

Abstract
Tobacco smoking is a risk factor for cancers of the liver and gastrointestinal (GI) tract, but the causal agents responsible for these cancers are uncertain. 2-Amino-9H-pyrido[2,3-b]indole (AαC) is an abundant heterocyclic aromatic amine present in tobacco smoke. AαC is a liver carcinogen and both a transgene mutagen and inducer of aberrant crypt foci in the colon of mice. We hypothesize that AαC may contribute to DNA damage and tumorigenesis in these organs of smokers. The potential of AαC to induce DNA adduct formation in liver, organs of the GI tract, lung, and urinary bladder, which are target organs of cancer in smokers, was examined using the C57BL/6 mouse as an animal model. AαC (400 or 800 ppm) and 2-amino-3,4-dimethylimidazo[4,5-f]quinoline (MeIQ) (300 ppm), a liver and colon carcinogen in C57BL/6 mice, were given in the diet for up to 12 weeks. Liquid chromatography/mass spectrometry was employed to measure DNA adducts. The major DNA adducts of both carcinogens were identified as deoxyguanosine-C8 adducts. The levels of formation of AαC- and MeIQ-DNA adducts were similar in liver and extrahepatic tissues when adjusted for dose. The highest levels of adducts occurred in liver, followed by urinary bladder, and then in cecum and colon; lower DNA adduct levels were formed in the lung and pancreas following 12 weeks of feeding. The high levels of AαC adduct formed in liver, GI tract, and bladder of C57BL/6 mice reinforce the notion that AαC may contribute to DNA damage and cancer of these organs in smokers.
Key Words: tobacco smoke, carcinogens, DNA adducts, heterocyclic aromatic amines.
Tobacco smoking is a risk factor for lung and urinary bladder cancer but also for cancers of the oral cavity, larynx, liver, pancreas, and digestive tract (IARC, 2004, 2012; Giovannucci, 2001). Tobacco-specific N-nitrosamines are thought to contribute to lung cancer, and aromatic amines are viewed as causal agents of bladder cancer in smokers, but the chemicals in tobacco associated with liver and digestive tract cancers are uncertain. A series of heterocyclic aromatic amines (HAAs) are formed during the cooking of meats and have been implicated as causal agents in the etiology of colorectal cancer (Sugimura et al., 2004). Most of these HAAs are present at negligible levels in mainstream tobacco smoke (< 1ng/cigarette) and unlikely to explain the elevated risk of liver and GI tract cancers in smokers. The HAA, 2-amino-9H-pyrido[4,3-b]indole (AαC), a pyrolysis product of protein, however, is present in main stream tobacco smoke at levels up to 258ng/cigarette (Zhang et al., 2011). These levels are 25- to 100-fold greater than the amounts of 4-aminobiphenyl (4-ABP) (Hoffmann, 1998), an aromatic amine that has been implicated in the pathogenesis of bladder cancer in smokers (IARC, 2004, 2012) Recently, AαC was detected in the urine of individuals of the Shanghai cohort study (Turesky et al., 2007). The number of cigarettes smoked per day was positively and significantly related to urinary levels of AαC, signifying that tobacco smoke is an important point source of AαC exposure.
4-ABP and 2-aminofluorene (2-AF), the most well studied among the carcinogenic aromatic amines (Kriek, 1992), are bladder and liver carcinogens in rodents (Poirier and Beland, 1992). In humans, one case-control study measured 4-ABP-DNA adducts, by immunohistochemistry methods, in hepatocytes of patients with hepatocellular carcinoma. A statistically significant increase in risk for hepatocellular carcinoma was reported with increasing levels of 4-ABP adducts (Wang et al., 1998). Because cigarette smoking is a major source of exposure to 4-ABP in humans, this molecular epidemiologic study has strengthened the notion that tobacco smoke is a hepatic carcinogen in humans. Epidemiologic studies have also consistently reported that smoking is risk factor for colorectal cancer (IARC, 2004, 2012; Giovannucci, 2001; Gong et al., 2012).
Apart from the endocyclic nitrogen atoms, AαC has the same chemical structure as 2-AF. AαC is a liver carcinogen in CDF1 mice (Sugimura et al., 2004) and both a potent lacI transgene colon mutagen and an inducer of aberrant crypt foci (ACF), early biomarkers of neoplasms, in the colon of C57BL/6 mice (Okonogi et al., 1997; Sugimura et al., 2004; Zhang et al., 1996). The genotoxic potential of AαC in humans is not known; however, AαC readily undergoes metabolic activation by human hepatocytes to form the DNA adduct N-(deoxyguanosin-8-yl)-dG-C8-AαC (dG-C8-AαC) (Nauwelaers et al., 2011), which is a mutagenic lesion (Turesky et al., 2009).
The ability of chemical carcinogens to form DNA adducts is regarded as one important factor in their carcinogenic potential (Jarabek et al., 2009). The C57BL/6 mouse has recently been used as a model for colorectal mutagenesis studies (Nagao et al., 2001), and this mouse strain is more susceptible to intestinal carcinogenesis than the commonly used CDF1 mouse model for several classes of genotoxicants, including HAAs (Fujita et al., 1999; Ochiai et al., 2002). There are no reports on dG-C8-AαC adduct formation in C57BL/6 mice. In this study, we investigated the potential of AαC to form DNA adducts in liver, organs of the digestive tract, lung, and urinary bladder, which are target organs of cancer in smokers, using the C57BL/6 mouse as an animal model. AαC was given in the diet for up to 12 weeks at dosages that induced lacI mutations in liver and colon and ACF in colon. The levels of AαC-DNA adduct formation were compared with those levels of adducts formed by 2-amino-3,4-dimethylimidazo[4,5-f]quinoline (MeIQ), an HAA formed in cooked meat and broiled fish (Kasai et al., 1980). MeIQ undergoes bioactivation to form N-(deoxyguanosin-8-yl)-dG-C8-MeIQ as the principal adduct (Tada et al., 1994), and MeIQ is a liver and colon carcinogen in C57BL/6 mice (Fujita et al., 1999). The structures of AαC, MeIQ, and the arylamines, 4-ABP, and 2-acetylaminofluorene (2-AAF), the N-acetylated derivative of 2-AF, are shown in Figure 1, and 4-ABP and 2-AAF are liver and bladder carcinogens in mice (Poirier and Beland, 1992).
Fig. 1.

Chemical structures of AαC, MeIQ, 4-ABP, and 2-AAF.
MATERIALS AND METHODS
Caution.
AαC and MeIQ and their derivatives are carcinogenic to rodents and should be handled accordingly.
Chemicals and reagents.
AαC and MeIQ were purchased from Toronto Research Chemicals (Toronto, ON, Canada). [13C10]-dG (isotopic purity 99%) was purchased from Cambridge Isotopes (Andover, MA). 5ʹ-[13C10 15N5]-Deoxyguanosine-5ʹ-phosphate (dGMP, isotopic purity 99%) was purchased from Sigma (St Louis, MO). DNase I (Type IV, from bovine pancreas), alkaline phosphatase (from Escherichia coli), and nuclease P1 (from Penicillium citrinum) were purchased from Sigma. Phosphodiesterase I (from Crotalus adamanteus venom) was purchased from Worthington Biochemical Corp. (Lakewood, NJ). All solvents used were high-purity B & J Brand from Honeywell Burdick and Jackson (Muskegon, MI).
Synthesis of DNA adducts of AαC and MeIQ.
5′-[13C10 15N5]-dGMP (5mg) underwent enzymatic hydrolysis with phosphodiesterase I and alkaline phosphatase, and the resulting [13C10 15N5]-dG was purified by HPLC. N-(Deoxyguanosin-8-yl)-AαC (dG-C8-AαC) and N-(deoxyguanosin-8-yl)-MeIQ (dG-C8-MeIQ) were prepared by reaction of their N-acetoxy-HAA derivatives with dG, [13C10]-dG, or [13C10 15N5]-dG (1mg/ml) in 100mM potassium phosphate buffer (pH 8.0) and purified by preparative HPLC (Bessette et al., 2009).
Oxidative studies on dG-C8-AαC and dG-C8-MeIQ.
dG-C8-AαC and dG-C8-MeIQ (50ng/0.1ml) were oxidized under aerobic conditions at 50°C in (A) 1N NaOH for 2h, followed by neutralization with an equal amount of 1N HCl, (B) TE buffer (50mM Tris-HCl and 10mM EDTA [pH 7.0] with H2O2 [175mM] for 20h, or (C) TE buffer (pH = 7.0) for 20h.
Animal studies: DNA adduct formation in liver and extrahepatic tissues.
Male and female C57BL/6J mice, 5–6 weeks of age, were purchased from Jackson Laboratories (Bar Harbor, ME). The use of these animals was in compliance with guidelines established by the National Institutes of Health Office of Laboratory Animal Welfare. Animals were housed four per cage with corn cob bedding in pathogen-free animal quarters of Research Animal Resources, University of Minnesota Academic Health Center. The mice were given a standard AIN-93G rodent diet containing 7% soybean oil (Dyets Inc., Bethlehem, PA) for 7days. One week after arrival, the mice were switched to AIN-93G-powdered diet containing 13% Primex and 10% soybean oil. A high fat diet was previously used to induce ACF in AαC-treated mice (Okonogi et al., 1997; Sugimura et al., 2004; Zhang et al., 1996). Mice of the treatment groups received AαC (400 or 800 ppm) or MeIQ (300 ppm) in the diet. These concentrations of carcinogens were previously used in long-term carcinogen studies and subchronic mutagenesis and ACF studies (Nagao et al., 2001; Sugimura et al., 2004; Zhang et al., 1996). The carcinogens were mixed with the diet as follows: for 1kg of diet, 180g of casein was added to 720g of AIN-93 G diet and mixed for 30min using a diet blender (Fleetwood food equipment, Philadelphia, PA). Subsequently, the carcinogens were added, and the mixture was further mixed for 30min. Finally, 100ml of soybean oil (Dyets Inc.) was added and mixed for another 30min. The diets were prepared every 4 weeks and stored in airtight plastic bags at 4°C. Chemical analysis, by LC/MS, showed that the carcinogens were stable in the diet over 4 weeks (R. Turesky, unpublished observations). The diet was administered using metal box feeders (Lab Products Inc., Seaford, DE). Fresh diet was provided every 3–4days. Food consumption and body weight were monitored, respectively, twice a week and weekly. The average estimated daily food intake for male and female mice was, respectively, 3.1 and 3.2g both for control and carcinogen-supplemented diets. The final body weights of the AαC- and MeIQ-treated animals were within 5% of the average body weights of untreated female mice and 10% of the average body weights of untreated male mice. The mice were euthanized by an overdose of carbon dioxide, and organs were harvested. The liver, lung, pancreas, and bladder were quickly excised, rinsed in cold saline, and snapped frozen in liquid nitrogen. The cecum and colon (ascending colon to the rectum) were cut longitudinally, washed in PBS, and then soaked for 15−20min in PBS containing 1.5 mM EDTA, 3000U heparin/l, DTT (80mg/l), and PMSF (40mg/l). Subsequently, the mucosal epithelial cell layer was scraped with a glass slide; epithelial cells were twice rinsed with chilled saline and then frozen in liquid nitrogen.
Isolation and enzymatic digestion of DNA.
The entire liver, lung, pancreas, cecum, and colon were homogenized in TE buffer. The nuclear pellets were obtained by centrifugation at 3000 × g for 10min. The equivalent of ~100mg tissue from nuclear pellets of liver, lung, and pancreas and the entire nuclear pellets obtained from the cecum, colon, and bladder homogenates were used for DNA isolation. The DNA from nuclear pellets was isolated by the chloroform/phenol extraction method (Gupta, 1993), except that the 2 mM ß-mercaptoethanol (ßME) was freshly added to the TE buffer.
DNA (10 μg) from animals, N = 4 per group, for all time points except for the 12-week time point, where there were two animals, was digested and analyzed in triplicate. For the case of bladder tissue, due to the limiting amount of material, DNA (2 µg) was digested and assayed in duplicate.
The internal standards [13C10]-dG-C8-AαC and [13C10 15N5]-dG-C8-MeIQ were added to DNA at a level of six adducts per 107 nucleotides prior to enzymatic digestion. The enzymatic hydrolysis of DNA was performed under conditions shown to be highly efficient in the recovery of HAA-DNA adducts (Nauwelaers et al., 2011).
Ultraperformance liquid chromatography-electrospray ionization/ multistage mass spectrometry analyses of DNA adducts.
Analysis was done with a NanoAcquity UPLC system (Waters Corporation, Milford, MA) interfaced with a linear quadrupole ion trap mass spectrometer (LTQ MS; Thermo Fisher, San Jose, CA), and an Advance CaptiveSpray source from Michrom Bioresources Inc. (Auburn, CA). A Waters Symmetry trap column (180 μm × 20mm, 5 μm particle size) was employed for online solid-phase enrichment. The analytical column was a C18 AQ (0.3×150mm, 3 μm particle size) from Michrom Bioresources (Auburn, CA). The DNA digests were injected onto the trap column and washed with 0.2% formic acid in 10% acetonitrile at a flow rate of 12 μl/min for 5min. Thereafter, the DNA adducts were back-flushed onto the C18 AQ column. A linear gradient was employed to resolve the DNA adducts, starting at 0.01% formic acid containing 10% acetonitrile and arriving at 0.01% formic acid in 95% acetonitrile at 20min. The flow rate was set at 5 μl/min.
Xcalibur version 2.1.0 software was used for data manipulations. The adducts were measured at the MS3 scan stage in the positive ionization mode. The MS3 transitions employed for quantitative measurements were dG-C8-AαC at m/z 449.1 → 333.1, [13C10]-dG-C8-AαC at m/z 459.1 → 338.1, dG-C8-MeIQ at m/z 478.1 → 362.1, and [13C10 15N5]-dG-C8-MeIQ at m/z 493.1 → 372.1. The total ion counts were employed for measurement at the MS3 scan stage. The mass spectral parameters were optimized as previously reported (Nauwelaers et al., 2011). The characterization of HAA-DNA adducts and oxidation products was also performed by data-dependent constant neutral loss scanning, followed by acquisition of the triple-stage MS3 (CNL/MS3) spectra of the aglycone adducts as previously reported (Bessette et al., 2009).
Statistical methods.
Statistical differences between mean values of DNA adducts formed in specific organs were compared by one-way ANOVA, followed by Bonferroni’s multiple comparison test for adduct levels in liver versus other organs. The comparison of mean levels of DNA adducts formed in liver of AαC- and MeIQ-treated mice at day 1 and day 7 was done by the Student’s t-test. All statistical hypothesis testing was performed at the α = 0.05 significance level, and all statistical tests were two sided. Analyses were done with GraphPad Prism 4 software (San Diego, CA).
RESULTS
Identification of dG-C8-AαC and dG-C8-MeIQ Adducts in Mouse Liver and Oxidative Degradation of dG-C8-AαC During Sample Workup
The bioactivation of AαC and MeIQ is carried out by cytochrome P450 (P450) enzymes. Oxidation of the exocyclic amine group produces genotoxic N-hydroxy-HAA metabolites. These metabolites can directly react with DNA or undergo further metabolism by phase II enzymes, such as N-acetyltransferase (NAT) or sulfotransferase (SULT) to form esters, which are unstable and undergo heterolytic cleavage to produce the proposed reactive nitrenium ion that binds covalently to DNA (Turesky and Le Marchand, 2011). The N-hydroxy metabolite of AαC also undergoes bioactivation by UDP-glucuronosyltransferases (UGT), a group of enzymes that are usually involved in detoxication of HAAs and aromatic amines (Tang et al., 2012). The structures of dG-C8-AαC and dG-C8-MeIQ are shown in Figure 2.
Fig. 2.

Bioactivation of AαC and MeIQ by P450 and phase II enzymes to form the DNA adducts.
Our initial findings showed that the levels of dG-C8-AαC formation were highly variable in liver, and the estimates of adducts were not reproducible. We speculated that dG-C8-AαC had undergone oxidation during the isolation of the DNA. The dG-C8 adduct of 2-AF, N-(deoxyguanosin-8-yl)-2-AF (dG-C8-AF), which is very similar in structure to dG-C8-AαC, undergoes facile aerial oxidation of the guanine ring to form the spirodiasteroisomeric adducts containing an iminoimidazolidine-one moiety (Shibutani et al., 1990). The guanyl moiety of dG-C8-AF can also undergo oxidation, followed by cleavage, to produce a ring-opened adduct containing the residual guanidino group of dG (Shibutani et al., 1990). We surmised that dG-C8-AαC also underwent oxidation to form the same types of ring-opened adducts (Fig. 3).
Fig. 3.
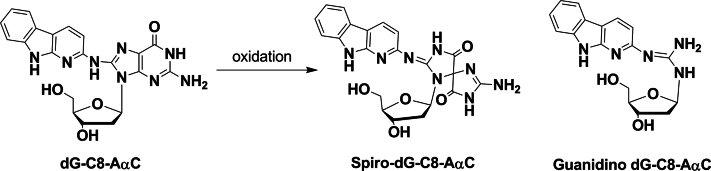
The proposed products formed from the aerial oxidation of dG-C8-AαC.
The ultraperformance liquid chromatography-electrospray ionization/multistage mass spectrometry (UPLC-ESI/MS3) chromatograms of liver DNA digest from an untreated animal and DNA from an AαC-treated animal either without or with ßME added as an antioxidant during the isolation of DNA were monitored for dG-C8-AαC and its putative oxidation products (Fig. 4). No adducts were detected in the chromatogram of the DNA digest from the untreated animal (Fig. 4A), whereas low levels of dG-C8-AαC and peaks attributed to the ring-opened oxidation products of dG-C8-AαC were observed in the chromatogram of DNA isolated without ßME (Fig. 4B). The addition of ßME (2mM) in the TE buffer prevented most of the oxidation of dG-C8-AαC and greatly suppressed the formation of the proposed oxidized adducts (Fig. 4C).
Fig. 4.

The UPLC-ESI/MS3 chromatogram of dG-C8 adducts of AαC and ring-opened oxidation products in liver DNA from (A) untreated mouse and DNA isolated with ßME, (B) mouse treated with AαC (800 ppm) for 4 weeks and DNA isolated without ßME, and (C) mouse treated with AαC (800 ppm) for 4 weeks and DNA isolated with ßME and (D) MS3 scan stage product ion spectra of dG-C8-AαC (m/z 449.1 > 333.1 >), the proposed oxidized spiro-dG-C8-AαC (m/z 465.1 > 349.1 >), the proposed ring-opened guanidino derivative of dG-C8-AαC (342.1 > 226.1 >), and the MS4 scan stage product ion spectrum of guanidino derivative of dG-C8-AαC (342.1 > 226.1 > 209.1 >).
The protonated molecular masses of the molecules are consistent with the formation of the spiroiminoimidazolidine-one ([M+H]+ at m/z 465.1) and ring-opened guanidino ([M+H]+ at m/z 342.1) products of dG-C8-AαC. The product ion spectrum at the MS2 scan stage of the proposed spiroiminoimidazolidine-one adduct ([M+H]+ at m/z 465.1) displayed a base peak ion at m/z 349.2, attributed to the loss of the deoxyribose (dR) moiety. The second generation product ion spectrum of adduct acquired on the ion at m/z 349.2 produced major fragment ions m/z at 290.2 and 252.1, attributed, respectively, to the losses of the guanidine (CH5N3) and 2-imino-3,4-dihydro-imidazol-4-one (C3H3N3O) moieties (Fig. 4D). The product ion spectrum of the proposed guanidino adduct ([M+H]+ at m/z 342.1) displayed a major ion at m/z 226.1, which is attributed to loss of dR. The second generation product ion spectrum of the adduct acquired on m/z 226.1 formed a base peak ion at m/z 209.1, attributed to loss of NH3 (Fig. 4D). The MS4 scan stage spectrum acquired on the ion at m/z 209.1 showed fragment ions at m/z 167.1 and 185.1, which are attributed to the loss of cyanamide (CH2N2) without or with a water cluster adducted to the deaminated AαC moiety. These product ion spectra are entirely consistent with the structures of the proposed ring-opened oxidized adducts of dG-C8-AαC. Synthetic dG-C8-AαC also underwent decomposition in NaOH or H2O2 to form the same two oxidized adducts. The CNL/MS3 data-dependent scanning experiments monitoring for the loss of dR (116.1Da) as a neutral species showed that these two ring-opened adducts were the sole oxidation products of dG-C8-AαC (Supplementary Fig. S-1).
Three other isomeric dG-AαC adducts were detected (labeled as peaks 1–3 in Fig. 4B). These adducts did not undergo noticeable oxidation during sample workup of DNA in the absence of ßME. Assuming similar ionization efficiencies, the levels of these isomeric dG-AαC adducts occur at several percentage or less of the level of dG-C8-AαC when DNA is isolated with ßME (Fig. 4C). The product ion spectra of these adducts are suggestive of dG-N2 adducts, but assignments of structures could not be unambiguously established. In contrast to the instability of dG-C8-AαC, dG-C8-MeIQ does not undergo facile oxidation. Comparable levels of dG-C8-MeIQ were measured in DNA samples, irrespective of the addition of ßME as an antioxidant during the isolation of DNA (Fig. 5). The data-dependent CNL/MS3 scan mode monitoring the loss of dR (116.1Da) as a neutral fragment was conducted on liver DNA, but only dG-C8-MeIQ was detected (data not shown). Representative UPLC-ESI/MS3 chromatograms of colon and bladder DNA adducts of AαC and MeIQ are shown in Supplementary figure S-2.
Fig. 5.

The UPLC-ESI/MS3 chromatogram of dG-C8-MeIQ in liver DNA from (A) untreated mouse and DNA isolated with βME, (B) mouse treated with MeIQ (300 ppm) for 4 weeks and DNA isolated without βME, and (C) mouse treated with MeIQ (300 ppm) for 4 weeks and DNA isolated with βME and and (D) MS3 stage product ion spectrum of dG-C8-MeIQ.
Kinetics DNA Adduct Formation of dG-C8-AαC and dG-C8-MeIQ in Liver and Extrahepatic Tissues of Mice
The levels of DNA adduct formation in liver, pancreas, lung, colon, cecum, and bladder are tabulated in Tables 1 and 2. The concentrations of adducts formed in the different tissues following 1 day and 1 week of feeding are shown in Figure 6, and the kinetics of adduct formation during the entire course of the study are depicted in Figure 7. The comparison of DNA adduct formation of AαC and MeIQ following a single day of treatment versus continuous feeding for 12 weeks revealed striking differences between adduct formation and persistence in certain tissues. After a single day of treatment, the liver contained the highest levels of AαC DNA adducts among all of the organs measured, followed by the colon and cecum. In contrast, MeIQ adduct formation was highest in colon and cecum, followed by liver, after 1 day of dosing. The liver became the primary organ for DNA adduct formation of MeIQ following 1 week of dose treatment. By the end of the 12-week feeding study, DNA adducts accumulated in a number of tissues: the urinary bladder contained the second highest levels of DNA adducts after the liver for both HAAs, followed by cecum, colon, pancreas, and lung in mice of both genders. Gender differences were observed between the mean adduct levels of AαC formed in liver during the first week of feeding: higher adduct levels were observed in female liver at 1 day and 1 week of the 400-ppm diet (p < 0.01) and at 1 week of the 800-ppm diet (p < 0.03). Higher adduct levels were also formed for MeIQ in female liver than in male liver at 1 week of feeding (p < 0.01). Given the small number of animals (N = 4) and large doses, these modest ~1.3-fold differences in hepatic DNA adduct formation between genders should be interpreted with caution.
Table 1.
DNA Adducts of AαC and MeIQ in Liver and Extrahepatic Tissues of Male C57BL/6 Mice
| Adducts/107 bases | ||||||
|---|---|---|---|---|---|---|
| Day 1 | Week 1 | Week 4 | Week 8 | Week 12 | ||
| Liver | AαC_400 ppm | 19.25±4.52 | 73.21±7.05 | 122.5±15.6 | 107.1±28.6 | 89.59±14.51 |
| AαC_800 ppm | 33.33±6.39 | 132.0±17.5 | 139.0±29.9 | 73.14±12.93 | 69.05±9.09 | |
| MeIQ_300 ppm | 2.40±0.14 | 11.45±1.71 | 28.32±5.54 | 43.79±12.50 | 42.30±1.24 | |
| Bladder | AαC_400 ppm | 3.75±0.91 | 8.35±1.51 | 9.76±2.27 | 9.28±2.33 | 10.01±2.19 |
| AαC_800 ppm | 10.43±0.55 | 16.25±2.75 | 15.77±4.23 | 19.79±3.55 | 24.92±2.70 | |
| MeIQ_300 ppm | 0.99±0.30 | 5.39±0.84 | 11.93±6.60 | 14.99±2.18 | 14.51±3.03 | |
| Colon | AαC_400 ppm | 4.60±0.80 | 4.80±0.68 | 1.77±0.37 | 2.31±0.49 | 1.81±0.23 |
| AαC_800 ppm | 10.18±0.86 | 8.05±1.49 | 3.90±0.43 | 5.13±0.80 | 3.50±0.12 | |
| MeIQ_300 ppm | 3.66±0.67 | 4.29±0.95 | 5.67±1.71 | 6.61±1.19 | 5.31±0.46 | |
| Cecum | AαC_400 ppm | 5.67±1.23 | 3.33±0.58 | 1.74±0.36 | 7.57±1.00 | 6.68±1.19 |
| AaC_800 ppm | 14.15±1.69 | 4.56±1.27 | 3.66±0.61 | 7.34±0.75 | 10.18±0.52 | |
| MeIQ_300 ppm | 2.37±0.68 | 1.94±0.52 | 2.62±0.62 | 4.37±0.92 | 4.59±0.95 | |
| Pancreas | AαC_400 ppm | 0.32±0.01 | 0.72±0.34 | 1.12±0.24 | 1.47±0.09 | 1.17±0.14 |
| AαC_800 ppm | 0.67±0.27 | 1.60±0.20 | 2.41±0.67 | 1.69±0.31 | 1.62±0.89 | |
| MeIQ_300 ppm | 0.37±0.04 | 1.25±0.31 | 2.69±0.21 | 4.07±0.80 | 6.96±0.99 | |
| Lung | AαC_400 ppm | 0.52±0.27 | 1.17±0.26 | 1.81±0.30 | 2.18±0.11 | 1.78±0.13 |
| AαC_800 ppm | 1.23±0.16 | 2.84±0.63 | 4.61±1.27 | 4.13±0.30 | 3.71±0.28* | |
| MeIQ_300 ppm | 0.22±0.02 | 0.84±0.16 | 2.40±0.08 | 4.31±0.63 | 4.76±0.64 | |
Notes. Data of each time point are the average and SD of four animals, except for week 12, where two animals were assayed. Each organ from each animal was assayed in triplicate, except for bladder, where two measurements were done, and *lung, where the average and SD were obtained from one animal with three independent measurements of DNA adducts.
Table 2.
DNA Adducts of AαC and MeIQ in Liver and Extrahepatic Tissues of Female C57BL/6 Mice
| Adducts/107 bases | ||||||
|---|---|---|---|---|---|---|
| Day 1 | Week 1 | Week 4 | Week 8 | Week 12 | ||
| Liver | AαC_400 ppm | 39.23±5.27 | 107.6±25.0 | 80.56±11.28 | 85.52±20.25 | 51.09±12.35 |
| AαC_800 ppm | 39.90±7.06 | 211.7±35.5 | 90.99±29.58 | 76.70±14.78 | 64.87±6.42 | |
| MeIQ_300 ppm | 1.76±0.75 | 16.94±1.43 | 31.83±13.71 | 59.86±2.15 | 55.77±7.02 | |
| Bladder | AαC_400 ppm | 2.00±0.60 | 6.63±2.38 | 8.45±1.38 | 8.20±0.76 | 8.49±1.74 |
| AαC_800 ppm | 2.91±0.62 | 8.86±1.25 | 17.26±2.71 | 20.77±1.01 | 16.68±2.55 | |
| MeIQ_300 ppm | 1.07±0.08 | 3.42±0.48 | 7.48±1.44 | 15.43±6.64 | 12.79±1.69 | |
| Colon | AαC_400 ppm | 4.06±1.13 | 5.41±1.08 | 2.28±0.48 | 2.35±0.53 | 2.89±0.87 |
| AαC_800 ppm | 6.79±0.61 | 10.56±1.23 | 4.82±1.11 | 4.45±0.92 | 3.00±0.24 | |
| MeIQ_300 ppm | 2.86±0.20 | 4.54±1.09 | 6.78±1.37 | 4.63±1.23 | 6.13±0.35 | |
| Cecum | AαC_400 ppm | 3.77±2.02 | 3.62±0.66 | 1.98±0.65 | 4.70±0.89 | 4.92±2.24 |
| AαC_800 ppm | 9.35±1.23 | 5.98±1.47 | 3.59±0.98 | 5.25±1.83 | 8.86±3.21 | |
| MeIQ_300 ppm | 2.71±0.34 | 2.03±0.87 | 3.42±0.68 | 4.71±0.38 | 5.80±0.74 | |
| Pancreas | AαC_400 ppm | 0.36±0.09 | 0.74±0.39 | 1.21±0.21 | 0.86±0.25 | 0.64±0.22 |
| AαC_800 ppm | 0.49±0.08 | 1.49±0.37 | 3.94±0.91 | 2.87±0.60 | 2.87±0.01 | |
| MeIQ_300 ppm | 0.46±0.10 | 1.29±0.17 | 3.16±1.29 | 5.75±1.24 | 7.13±1.01 | |
| Lung | AαC_400 ppm | 0.45±0.19 | 0.97±0.10 | 1.49±0.45 | 1.95±0.29 | 1.41±0.06 |
| AαC_800 ppm | 0.94±0.19 | 2.81±0.66 | 5.89±1.64 | 4.62±0.30 | 4.47±0.68 | |
| MeIQ_300 ppm | 0.22±0.05 | 0.91±0.12 | 1.82±0.31 | 4.14±0.81 | 4.83±0.87 | |
Notes. Data of each time point are the average and SD of four animals, except for week 12, where two animals were assayed. Each organ from each animal was assayed in triplicate, except for bladder, where two measurements were done.
Fig. 6.

DNA adduct formation of AαC and MeIQ in liver, bladder, colon, cecum, pancreas, and lung of male and female C57BL/6 mice following 1 day and 1 week of dose treatment. The one-way ANOVA p values for the mean DNA adduct levels of AαC and MeIQ in liver and extrahepatic tissues of both genders for both doses and time points were p < 0.001. Bonferroni’s multiple comparison test revealed that the mean level of dG-C8-AαC was elevated and significantly different in liver versus all other organs for both genders at day 1 and week 1 (p < 0.001). The mean level of dG-C8-MeIQ was also elevated and significantly different in liver versus all other organs at week 1(p < 0.001). The mean levels of dG-C8-MeIQ in colon of males and colon and cecum of females were significantly higher than the adduct level in liver, whereas lower levels of dG-C8-MeIQ were formed in pancreas, lung, and bladder (females only) than in liver of both genders at day 1 (**p < 0.01; ***p < 0.001).
Fig. 7.

Kinetics of DNA adduct formation of AαC and MeIQ over 12 weeks in liver, bladder, colon, cecum, pancreas, and lung of male and female C57BL/6 mice. Data of each time point are the average and SD of four animals, except for week 12, where two animals were assayed. Data of each animal were assayed in triplicate, except for bladder, where two independent measurements were done.
The level of dG-C8-AαC reached an optimum in liver by 1 week and 4 weeks of feeding, respectively, in female and mice. Thereafter, the adduct levels declined over time, particularly in the high dose–treated animals. The level of dG-C8-MeIQ in liver reached a peak by 8 weeks of feeding and remained constant for the duration of the study (Fig. 7). The dG-C8-AαC adduct reached a steady-state level in the urinary bladder by ~1 week and 4 weeks of treatment, respectively, for male and female mice (Fig. 7). The level of dG-C8-MeIQ adduct formation in bladder was lower than the level of dG-C8-AαC during the first week of treatment, but surpassed the concentration of dG-C8-AαC, when adjusted for dose, after 1 month of feeding.
The cecum contained comparable levels of DNA adducts for both AαC and MeIQ (Fig. 7). The level of dG-C8-AαC was greater than the amount of dG-C8-MeIQ in the colon during the first week of treatment, but the amount of dG-C8-MeIQ surpassed the level of dG-C8-AαC during prolonged dosing. The adduct levels of both carcinogens varied by about two- to threefold in the large intestine during the 12-week feeding.
Pancreas and lung tissue harbored the lowest levels of DNA adducts. When adjusted for dose, dG-C8-MeIQ adduct formation was greater than dG-C8-AαC at most time points. dG-C8-AαC adduct formation reached a steady state at ~1 month in both organs, whereas the levels of dG-C8-MeIQ continued to increase in pancreas and lung throughout the 12-week feeding.
DISCUSSION
HAAs and aromatic amines undergo bioactivation or detoxication by P450 enzymes and phase II enzymes, including NAT, SULT, and UGT expressed in liver and extrahepatic tissues, but with markedly different enzyme kinetic parameters (Kadlubar and Beland, 1985; Turesky and Le Marchand, 2011). Differences in enzyme biotransformation pathways are likely to affect relative levels of adducts formed and influence the potential target tissues of tumorigenesis by HAAs and aromatic amines (Kadlubar and Beland, 1985; Turesky and Le Marchand, 2011). Several aromatic amines present in tobacco smoke have been implicated in the pathogenesis of bladder cancer in smokers (IARC, 2012). Interestingly, some epidemiology studies have reported that high intakes of red meat, well-done meat, and consumption of HAAs is associated with an increased risk in the development of bladder cancer ((Lin et al., 2012) and references within).
Clear differences in the kinetics of DNA adduct formation were observed for both AαC and MeIQ. In the case of AαC, the dG-C8-AαC adduct is preferentially removed in the liver compared with other tissues; however, the efficiency of adduct removal/repair may be concentration dependent or require a threshold level of DNA adducts. Indeed, the rate of removal of dG-C8-AαC in liver was greater in mice dosed with 800 ppm AαC than in mice treated with 400 ppm of AαC, and comparable levels of dG-C8-AαC were present in the liver at both dosages at the end of the 12-week feeding. Alternatively, subchronic administration of AαC may have altered the expression of carcinogen metabolism enzymes in liver and diminished the P450-mediated N-oxidation or augmented phase II detoxication pathways, resulting in diminished hepatic DNA adduct levels over time. In contrast to dG-C8-AαC, the adduct concentrations of dG-C8-MeIQ reached a steady-state level in the liver, bladder, pancreas, and lung either by 4 weeks or continued to increase during the course of the study.
The amounts of AαC and MeIQ adducts were higher in the colon and cecum compared with other extrahepatic tissues examined during the first week of feeding. Because of the rapid cell turnover in cecum and colon (Westra et al., 1985), DNA adducts of AαC and MeIQ did not accumulate in the large intestine (Fig. 7). However, dG-C8-AαC or dG-C8-MeIQ adducts can induce mutations during the high rate of DNA synthesis and cell division taking place in the stem cells of the large intestine, and these cells may rapidly proliferate to produce ACF and tumors more readily than the cells of the liver or other slowly dividing tissues containing DNA adducts of AαC and MeIQ (Nagao et al., 2001).
Female C57BL/6 mice develop tumors of the colon, cecum, and liver when given a diet containing 300 ppm of MeIQ (Fujita et al., 1999). Carcinogenicity studies have not been conducted with AαC in C57BL/6 mice, but CDF1 mice developed liver and blood vessel tumors when given a diet containing 800 ppm of AαC (Sugimura et al., 2004). The Big Blue mouse has the genetic background of C57BL/6, and both MeIQ and AαC induced high levels of mutants in the lacI transgene in the liver and colon of this mouse strain (Nagao et al., 2001; Zhang et al., 1996). The majority of the lacI mutations reported in the colon were base substitutions. Approximately 50% of the total number of mutations induced by both HAA adducts were G:C to T:A transversions although the mutations occurred at different sequence contexts of the lacI gene (Nagao et al., 2001; Zhang et al., 1996). C57BL/6 mice also form ACF when treated with AαC (500 or 800 ppm), and more than half the ACF clustered in the region about 20–40% of the distance from the ileocecal portion of the large intestine (Okonogi et al., 1997). Under a similar dosing regimen, our LC/MS measurements show that dG-C8-AαC adduct formation ranged between ~several adducts up to 14 adducts per 107 DNA bases in cecum and colon during the 12-week feeding study. Thus, this level of dG-C8-AαC adduct formation appears sufficient to induce lacI transgene mutations and ACF in the colon of C57BL/6 mice.
The bladder contained the second highest levels of DNA adducts, following the liver, in mice treated subchronically with either AαC or MeIQ. The high level of DNA adducts formed in bladder was unexpected because studies conducted on other structurally related HAAs in mice or nonhuman primates reported very low levels of adduct formation in bladder in comparison to other organs (Nerurkar et al., 1995; Snyderwine et al., 1988, 1994). Some aromatic amines cause bladder and liver cancer in rodents.
Subchronic dosing studies showed that 4-ABP and 2-AAF formed up to ~10-fold higher levels of DNA adducts in liver and bladder of BALB/c mice (Poirier and Beland, 1992) than the adduct levels formed in these organs of C57BL/6 mice treated with AαC or MeIQ. However, the metabolism of these structurally related class of chemicals and the covalent binding potencies may be different in these mouse strains. In the case of 4-ABP, the carcinogen was administered in the water supply rather than the diet, and the bioavailability of 4-ABP may be higher than when given in the diet. The levels of 4-ABP and 2-AAF DNA formation were linearly correlated to dose and tumorigenesis in liver of BALB/c mice although the relationship was markedly nonlinear in bladder. The investigators noted that DNA adduct formation was insufficient for bladder tumorigenesis, and other factors, such as cell proliferation, were required to develop bladder tumors (Poirier and Beland, 1992). Moreover, gender differences in bladder tumor formation were observed, and female mice were refractory toward 4-ABP treatment (Poirier et al., 1995). Cell proliferation has also been reported to be a critical factor in the development of cancer of the colon and prostate in rodents exposed to the structurally related HAA 2-amino-1-methyl- 6-phenylimidazo[4,5-b]pyridine (PhIP) (Cheung, et al., 2011, Li, et al., 2012).
MeIQ was one of the first of the HAAs discovered in cooked foods. MeIQ was identified in well-done broiled sardines and beef extract (Kasai et al., 1980), but MeIQ has rarely been found at appreciable levels in commonly cooked meats, poultry, or fish staples of the western diet (Felton et al., 2000), and the contribution of MeIQ to the daily burden of exposure to HAAs appears to be minor. In contrast to MeIQ, the exposure to AαC is widespread. AαC arises in mainstream tobacco smoke (Zhang et al., 2011) and diesel exhausts (Manabe et al., 1991), and AαC can form in meats cooked very well done (Holder et al., 1997). However, cigarette smoking is thought to be a major point source of exposure to AαC (Turesky et al., 2007). Although the lung contained the lowest levels of AαC-DNA adducts of the organs measured in mice when AαC was given as part of the diet, the route of administration may greatly affect the level of AαC-DNA adduct formation in different organs. A future study will examine AαC-DNA adduct formation in lung and other organs via inhalation to better simulate exposure to AαC from smoking.
DNA adducts are thought to play a critical role in the development of cancer by genotoxic carcinogens (Jarabek et al., 2009), but DNA adduct formation alone is insufficient for tumorigenesis (Poirier and Beland, 1992). As reported by Nagao et al. (2001), the cell turnover kinetics of mutated cells might be different in different organs. Also, differences in transcription-coupled repair capacity among different organs and the number of genetic alterations required for cancer development may be different in different organs. All of these factors are likely to impact the incidence of tumorigenesis in different organs. Given the high levels of DNA adduct formation by AαC, combined with the induction of lacI mutations in liver and colon, and the occurrence of ACF in colon of C57BL/6 mice, long-term studies are warranted to determine whether liver, the GI tract, or possibly bladder are target organs of AαC-mediated carcinogenicity in this mouse strain.
Epidemiologic studies conducted over the past two decades have consistently shown that tobacco smoking is a risk factor for cancers of the GI tract (IARC, 2004, 2012; Giovannucci, 2001; Gong et al., 2012). There is also mounting evidence that tobacco smoke is an independent risk factor for hepatocellular carcinoma, the predominant form of human liver cancer (IARC, 2012). Moreover, the risk of developing colorectal cancer is increased in smokers who harbor rapid phenotype for both NAT2 and P450 1A2 (Le Marchand et al., 2002; Nöthlings et al., 2009); both enzymes are involved in the bioactivation of AαC (Turesky and Le Marchand, 2011). The high levels of AαC present in tobacco coupled with its efficient bioactivation by liver and extrahepatic tissues of the C57BL/6 mouse and by human liver to form DNA adducts (Nauwelaers et al., 2011; Tang et al., 2012) provide a biochemical mechanism and a plausible role for AαC in tobacco-associated cancers of the liver and digestive tract of smokers. Molecular epidemiology studies investigating the role of AαC in tobacco-associated cancers are warranted.
SUPPLEMENTARY DATA
Supplementary data are available online at http://toxsci.oxfordjournals.org/.
FUNDING
National Cancer Institute at the National Institutes of Health (R01 CA134700 to R.J.T.).
Supplementary Material
REFERENCES
- Bessette E. E., Goodenough A. K., Langouët S., Yasa I., Kozekov I. D., Spivack S. D., Turesky R. J. (2009). Screening for DNA adducts by data-dependent constant neutral loss-triple stage mass spectrometry with a linear quadrupole ion trap mass spectrometer. Anal. Chem. 81, 809–819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung C., Loy S., Li G. X., Liu A. B., Yang C. S. (2011). Rapid induction of colon carcinogenesis in CYP1A-humanized mice by 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine and dextran sodium sulfate. Carcinogenesis 32, 233–239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felton J. S., Jagerstad M., Knize M. G., Skog K., Wakabayashi K. (2000). Contents in foods, beverages and tobacco. In Food Borne Carcinogens Heterocyclic Amines (Nagao M., Sugimura T., Eds.), pp. 31–71 John Wiley & Sons Ltd, Chichester, England: [Google Scholar]
- Fujita H., Nagano K., Ochiai M., Ushijima T., Sugimura T., Nagao M., Matsushima T. (1999). Difference in target organs in carcinogenesis with a heterocyclic amine, 2-amino-3,4-dimethylimidazo[4,5-f]quinoline, in different strains of mice. Jpn. J. Cancer Res. 90, 1203–1206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giovannucci E. (2001). An updated review of the epidemiological evidence that cigarette smoking increases risk of colorectal cancer. Cancer Epidemiol. Biomarkers Prev. 10, 725–731 [PubMed] [Google Scholar]
- Gong J., Hutter C., Baron J. A., Berndt S., Caan B., Campbell P. T., Casey G., Chan A. T., Cotterchio M., Fuchs C. S., et al. (2012). A pooled analysis of smoking and colorectal cancer: Timing of exposure and interactions with environmental factors. Cancer Epidemiol. Biomarkers Prev. 21, 1974–1985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta R. C. (1993). 32P-postlabelling analysis of bulky aromatic adducts. IARC Sci. Publ. 124, 11–23 [PubMed] [Google Scholar]
- Hoffmann D. (1998). Letters to the editor: Tobacco smoke components. Beiträge zur Tabakforschung Int. 18, 49–52 [Google Scholar]
- Holder C. L., Preece S. W., Conway S. C., Pu Y. M., Doerge D. R. (1997). Quantification of heterocyclic amine carcinogens in cooked meats using isotope dilution liquid chromatography/atmospheric pressure chemical ionization tandem mass spectrometry. Rapid Commun. Mass Spectrom. 11, 1667–1672 [DOI] [PubMed] [Google Scholar]
- International Agency for Research on Cancer (IARC) (2004). Tobacco Smoke and Involuntary Smoking. Monographs on the Evaluation of Carcinogenic Risks to Humans. International Agency for Research on Cancer, Lyon, France: [Google Scholar]
- International Agency for Research on Cancer (IARC) (2012). Personal Habits and Indoor Combustions. International Agency for Research on Cancer, Lyon, France: [Google Scholar]
- Jarabek A. M., Pottenger L. H., Andrews L. S., Casciano D., Embry M. R., Kim J. H., Preston R. J., Reddy M. V., Schoeny R., Shuker D., et al. (2009). Creating context for the use of DNA adduct data in cancer risk assessment: I. Data organization. Crit. Rev. Toxicol. 39, 659–678 [DOI] [PubMed] [Google Scholar]
- Kadlubar F. F., Beland F. A. (1985). Chemical properties of ultimate carcinogenic metabolites of arylamines and arylamides. In Polycyclic Hydrocarbons and Carcinogenesis (Harvey R. G., Ed.), pp. 332–370 American Chemical Society, Washington, DC: [Google Scholar]
- Kasai H., Yamaizumi K., Wakabayashi K., Nagao M., Sugimura T., Yokoyama T., Miyazawa T., Nishimura S. (1980). Structure and chemical synthesis of Me-IQ, a potent mutagen isolated from broiled fish. Chem. Lett. 9, 1391–1394 [Google Scholar]
- Kriek E. (1992). Fifty years of research on N-acetyl-2-aminofluorene, one of the most versatile compounds in experimental cancer research. J. Cancer Res. Clin. Oncol. 118, 481–489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Marchand L., Hankin J. H., Pierce L. M., Sinha R., Nerurkar P. V., Franke A. A., Wilkens L. R., Kolonel L. N., Donlon T., Seifried A., et al. (2002). Well-done red meat, metabolic phenotypes and colorectal cancer in Hawaii. Mutat. Res. 506-507, 205–214 [DOI] [PubMed] [Google Scholar]
- Li G., Wang H., Liu A. B., Cheung C., Reuhl K. R., Bosland M. C., Yang C. S. (2012). Dietary carcinogen 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine-induced prostate carcinogenesis in CYP1A-humanized mice. Cancer Prev. Res. (Phila). 5, 963–972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin J., Forman M. R., Wang J., Grossman H. B., Chen M., Dinney C. P., Hawk E. T., Wu X. (2012). Intake of red meat and heterocyclic amines, metabolic pathway genes and bladder cancer risk. Int. J. Cancer. 131, 1892–1903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manabe S., Izumikawa S., Asakuno K., Wada O., Kanai Y. (1991). Detection of carcinogenic amino-alpha-carbolines and amino-gamma- carbolines in diesel-exhaust particles. Environ. Pollut. 70, 255–265 [DOI] [PubMed] [Google Scholar]
- Nagao M., Ochiai M., Okochi E., Ushijima T., Sugimura T. (2001). LacI transgenic animal study: Relationships among DNA-adduct levels, mutant frequencies and cancer incidences. Mutat. Res. 477, 119–124 [DOI] [PubMed] [Google Scholar]
- Nauwelaers G., Bessette E. E., Gu D., Tang Y., Rageul J., Fessard V., Yuan J. M., Yu M. C., Langouët S., Turesky R. J. (2011). DNA adduct formation of 4-aminobiphenyl and heterocyclic aromatic amines in human hepatocytes. Chem. Res. Toxicol. 24, 913–925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nerurkar P. V., Schut H. A., Anderson L. M., Riggs C. W., Snyderwine E. G., Thorgeirsson S. S., Weber W. W., Rice J. M., Levy G. N. (1995). DNA adducts of 2-amino-3-methylimidazo[4,5-f]quinoline (IQ) in colon, bladder, and kidney of congenic mice differing in Ah responsiveness and N-acetyltransferase genotype. Cancer Res. 55, 3043–3049 [PubMed] [Google Scholar]
- Nöthlings U., Yamamoto J. F., Wilkens L. R., Murphy S. P., Park S. Y., Henderson B. E., Kolonel L. N., Le Marchand L. (2009). Meat and heterocyclic amine intake, smoking, NAT1 and NAT2 polymorphisms, and colorectal cancer risk in the multiethnic cohort study. Cancer Epidemiol. Biomarkers Prev. 18, 2098–2106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ochiai M., Imai H., Sugimura T., Nagao M., Nakagama H. (2002). Induction of intestinal tumors and lymphomas in C57BL/6N mice by a food-borne carcinogen, 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine. Jpn. J. Cancer Res. 93, 478–483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okonogi H., Ushijima T., Shimizu H., Sugimura T., Nagao M. (1997). Induction of aberrant crypt foci in C57BL/6N mice by 2-amino-9H-pyrido[2,3-b]indole (A alphaC) and 2-amino-3,8-dimethylimidazo[4,5-f]quinoxaline (MeIQx). Cancer Lett. 111, 105–109 [DOI] [PubMed] [Google Scholar]
- Poirier M. C., Beland F. A. (1992). DNA adduct measurements and tumor incidence during chronic carcinogen exposure in animal models: Implications for DNA adduct-based human cancer risk assessment. Chem. Res. Toxicol. 5, 749–755 [DOI] [PubMed] [Google Scholar]
- Poirier M. C., Fullerton N. F., Smith B. A., Beland F. A. (1995). DNA adduct formation and tumorigenesis in mice during the chronic administration of 4-aminobiphenyl at multiple dose levels. Carcinogenesis 16, 2917–2921 [DOI] [PubMed] [Google Scholar]
- Shibutani S., Gentles R. G., Iden C. R., Johnson F. (1990). Facile aerial oxidation of the DNA-base adduct N-(2’-deoxyguanosin-8-yl)-2-aminofluorene [dG(C8)AF]. J. Am. Chem. Soc. 112, 5667–5668 [Google Scholar]
- Snyderwine E. G., Schut H. A., Sugimura T., Nagao M., Adamson R. H. (1994). DNA adduct levels of 2-amino-1-methyl-6-phenylimidazo-[4,5-b]pyridine (PhIP) in tissues of cynomolgus monkeys after single or multiple dosing. Carcinogenesis 15, 2757–2761 [DOI] [PubMed] [Google Scholar]
- Snyderwine E. G., Yamashita K., Adamson R. H., Sato S., Nagao M., Sugimura T., Thorgeirsson S. S. (1988). Use of the 32P-postlabeling method to detect DNA adducts of 2-amino-3-methylimidazolo[4,5-f]quinoline (IQ) in monkeys fed IQ: Identification of the N-(deoxyguanosin-8-yl)-IQ adduct. Carcinogenesis 9, 1739–1743 [DOI] [PubMed] [Google Scholar]
- Sugimura T., Wakabayashi K., Nakagama H., Nagao M. (2004). Heterocyclic amines: Mutagens/carcinogens produced during cooking of meat and fish. Cancer Sci. 95, 290–299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tada A., Ochiai M., Wakabayashi K., Nukaya H., Sugimura T., Nagao M. (1994). Identification of N-(deoxyguanosin-8-yl)-2-amino-3,4-dimethylimidazo[4,5-f]quinoline (dG-C8-MeIQ) as a major adduct formed by MeIQ with nucleotides in vitro with DNA in vivo. Carcinogenesis 15, 1275–1278 [DOI] [PubMed] [Google Scholar]
- Tang Y., LeMaster D. M., Nauwelaërs G., Gu D., Langouët S., Turesky R. J. (2012). UDP-glucuronosyltransferase-mediated metabolic activation of the tobacco carcinogen 2-amino-9H-pyrido[2,3-b]indole. J. Biol. Chem. 287, 14960–14972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turesky R. J., Bendaly J., Yasa I., Doll M. A., Hein D. W. (2009). The impact of NAT2 acetylator genotype on mutagenesis and DNA adducts from 2-amino-9H-pyrido[2,3-b]indole. Chem. Res. Toxicol. 22, 726–733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turesky R. J., Le Marchand L. (2011). Metabolism and biomarkers of heterocyclic aromatic amines in molecular epidemiology studies: Lessons learned from aromatic amines. Chem. Res. Toxicol. 24, 1169–1214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turesky R. J., Yuan J. M., Wang R., Peterson S., Yu M. C. (2007). Tobacco smoking and urinary levels of 2-amino-9H-pyrido[2,3-b]indole in men of Shanghai, China. Cancer Epidemiol. Biomarkers Prev. 16, 1554–1560 [DOI] [PubMed] [Google Scholar]
- Wang L. Y., Chen C. J., Zhang Y. J., Tsai W. Y., Lee P. H., Feitelson M. A., Lee C. S., Santella R. M. (1998). 4-Aminobiphenyl DNA damage in liver tissue of hepatocellular carcinoma patients and controls. Am. J. Epidemiol. 147, 315–323 [DOI] [PubMed] [Google Scholar]
- Westra J. G., Flammang T. J., Fullerton N. F., Beland F. A., Weis C. C., Kadlubar F. F. (1985). Formation of DNA adducts in vivo in rat liver and intestinal epithelium after administration of the carcinogen 3,2’-dimethyl-4-aminobiphenyl and its hydroxamic acid. Carcinogenesis 6, 37–44 [DOI] [PubMed] [Google Scholar]
- Zhang L., Ashley D. L., Watson C. H. (2011). Quantitative analysis of six heterocyclic aromatic amines in mainstream cigarette smoke condensate using isotope dilution liquid chromatography-electrospray ionization tandem mass spectrometry. Nicotine Tob. Res. 13, 120–126 [DOI] [PubMed] [Google Scholar]
- Zhang X. B., Felton J. S., Tucker J. D., Urlando C., Heddle J. A. (1996). Intestinal mutagenicity of two carcinogenic food mutagens in transgenic mice: 2-Amino-1-methyl-6-phenylimidazo[4,5-b]pyridine and amino(alpha)carboline. Carcinogenesis 17, 2259–2265 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.