, COX-2, or arginase-1. Binding was visualized using a peroxidase DAB substrate kit. One representative section from three rats per treatment group is shown (original magnification, ×20).
Effects of Ozone on Pulmonary Mechanics
In our next series of studies, we determined if structural and inflammatory changes induced by ozone were associated with alterations in pulmonary mechanics. No significant differences in baseline lung function were noted between air- and ozone-exposed animals (data not shown). Upon challenge with methacholine, dose-dependent increases in total lung resistance were observed in air-exposed animals, with concomitant decreases in total lung compliance, end tidal volume, and static lung compliance (Fig. 6). Whereas exposure of animals to ozone had no significant effect on this pattern of response to methacholine, increases in the methacholine dependency of the changes in lung resistance were observed, along with blunting of changes in compliance, end tidal volume, and static compliance (Fig. 6). To elucidate potential mechanisms underlying these alterations, pulmonary impedance spectra generated using a forced oscillation technique were analyzed using an empirical model (Groves et al., 2012). Dose-dependent increases in resistance (RRS) and elastance (ERS) spectra were observed in both control and ozone-exposed rats in response to methacholine (Fig. 7, upper and middle panels). At higher doses of methacholine (48 and 96mg/ml), RRS and ERS spectra were significantly elevated in animals exposed to ozone, compared with control animals. Whereas differences in the RRS spectra between control and ozone-exposed rats at the higher methacholine doses occurred predominantly at lower frequencies (0–5 Hz), as exemplified by elevated values of a, increases in the ERS spectra were mainly observed at frequencies above 2 Hz, as reflected by increase in ΔE (Fig. 7, lower panels). In contrast, no changes were observed in the high-frequency resistance measure, b (not shown).
FIG. 6.

Effects of ozone on methacholine-induced pulmonary mechanics. Total lung resistance, total lung compliance, static compliance, and end tidal volume were evaluated in response to increasing doses of methacholine 24h following exposure of rats to air or ozone. Values were normalized and expressed as percentage change from baseline. Each point is the mean ± SE (n = 6 rats). *Significantly different (p ≤ 0.05) from air-exposed animals.
FIG. 7.

Effects of ozone on methacholine-induced resistance and elastance spectra. Total respiratory resistance (RRS) and elastance (ERS) were evaluated in response to increasing doses of methacholine (0–96mg/ml), 24h following exposure of rats to air or ozone. Data are cm H2O/ml/s (for RRS) and cm H2O/ml (for ERS). Top and middle panels: Best fit lines of the primary model fitting of the ZL spectra to the constant-phase model. Each point is the average value of three to four rats per treatment. Bottom panels: a, low-frequency resistance; ΔE, frequency dependence of elastance. *Significantly different (p ≤ 0.05) from air-exposed animals.
DISCUSSION
Epithelial cells in the lower airways are susceptible to injury following exposure to inhaled ozone. Thus, loss of ciliary function, increased airway epithelial permeability, and defects in mucociliary clearance have been described (Mustafa, 1990). In this study, we further characterized the response of the bronchiolar epithelium to ozone, assessing structural alterations, oxidative stress and expression of inflammatory proteins, and pulmonary mechanics.
Following ozone exposure, significant morphological alterations were observed in the bronchiolar epithelium including desquamation and hypercellularity, loss of cilia, necrotic debris in the lumen, and bronchiolar interstitial inflammation. Similar changes have been described in the terminal bronchioles of mice 48h after ozone exposure (Cho et al., 2001; Longphre et al., 1999). Our findings that injury to the bronchiolar epithelium occurs within 3h of ozone exposure are novel and consistent with reports that these cells are highly sensitive to oxidative injury (Broeckaert et al., 2003; Mustafa, 1990). The fact that histological and structural alterations persist in bronchiolar regions for at least 72h post-exposure suggests a mechanism underlying prolonged alterations in pulmonary mechanics, despite resolution of inflammation and injury in the lower lung (Groves et al., 2012).
CCSP levels have been reported to be reduced in various pulmonary diseases, consistent with its role in the maintenance of lung homeostasis (Wong et al., 2009). CCSP has also been shown to have anti-inflammatory functions. Ozone exposure resulted in a decrease in constitutive BAL CCSP levels. The observation that this response correlated with bronchiolar epithelial damage suggests that alterations in CCSP may contribute to ozone-induced injury. This is supported by findings that loss of CCSP results in hypersensitivity of mice to ozone, as well as to cigarette smoke and particulate matter (Plopper et al., 2006; Watson et al., 2001; Williams et al., 2012).
In previous studies, we demonstrated that ozone induces oxidative stress in alveolar macrophages, as measured by the appearance of 8-OHdG and expression of Ym1 and HO-1 (Sunil et al., 2012a). This study shows a similar response to ozone in the terminal bronchioles, suggesting that bronchiolar epithelial cells are also a target for oxidative DNA damage. This is in accord with findings of 8-OHdG in bronchiolar epithelium of mice exposed to cigarette smoke, which is known to cause oxidative stress (Aoshiba et al., 2003). Ym1 has been identified in proximal airway epithelium in allergic inflammation (Homer et al., 2006); moreover, treatment of mice with anti-Ym1/2 antibody attenuates the development of airway inflammation (Cai et al., 2009). These findings, along with reports of increases in lung Ym1 after exposure to diesel exhaust (Song et al., 2008) or vesicants (Sunil et al., 2011, 2012b), suggest that it may be a useful biomarker of oxidative epithelial cell injury. Increased expression of HO-1 was also observed in bronchiolar epithelium after ozone intoxication at 3–6h and then again at 48h. This biphasic response may reflect distinct functions of HO-1 in bronchiolar epithelium. Thus, while early in the pathogenic response, bronchiolar epithelial cells upregulate HO-1 to mitigate oxidative stress and DNA damage; at later times, they are involved in suppressing inflammation and initiating wound repair (Lee et al., 2009). This may be due to HO-1-mediated suppression of adhesion molecules important in inflammatory cell recruitment and/or attenuation of chemokine production (Zampetaki et al., 2003).
Following ozone exposure, we found evidence of both apoptosis and autophagy in the bronchiolar epithelium, consistent with oxidative stress. Hence, increases in early markers of apoptosis (cleaved caspase-9) and autophagy (beclin-1) were observed at 3 and 24h, respectively, post-ozone exposure. Analogous increases in cleaved caspase-9 have been observed in cultured human bronchial epithelial cells following exposure to particulate matter (Kamdar et al., 2008) and in airway epithelial cells from asthmatic patients (Comhair et al., 2005), conditions associated with oxidative stress. Apoptosis and autophagy are important in protection against oxidative stress and in the maintenance of cellular homeostasis (Lin and Thomas, 2010; Ryter and Choi, 2010), and they may have a similar function in the bronchiolar epithelium following ozone intoxication. We also observed increased expression of galectin-3 in bronchiolar epithelium 3–72h after ozone exposure. Galectin-3 induces apoptosis in epithelial cells, a process thought to be important in re-epithelialization during wound repair (Cao et al., 2002). Our findings that galectin-3 expression is rapid (within 3h) and prolonged (up to 72h) suggest that it may be involved in both protection against oxidative stress and wound repair.
COX-2 and iNOS are enzymes involved in the generation of cytotoxic eicosanoids and reactive nitrogen species (RNS). Previously, we reported that ozone exposure results in increased expression of COX-2 and iNOS in alveolar macrophages and Type II cells (Fakhrzadeh et al., 2002; Sunil et al., 2012a). This study demonstrates that COX-2 and iNOS are also upregulated in bronchiolar epithelial cells after ozone inhalation, consistent with the idea that these cells can synthesize RNS and eicosanoids and promote oxidative stress. Increases in COX-2 and iNOS expression have been described in the airway epithelium of asthmatic patients (Redington et al., 2001); iNOS has also been noted in bronchial ciliated cells and in nasal epithelium after smoke inhalation (Cox et al., 2009). Our findings of increased COX-2 and iNOS protein expression in bronchiolar epithelial cells provide additional support for the idea that these cells are important in the inflammatory response to ozone. Of note, significant levels of constitutive COX-2 expression were evident in the bronchiolar epithelium, which may be important in the maintenance of airway tone, as well as in protection against inflammation and injury (Fukunaga et al., 2005; Safholm et al., 2012). Evidence suggests that arginase-1 is involved in airway inflammation and hyperresponsiveness (Maarsingh et al., 2008). Consistent with these activities, we found that arginase-1 was rapidly upregulated in bronchiolar epithelium following ozone exposure. Arginase-1 has been reported to be increased in lung tissue from asthmatic patients (North et al., 2009); moreover, inhibition of arginase attenuated methacholine responsiveness of the central airways. It remains to be determined if arginase-1 similarly contributes to airway hypersensitivity following ozone exposure.
Methacholine is a potent bronchoconstrictor used to assess airway wall stiffness and parenchymal elasticity (Bates and Lauzon, 2007). In both control and ozone-treated animals, dose-dependent increases in total lung resistance were observed following methacholine challenge, with concomitant decreases in total lung compliance, end tidal volume, and static lung compliance. Interestingly, exposure of animals to ozone resulted in opposing effects on the methacholine-dependency of lung resistance and compliance. Thus, while increases in the absolute change in methacholine-dependent lung resistance were observed, decreases in compliance and static compliance were blunted, compared with control animals. Our data show no major differences between control and ozone-exposed rats in methacholine dose responsiveness, which is in contrast to prior studies (Savov et al., 2004; Shore et al., 2001) These earlier studies employed unrestrained plethysmography, a technique shown to be unreliable in the assessment of airway resistance (Lundblad, 2012). Thus, differences in our findings may be due to distinct methods used to assess lung mechanics.
Forced oscillation measurements have been used to differentiate between tissue and airway components of lung function. Traditionally, these data are fit to a constant-phase model to give estimates of certain aspects of lung function (Bates and Lutchen, 2005). However, following ozone intoxication, we found that the outcome impedance data did not fit the constant-phase model with sufficient stringency. This likely results from increased heterogeneity occurring as a response to injury. Therefore, the data from the resistance and elastance spectra were fit to an empirical model previously developed for heterogeneous lung injury (Groves et al., 2012). As predicted, methacholine challenge resulted in an increase in both the resistance and elastance spectra. Unexpectedly, however, changes in the resistance spectra were restricted to the low frequency range, estimated by the parameter a. These data support the idea that changes in the resistance spectra cannot be attributed to simple smooth muscle hyperreactivity, as such effects would be observed at high frequency. The parameter b in the empirical model, a measure of high-frequency lung resistance, was unaltered by methacholine or ozone treatment. As resistance in the high frequency range is attributed to airway caliber, these data suggest that ozone exposure produces minimal changes in resting smooth muscle tone and does not alter its intrinsic sensitivity to methacholine. Low frequency increases in resistance are thought to be due to collapse of small airways; however, such changes would be expected to produce large increases in the inherent stiffness of the lung, which were not observed, as estimated by the parameter E0, which was unaltered by ozone treatment. Rather, an increase in frequency-dependent elastance ΔE was noted, a response exacerbated by ozone exposure; moreover, this increase matched changes in low-frequency resistance a. These data indicate that ozone induces changes in the airflow through the conducting circuit, which are exacerbated by smooth muscle contraction. In other words, ozone exposure results in increases in airway stiffness, as well as resistance to airflow. This is supported by findings that the observed changes in pulmonary mechanics following ozone exposure correlate with pronounced airway epithelial hyperplasia and airway wall inflammation.
In summary, this study demonstrates that acute exposure of rats to ozone results in persistent injury and oxidative stress in the bronchiolar epithelium. This is associated with apoptosis and autophagy and increased expression of cytotoxic/proinflammatory proteins. Our findings that these pathogenic responses are correlated with increases in resistance and elastance in response to methacholine challenge suggest that the bronchiolar epithelium is important not only in ozone-induced inflammation but also in altered pulmonary mechanics. Further studies on the contribution of the bronchiolar epithelium to tissue remodeling and repair may provide new therapeutic options for populations at increased risk for ozone-induced acute lung injury.
FUNDING
National Institutes of Health (R01ES004738, R01GM034310, R01CA132624, R01HL086621, U54AR055073, and P30ES05022).
ACKNOWLEDGMENTS
The authors would like to thank Sherritta Ridgely, DVM, PhD, for histological evaluation of HE-stained sections, and Jianliang Shen, MS, for help with animals.
REFERENCES
- Aoshiba K., Koinuma M., Yokohori N., Nagai A. (2003). Immunohistochemical evaluation of oxidative stress in murine lungs after cigarette smoke exposure. Inhal. Toxicol. 15, 1029–1038 [DOI] [PubMed] [Google Scholar]
- Bates J. H., Lauzon A. M. (2007). Parenchymal tethering, airway wall stiffness, and the dynamics of bronchoconstriction. J. Appl. Physiol. 102, 1912–1920 [DOI] [PubMed] [Google Scholar]
- Bates J. H., Lutchen K. R. (2005). The interface between measurement and modeling of peripheral lung mechanics. Respir. Physiol. Neurobiol. 148, 153–164 [DOI] [PubMed] [Google Scholar]
- Broeckaert F., Clippe A., Wattiez R., Falmagne P., Bernard A. (2003). Lung hyperpermeability, Clara-cell secretory protein (CC16), and susceptibility to ozone of five inbred strains of mice. Inhal. Toxicol. 15, 1209–1230 [DOI] [PubMed] [Google Scholar]
- Cai Y., Kumar R. K., Zhou J., Foster P. S., Webb D. C. (2009). Ym1/2 promotes Th2 cytokine expression by inhibiting 12/15(S)-lipoxygenase: Identification of a novel pathway for regulating allergic inflammation. J. Immunol. 182, 5393–5399 [DOI] [PubMed] [Google Scholar]
- Cao Z., Said N., Amin S., Wu H. K., Bruce A., Garate M., Hsu D. K., Kuwabara I., Liu F. T., Panjwani N. (2002). Galectins-3 and -7, but not galectin-1, play a role in re-epithelialization of wounds. J. Biol. Chem. 277, 42299–42305 [DOI] [PubMed] [Google Scholar]
- Cho H. Y., Zhang L. Y., Kleeberger S. R. (2001). Ozone-induced lung inflammation and hyperreactivity are mediated via tumor necrosis factor-alpha receptors. Am. J. Physiol. Lung Cell Mol. Physiol. 280, L537–L546 [DOI] [PubMed] [Google Scholar]
- Comhair S. A., Xu W., Ghosh S., Thunnissen F. B., Almasan A., Calhoun W. J., Janocha A. J., Zheng L., Hazen S. L., Erzurum S. C. (2005). Superoxide dismutase inactivation in pathophysiology of asthmatic airway remodeling and reactivity. Am. J. Pathol. 166, 663–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox R. A., Jacob S., Oliveras G., Murakami K., Enkhbaatar P., Traber L., Schmalstieg F. C., Herndon D. N., Traber D. L., Hawkins H. K. (2009). Pulmonary expression of nitric oxide synthase isoforms in sheep with smoke inhalation and burn injury. Exp. Lung Res. 35, 104–118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fakhrzadeh L., Laskin J. D., Laskin D. L. (2002). Deficiency in inducible nitric oxide synthase protects mice from ozone-induced lung inflammation and tissue injury. Am. J. Respir. Cell Mol. Biol. 26, 413–419 [DOI] [PubMed] [Google Scholar]
- Fukunaga K., Kohli P., Bonnans C., Fredenburgh L. E., Levy B. D. (2005). Cyclooxygenase 2 plays a pivotal role in the resolution of acute lung injury. J. Immunol. 174, 5033–5039 [DOI] [PubMed] [Google Scholar]
- Groves A. M., Gow A. J., Massa C. B., Laskin J. D., Laskin D. L. (2012). Prolonged injury and altered lung function after ozone inhalation in mice with chronic lung inflammation. Am. J. Respir. Cell Mol. Biol. 47, 776–783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hackett T. L., Knight D. A. (2007). The role of epithelial injury and repair in the origins of asthma. Curr. Opin. Allergy Clin. Immunol. 7, 63–68 [DOI] [PubMed] [Google Scholar]
- Hirai T., McKeown K. A., Gomes R. F., Bates J. H. (1999). Effects of lung volume on lung and chest wall mechanics in rats. J. Appl. Physiol. 86, 16–21 [DOI] [PubMed] [Google Scholar]
- Homer R. J., Zhu Z., Cohn L., Lee C. G., White W. I., Chen S., Elias J. A. (2006). Differential expression of chitinases identify subsets of murine airway epithelial cells in allergic inflammation. Am. J. Physiol. Lung Cell Mol. Physiol. 291, L502–L511 [DOI] [PubMed] [Google Scholar]
- Kamdar O., Le W., Zhang J., Ghio A. J., Rosen G. D., Upadhyay D. (2008). Air pollution induces enhanced mitochondrial oxidative stress in cystic fibrosis airway epithelium. FEBS Lett. 582, 3601–3606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laskin D. L., Sunil V. R., Gardner C. R., Laskin J. D. (2011). Macrophages and tissue injury: Agents of defense or destruction? Annu. Rev. Pharmacol. Toxicol. 51, 267–288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee I. T., Luo S. F., Lee C. W., Wang S. W., Lin C. C., Chang C. C., Chen Y. L., Chau L. Y., Yang C. M. (2009). Overexpression of HO-1 protects against TNF-alpha-mediated airway inflammation by down-regulation of TNFR1-dependent oxidative stress. Am. J. Pathol. 175, 519–532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin J. L., Thomas P. S. (2010). Current perspectives of oxidative stress and its measurement in chronic obstructive pulmonary disease. COPD. 7, 291–306 [DOI] [PubMed] [Google Scholar]
- Longphre M., Zhang L., Harkema J. R., Kleeberger S. R. (1999). Ozone-induced pulmonary inflammation and epithelial proliferation are partially mediated by PAF. J. Appl. Physiol. 86, 341–349 [DOI] [PubMed] [Google Scholar]
- Lundblad L. K. (2012). Issues determining direct airways hyperresponsiveness in mice. Front. Physiol. 3, 408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maarsingh H., Pera T., Meurs H. (2008). Arginase and pulmonary diseases. Naunyn Schmiedebergs Arch. Pharmacol. 378, 171–184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mustafa M. G. (1990). Biochemical basis of ozone toxicity. Free Radic. Biol. Med. 9, 245–265 [DOI] [PubMed] [Google Scholar]
- North M. L., Khanna N., Marsden P. A., Grasemann H., Scott J. A. (2009). Functionally important role for arginase 1 in the airway hyperresponsiveness of asthma. Am. J. Physiol. Lung Cell Mol. Physiol. 296, L911–L920 [DOI] [PubMed] [Google Scholar]
- Pilette C., Colinet B., Kiss R., André S., Kaltner H., Gabius H. J., Delos M., Vaerman J. P., Decramer M., Sibille Y. (2007). Increased galectin-3 expression and intra-epithelial neutrophils in small airways in severe COPD. Eur. Respir. J. 29, 914–922 [DOI] [PubMed] [Google Scholar]
- Plopper C. G., Mango G. W., Hatch G. E., Wong V. J., Toskala E., Reynolds S. D., Tarkington B. K., Stripp B. R. (2006). Elevation of susceptibility to ozone-induced acute tracheobronchial injury in transgenic mice deficient in Clara cell secretory protein. Toxicol. Appl. Pharmacol. 213, 74–85 [DOI] [PubMed] [Google Scholar]
- Proud D., Leigh R. (2011). Epithelial cells and airway diseases. Immunol. Rev. 242, 186–204 [DOI] [PubMed] [Google Scholar]
- Pryor W. A., Church D. F. (1991). Aldehydes, hydrogen peroxide, and organic radicals as mediators of ozone toxicity. Free Radic. Biol. Med. 11, 41–46 [DOI] [PubMed] [Google Scholar]
- Puchelle E., Zahm J. M., Tournier J. M., Coraux C. (2006). Airway epithelial repair, regeneration, and remodeling after injury in chronic obstructive pulmonary disease. Proc. Am. Thorac. Soc. 3, 726–733 [DOI] [PubMed] [Google Scholar]
- Redington A. E., Meng Q. H., Springall D. R., Evans T. J., Créminon C., Maclouf J., Holgate S. T., Howarth P. H., Polak J. M. (2001). Increased expression of inducible nitric oxide synthase and cyclo-oxygenase-2 in the airway epithelium of asthmatic subjects and regulation by corticosteroid treatment. Thorax. 56, 351–357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryter S. W., Choi A. M. (2010). Autophagy in the lung. Proc. Am. Thorac. Soc. 7, 13–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Safholm J., Dahlen S. E., Delin I., Maxey K., Stark K., Cardell L. O., Adner M. (2012). Prostaglandin E2 maintains the tone of the guinea pig trachea through a balance between activation of contractile EP1 receptors and relaxant EP2 receptors. Br. J. Pharmacol. 168, 794–806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savov J. D., Whitehead G. S., Wang J., Liao G., Usuka J., Peltz G., Foster W. M., Schwartz D. A. (2004). Ozone-induced acute pulmonary injury in inbred mouse strains. Am. J. Respir. Cell Mol. Biol. 31, 69–77 [DOI] [PubMed] [Google Scholar]
- Shore S. A., Schwartzman I. N., Le Blanc B., Murthy G. G., Doerschuk C. M. (2001). Tumor necrosis factor receptor 2 contributes to ozone-induced airway hyperresponsiveness in mice. Am. J. Respir. Crit. Care Med. 164, 602–607 [DOI] [PubMed] [Google Scholar]
- Song H. M., Jang A. S., Ahn M. H., Takizawa H., Lee S. H., Kwon J. H., Lee Y. M., Rhim T. Y., Park C. S. (2008). Ym1 and Ym2 expression in a mouse model exposed to diesel exhaust particles. Environ. Toxicol. 23, 110–116 [DOI] [PubMed] [Google Scholar]
- Sunil V. R., Patel-Vayas K., Shen J., Gow A. J., Laskin J. D., Laskin D. L. (2011). Role of TNFR1 in lung injury and altered lung function induced by the model sulfur mustard vesicant, 2-chloroethyl ethyl sulfide. Toxicol. Appl. Pharmacol. 250, 245–255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sunil V. R., Patel-Vayas K., Shen J., Laskin J. D., Laskin D. L. (2012a). Classical and alternative macrophage activation in the lung following ozone-induced oxidative stress. Toxicol. Appl. Pharmacol. 263, 195–202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sunil V. R., Shen J., Patel-Vayas K., Gow A. J., Laskin J. D., Laskin D. L. (2012b). Role of reactive nitrogen species generated via inducible nitric oxide synthase in vesicant-induced lung injury, inflammation and altered lung functioning. Toxicol. Appl. Pharmacol. 261, 22–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tesfaigzi Y. (2003). Processes involved in the repair of injured airway epithelia. Arch. Immunol. Ther. Exp. (Warsz.). 51, 283–288 [PubMed] [Google Scholar]
- Watson T. M., Reynolds S. D., Mango G. W., Boe I. M., Lund J., Stripp B. R. (2001). Altered lung gene expression in CCSP-null mice suggests immunoregulatory roles for Clara cells. Am. J. Physiol. Lung Cell Mol. Physiol. 281, L1523–L1530 [DOI] [PubMed] [Google Scholar]
- Williams K. M., Franzi L. M., Last J. A. (2012). Cell-specific oxidative stress and cytotoxicity after wildfire coarse particulate matter instillation into mouse lung. Toxicol. Appl. Pharmacol. 266, 48–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong A. P., Keating A., Waddell T. K. (2009). Airway regeneration: The role of the Clara cell secretory protein and the cells that express it. Cytotherapy. 11, 676–687 [DOI] [PubMed] [Google Scholar]
- Zampetaki A., Minamino T., Mitsialis S. A., Kourembanas S. (2003). Effect of heme oxygenase-1 overexpression in two models of lung inflammation. Exp. Biol. Med. (Maywood). 228, 442–446 [DOI] [PubMed] [Google Scholar]